

Abstract
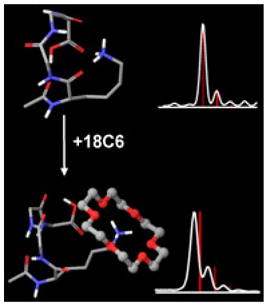
The ability of 18-crown-6 (18C6) to form noncovalent complexes with cationic groups in the gas phase has been leveraged in numerous, largely orthogonal mass spectrometry-based applications. Although the fundamental interaction between 18C6 and a charged group in the gas phase is quite strong, the strength of attachment of 18C6 to large molecules is more difficult to predict because intramolecular binding of the cation can be competitive. Herein, we demonstrate in experiments with model peptides that 18C6 adducts are not strongly attached to flexible molecules with numerous potential hydrogen bonding sites. 18C6 adduct stability is increased if intramolecular charge complexation is inhibited by sterics or competitive binding. It is demonstrated with molecular mechanics that significant structural changes occur upon loss of 18C6 in model peptides. Examination of the loss of 18C6 adducts from proteins following collisional activation reveals that lower charge states lose the most 18C6. The degree of 18C6 adduct stability may reflect the degree of structural reorganization that occurs following collisional activation, suggesting that lower charge states represent structures that are not similar to gas phase idealized states. In this regard, 18C6 may serve the function of protecting solution phase protein structure. Collisional activation of holomyoglobin with 18C6 adducts attached reveals that heme loss occurs primarily after 18C6 loss, further supporting the notion that 18C6 protects native structure by solvating charged sites.
Keywords: 18-Crown-6, Noncovalent, Crown ether, SNAPP
Introduction
Although the first examples were synthesized in the 1930s, crown ethers only came to the forefront following recognition of their cation binding properties by Pederson in the late 1960s [1]. Crown ethers are cyclic oligomers, most typically constituted of ethylene oxide unit repeats. The nomenclature of crown ethers refers to the total number of atoms comprising the ring followed by the number of heteroatoms that are present. For example, 18-crown-6 (18C6) is an 18 atom ring with six oxygen atoms. The cyclic arrangement of crown ethers creates electronegative cavities that are well-suited for binding to cations. Crown ethers are freely soluble in both polar and nonpolar solvents, making them well-suited for phase transfer catalysis [2]. Crown ethers have also been used for cation recognition [3], in separations [4], and for gas-phase experiments [5–7].
18C6 in particular has found use in mass spectrometry (MS)-based experiments because of its ability to bind protonated primary amines via the formation of three specific hydrogen bonds between every other oxygen atom [8]. Attachment of 18C6 to protonated groups has been used to investigate mechanistic aspects of hydrogen/deuterium exchange [9], examine protein structure [10], generate radical peptides [11], in conjunction with ion mobility [12], for charge stripping [13], and for spectroscopy [14]. These experiments are typically conducted with electrospray ionization, where 18C6 is added to the solution to be electrosprayed. Under sufficiently gentle ionization conditions, noncovalent 18C6 adducts are formed and can be observed in the final mass spectra. It is important to point out that with 18C6, observation of a noncovalent adduct in the gas phase does not necessarily imply that the complex was present in solution. For example, the dissociation constant for 18C6 with protonated primary amines in water is quite low (in the high millimolar range) [15], yet abundant adducts can be observed from aqueous solutions of peptides or proteins that contain 18C6. It has been proposed that complexation occurs during the electrospray process, where the effective concentration of 18C6 increases dramatically during droplet evaporation and drives complex formation [16].
Once in the gas phase, the fundamental interaction between 18C6 and several biologically relevant protonated groups is quite strong [17–19]. For example, the bond dissociation energy for protonated butyl amine (a lysine mimic) and 18C6 is 223 kJ/mol, which represents a significant fraction of the energy required to break a typical covalent bond [18]. However, the bond dissociation energies of small molecules cannot be straightforwardly used to estimate the binding of 18C6 to similar functional groups in more complex molecules such as peptides or proteins. For these larger molecules, intramolecular binding sites that can competitively interact with charged groups are frequently available and can significantly reduce the effective binding energy of the crown. Entropy also becomes potentially more important when intramolecular charge solvation is possible because the noncovalent bonding of two molecules is always entropically unfavorable. The stability of the 18C6 adducts in larger molecules therefore depends on competition between intramolecular and intermolecular solvation of charged sites, keeping in mind that optimal configurations for each may be separated by significant kinetic barriers.
