

Abstract
To obtain a landscape of gross genetic alterations in small cell lung cancer (SCLC), genome-wide copy number analysis and whole-transcriptome sequencing were performed in 58 and 42 SCLCs, respectively. Focal amplification of known oncogene loci, MYCL1 (1p34.2), MYCN (2p24.3), and MYC (8q24.21), was frequently and mutually exclusively detected. MYCL1 and MYC were co-amplified with other regions on either the same or the different chromosome in several cases. In addition, the 9p24.1 region was identified as being amplified in SCLCs without amplification of MYC family oncogenes. Notably, expression of the KIAA1432 gene in this region was significantly higher in KIAA1432 amplified cells than in non-amplified cells, and its mRNA expression showed strong correlations with the copy numbers. Thus, KIAA1432 is a novel gene activated by amplification in SCLCs. By whole-transcriptome sequencing, a total of 60 fusion transcripts, transcribed from 95 different genes, were identified as being expressed in SCLC cells. However, no in-frame fusion transcripts were recurrently detected in ≥2 SCLCs, and genes in the amplified regions, such as PVT1 neighboring MYC and RLF in MYCL1 amplicons, were recurrently fused with genes in the same amplicons or with those in different amplicons on either the same or different chromosome. Thus, it was indicated that amplification and fusion of several genes on chromosomes 1 and 8 occur simultaneously but not sequentially through chromothripsis in the development of SCLC, and amplification rather than fusion of genes plays an important role in its development.
INTRODUCTION
Lung cancer is the leading cause of cancer death worldwide, and accounts for 18% of total cancer deaths in a year (Jemal et al., 2011). In particular, most of small cell lung cancer (SCLC) cases are diagnosed after metastatic spread of the diseases, and only 5% of SCLC patients survive beyond 5 years after diagnosis (Worden and Kalemkerian, 2000; Cooper and Spiro, 2006). Therefore, for the improvement of patients’ outcome in this disease, it is necessary to identify druggable targets that are activated by genetic alterations in SCLC cells. However, since only a limited fraction of SCLC cases are treated by surgery and most of them are treated by chemotherapy and/or radiotherapy, tumor tissues are rarely available for molecular analysis. For this reason, only a few activating genetic alterations have been identified to date in SCLC cells, including amplification of the MYC family oncogenes, MYCL1 (1p34), MYCN (2p24), and MYC (8q24) (Wistuba et al., 2001). Recently, whole-genome profiling has been applied to further obtain information about copy number alterations, point mutations, and fusions in SCLCs (Kim et al., 2006; Campbell et al., 2008; Pleasance et al., 2010; Voortman et al., 2010; Dooley et al., 2011; Peifer et al., 2012; Rudin et al., 2012). The results indicated that copy number gains occur in various chromosomal regions, including the JAK2 (9p24), FGFR1 (8p12), TNFRSF4 (1p36), DAD1 (14q11), BCL2L1 (20q11), BCL2L2 (14q11), FAK (8q24), NF1B (9p23), and SOX2 (3q26) genes, in SCLCs. As for the gene fusions, the PVT1 gene that is immediately downstream of the MYC gene at 8q24 and the CHD7 gene at 8q12 with copy number alterations were found to be fused in the H2171 and Lu135 cell lines (Campbell et al., 2008; Pleasance et al., 2010). However, since most genetic studies in SCLC have been done using cultured cell lines, genetic alterations accumulated in fresh SCLCs in vivo are still unclear.
In this study, to obtain a landscape of gross genetic alterations in both fresh tumors and cell lines, genome-wide copy number analysis was performed for 33 fresh tumors and 25 cell lines to identify genes amplified in SCLCs. In parallel, whole-transcriptome sequencing was performed for 19 fresh tumors and 23 cell lines to identify fusion genes expressed in SCLCs. By copy number analysis, a novel chromosomal region amplified in a mutually exclusive manner with MYC family genes was identified, and genes overexpressed accompanied by gene amplification in this region was further identified. By combining the results of copy number analysis with those of whole-transcriptome sequencing, it was further revealed that fusion transcripts were often expressed from genes in several amplified regions, suggesting that amplification and fusion of genes occur simultaneously but not independently by chromothripsis in the development of SCLC.
MATERIALS AND METHODS
Patients and Tissues
Sixty-two tumors and corresponding non-cancerous tissues were obtained at surgery or autopsy from 1985 to 2010 at the National Cancer Center Hospital, Tokyo, Saitama Medical University, Saitama, and University of Tsukuba, Ibaraki, Japan (Supporting Information Table S1A). Genomic DNA was extracted with a QIAamp DNA mini kit (Qiagen, Hilden, Germany). Total RNA was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA), purified by an RNeasy kit (Qiagen), and reverse-transcribed to cDNA by using the SuperScript III First-Strand Synthesis System (Invitrogen) with random hexamers according to the manufacturer’s instructions. This study was performed under the approval of the Institutional Review Board of the National Cancer Center.
Cell Lines
Twenty-five cell lines were used in this study (Supporting Information Table S1B). HCC33, N417, H69, H82, H1607, H1963, H2107, H2141, and H2171 were obtained from Dr. J. D. Minna (University of Texas Southwestern, Dallas), H526 and H841 from Dr. C. C. Harris (NCI, Bethesda), Ms18 from Dr. E. Shimizu (Tottori University, Tottori, Japan), and Lu-series from Dr. T. Terasaki (National Cancer Center, Tokyo, Japan). Other cell lines were obtained from the American Type Culture Collection or the Japanese Collection of Research Bioresources. Genomic DNA was extracted as described previously (Iwakawa et al., 2011). Poly-A(+) RNA was extracted with a Fast Track mRNA isolation kit (Invitrogen) and reverse-transcribed to cDNA as described above.
Genome-wide Copy Number Analysis
Copy number analysis was performed using SNP-Chips for human 250K Nsp SNP arrays (Affymetrix, Inc., Santa Clara, CA). Methods used for the analysis were previously described (Nakanishi et al., 2009; Iwakawa et al., 2011). Copy numbers were determined using the Copy Number Analyzer for Affymetrix GeneChip Mapping Array (CNAG) software (Nannya et al., 2005; Yamamoto et al., 2007).
Whole-transcriptome Sequencing
cDNA libraries for RNA sequencing were prepared using the mRNA-Seq Sample Prep Kit (Illumina, San Diego, CA) according to the manufacturer’s protocol. Briefly, poly-A(+) RNA purified from 4 μg of total RNA extracted from tumors or 0.1 μg of poly-A(+) RNA extracted from cell lines was fragmented in a fragmentation buffer, and used for double-stranded cDNA synthesis. After ligation of the paired-end (PE) adapter, a fraction of 300–350 bp was gel-purified and amplified with PCR. The resulting libraries were subjected to the PE sequencing of 50-bp reads on the Genome Analyzer IIx (GAIIx) (Illumina).
Detection of Fusion Transcripts
PE reads derived from fusion transcripts were searched for as recently described (Kohno et al., 2012). Briefly, PE reads were mapped on human reference RNA sequences deposited in the RefSeq database using the BOWTIE program (version 0.12.5), and PE reads in which both reads were mapped on different RNA sequences were assembled to “clusters”. Paired-clusters consisting of ≥10 PE reads in each sample, for which PE reads did not appear in any of three non-cancerous lung tissues, were picked up. Paired-clusters mapped within a gene region or a neighboring-gene region (≥100 kb in the genome and the same strand) were removed due to the possibility of alternative splicing and read-through transcription. Junction reads encompassing the fusion boundaries were searched using the MapSplice (version 1.14.1) software with modifications. Transcripts that were supported by ≥10 PE reads and ≥10 junction reads were defined as gene fusions.
Reverse Transcription (RT)-PCR and Sanger Sequencing
cDNA was amplified by PCR using KAPA Taq DNA Polymerase (KAPA Biosystems, Woburn, MA). PCR products were directly sequenced in both directions using the BigDye Termination kit and an ABI 3130xl DNA Sequencer (Applied Biosystems, Foster City, CA).
Real-time RT-/Genomic-PCR
The amount of mRNA was quantified using TaqMan Gene Expression Assays (Applied Biosystems). The copy number of gene was determined by TaqMan Copy Number Assay (Applied Biosystems). Primers are listed in Supporting Information Table S1C. HPRT1 and RPPH1 were used as references for mRNA and copy number analyses, respectively. Real-time PCR was performed using the ABI 7900HT real-time PCR system (Applied Biosystems). Data was analyzed by ABI RQ Manager v1.2 for mRNA analysis and ABI Prism 7900HT Sequence Detection Software v2.3 for copy number analysis.