In this manuscript, we examine the potential of 18C6 to act as “solvent” for charged residues in the gas phase. It is found that for systems with significant structural flexibility and available hydrogen binding sites, 18C6 adducts are kinetically trapped unstable complexes. Very mild excitation leads to prompt loss of the 18C6 adduct and rearrangement of peptide structure to accommodate the charged group. In more rigidly constrained systems where optimal charge solvation is either not feasible or protected by large kinetic barriers, 18C6 adduct stability is significantly enhanced. Molecular mechanics calculations on several small glycine oligomers are used to examine the structures of specific examples of unstable and stable 18C6 adducts. Collisional activation of multi-adduct proteins reveals that adduct stability increases significantly with increasing charge state, which may reflect the degree of structural reorganization that has taken place in transit into the gas phase.
Experimental
Materials and Peptide Synthesis
All the organic solvents were purchased from Sigma-Aldrich (St. Louis, MO, USA) and used without further purification. All amino acids and resin were purchased from Ana Spec (Fremont, CA, USA). Cytochrome c and myoglobin were purchased from Sigma-Aldrich. 18-Crown-6 was purchased from Alfa Aesar (Pelham, NH, USA). Water was purified to 18.2 MΩ using a Millipore 147 (Billerica, MA, USA) Direct-Q system.
The GG, KG, RG peptide series were synthesized manually using standard Fmoc procedures with Wang resin as the solid support [20]. Amino acids with protected side chains were used when needed. The N-terminus of KG and RG peptide series were acetylated by acetic acid before the cleavage from beads (indicated by Ac- prior to the peptide sequence).
Mass Spectrometry
Mass spectra were recorded with a Finnigan LTQ ion trap mass spectrometer (Thermo Fisher Scientific, San Jose, CA, USA) with a standard ESI source. The concentrations for all peptides and proteins were 10 μM. All samples were prepared in 50/50 water/methanol except for holo myoglobin; 18C6 at 5~10 times the peptide/protein concentration was added to the sample before electrospraying. The isolation window width was set to 10 Da for the peptidecrown adduct peak and 10 Da for the protein-adduct peak. This wide isolation window is needed to avoid collisional heating of the ions during isolation. The width is also 10 Da for ultrazoom scan mode (slower scan speed). The activation voltage (V) was converted from normalized collisional energy (NCE) using the Equation (1):
| (1) |
where tick amp slope and tick amp intercept are instrument-specific parameters, equal to 0.000026 and 0.010236. The degrees of freedom (DOF) for an N-atom molecule can be obtained with: DOF=3 N-6.
Molecular Mechanics Calculations
Maestro and MacroModel (Schrodinger, Inc., San Diego, CA, USA) were used to build models and perform conformational searches. For all the calculations, the OPLS atomic force field was used, with no solvent. The initial peptide structures were alpha helical with the positive charge on the N-terminus for GGGG and the lysine side chain for Ac-KGGG. For the GGGG complex with 18C6, the 18C6 molecule was manually placed next to the N-terminus. For Ac-KGGG-18C6 complex, the lysine side chain was rotated away from the peptide backbone into a position where 18C6 could interact. A torsional sampling conformational search algorithm (MCMM) was used to search conformational space and identify low energy structures. Fifty thousand structures were sampled in each conformational search and 1000 low energy structures were saved. The lowest energy structures obtained in each case were used to calculate binding energies (ΔH between isolated and complexed, minimized structures).
Results and Discussion
Our group has conducted many experiments utilizing attachment of 18C6 or 18C6-based molecules to generate noncovalent complexes in the gas phase for subsequent examination by MS. The vast majority of these experiments have been conducted in ion trap instruments where we have noticed on some occasions that the masses of the complexes deviate significantly (>0.2 Da at normal scan speed) from the expected masses. In all cases, collisional activation of the peak results in loss of a molecule corresponding nominally to the mass of 18C6 and generation of the peptide with the correct mass, which suggests that the complexes are correctly assigned but appear at the wrong m/z for some reason. It has been well-documented that fragile ions can fragment during resonant excitation in ion traps, leading to peak broadening and mass shifts [21–23]. The LTQ is a forward scanning instrument, which means that it ejects ions of lower mass first. Fragile ions ejected prematurely during this process will, therefore, broaden and mass shift towards the direction of lower m/z.