Microarray Experiments and Data Processing
Two micrograms of total RNA were labeled using a 5X MEGAscript T7 kit (Ambion, Inc., Austin, Texas) and analyzed by U133Plus2.0 arrays (Affymetrix), and data was processed by the MAS5 algorithm as described previously (Okayama et al., 2012).
RESULTS
Amplified Genes Identified by Genome-wide Copy Number Analysis
A total of 58 SCLCs, consisted of 33 fresh tumors and 25 cell lines (Supporting Information Table S1A, B), were subjected to 250K SNP array analysis, and all genomic regions with ≥ 5 copies in ≥ 5 consecutive SNP loci were first picked up as the amplified regions in the SCLC genomes. However, by these criteria, whole chromosomes or whole chromosomal arms were more frequently picked up than focal chromosomal regions in various chromosomes among various tumors and cell lines. Therefore, amplified chromosomal regions defined as segments of ≥5 consecutive SNP loci with estimated copy numbers of ≥6 were next picked up from each SCLC. Ten amplified regions were identified on chromosomes 1p, 8q, 9p, 12p, and 19p in 7 of the 33 fresh tumors (Supporting Information Table S2). Sizes of amplified regions ranged from 0.05 to 3.61 Mb (mean ± SD = 1.06 ± 1.25 Mb), and 110 genes were mapped in these regions. Forty-seven amplified regions were identified on chromosomes 1p, 2p, 8q, 9p, 12p, 14q, 17q, and 20q in 13 of the 25 cell lines (Supporting Information Table S2). Sizes of amplified regions ranged from 0.08 to 4.22 Mb (mean ± SD = 0.67 ± 0.81 Mb), and 211 genes were mapped in these regions. Therefore, various chromosomal regions were identified as being focally amplified by the criteria of copy numbers ≥6, sizes of amplified regions were similar in fresh tumors and cell lines, and the several amplified regions in fresh tumors overlapped with those in cell lines (Supporting Information Tables S2). Accordingly, commonly amplified regions were determined by comparison of amplified regions among all the 58 SCLCs, including both fresh tumors and cell lines. Eight regions on chromosomes 1p, 2p, 8q, and 9p were commonly (≥2 SCLCs) amplified in these SCLCs (Table 1). Sizes of the regions ranged from 0.03 to 0.77 Mb (mean ± SD = 0.25 ± 0.23 Mb), and 34 genes were mapped in these regions. Three of the 8 regions contained MYC family oncogenes, MYCL1, MYCN, and MYC, respectively, known to be frequently amplified in SCLCs (Wistuba et al., 2001; Kim et al., 2006; Voortman et al., 2010; Larsen and Minna, 2011).
TABLE 1.
Chromosomal Regions and Genes Commonly Amplified in Small Cell Lung Cancers
| Sample name | ||||||||
|---|---|---|---|---|---|---|---|---|
| No | Cytoband | Start (Mb) | End (Mb) | Size (Mb) | Gene | Fresh tumor | Cell line | No. of amplified SCLCs |
| 1 | 1p34.3 | 37.37 | 38.19 | 0.77 | LOC728431, ZC3H12A, MEAF6, SNIP1, DNALI1, GNL2, RSPO1, C1orf109, CDCA8, EPHA10, MANEAL, YRDC, C1orf122, MTF1, INPP5B, SF3A3, FHL3, UTP11L, POU3F1 | SM09-008T | H1184 | |
| H510 | ||||||||
| 3 | ||||||||
| 2 | 1p34.2 | 39.96 | 40.05 | 0.09 | TRIT1, MYCL1 | SM09-012T | H1184 | 6 |
| H1963 | ||||||||
| H510 | ||||||||
| HCC33 | ||||||||
| H2141 | ||||||||
| 3 | 2p24.3 | 15.98 | 16.16 | 0.18 | MYCNOS, MYCN | - | H526 | 2 |
| H69 | ||||||||
| 4 | 8q12.2 | 62.01 | 62.18 | 0.17 | LOC100130298 | - | H2171 | 3 |
| Lu135 | ||||||||
| N417 | ||||||||
| 5 | 8q12.2 | 62.36 | 62.39 | 0.03 | CLVS1 | - | H2171 | 3 |
| Lu135 | ||||||||
| N417 | ||||||||
| 6 | 8q24.21 | 128.75 | 128.88 | 0.13 | MYC, MIR1204, PVT1 | H2171 | 6 | |
| SM09-011T1 | H446 | |||||||
| SM09-019T | H82 | |||||||
| N417 | ||||||||
| 7 | 9p24.1 | 5.35 | 5.78 | 0.43 | PLGRKT, CD274, PDCD1LG2, KIAA1432, ERMP1 | R-513T | H1607 | 3 |
| SM09-010T | ||||||||
| 8 | 9p23 | 13.27 | 13.45 | 0.18 | FLJ41200 | - | H1607 | 2 |
| H446 | ||||||||
Candidate target genes are described in bold.
MYCL1 and MYCN were co-amplified with TRIT1 at 1p34.2 and MYCNOS at 2p24.3, respectively, in 6 and 2 SCLCs. In the 8q24.21 region, MYC, MIR1204, and PVT1 were co-amplified in 6 SCLCs. Copy number breakpoints of amplified regions at 8q24.21 were mapped in the PVT1 gene in five of the six SCLCs (Supporting Information Tables S2; Supporting Information Fig. S1). The 1p34.3 region was co-amplified with MYCL1 at 1p34.2, and the 8q12.2 regions were co-amplified with MYC at 8q24.21, respectively, in several SCLCs (Supporting Information Fig. S2). Therefore, occurrence of complicated intrachromosomal rearrangements was suggested in the process of MYCL1 and MYC amplification, resulting in the co-amplification of several other genes on chromosomes 1 and 8, respectively.
In addition to the regions on chromosomes 1, 2, and 8, two novel commonly amplified regions were identified on chromosome 9p, 9p23, and 9p24.1 (Table 1; Fig. 1). The 9p23 region including FLJ41200 was amplified in 2 SCLCs, whereas the 9p24.1 region, including PLGRKT, CD274, PDCD1LG2, KIAA1432, and ERMP1, was amplified in three SCLCs. Both regions were co-amplified in the H1607 cell line, whereas only the 9p23 region was amplified in the H446 cell line and only the 9p24.1 region was amplified in two fresh tumors (Supporting Information Fig. S2). On chromosome 9p, NFIB at 9p23 and JAK2 at 9p24.1 were reported to be amplified in SCLC (Voortman et al., 2010; Dooley et al., 2011). However, NFIB and JAK2 were amplified only in one SCLC, respectively. Therefore, these two genes were not mapped in the commonly amplified regions on chromosome 9p (Supporting Information Table S2; Table 1; Fig. 1).
Figure 1.

Copy number plots of commonly amplified regions on chromosome 9p in four SCLCs. Copy numbers determined by 250K SNP array analysis are indicated by bars in colors. Genes mapped in two commonly amplified regions were aligned according to the BLAST human sequences (Build 37.3) in the NCBI database (http://www.ncbi.nlm.nih.gov/).
Expression of Amplified Genes on Chromosome 9p
Five genes, PLGRKT, CD274, PDCD1LG2, KIAA1432, and ERMP1, were mapped in the commonly amplified region at 9p24.1 (Table 1). To determine which genes were overexpressed by gene amplification, their mRNA expression was profiled in 19 fresh tumors (Supporting Information Fig. S3A). These five genes were amplified in one of the 19 tumors, SM09-010T. Expression of PLGRKT, CD274 and KIAA1432, but not of PDCD1LG2 and ERMP1, was distinctly high in SM09-010T (Supporting Information Fig. S3B). Therefore, PLGRKT, CD274, and KIAA1432 could be overexpressed by gene amplification in SCLCs.
To further determine genes whose expression is associated with copy numbers on chromosome 9p, we next performed the association study of gene expression with gene copy number in 55 SCLCs, including 30 fresh tumors and 25 cell lines (Supporting Information Table S1A, B). In addition to PLGRKT, CD274 and KIAA1432 at 9p24.1 and FLJ41200 at 9p23 as genes commonly amplified in SCLCs, NFIB at 9p23 and JAK2 at 9p24.1 were also subjected to the analysis. Expression of CD274 and KIAA1432 in amplified cells was significantly higher than that in non-amplified cells (P < 0.05) (Fig. 2A), and five genes except FLJ41200 showed significant associations between the levels of mRNA expression and copy numbers (P < 0.05) (Fig. 2B). Notably, KIAA1432 showed the strongest association between them (P = 1.04E-06). Therefore, KIAA1432 is the strongest target activated by gene amplification on chromosome 9p in SCLC. If there is another target in the 9p23 region, NF1B is more likely to be the one than FLJ41200.
Figure 2.