Figure 1a illustrates isolation windows for a series of Ac-KGx (x=0–5) peptides that have been complexed with 18C6. The predicted masses and relative intensities for isotopic peaks are shown for each complex as red lines. The agreement between the expected and observed mass is very good for the 18C6 complex with Ac-K; however, there is a clear trend of increasingly large mass shifts towards lower m/z that is accompanied by peak broadening as the number of glycine residues increases. These results are consistent with decreasing complex stability as more glycine residues are added to the peptide. The polyglycine part of the backbone is flexible and contains numerous hydrogen bonding sites that can easily interact with the flexible lysine side chain. The results in Figure 1a strongly suggest that the 18C6 adducts represent kinetically trapped structures that rapidly dissociate upon very minimal activation, leading to the observed mass shifts and broadening. Evaluation of identical samples with time-of-flight MS, which does not activate ions during detection (see Supporting Information), yields masses and isotope patterns that agree well with predicted values. In Figure 1b, the precursor ion survival yields are shown as a function of activation voltage normalized by the number of vibrational degrees of freedom [24, 19]. The trends in peptide complex stability are nearly identical to those observed by mass shifting in Figure 1a (a quantitative comparison is shown in the Supporting Information).
Figure 1.
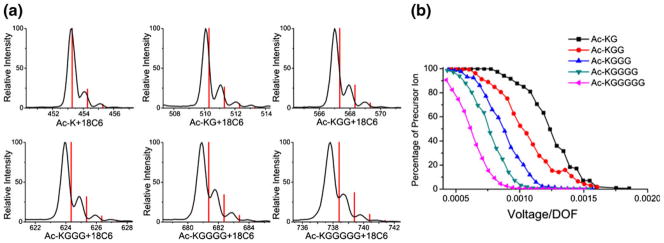
(a) Isolation windows for a series of Ac-KGx (x=0 to 5) peptides that have been complexed with 18C6. The predicted masses and isotope distributions are shown as red lines. The mass shifts increase as more glycine residues are added to the peptide. (b) Precursor ion survival as a function of excitation voltage/degrees of freedom. Again, additional glycine residues lead to decreased stability
A similar set of experiments were conducted with Ac-RGx (x=0–5) and Gx (x=2–5) peptides. The results are summarized for two different scan speeds in Figure 2, which also contains the results from the Ac-KGx (x=0–5) peptides for comparison. The lower scan speed will lead to excitation of the ions closer to the true m/z, and is observed to reduce the mass shifting significantly though the same trends are observed in both Figure 2a and b. Arginine is the most basic residue and is therefore also a potential target for complexation by 18C6 at the protonated guanidinyl side chain. Previous results have suggested that arginine complexation with 18C6 is less favorable than with lysine [8, 17]. The results in Figure 2 are consistent with this finding. A significant mass deviation is observed even for the complex with acetylated arginine itself. Overall, the trend is similar to that observed for the Ac-KGx peptides, although the degree of mass shifting and peak broadening is greater for arginine than it is for lysine (see Supporting Information). Given that the inherent binding energy of arginine to 18C6 is weaker than it is for lysine, it is logical that competitive intramolecular binding is able to out-compete retention of the 18C6 adduct more easily.
Figure 2.
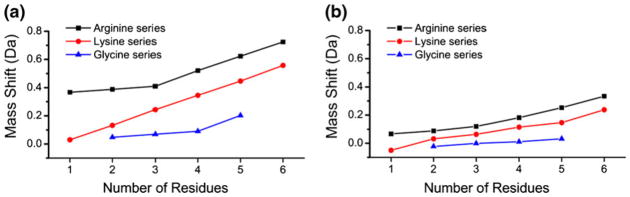
(a) Mass shifts for the primary isotopic peaks obtained from isolation windows of 18C6 adducts with Ac-RGx, Ac-KGx, and Gx peptides. Reduced intramolecular charge solvation in polyglycine leads to greater 18C6 adduct stability and smaller mass shifts. (b) The magnitude of the mass shifting is reduced if the scan speed of the instrument is decreased
For polyglycine, 18C6 attachment most likely occurs at the protonated N-terminus. Interestingly, very minimal mass shifts (see Figure 2) or peak broadening (see Supporting Information) are observed for most of the polyglycine peptides. Even for GGGGG, only minimal mass shifting is noticed. The fundamental binding energy for 18C6 to the N-terminus (223 kJ/mol) [17] is comparable to that for the side chain of lysine (based on n-butyl amine, 223 kJ/mol) [18], meaning that there must be another explanation for the enhanced stability. Although the polyglycine backbone is fairly flexible, it is not sufficiently long to effectively solvate a charge at the protonated N-terminus via multidentate interactions as are present with 18C6. It appears that these steric constraints inhibit competitive complexation of the charge by the peptide backbone, which leads to enhanced retention of the 18C6 adduct. It has been demonstrated previously that changes in the polarizability of small molecules may influence crown binding strengths [17, 18], though we anticipate such effects to be secondary to hydrogen bonding capacity for these larger systems. At the slower scan speed in Figure 2b, the mass shifts are negligible for the polyglycines.
We employed molecular dynamics calculations to explore the structures and energetics of 18C6 complexes with Ac-KGGG and GGGG. The lowest energy structures that we were able to obtain for each peptide alone and complexed with 18C6 are shown in Figure 3. When attached to 18C6, the optimal structures have the protonated amines in both peptides forming three hydrogen bonds with 18C6, indicating a strong interaction between the charged site and the crown. However, solvation of the charged group in the absence of 18C6 is much less comparable between the two peptides. For Ac-KGGG, the peptide backbone and flexible side chain are able to rearrange in such a way that three relatively strong hydrogen bonds again stabilize the charged sited. For GGGG, only a single strong hydrogen bond is formed with an additional hydrogen bond that is weakened by larger separation and an unfavorable OHN interaction angle. The calculated binding energies for the two complexes are −176 kJ/mol for Ac-KGGG and −221 kJ/mol for GGGG. It is unlikely that molecular dynamics can be relied upon to accurately quantify these binding energies; however, the magnitude of the difference suggests that the trend would likely hold up even with higher level calculations. The trend in binding energy obtained by the calculations is also in agreement with experiment.
Figure 3.
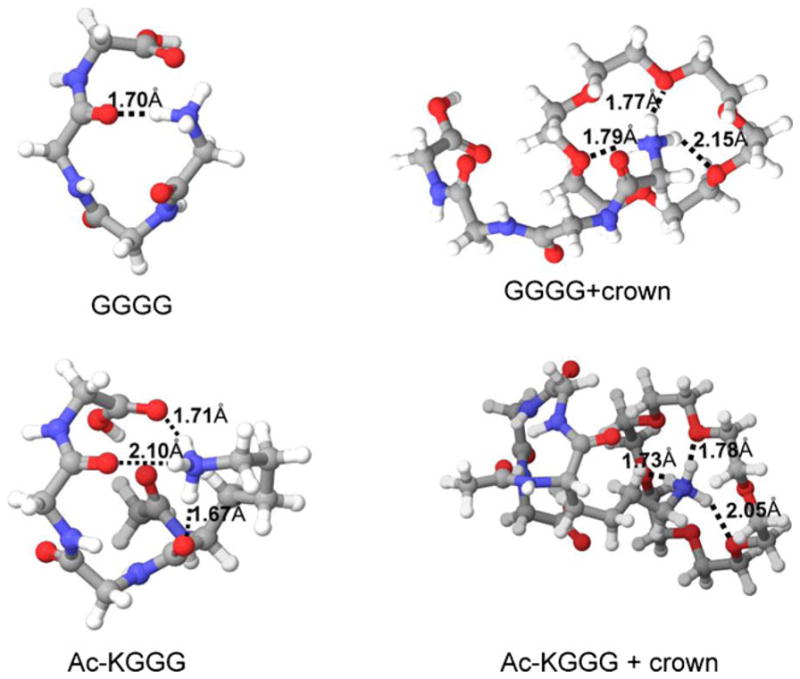
Lowest energy structures for GGGG, GGGG-18C6, Ac–KGGG, and Ac-KGGG–18C6 from molecular dynamics conformational searches. The hydrogen bond distances between H and O atoms are shown for each structure. The peptide structures change significantly when 18C6 is attached. The interactions between peptide backbone and side chain are stronger for Ac-KGGG than for GGGG
The results in Figures 1, 2, and 3 suggest that 18C6 adduct stability is a sensitive probe of local conformational flexibility and hydrogen bonding capacity. We next examined 18C6 adduct stability in proteins, which frequently attach multiple 18C6 adducts. In Figure 4, spectra obtained by collisional activation of four-adduct peaks at various excitation energies are shown for the +9 and +14 charge states of cytochrome c. For the +9 charge state, a single step of activation is sufficient to produce the bare protein. In fact, once sufficient energy is used to completely deplete the precursor ion, the bare protein is by far the most dominant product. This is a very interesting result that requires careful consideration. In an ion trap, activation is achieved by multiple, low-energy collisions. As a result, the lowest energy dissociation pathways are typically observed. Furthermore, excitation is resonant, meaning that once a single 18C6 adduct is lost, no further energy will be pumped into the ion. Generally, this will lead to loss of a single 18C6 adduct from a multiple adduct complex following collisional activation because this represents