Association of copy numbers with expression levels in the 9p23-24 genes in SCLC cells. Levels of mRNA expression were quantified as ΔCt values using the HPRT1 gene as a control. Levels relative to normal lung were calculated using Human Lung Poly-A(+) RNA (Clontech) as the calibrator. (A) Levels of mRNA expression (log2) quantified by real-time RT-PCR in amplified (+) and not amplified (–) SCLCs. P-values by Student’s T-test for differences are shown. (B) Correlation of copy number ratios by real-time genomic-PCR with mRNA expression levels by real-time RT-PCR among 55 SCLCs.
Copy Numbers of Amplified Genes Defined by Real-time Genomic-PCR
To further investigate the prevalence and specificity of gene amplification in the chromosome 1, 2, 8, and 9 regions in SCLCs, 87 SCLCs comprised of 62 tumors and 25 cell lines were subjected to real-time genomic-PCR analyses. Among them, 33 tumors and 25 cell lines were also subjected to 250K SNP array analyses (Supporting Information Table S1A, B). Three MYC family genes and six genes on chromosome 9p were analyzed, and criteria of gene amplification by real-time genomic-PCR were defined as DNA copy number ratios ≥3 that was equivalent to the copy number ≥6. Five of the 33 tumors and 12 of the 25 cell lines showed amplification of 1–5 of the 9 genes by 250K SNP array analysis (Supporting Information Tables S2). Except for two SCLCs, the H1184 cell line with MYCL1 amplification and the SM09-011T1 tumor with MYC amplification, amplification of genes defined by 250K SNP arrays was consistently detected by real-time genomic-PCR (Supporting Information Table S3). On the other hand, in seven SCLCs without amplification by 250K SNP array analyses, 1–6 genes were judged as being amplified by real-time genomic-PCR analyses. Inconsistencies of the results were due to the following reasons. Firstly, only a small region including MYC covered by two SNP markers was amplified in the Lu135 cell line; therefore, this region was not defined as being amplified by SNP array analysis, but defined as being amplified by real-time genomic-PCR analysis. Secondly, in R-511T and SM09-006T, copy number loss of the reference locus, RPPH1, on chromosome 14 enhanced the degree of amplification in several genes by real-time genomic-PCR analysis. Thirdly, in H2195 and R-506M1, it was difficult to define the copy numbers possibly due to the heterogeneity of aneuploid cells and the presence of contaminated non-cancerous cells, respectively. Therefore, in these SCLCs, genes with copy number ratios ≥3 by real-time genomic-PCR was defined as five copies by SNP array analysis.
Even with some inconsistencies between the results of SNP array analyses and those of real-time genomic-PCR analyses, the association between them was highly significant (P = 5.58E-07 by Fisher’s exact test). Therefore, we then investigated the prevalence and specificity of gene amplification based on the data obtained by real-time genomic-PCR analysis. Amplification of these genes was detected in 12 of the 62 fresh tumors (19.4%) and 13 of the 25 cell lines (52.0%) (Table 2). Three MYC family genes were amplified in a mutually exclusive manner in 16 SCLCs (18.4%). Genes on chromosome 9p were amplified in 10 SCLCs (11.5%). Notably, NF1B/FLJ41200 at 9p23 and JAK2/CD274/PLGRKT/KIAA1432 at 9p24.21 were not co-amplified in 8 of the 10 SCLCs, indicating that amplification of these two regions occurred independently in most SCLCs. Therefore, the presence of target genes for amplification in each region was highly suggested. For this reason, specificities of 9p23 amplification and 9p24.21 amplification were independently analyzed among the 87 SCLCs. Importantly, four genes in the 9p24.1 region were amplified in a mutually exclusive manner with MYC family genes, whereas the MYC gene was co-amplified in one of three SCLCs with NF1B amplification, consistent with the results of 250K SNP array analyses (Supporting Information Fig. S2).
TABLE 2.
Occurrence of Gene Amplification in a Mutually Exclusive Manner in Small Cell Lung Cancers
| 1p34.2 | 2p24.3 | 8q24.21 | 9p23 | 9p24.1 | |||||
|---|---|---|---|---|---|---|---|---|---|
| Sample name | MYCL1 | MYCN | MYC | NFIB | FLJ41200 | JAK2 | PLGRKT | CD274 | KIAA1432 |
| H1963 | + | – | – | – | – | – | – | – | – |
| HCC33 | + | – | – | – | – | – | – | – | – |
| H510 | + | – | – | – | – | – | – | – | – |
| H2141 | + | – | – | – | – | – | – | – | – |
| SM09-012T | + | – | – | – | – | – | – | – | – |
| S391T | + | – | – | – | – | – | – | – | – |
| H69 | – | + | – | – | – | – | – | – | – |
| H526 | – | + | – | – | – | – | – | – | – |
| 1591T | – | + | – | – | – | – | – | – | – |
| R-511T | – | + | – | – | – | – | – | – | – |
| H82 | – | – | + | – | – | – | – | – | – |
| N417 | – | – | + | – | – | – | – | – | – |
| Lu135 | – | – | + | – | – | – | – | – | – |
| H2171 | – | – | + | – | – | – | – | – | – |
| SM09-019T | – | – | + | – | – | – | – | – | – |
| H446 | – | – | + | + | + | – | – | – | – |
| H2195 | – | – | – | + | + | – | – | – | – |
| SM09-008T | – | – | – | – | + | – | – | – | – |
| SM09-006T | – | – | – | + | + | + | + | + | + |
| H1607 | – | – | – | – | + | + | + | + | + |
| R-506M1 | – | – | – | – | – | + | + | + | + |
| SM09-010T | – | – | – | – | – | – | + | + | + |
| R-513T | – | – | – | – | – | – | + | + | + |
| SM09-004T | – | – | – | – | – | – | + | + | – |
| 1491M | – | – | – | – | – | – | – | – | + |
| Amplification rate in 87 SCLCs (%) | 6.9 | 4.6 | 6.9 | 3.4 | 5.7 | 3.4 | 6.9 | 6.9 | 6.9 |
Identification of Fusion Transcripts by Whole-transcriptome Sequencing
Forty-two SCLCs, consisted of 19 fresh tumors and 23 cell lines, were subjected to whole-transcriptome sequencing to identify fusion genes expressed in SCLCs (Supporting Information Tables S1A, B and S4). Total read counts ranged from 74,378,482 to 93,612,490, and their average was 86,529,758. A total of 60 fusion transcripts, transcribed from a portion of 95 genes, were identified as being expressed by the criteria of ≥10 paired-end (PE) reads for each transcript (Table 3). Twenty-two of them (36.7%) were in-frame; thus, were predicted to produce fusion proteins. There was no set of 5′-3′ fusion transcript recurrently detected in ≥2 SCLCs. However, two of the 5′ partner genes, PVT1 and RLF, were detected recurrently in ≥2 SCLCs (14 pairs in 7 SCLCs). PVT1 was detected as the 5′ partner gene of seven fusion pairs with different 3′ partner genes in five SCLCs. Previously, PVT1 was shown to be fused with CHD7 in H2171 and Lu135 (Campbell et al., 2008; Pleasance et al., 2010). In this study, PVT1-CHD7 was detected in H2171 with ≥10 PE reads but was not in Lu135. RLF was detected as the 5′ partner gene of 7 fusion pairs with different 3′ partner genes in 2 SCLCs. RLF was reported as being a fusion gene with MYCL1 expressed in SCLC cells (Makela et al., 1991a, 1991b, 1995), and the RLF-MYCL1 fusion was also detected in H1963 with ≥10 PE reads in this study. Three of the five fusion pairs having RLF as the 5′ partner gene, RLF-MYCL1, RLF-SMAP2 and RLF-FAM132A, were predicted to produce fusion proteins. The remaining 46 fusion transcripts were detected in a single SCLC, respectively, and 19 of them were predicted to produce fusion proteins. Therefore, none of the 22 fusion transcripts predicted to produce fusion proteins was expressed recurrently in multiple SCLC cases.
TABLE 3.