the lowest energy dissociation pathway. For example, excitation of [KKKKK+ 4(18C6)+4H]4+ yields dominant loss of a single 18C6 (see Supporting Information). Therefore, in order for all four 18C6 adducts to be lost simultaneously, the complex must acquire sufficient energy to lose all four adducts prior to the loss of a single 18C6, or the acquisition of energy must somehow result in significantly lower binding strength of the adducts to the protein. The second scenario is plausible if activation leads to significant structural perturbation of the protein, leading to weakening of 18C6 binding for all four adducts. In this case, the rearrangement would presumably be from a kinetically trapped solution phase-like structure to the preferred gas phase structure. Alternatively, proteins are very large molecules and can potentially store sufficient energy to eventually lose four adducts prior to the loss of the first adduct. Given the rapid cooling that occurs in ion traps (as illustrated by difficulty in carrying out ion activation by infrared radiation) [25, 26], we find this possibility to be less likely.
Figure 4.
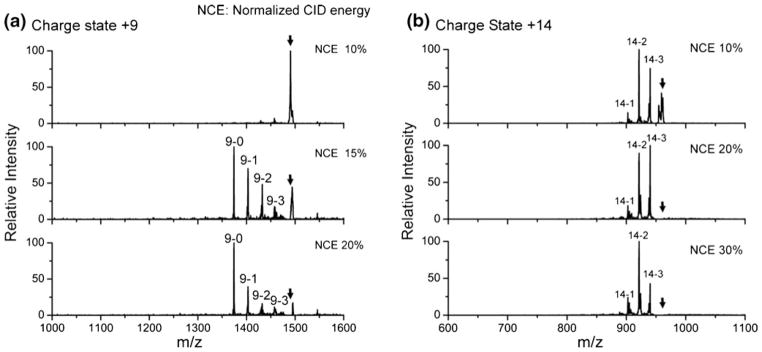
CID spectra of cytochrome c with four-adduct peaks at various excitation energies. (a) +9 charge state; (b) +14 charge state. The black arrows represent precursor ions. Peaks are labeled by “charge state-number of crown adducts;” 15 % NCE is sufficient to produce the bare protein for +9 charge state whereas the activation of +14 charge state mainly lead to the loss of two crowns even at higher activation energies
In contrast, for the +14 charge state, activation leads primarily to the loss of one or two 18C6 adducts regardless of activation energy. In this case, it is difficult to distinguish between a situation where modest structural rearrangement could occur and facilitate loss of two 18C6 adducts or the case where a large protein might store sufficient energy to accommodate loss of two crowns. Nevertheless, activation of the three adduct or two adduct peaks (following loss of one or two crowns) does not lead to preferential formation of the naked protein (see Supporting Information), suggesting that significant structural rearrangement is unlikely and that a different mechanism is responsible for 18C6 adduct loss in the higher charge state. Since higher charge states consist of elongated structures [27] that likely bear little resemblance to solution phase structures, it is less likely that these structures would be kinetically trapped and prone to undergo significant structural reorganization following collisional activation. This may account for the observation that 18C6 adducts are more stable on higher charge state proteins. Furthermore, as additional charges are added to a protein, the capacity of the protein for self-solvation will decrease, which should also lead to increased 18C6 adduct stability.
An interesting possibility arises from consideration of these results, namely, the degree of loss of 18C6 adducts may reflect the degree to which a protein has undergone structural reorganization to accommodate the gas phase environment. Greater loss of 18C6 would then presumably indicate that less reorganization occurred during transit into the gas phase and, therefore, greater similarity with solution phase structure (at least prior to loss of the crowns). One possible benefit that could occur from addition of 18C6 is that it may serve as solvent replacement during the transition of the protein from solution into the gas phase, allowing the protein to retain a greater degree of structural resemblance to the solution phase structure. If so, 18C6 adduct formation may represent a method for essentially preserving solution phase structures via pseudo-solvation. Indeed, recent work examining collision cross sections from ion mobility experiments concluded that 18C6 can micro-solvate charge sites and help to preserve solution phase structure for certain charge states [28].
The possibility of native structure retention is examined further in Figure 5, where the results from collisional activation of holomyoglobin in the +10 and +11 charge states with four 18C6 adducts are shown. Holomyoglobin has a noncovalently attached heme group that is known to be labile and easily lost in MS experiments [29]. Interestingly, the results in Figure 5 show that heme loss generally occurs only after 18C6 is lost. For example in Figure 5a, the 10–0 (where 10 indicates charge state and 0 indicates the number of 18C6 adducts) apo peak is not accompanied by any 10–1 or higher order 18C6 adduct peaks, indicating that it originated only from the 10–0 holo peak. Similarly, in Figure 5b, the 10–0 apo peak is not accompanied by any 18C6 adducts. The 11–0 apo peak does have an accompanying 11–1 adduct peak, but the intensity of this peak is significantly lower than the 11–1 holo peak. All of these results are consistent with preferential loss of 18C6 over loss of heme (the key indicator for loss of native-like structure). This is further support that 18C6 can behave in a protective fashion, solvating charged side chains and preserving native structures. For both the +10 and +11 data, loss of all four adducts is observed in a single activation step, which is also consistent with our analysis of the data in Figure 4 and with significant structural reorganization and possible retention of solution phase structure prior to activation.
Figure 5.
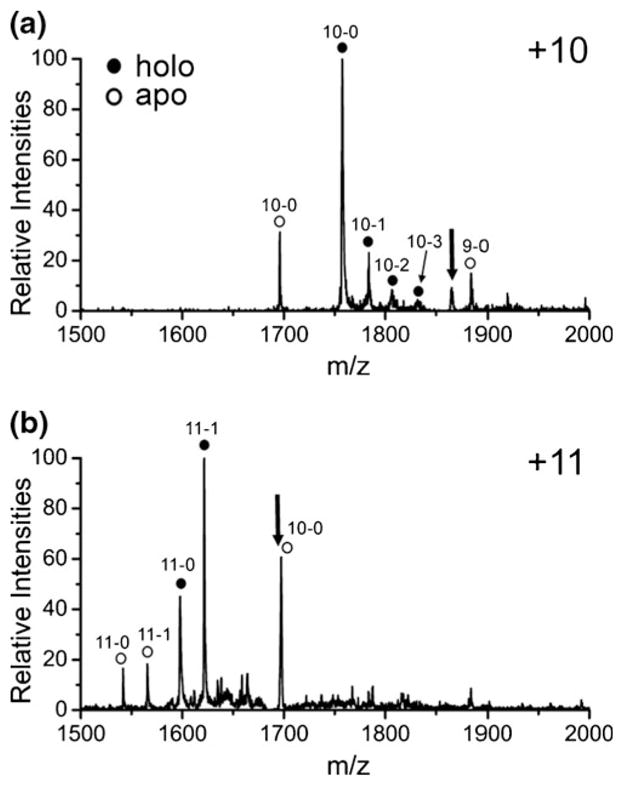
CID of holomyoglobin with four crown adducts for the (a) +10 and (b) +11 charge states. Heme loss occurs primarily after 18C6 is lost, suggesting a protective function. Numbers refer to charge state and number of 18C6 adducts (i.e., 11–1 is the +11 charge state with a single 18C6 adduct. The black arrows represent the precursor ions
Conclusions
Although the fundamental interaction between 18C6 and protonated amines in the gas phase is quite strong, it is clear that in complex molecules this interaction can be significantly weakened by competitive intramolecular charge solvation. The structures of small model peptides which have been examined in detail undergo significant rearrangement following loss of 18C6 in the gas phase. This observation is logical, given that hydrogen bonds with charged groups are among the strongest noncovalent forces present in the gas phase. Examination of proteins reveals that 18C6 adducts on lower charge states are weakly bound and easily lost upon collisional activation. In contrast, higher charge states exhibit greater 18C6 adduct retention. These results are consistent with the idea that proteins in lower charge states have greater resemblance to solution phase structures and therefore undergo more structural rearrangement that facilitates loss of 18C6. If this is the case, 18C6 may serve the function of solvating side chains and protecting solution phase structure. Experiments with holomyoglobin support this idea because the labile heme group is observed to be lost primarily after 18C6 adducts. These results suggest that the potential of 18C6 as a solvent replacement should be investigated further.