Fusion Transcripts Expressed in Small Cell Lung Cancer Cells Identified by Whole-transcriptome Sequencing
| 5’ partner gene | 3’ partner gene | Frame | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No | Sample | #PE reads | #junction reads | Gene | mRNA No | Chr | Amp. | Gene | mRNA No | Chr | Amp. | Distance on genome (bp) | In frame |
| I. 5’-transcripts detected in ≥ 2 cases | |||||||||||||
| 1 | H82 | 408 | 239 | PVT1 | NR_003367.1 | 8 | + | MYH7 | NM_000257.2 | 14 | + | ||
| 2 | H2171 | 345 | 177 | PVT1 | NR_003367.1 | 8 | + | CHD7 | NM_017780.3 | 8 | + | 67027313 | |
| 3 | H2171 | 219 | 20 | PVT1 | NR_003367.1 | 8 | + | SLC7A7 | NM_001126105.2 | 14 | 105507749 | ||
| 4 | H2171 | 114 | 17 | PVT1 | NR_003367.1 | 8 | + | CCNB1IP1 | NM_021178.3 | 14 | NA | ||
| 5 | H2107 | 37 | 13 | PVT1 | NR_003367.1 | 8 | NOL4 | NM_001198546.1 | 18 | NA | |||
| 6 | N417 | 34 | 82 | PVT1 | NR_003367.1 | 8 | + | CLVS1 | NM_173519.2 | 8 | + | 66392576 | |
| 7 | H446 | 12 | 26 | PVT1 | NR_003367.1 | 8 | + | LY6H | NM_001130478.1 | 8 | 15125833 | ||
| 8 | HCC33 | 525 | 128 | RLF | NM_012421.3 | 1 | + | UBE2J2 | NM_058167.2 | 1 | 39417806 | ||
| 9 | H1963 | 391 | 562 | RLF | NM_012421.3 | 1 | + | MYCL1 | NM_001033081.2 | 1 | + | 259353 | + |
| 10 | H1963 | 194 | 68 | RLF | NM_012421.3 | 1 | + | COL9A2 | NM_001852.3 | 1 | + | 59570 | |
| 11 | H1963 | 118 | 68 | RLF | NM_012421.3 | 1 | + | BCL2L1 | NM_001191.2 | 20 | + | NA | |
| 12 | H1963 | 112 | 124 | RLF | NM_012421.3 | 1 | + | HM13 | NM_030789.2 | 20 | + | NA | |
| 13 | H1963 | 43 | 163 | RLF | NM_012421.3 | 1 | + | SMAP2 | NM_001198978.1 | 1 | + | 133135 | + |
| 14 | HCC33 | 33 | 132 | RLF | NM_012421.3 | 1 | + | FAM132A | NM_001014980.2 | 1 | 39444938 | + | |
| II. Fusion transcripts detected in a single case | |||||||||||||
| 15 | H1963 | 2168 | 1627 | TPX2 | NM_012112.4 | 20 | + | HM13 | NM_030789.2 | 20 | + | 169703 | + |
| 16 | SM09-012T | 290 | 335 | CAP1 | NM_001105530.1 | 1 | + | MACF1 | NM_012090.4 | 1 | + | 991332 | |
| 17 | H1963 | 226 | 52 | BCL2L1 | NM_138578.1 | 20 | + | HM13 | NM_030789.2 | 20 | + | 94884 | |
| 18 | HCC33 | 193 | 83 | TRIT1 | NM_017646.4 | 1 | + | EP400 | NM_015409.4 | 12 | + | NA | |
| 19 | H1963 | 125 | 45 | BCL2L1 | NM_138578.1 | 20 | + | DEM1 | NM_022774.1 | 1 | + | NA | + |
| 20 | H2171 | 122 | 56 | ENO2 | NM_001975.2 | 12 | ACRBP | NM_032489.2 | 12 | + | |||
| 21 | H1963 | 99 | 99 | BCL2L1 | NM_138578.1 | 20 | + | RIMS3 | NM_014747.2 | 1 | + | NA | |
| 22 | SM09-004T | 82 | 101 | WAC | NM_016628.4 | 10 | GPR158 | NM_020752.2 | 10 | 2931268 | |||
| 23 | Lu139 | 79 | 45 | CSMD3 | NM_052900.2 | 8 | MYC | NM_002467.4 | 8 | 14299072 | |||
| 24 | H1184 | 54 | 88 | RERE | NM_001042681.1 | 1 | SLC2A5 | NM_001135585.1 | 1 | 223728 | + | ||
| 25 | H69 | 50 | 44 | FOXK2 | NM_004514.3 | 17 | HEXDC | NM_173620.2 | 17 | 77073 | |||
| 26 | H1963 | 50 | 17 | BCL2L1 | NM_138578.1 | 20 | + | ZNF684 | NM_152373.3 | 1 | + | NA | + |
| 27 | N417 | 42 | 30 | NCOR2 | NM_001077261.3 | 12 | SCARB1 | NM_005505.4 | 12 | 210164 | |||
| 28 | HCC33 | 41 | 71 | UBE4B | NM_001105562.2 | 1 | TBCB | NM_001281.2 | 19 | NA | + | ||
| 29 | SM09-010T | 41 | 43 | KIAA1432 | NM_001135920.2 | 9 | + | JAK2 | NM_004972.3 | 9 | 501144 | + | |
| 30 | H1963 | 41 | 31 | ZMPSTE24 | NM_005857.4 | 1 | + | MFSD2A | NM_001136493.1 | 1 | + | 288105 | |
| 31 | H510 | 38 | 37 | SMEK1 | NM_032560.4 | 14 | HEATR3 | NM_182922.2 | 16 | NA | + | ||
| 32 | SM09-016T | 36 | 48 | NAV2 | NM_001111018.1 | 11 | LOC494141 | NR_026563.1 | 11 | 1137161 | |||
| 33 | Lu135 | 32 | 23 | LRRC45 | NM_144999.2 | 17 | GCGR | NM_000160.3 | 17 | 209390 | |||
| 34 | H510 | 30 | 67 | TWSG1 | NM_020648.5 | 18 | PIK3C3 | NM_002647.2 | 18 | 30132781 | |||
| 35 | H69 | 30 | 34 | PAWR | NM_002583.2 | 12 | GNS | NM_002076.3 | 12 | 14832520 | |||
| 36 | H1184 | 25 | 20 | SF3A3 | NM_006802.2 | 1 | + | GNL2 | NM_013285.2 | 1 | + | 361067 | + |
| 37 | H209 | 24 | 19 | CREBBP | NM_001079846.1 | 16 | SLX4 | NM_032444.2 | 16 | 113472 | |||
| 38 | SM09-016T | 24 | 14 | RASA2 | NM_006506.2 | 3 | NICN1 | NM_032316.3 | 3 | 91739168 | + | ||
| 39 | H1963 | 22 | 48 | SMAP2 | NM_022733.2 | 1 | + | MYCL1 | NM_001033081.2 | 1 | + | 472040 | |
| 40 | SM09-016T | 22 | 13 | SFMBT1 | NM_001005158.2 | 3 | AP2A2 | NM_001242837.1 | 11 | NA | |||
| 41 | H510 | 21 | 12 | PTK2 | NM_001199649.1 | 8 | PKHD1L1 | NM_177531.4 | 8 | 31125439 | |||
| 42 | H526 | 19 | 31 | SLC25A36 | NM_001104647.1 | 3 | PLSCR1 | NM_021105.2 | 3 | 5534191 | |||
| 43 | H526 | 19 | 20 | XPR1 | NM_001135669.1 | 1 | TRMT1L | NM_001202423.1 | 1 | 4227805 | + | ||
| 44 | H2195 | 18 | 22 | HMBOX1 | NM_001135726.1 | 8 | ZFAND3 | NM_021943.2 | 6 | NA | + | ||
| 45 | SM09-014T | 18 | 10 | CRLS1 | NM_001127458.1 | 20 | KCNK17 | NM_001135111.1 | 6 | NA | |||
| 46 | H510 | 17 | 14 | PHF15 | NM_015288.4 | 5 | UBE2B | NM_003337.2 | 5 | 133999 | |||
| 47 | H1963 | 17 | 10 | BCL2L1 | NM_138578.1 | 20 | + | ZNF643 | NM_023070.2 | 1 | + | NA | + |
| 48 | SM09-016T | 15 | 40 | ATP5L | NM_006476.4 | 11 | TEAD1 | NM_021961.5 | 11 | 105305820 | |||
| 49 | SM09-014T | 14 | 23 | NUDCD1 | NM_001128211.1 | 8 | SYBU | NM_001099743.1 | 8 | 244247 | + | ||
| 50 | Lu134 | 14 | 17 | SPG11 | NM_001160227.1 | 15 | SORD | NM_003104.5 | 15 | 359425 | + | ||
| 51 | SM09-016T | 14 | 17 | NGLY1 | NM_001145293.1 | 3 | CCKBR | NM_176875.3 | 11 | NA | + | ||
| 52 | SM09-016T | 13 | 21 | DLEC1 | NM_007335.2 | 3 | ODZ4 | NM_001098816.2 | 11 | NA | |||
| 53 | H1963 | 13 | 19 | PPT1 | NM_000310.3 | 1 | + | BCL2L1 | NM_001191.2 | 20 | + | NA | |
| 54 | H128 | 13 | 12 | NAIP | NM_004536.2 | 5 | OCLN | NM_001205254.1 | 5 | 1414179 | |||
| 55 | H1963 | 12 | 10 | BCL2L1 | NM_138578.1 | 20 | + | BMP8B | NM_001720.3 | 1 | + | NA | |
| 56 | H526 | 12 | 10 | CIT | NM_001206999.1 | 12 | RFC5 | NM_001130112.2 | 12 | 1653559 | + | ||
| 57 | H1963 | 11 | 22 | BCL2L1 | NM_138578.1 | 20 | + | RLF | NM_012421.3 | 1 | + | NA | |
| 58 | SM09-018T | 10 | 22 | NFIX | NM_002501.2 | 19 | GATAD2A | NM_017660.3 | 19 | 6287032 | + | ||
| 59 | SM09-016T | 11 | 19 | STAG1 | NM_005862.2 | 3 | STXBP5L | NM_014980.2 | 3 | 14912391 | |||
| 60 | H2171 | 10 | 12 | PICALM | NM_001008660.2 | 11 | CCDC81 | NM_001156474.1 | 11 | 304853 | + | ||
Amplification of Genes with Fusions
We next investigated the copy numbers of 95 genes in the 60 fusion pairs identified by whole-transcriptome sequencing (Table 3). Twenty-eight of the 5′ partner genes detected in 9 SCLCs and 22 of the 3′ partner genes detected in seven SCLCs were mapped in the amplified regions.