Supplementary Material
Acknowledgments
The authors thank NIH for funding (R01 GM084106) and John Syka and Jae Schwartz for helpful discussions.
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s13361-013-0684-z) contains supplementary material, which is available to authorized users.
References
- 1.Gokel GW, Leevy WM, Weber ME. Crown ethers: sensors for ions and molecular scaffolds for materials and biological models. Chem Rev. 2004;104:2723–2750. doi: 10.1021/cr020080k. [DOI] [PubMed] [Google Scholar]
- 2.Landini D, Montanar F, Pirisi FM. Crown ethers as phase-transfer catalysts in 2-phase reactions. J Chem Soc Chem Commun. 1974;21:879–880. [Google Scholar]
- 3.De Silva AP, Desilva SA. Fluorescent signaling crown ethers-switching on of fluorescence by alkali-metal ion recognition and binding in situ. J Chem Soc Chem Commun. 1986;23:1709–1710. [Google Scholar]
- 4.Kuhn R, Stoecklin F, Erni F. Chiral separations by host–guest complexation with cyclodextrin and crown-ether in capillary zone electrophoresis. Chromatographia. 1992;33:32–36. [Google Scholar]
- 5.Maleknia S, Brodbelt J. Gas-phase selectivities of crown ethers for alkali–metal ion complexation. J Am Chem Soc. 1992;114:4295–4298. [Google Scholar]
- 6.Chu IH, Zhang H, Dearden DV. Macrocyclic chemistry in the gas-phase—intrinsic cation affinities and complexation rates for alkalimetal cation complexes of crown-ethers and glymes. J Am Chem oc. 1993;115:5736–5744. [Google Scholar]
- 7.More MB, Ray D, Armentrout PB. Intrinsic affinities of alkali cations for 15-crown-5 and 18-crown-6: bond dissociation energies of gas-phase M+–crown ether complexes. J Am Chem Soc. 1999;121:417–423. [Google Scholar]
- 8.Julian RR, Beauchamp JL. Site specific sequestering and stabilization of charge in peptides by supramolecular adduct formation with 18-crown-6 ether by way of electrospray ionization. Int J Mass Spectrom. 2001;210:613–623. [Google Scholar]
- 9.Lee SW, Lee HN, Kim HS, Beauchamp JL. Selective binding of crown ethers to protonated peptides can be used to probe mechanisms of H/D exchange and collision-induced dissociation reactions in the gas phase. J Am Chem Soc. 1998;120:5800–5805. [Google Scholar]
- 10.Ly T, Julian RR. Using ESI-MS to probe protein structure by site-specific noncovalent attachment of 18-crown-6. J Am Soc Mass Spectrom. 2006;17:1209–1215. doi: 10.1016/j.jasms.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 11.Sun QY, Nelson H, Ly T, Stoltz BM, Julian RR. Side chain chemistry mediates backbone fragmentation in hydrogen deficient peptide radicals. J Proteome Res. 2009;8:958–966. doi: 10.1021/pr800592t. [DOI] [PubMed] [Google Scholar]
- 12.Bohrer BC, Clemmer DE. Shift reagents for multidimensional ion mobility spectrometry-mass spectrometry analysis of complex peptide mixtures: evaluation of 18-Crown-6 ether complexes. Anal Chem. 2011;83:5377–5385. doi: 10.1021/ac200892r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pagel K, Hyung SJ, Ruotolo BT, Robinson CV. Alternate dissociation pathways identified in charge-reduced protein complex ions. Anal Chem. 2010;82(12):5363–5372. doi: 10.1021/ac101121r. [DOI] [PubMed] [Google Scholar]
- 14.Kupser P, Pagel K, Oomens J, Polfer N, Koksch B, Meijer G, von Helden G. Amide-I and -II vibrations of the cyclic beta-sheet model peptide gramicidin S in the gas phase. J Am Chem Soc. 2010;132:2085–2093. doi: 10.1021/ja909842j. [DOI] [PubMed] [Google Scholar]