In four of the five SCLCs expressing fusion transcripts with PVT1 as the 5′ partner gene, the 5′ portions of the PVT1 gene at 8q24.21 were amplified, indicating that chromosomal breaks had occurred in the PVT1 locus (Supporting Information Fig. S1). Three of seven genes fused with PVT1 were also amplified. Therefore, it was indicated that chromosomal breaks often occur in the PVT1 locus during the process of MYC amplification. For this reason, we further searched for PVT1 fusion transcripts expressed in the H2171, Lu135, and N417 cell lines, which showed co-amplification of three regions on chromosome 8q (Table 1). In addition to PVT1-CHD7 (PE reads = 345), PVT1-SLC7A7 (PE read = 219), and PVT1-CCNB1IP1 (PE reads = 114) were detected in H2171. As described above, no PVT1 fusion was detected in Lu135. PVT1-CLVS1 (PE read = 34) and PVT1-ASPH (PE reads = 27, junction reads = 9) were detected in N417.
RLF, detected as the 5′ partner gene of seven fusion pairs in two cell lines, H1963 and HCC33, was amplified in both cell lines (Table 3). Three 3′ partner genes, MYCL1, COL9A2, and SMAP2, fused with RLF in H1963 were also mapped to 1p34.2 and amplified. Two other 3′ partner genes, BCL2L1 and HM13, fused with RLF in H1963 and mapped to 20q11.21, were also amplified. The remaining two 3′ partner genes, UBE2J2 and FAM132A at 1p36.33, were also amplified in HCC33 with consecutive 2 SNPs. Therefore, production of fusion transcripts with RLF was always accompanied by amplification of both the 5′ and 3′ genes, indicating that those genes had fused in the process of MYCL1 amplification.
These results strongly indicate that amplification of several regions on chromosomes 1 and 8 simultaneously but not sequentially occurs in SCLC cells, and further support that complicated intrachromosomal rearrangements occur in the process of MYCL1 or MYC amplification, resulting in the co-amplification and fusion of several genes on chromosomes 1 and 8. Therefore, the PVT1 and RLF loci would be hotspots of chromosomal breaks in the process of gene amplification in SCLC cells.
SCLCs with Expression of Multiple Fusion Transcripts
Sixty fusion transcripts were detected in 23 of the 42 SCLCs (Table 3, Supporting Information Table S4), indicating the presence of SCLCs expressing multiple fusion transcripts. Indeed, sixteen fusion pairs consisted of 15 genes were identified in H1963 (Supporting Information Fig. S4A). Twelve of the 15 genes, including MYCL1, were mapped to the 1p34.2 amplicon, and the remaining 3 genes were mapped to the 20q11.21 amplicon. Seven fusions were intrachromosomal among genes at 1p34.2 or 20q11.21, while the other nine fusions were interchromosomal between genes at 1p34.2 and genes at 20q11.21. Therefore, in H1963, complicated chromosomal rearrangements were likely to have occurred in the process of MYCL1 amplification.
Seven fusion pairs consisted of 14 genes were identified in SM09-016T (Supporting Information Fig. S4B). Seven and 7 of the 14 genes were mapped to chromosomes 3 and 11, respectively. The 5′ and 3′ partner genes for four of the seven fusions were mapped to the same chromosomes, while those for the remaining three fusions were mapped to different chromosomes. Therefore, these genes were fused by either intrachromosomal or interchromosomal rearrangements. Interestingly, no fused genes were amplified in SM09-016T, indicating that complicated chromosomal rearrangements had occurred without gene amplification. Two to five fusion transcripts were detected in eight other SCLCs. Among 24 fusions detected in these SCLCs, 18 of them were intrachromosomal and the remaining six were interchromosomal. Eight of 5′ partner genes and four of 3′ partner genes were mapped to the amplified regions. Therefore, intrachromosomal rearrangements seemed to occur preferentially in SCLC cells irrespective of the process of gene amplification.
KIAA1432-JAK2 Fusion Detected in a SCLC with 9p24.1 Amplification
Interestingly, a KIAA1432–JAK2 fusion transcript was detected in SM09-010T with amplification of the KIAA1432 gene at 9p24.1 (Table 3; Fig. 3A). Furthermore, three (4.6%) of 65 SCLCs analyzed by RT-PCR were shown to express KIAA1432–JAK2 fusion transcripts. However, only one of them expressed in SM09-010T was predicted to produce a fusion protein, although the tyrosine kinase domain was disrupted by fusion (Fig. 3B).
Figure 3.
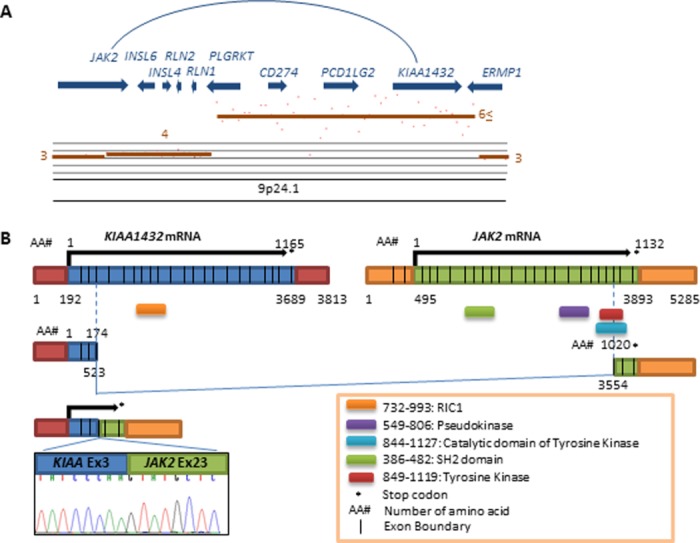
A KIAA1432-JAK2 fusion detected in the SM09-010T tumor with 9p24.21 amplification. (A) Copy number plots at 9p24.1 by 250K SNP array. (B) Schematic presentation of wild-type KIAA1432 and JAK2 proteins, and the KIAA1432-JAK2 fusion protein. Electrophoregram for Sanger sequencing of cDNA for the KIAA1432–JAK2 fusion transcript is shown below.
DISCUSSION
The purpose of this study was to identify genes activated by amplification and/or fusion in SCLC. By a copy number analysis, 34 genes were identified as being frequently amplified in SCLCs. In concordance with previous studies, three MYC family genes were frequently amplified in SCLCs (Wistuba et al., 2001; Kim et al., 2006; Voortman et al., 2010; Larsen and Minna, 2011; Sos et al., 2012). Recently, several MYC inhibitors, including Omomyc, BET bromodomain inhibitor and Aurora kinase inhibitor, have been reported (Soucek et al., 2008; Delmore et al., 2011; Sos et al., 2012); therefore, the MYC family gene products could be druggable targets in SCLC cells with their activation.
Co-amplification of PVT1 and MIR1204 with MYC has been reported in several types of cancers, and their oncogenic roles have been also suggested (Guan et al., 2007; Huppi et al., 2008; Haverty et al., 2009; Schiffman et al., 2010; Sircoulomb et al., 2010). However, in this study, only the 5′ portion of the PVT1 gene including exon 1 was commonly amplified, and amplified PVT1 genes were often fused with other genes. Since the 3′ partner genes in five PVT1 fusions were different from each other, biological significance of PVT1 amplification and/or fusion is unclear at present. The PVT1 locus could be a hotspot of chromosomal breaks in the process of MYC amplification in SCLCs. Since MIR1204 was always co-amplified with MYC, involvement of MIR1204 in the development of SCLC cannot be excluded. Recently, PVT1-MYC fusions were detected in >60% of medulloblastomas with MYC amplification, and these fusions also involved PVT1 exon1 and MIR1204 (Northcott et al., 2012). These results further support that the PVT1 locus is a hotspot of chromosomal breaks in the process of MYC amplification, although no PVT1-MYC fusions were detected in SCLCs.
In addition to MYCL1 and MYC, several regions on either the same or different chromosomes were co-amplified in SCLCs. Furthermore, several genes, especially RLF and PVT1 in the MYCL1 and MYC amplicons, respectively, were fused with genes in different amplified regions. These results strongly indicate that amplification of several regions on chromosomes 1 and 8 occurred simultaneously but not independently/sequentially in these SCLCs. Therefore, several genes co-amplified with MYCL1 and MYC were likely to be rearranged and amplified together with MYCL1 and MYC by a massive genomic rearrangement acquired in a single catastrophic event. Recently, a new mechanism for genetic instability in cancer cells, chromothripsis, was proposed by Stephens et al. (2011). In chromothripsis, tens to hundreds of chromosomal rearrangements involving localized genomic regions can be acquired in a one-off cellular catastrophe. Indeed, CHD7 at 8q12 was shown to be rearranged in three SCLC cell lines (Campbell et al., 2008; Pleasance et al., 2010). In this study, amplified genes in SCLC cells often showed fusions with genes in the same amplicons, different amplicons on the same chromosome, or different amplicons on different chromosomes. These results strongly indicate that amplification of MYCL1 and MYC often occurs through chromothripsis in SCLCs, although the presence of tens to hundreds of chromosomal rearrangements in particular genomic regions should be confirmed by whole genome sequencing. Therefore, target genes of amplification on these chromosomes would be MYCL1 and MYC, respectively, even though multiple regions on chromosomes 1 and 8 were commonly amplified in SCLCs.
Two regions on chromosome 9p were also commonly amplified in SCLCs. Notably, amplification at 9p24.1 tended to occur in SCLCs without amplification of MYC family genes. In contrast, the 9p23 region including NF1B was co-amplified with MYC in H446. Previously, Nf1b in a mouse SCLC model was shown to be frequently co-amplified with Mycl1 (Dooley et al., 2011), consistent with the present results. However, 9p24.1 and 9p23 were independently amplified in most SCLCs. Therefore, these regions were unlikely to be amplified by chromothripsis, and the 9p23 and 9p24.1 regions may contain independent target genes, respectively. Expression analyses revealed that NF1B at 9p23 and KIAA1432 at 9p24.1 were overexpressed by gene amplification in SCLCs, thus, were strong candidates of genes activated by amplification in SCLCs. Recently, KIAA1432 was reported to be also amplified and overexpressed in breast cancer, thus, is a target gene of amplification not only in SCLC but also in breast cancer (Wu et al., 2012). KIAA1432 encodes a partner protein, CIP150, of connexin 43 (Cx43) (Akiyama et al., 2005). Cx43, a structural protein in the gap junction, has been reported as being a tumor suppressor inactivated in several cancers (Li et al., 2008; Naus and Laird, 2010; Plante et al., 2011). Therefore, it is possible that CIP150 encoded by KIAA1432 is involved in the regulation of Cx43 activities and its overexpression may play a role in SCLC development. Further functional studies are needed to clarify the biological significance of KIAA1432 amplification in SCLC development. Interestingly, a KIAA1432-JAK2 fusion was identified in a case with KIAA1432 amplification. Various fusions with JAK2 have been reported in hematological malignancies (Van Roosbroeck et al., 2011). These fusions contained the whole tyrosine kinase domain and lead to constitutive phosphorylation of the kinase (Lacronique et al., 1997; Griesinger et al., 2005; Poitras et al., 2008; Nebral et al., 2009; Van Roosbroeck et al., 2011). However, the kinase domain was disrupted by the KIAA1432-JAK2 fusion identified in this study. Therefore, it is unlikely that JAK2 is activated by fusion with KIAA1432 in SCLCs. There might be hotspots of chromosomal breakpoints in the JAK2 and KIAA1432 loci in the process of 9p24.1 amplification in SCLCs.
During the preparation of this manuscript, the results of comprehensive and integrative genome analyses on SCLCs were reported by two groups (Peifer et al., 2012; Rudin et al., 2012). Frequent amplification of the SOX2 (copy number ≥4) and FGFR1 (copy number ≥3.5) genes at 3q26.3-q27 and 8p12, respectively, were shown in their articles. When we used the same criteria (copy number ≥4), the SOX2 and FGFR1 genes were amplified in 21 (36.0%) and 7 (12.1%), respectively, of 58 SCLCs subjected to 250K SNP array analysis. However, in this study, by using the criteria of copy number ≥6 for detection of focally amplified genes, neither SOX2 nor FGFR1 were picked up as the amplified genes in SCLCs, because extents of amplification for the SOX2 and FGFR1 genes were not so high as those for MYC family genes and 9p genes. Therefore, in our criteria, genes with activation by low degree of amplification (3–5 copies) were overlooked. However, genes with high degree of amplification (copy number ≥6) were successfully and efficiently picked up from the SCLC genomes. A recurrent RLF-MYCL1 fusion was reported in one article (Rudin et al., 2012). The RLF-MYCL1 fusion was also identified in H1963 in this study, but both RLF and MYCL1 were fused with several other genes in this cell line, indicating the occurrence of chromothripsis in the production of those fusions in SCLCs.
We should also point out here that statuses of MYC family gene amplification in some cell lines defined in this study were not the same as those reported previously. To depict such inconsistencies more critically and clearly, we prepared Supporting Information Table S5, in which the statuses for MYC family amplification defined in this study were summarized together with those in three other studies (Kim et al., 2006; Voortman et al., 2010; Sos et al., 2012) for each of all the 25 cell lines analyzed in this study. In 18 of the 25 cell lines, statuses of MYC family gene amplification were also defined in 1–3 of the other studies. In 14 of the 18 cell lines, statuses of MYC family gene amplification were consistent among studies. However, in the H69 cell line, MYCN amplification was detected in three of the four studies, and in the remaining three cell lines, H128, H187, and H2107, either MYC or MYCL1 amplification was detected only in one of three or four studies. These inconsistencies would be due to the differences in the criteria of gene amplification among the four studies and also could be due to the differences in the methods as well as the platforms used for assessing copy numbers of each gene among them.
In this study, we did not refer to somatic mutations that could be detected by whole-transcriptome sequencing, because genes with somatic mutations that are highly expressed in the cells can be only detected by whole-transcriptome sequencing. In our preliminary results, various types of mutations detected by genome sequencing were not detected by whole transcriptome sequencing possibly due to the low levels or absence of expression. In addition, due to the differences in the level of mRNA expression among genes analyzed, total read counts of transcripts for sequencing varied among genes in each sample. Therefore, it was difficult to obtain conclusive results for the presence of mutations by whole-transcriptome sequencing only. To obtain more convincing results, we have to confirm the presence of mutations by using several types of genome sequencing, such as direct sequencing and whole exome/genome sequencing. Accordingly, in this study, we did not present the data for possible somatic mutations detected by whole-transcriptome sequencing. In contrast, the presence of amplified or fused genes could be easily confirmed by PCR analysis. Therefore, in this study, we attempted to compile the list of genes that were activated by amplification and/or fusion in SCLC cells. Further studies are now in progress.
Supplementary material
Additional Supporting Information may be found in the online version of this article.