- 15.Buschmann HJ, Schollmeyer E, Mutihac L. The complexation of the ammonium ion by 18-crown-6 in different solvents and by noncyclic ligands, crown ethers, and cryptands in methanol. Supramol Sci. 1998;5:139–142. [Google Scholar]
- 16.Hamdy OM, Julian RR. Reflections on charge state distributions, protein structure, and the mystical mechanism of electrospray ionization. J Am Soc Mass Spectrom. 2012;23:1–6. doi: 10.1007/s13361-011-0284-8. [DOI] [PubMed] [Google Scholar]
- 17.Chen Y, Rodgers MT. Structural and energetic effects in the molecular recognition of amino acids by 18-crown-6. J Am Chem Soc. 2012;134:5863–5875. doi: 10.1021/ja211021h. [DOI] [PubMed] [Google Scholar]
- 18.Chen Y, Rodgers MT. Structural and energetic effects in the molecular recognition of protonated peptidomimetic bases by 18-crown-6. J Am Chem Soc. 2012;134:2313–2324. doi: 10.1021/ja2102345. [DOI] [PubMed] [Google Scholar]
- 19.David WM, Brodbelt JS. Threshold dissociation energies of protonated amine/polyether complexes in a quadrupole ion trap. J Am Soc Mass Spectrom. 2003;14(4):383–392. doi: 10.1016/S1044-0305(03)00070-9. [DOI] [PubMed] [Google Scholar]
- 20.Chan WC, White PD. Fmoc Solid Phase Peptide Synthesis. Oxford University Press; New York: 2000. pp. 9–74. [Google Scholar]
- 21.McClellan JE, Murphy JP, Mulholland JJ, Yost RA. Effects of fragile ions on mass resolution and on isolation for tandem mass spectrometry in the quadrupole ion trap mass spectrometer. Anal Chem. 2002;74:402–412. doi: 10.1021/ac015610b. [DOI] [PubMed] [Google Scholar]
- 22.Murphy JP, Yost RA. Origin of mass shifts in the quadrupole ion trap: dissociation of fragile ions observed with a hybrid ion trap/mass filter instrument. Rapid Commun Mass Spectrom. 2000;14:270–273. doi: 10.1002/(SICI)1097-0231(20000229)14:4<270::AID-RCM875>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 23.Cowan DA, Kiman AT, Kubli-Garfias C, Welchman HJ. Ion trap MS/MS of intact testosterone and epitestosterone conjugates—adducts, fragile ions, and the advantages of derivatization. Steroids. 2008;73:621–628. doi: 10.1016/j.steroids.2008.01.026. [DOI] [PubMed] [Google Scholar]
- 24.Huffman CL, Williams ML, Benoist DM, Overstreet RE, Jellen-McCullough EE. Dependence of collision-induced dissociation energy on molecular degrees of freedom as a means to assess relative binding affinity in multivalent complexes. Rapid Commun Mass Spectrom. 2011;25:2299–2306. doi: 10.1002/rcm.5124. [DOI] [PubMed] [Google Scholar]
- 25.Brodbelt JS. Shedding light on the Frontier of photodissociation. J Am Soc Mass Spectrom. 2011;22:197–206. doi: 10.1007/s13361-010-0023-6. [DOI] [PubMed] [Google Scholar]
- 26.Newsome GA, Glish GL. Improving IRMPD in a quadrupole ion trap. J Am Soc Mass Spectrom. 2009;20:1127–1131. doi: 10.1016/j.jasms.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 27.Uetrecht C, Rose RJ, van Duijn E, Lorenzen K, Heck AJR. Ion mobility mass spectrometry of proteins and protein assemblies. Chem Soc Rev. 2009;39:1633–1655. doi: 10.1039/b914002f. [DOI] [PubMed] [Google Scholar]
- 28.Warnke S, von Helden G, Pagel K. Protein structure in the gas phase: the influence of side-chain microsolvation. J Am Chem Soc. 2013;135:1177–1180. doi: 10.1021/ja308528d. [DOI] [PubMed] [Google Scholar]
- 29.Babu KR, Douglas DJ. Methanol-induced conformations of myoglobin at pH 4.0. Biochemistry. 2000;39:14702–14710. doi: 10.1021/bi001265t. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.