REFERENCES
- Akiyama M, Ishida N, Ogawa T, Yogo K, Takeya T. Molecular cloning and functional analysis of a novel Cx43 partner protein CIP150. Biochem Biophys Res Commun. 2005;335:1264–1271. doi: 10.1016/j.bbrc.2005.08.019. [DOI] [PubMed] [Google Scholar]
- Campbell PJ, Stephens PJ, Pleasance ED, O’Meara S, Li H, Santarius T, Stebbings LA, Leroy C, Edkins S, Hardy C, Teague JW, Menzies A, Goodhead I, Turner DJ, Clee CM, Quail MA, Cox A, Brown C, Durbin R, Hurles ME, Edwards PA, Bignell GR, Stratton MR, Futreal PA. Identification of somatically acquired rearrangements in cancer using genome-wide massively parallel paired-end sequencing. Nat Genet. 2008;40:722–729. doi: 10.1038/ng.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper S, Spiro SG. Small cell lung cancer: Treatment review. Respirology. 2006;11:241–248. doi: 10.1111/j.1440-1843.2006.00850.x. [DOI] [PubMed] [Google Scholar]
- Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, Chesi M, Schinzel AC, McKeown MR, Heffernan TP, Vakoc CR, Bergsagel PL, Ghobrial IM, Richardson PG, Young RA, Hahn WC, Anderson KC, Kung AL, Bradner JE, Mitsiades CS. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146:904–917. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dooley AL, Winslow MM, Chiang DY, Banerji S, Stransky N, Dayton TL, Snyder EL, Senna S, Whittaker CA, Bronson RT, Crowley D, Barretina J, Garraway L, Meyerson M, Jacks T. Nuclear factor I/B is an oncogene in small cell lung cancer. Genes Dev. 2011;25:1470–1475. doi: 10.1101/gad.2046711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griesinger F, Hennig H, Hillmer F, Podleschny M, Steffens R, Pies A, Wormann B, Haase D, Bohlander SK. A BCR-JAK2 fusion gene as the result of a t(9;22)(p24;q11.2) translocation in a patient with a clinically typical chronic myeloid leukemia. Genes Chromosomes Cancer. 2005;44:329–333. doi: 10.1002/gcc.20235. [DOI] [PubMed] [Google Scholar]
- Guan Y, Kuo WL, Stilwell JL, Takano H, Lapuk AV, Fridlyand J, Mao JH, Yu M, Miller MA, Santos JL, Kalloger SE, Carlson JW, Ginzinger DG, Celniker SE, Mills GB, Huntsman DG, Gray JW. Amplification of PVT1 contributes to the pathophysiology of ovarian and breast cancer. Clin Cancer Res. 2007;13:5745–5755. doi: 10.1158/1078-0432.CCR-06-2882. [DOI] [PubMed] [Google Scholar]
- Haverty PM, Hon LS, Kaminker JS, Chant J, Zhang Z. High-resolution analysis of copy number alterations and associated expression changes in ovarian tumors. BMC Med Genomics. 2009;2:21. doi: 10.1186/1755-8794-2-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huppi K, Volfovsky N, Runfola T, Jones TL, Mackiewicz M, Martin SE, Mushinski JF, Stephens R, Caplen NJ. The identification of microRNAs in a genomically unstable region of human chromosome 8q24. Mol Cancer Res. 2008;6:212–221. doi: 10.1158/1541-7786.MCR-07-0105. [DOI] [PubMed] [Google Scholar]
- Iwakawa R, Kohno T, Kato M, Shiraishi K, Tsuta K, Noguchi M, Ogawa S, Yokota J. MYC amplification as a prognostic marker of early-stage lung adenocarcinoma identified by whole genome copy number analysis. Clin Cancer Res. 2011;17:1481–1489. doi: 10.1158/1078-0432.CCR-10-2484. [DOI] [PubMed] [Google Scholar]
- Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- Kim YH, Girard L, Giacomini CP, Wang P, Hernandez-Boussard T, Tibshirani R, Minna JD, Pollack JR. Combined microarray analysis of small cell lung cancer reveals altered apoptotic balance and distinct expression signatures of MYC family gene amplification. Oncogene. 2006;25:130–138. doi: 10.1038/sj.onc.1208997. [DOI] [PubMed] [Google Scholar]
- Kohno T, Ichikawa H, Totoki Y, Yasuda K, Hiramoto M, Nammo T, Sakamoto H, Tsuta K, Furuta K, Shimada Y, Iwakawa R, Ogiwara H, Oike T, Enari M, Schetter AJ, Okayama H, Haugen A, Skaug V, Chiku S, Yamanaka I, Arai Y, Watanabe S, Sekine I, Ogawa S, Harris CC, Tsuda H, Yoshida T, Yokota J, Shibata T. KIF5B-RET fusions in lung adenocarcinoma. Nat Med. 2012;18:375–377. doi: 10.1038/nm.2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacronique V, Boureux A, Valle VD, Poirel H, Quang CT, Mauchauffe M, Berthou C, Lessard M, Berger R, Ghysdael J, Bernard OA. A TEL-JAK2 fusion protein with constitutive kinase activity in human leukemia. Science. 1997;278:1309–1312. doi: 10.1126/science.278.5341.1309. [DOI] [PubMed] [Google Scholar]
- Larsen JE, Minna JD. Molecular biology of lung cancer: Clinical implications. Clin Chest Med. 2011;32:703–740. doi: 10.1016/j.ccm.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Zhou Z, Welch DR, Donahue HJ. Expressing connexin 43 in breast cancer cells reduces their metastasis to lungs. Clin Exp Metastasis. 2008;25:893–901. doi: 10.1007/s10585-008-9208-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makela TP, Kere J, Winqvist R, Alitalo K. Intrachromosomal rearrangements fusing L-myc and rlf in small-cell lung cancer. Mol Cell Biol. 1991a;11:4015–4021. doi: 10.1128/mcb.11.8.4015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makela TP, Saksela K, Evan G, Alitalo K. A fusion protein formed by L-myc and a novel gene in SCLC. EMBO J. 1991b;10:1331–1335. doi: 10.1002/j.1460-2075.1991.tb07652.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makela TP, Hellsten E, Vesa J, Hirvonen H, Palotie A, Peltonen L, Alitalo K. The rearranged L-myc fusion gene (RLF) encodes a Zn-15 related zinc finger protein. Oncogene. 1995;11:2699–2704. [PubMed] [Google Scholar]
- Nakanishi H, Matsumoto S, Iwakawa R, Kohno T, Suzuki K, Tsuta K, Matsuno Y, Noguchi M, Shimizu E, Yokota J. Whole genome comparison of allelic imbalance between noninvasive and invasive small-sized lung adenocarcinomas. Cancer Res. 2009;69:1615–1623. doi: 10.1158/0008-5472.CAN-08-3218. [DOI] [PubMed] [Google Scholar]
- Nannya Y, Sanada M, Nakazaki K, Hosoya N, Wang L, Hangaishi A, Kurokawa M, Chiba S, Bailey DK, Kennedy GC, Ogawa S. A robust algorithm for copy number detection using high-density oligonucleotide single nucleotide polymorphism genotyping arrays. Cancer Res. 2005;65:6071–6079. doi: 10.1158/0008-5472.CAN-05-0465. [DOI] [PubMed] [Google Scholar]
- Naus CC, Laird DW. Implications and challenges of connexin connections to cancer. Nat Rev Cancer. 2010;10:435–441. doi: 10.1038/nrc2841. [DOI] [PubMed] [Google Scholar]
- Nebral K, Denk D, Attarbaschi A, Konig M, Mann G, Haas OA, Strehl S. Incidence and diversity of PAX5 fusion genes in childhood acute lymphoblastic leukemia. Leukemia. 2009;23:134–143. doi: 10.1038/leu.2008.306. [DOI] [PubMed] [Google Scholar]
- Northcott PA, Shih DJ, Peacock J, Garzia L, Sorana Morrissy A, Zichner T, Stutz AM, Korshunov A, Reimand J, Schumacher SE, Beroukhim R, Ellison DW, Marshall CR, Lionel AC, Mack S, Dubuc A, Yao Y, Ramaswamy V, Luu B, Rolider A, Cavalli FM, Wang X, Remke M, Wu X, Chiu RY, Chu A, Chuah E, Corbett RD, Hoad GR, Jackman SD, Li Y, Lo A, Mungall KL, Ming Nip K, Qian JQ, Raymond AG, Thiessen N, Varhol RJ, Birol I, Moore RA, Mungall AJ, Holt R, Kawauchi D, Roussel MF, Kool M, Jones DT, Witt H, Fernandez LA, Kenney AM, Wechsler-Reya RJ, Dirks P, Aviv T, Grajkowska WA, Perek-Polnik M, Haberler CC, Delattre O, Reynaud SS, Doz FF, Pernet-Fattet SS, Cho BK, Kim SK, Wang KC, Scheurlen W, Eberhart CG, Fevre-Montange M, Jouvet A, Pollack IF, Fan X, Muraszko KM, Yancey Gillespie G, Di Rocco C, Massimi L, Michiels EM, Kloosterhof NK, French PJ, Kros JM, Olson JM, Ellenbogen RG, Zitterbart K, Kren L, Thompson RC, Cooper MK, Lach B, McLendon RE, Bigner DD, Fontebasso A, Albrecht S, Jabado N, Lindsey JC, Bailey S, Gupta N, Weiss WA, Bognar L, Klekner A, Van Meter TE, Kumabe T, Tominaga T, Elbabaa SK, Leonard JR, Rubin JB, Liau LM, Van Meir EG, Fouladi M, Nakamura H, Cinalli G, Garami M, Hauser P, Saad AG, Iolascon A, Jung S, Carlotti CG, Vibhakar R, Shin Ra Y, Robinson S, Zollo M, Faria CC, Chan JA, Levy ML, Sorensen PH, Meyerson M, Pomeroy SL, Cho YJ, Bader GD, Tabori U, Hawkins CE, Bouffet E, Scherer SW, Rutka JT, Malkin D, Clifford SC, Jones SJ, Korbel JO, Pfister SM, Marra MA, Taylor MD. Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nature. 2012;488:49–56. doi: 10.1038/nature11327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okayama H, Kohno T, Ishii Y, Shimada Y, Shiraishi K, Iwakawa R, Furuta K, Tsuta K, Shibata T, Yamamoto S, Watanabe S, Sakamoto H, Kumamoto K, Takenoshita S, Gotoh N, Mizuno H, Sarai A, Kawano S, Yamaguchi R, Miyano S, Yokota J. Identification of genes upregulated in ALK-positive and EGFR/KRAS/ALK-negative lung adenocarcinomas. Cancer Res. 2012;72:100–111. doi: 10.1158/0008-5472.CAN-11-1403. [DOI] [PubMed] [Google Scholar]
- Peifer M, Fernandez-Cuesta L, Sos ML, George J, Seidel D, Kasper LH, Plenker D, Leenders F, Sun R, Zander T, Menon R, Koker M, Dahmen I, Muller C, Di Cerbo V, Schildhaus HU, Altmuller J, Baessmann I, Becker C, de Wilde B, Vandesompele J, Bohm D, Ansen S, Gabler F, Wilkening I, Heynck S, Heuckmann JM, Lu X, Carter SL, Cibulskis K, Banerji S, Getz G, Park KS, Rauh D, Grutter C, Fischer M, Pasqualucci L, Wright G, Wainer Z, Russell P, Petersen I, Chen Y, Stoelben E, Ludwig C, Schnabel P, Hoffmann H, Muley T, Brockmann M, Engel-Riedel W, Muscarella LA, Fazio VM, Groen H, Timens W, Sietsma H, Thunnissen E, Smit E, Heideman DA, Snijders PJ, Cappuzzo F, Ligorio C, Damiani S, Field J, Solberg S, Brustugun OT, Lund-Iversen M, Sanger J, Clement JH, Soltermann A, Moch H, Weder W, Solomon B, Soria JC, Validire P, Besse B, Brambilla E, Brambilla C, Lantuejoul S, Lorimier P, Schneider PM, Hallek M, Pao W, Meyerson M, Sage J, Shendure J, Schneider R, Buttner R, Wolf J, Nurnberg P, Perner S, Heukamp LC, Brindle PK, Haas S, Thomas RK. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat Genet. 2012;44:1104–1110. doi: 10.1038/ng.2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plante I, Stewart MK, Barr K, Allan AL, Laird DW. Cx43 suppresses mammary tumor metastasis to the lung in a Cx43 mutant mouse model of human disease. Oncogene. 2011;30:1681–1692. doi: 10.1038/onc.2010.551. [DOI] [PubMed] [Google Scholar]
- Pleasance ED, Stephens PJ, O’Meara S, McBride DJ, Meynert A, Jones D, Lin ML, Beare D, Lau KW, Greenman C, Varela I, Nik-Zainal S, Davies HR, Ordonez GR, Mudie LJ, Latimer C, Edkins S, Stebbings L, Chen L, Jia M, Leroy C, Marshall J, Menzies A, Butler A, Teague JW, Mangion J, Sun YA, McLaughlin SF, Peckham HE, Tsung EF, Costa GL, Lee CC, Minna JD, Gazdar A, Birney E, Rhodes MD, McKernan KJ, Stratton MR, Futreal PA, Campbell PJ. A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature. 2010;463:184–190. doi: 10.1038/nature08629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poitras JL, Dal Cin P, Aster JC, Deangelo DJ, Morton CC. Novel SSBP2-JAK2 fusion gene resulting from a t(5;9)(q14.1;p24.1) in pre-B acute lymphocytic leukemia. Genes Chromosomes Cancer. 2008;47:884–889. doi: 10.1002/gcc.20585. [DOI] [PubMed] [Google Scholar]
- Rudin CM, Durinck S, Stawiski EW, Poirier JT, Modrusan Z, Shames DS, Bergbower EA, Guan Y, Shin J, Guillory J, Rivers CS, Foo CK, Bhatt D, Stinson J, Gnad F, Haverty PM, Gentleman R, Chaudhuri S, Janakiraman V, Jaiswal BS, Parikh C, Yuan W, Zhang Z, Koeppen H, Wu TD, Stern HM, Yauch RL, Huffman KE, Paskulin DD, Illei PB, Varella-Garcia M, Gazdar AF, de Sauvage FJ, Bourgon R, Minna JD, Brock MV, Seshagiri S. Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nat Genet. 2012;44:1111–1116. doi: 10.1038/ng.2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiffman JD, Hodgson JG, VandenBerg SR, Flaherty P, Polley MY, Yu M, Fisher PG, Rowitch DH, Ford JM, Berger MS, Ji H, Gutmann DH, James CD. Oncogenic BRAF mutation with CDKN2A inactivation is characteristic of a subset of pediatric malignant astrocytomas. Cancer Res. 2010;70:512–519. doi: 10.1158/0008-5472.CAN-09-1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sircoulomb F, Bekhouche I, Finetti P, Adelaide J, Ben Hamida A, Bonansea J, Raynaud S, Innocenti C, Charafe-Jauffret E, Tarpin C, Ben Ayed F, Viens P, Jacquemier J, Bertucci F, Birnbaum D, Chaffanet M. Genome profiling of ERBB2-amplified breast cancers. BMC Cancer. 2010;10:539. doi: 10.1186/1471-2407-10-539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sos ML, Dietlein F, Peifer M, Schottle J, Balke-Want H, Muller C, Koker M, Richters A, Heynck S, Malchers F, Heuckmann JM, Seidel D, Eyers PA, Ullrich RT, Antonchick AP, Vintonyak VV, Schneider PM, Ninomiya T, Waldmann H, Buttner R, Rauh D, Heukamp LC, Thomas RK. A framework for identification of actionable cancer genome dependencies in small cell lung cancer. Proc Natl Acad Sci U S A. 2012;109:17034–17039. doi: 10.1073/pnas.1207310109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soucek L, Whitfield J, Martins CP, Finch AJ, Murphy DJ, Sodir NM, Karnezis AN, Swigart LB, Nasi S, Evan GI. Modelling Myc inhibition as a cancer therapy. Nature. 2008;455:679–683. doi: 10.1038/nature07260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, Pleasance ED, Lau KW, Beare D, Stebbings LA, McLaren S, Lin ML, McBride DJ, Varela I, Nik-Zainal S, Leroy C, Jia M, Menzies A, Butler AP, Teague JW, Quail MA, Burton J, Swerdlow H, Carter NP, Morsberger LA, Iacobuzio-Donahue C, Follows GA, Green AR, Flanagan AM, Stratton MR, Futreal PA, Campbell PJ. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. 2011;144:27–40. doi: 10.1016/j.cell.2010.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Roosbroeck K, Cox L, Tousseyn T, Lahortiga I, Gielen O, Cauwelier B, De Paepe P, Verhoef G, Marynen P, Vandenberghe P, De Wolf-Peeters C, Cools J, Wlodarska I. JAK2 rearrangements, including the novel SEC31A-JAK2 fusion, are recurrent in classical Hodgkin lymphoma. Blood. 2011;117:4056–4064. doi: 10.1182/blood-2010-06-291310. [DOI] [PubMed] [Google Scholar]
- Voortman J, Lee JH, Killian JK, Suuriniemi M, Wang Y, Lucchi M, Smith WI, Jr, Meltzer P, Giaccone G. Array comparative genomic hybridization-based characterization of genetic alterations in pulmonary neuroendocrine tumors. Proc Natl Acad Sci U S A. 2010;107:13040–13045. doi: 10.1073/pnas.1008132107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wistuba II, Gazdar AF, Minna JD. Molecular genetics of small cell lung carcinoma. Semin Oncol. 2001;28:3–13. [PubMed] [Google Scholar]
- Worden FP, Kalemkerian GP. Therapeutic advances in small cell lung cancer. Expert Opin Investig Drugs. 2000;9:565–579. doi: 10.1517/13543784.9.3.565. [DOI] [PubMed] [Google Scholar]
- Wu J, Liu S, Liu G, Dombkowski A, Abrams J, Martin-Trevino R, Wicha MS, Ethier SP, Yang ZQ. Identification and functional analysis of 9p24 amplified genes in human breast cancer. Oncogene. 2012;31:333–341. doi: 10.1038/onc.2011.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto G, Nannya Y, Kato M, Sanada M, Levine RL, Kawamata N, Hangaishi A, Kurokawa M, Chiba S, Gilliland DG, Koeffler HP, Ogawa S. Highly sensitive method for genomewide detection of allelic composition in nonpaired, primary tumor specimens by use of affymetrix single-nucleotide-polymorphism genotyping microarrays. Am J Hum Genet. 2007;81:114–126. doi: 10.1086/518809. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.