

Summary
Aims
Neurologic impairment following ischemic injury complicates the quality of life for stroke survivors. Xenon (Xe) has favorable neuroprotective properties to modify stroke. Xe delivery is hampered by a lack of suitable administration strategies. We have developed Xe‐containing echogenic liposomes (Xe‐ELIP) for systemic Xe delivery. We investigated the time window for Xe‐ELIP therapeutic effect and the most efficacious dose for neuroprotection. Molecular mechanisms for Xe neuroprotection were investigated.
Methods
Xenon‐containing echogenic liposomes were created by a previously developed pressurization‐freezing method. Following right middle cerebral artery occlusion (2 h), animals were treated with Xe‐ELIP at 2, 3, or 5 h to determine time window of therapeutic effect. The neuroprotectant dosage for optimal effect was evaluated 3 h after stroke onset. Expression of brain‐derived neurotrophic factor (BDNF), protein kinase B (Akt), and mitogen‐activated protein kinases (MAPK) was determined.
Results
Xenon‐containing echogenic liposomes administration for up to 5 h after stroke onset reduced infract size. Treatment groups given 7 and 14 mg/kg of Xe‐ELIP reduced infarct size. Behavioral outcomes corresponded to changes in infarct volume. Xe‐ELIP treatment reduced ischemic neuronal cell death via activation of both MAPK and Akt. Elevated BDNF expression was shown following Xe‐ELIP delivery.
Conclusion
This study demonstrates the therapeutic efficacy of Xe‐ELIP administered within 5 h after stroke onset with an optimal dosage range of 7–14 mg/kg for maximal neuroprotection.
Keywords: Cerebral ischemia, Liposomes, Neuroprotection, Stroke, Xenon
Introduction
Stroke is the fourth leading cause of death and the most common cause of adult disabilities in the United States 1. Ischemic stroke occurs when a cerebral artery is occluded obstructing blood flow to a portion of the brain 2. The cerebral ischemic area is composed of a zone of irreversibly damaged tissue known as the core, and a surrounding region of less severe ischemic and reversible tissue known as the penumbra 3. The penumbra can turn into irreversible ischemic tissue through a complex cascade of cellular events. The primary focus of neuroprotective therapies is to prevent irreversible neuronal damage in the ischemic penumbra.
Although multiple factors are involved when trying to provide neuroprotection, one of the fundamental factors is the excitotoxic effect caused by excessive extracellular glutamate accumulation. Strategies to block glutamate receptors have become an important strategy in neuroprotection. A number of N‐Methyl‐D‐aspartic acid (NMDA)‐receptor blockers have been reported for effective neuroprotection in animal studies, but have failed in clinical trials. Primary reasons are low beneficial effects, severe adverse hemodynamic effects 4, or importantly too late therapeutic delivery following stroke onset to result in a benefit 5. Thus, the greatest therapeutic potential lays upon agents that have low side effects, directly target brain tissue, and that can be given to patients early after stroke onset.
The use of medical gases as therapeutics holds great promise for tissue protection 6, 7, 8. Unlike other therapeutics, medical gases are small molecules that easily diffuse into target tissue, easily cross the blood brain barrier (BBB), and easily get them to all parts of the brain. Xe, a pleiotypic cytoprotective gas, has unique advantages including little clinically adverse effects 9, 10. Xe rapidly diffuses across the blood–brain barrier and across cell membranes due to its low blood‐gas partition coefficient 11, 12. Although medical gases are usually delivered by inhalation, administration of Xe by inhalation is rather expensive and complicated requiring intubation and ventilation with a large Xe concentration. Administration of Xe by inhalation reduces the maximum fraction of inspired oxygen 4 and can only be given in a hospital setting, for fear of hypoxic tissue damage.
Building on our previous work encapsulating gases into liposomes 13, 14, 15, 16, 17, 18, we have developed a therapeutic delivery system using ultrasound‐controlled Xe release for effective Xe delivery into the brain for stroke treatment 19. In this study, we determined the time window of injection for therapeutic efficacy and the most efficacious neuroprotective dose of Xe‐containing echogenic liposomes (Xe‐ELIP) in an in vivo model of focal brain ischemia. Molecular signal transduction responsible for Xe protection was investigated. These two parameters, effective time of neuroprotection and optimal neuroprotective dose range, provide the groundwork for developing clinical trials in patients with acute stroke.
Methods
Preparation of Xe‐ELIP
Liposomes were composed of 1,2‐dipalmitoyl‐sn‐glycero‐3‐phosphocholine (DPPC; Avanti Polar Lipids, Alabaster, Ala), Egg phosphocholine (Egg‐PC; Avanti Polar Lipids), 1,2‐dipalmitoyl‐sn‐glycero‐3‐phosphoethanolamine‐N‐[methoxy(polyethylene glycol)‐2000] (PEG2000 PE) 1,2‐dipalmitoyl‐sn‐glycero‐3‐phospho‐(1′‐rac‐glycerol; DPPG), and cholesterol (Sigma, St Louis, MO, USA) at a molar ratio of 43:28:6:8:15. Xe‐ELIP were prepared by the previously described pressurization‐freeze method 19.
Animal Model with Middle Cerebral Artery Occlusion (MCAO)
All animal studies were approved by the Animal Welfare Committee at The University of Texas Health Science Center at Houston. Randomly selected male Sprague‐Dawley rats (260–280 g; Harlan Laboratories Inc., Indianapolis, IN, USA) were used. Cerebral ischemia was induced by occluding the right middle cerebral artery (MCA) for 2 h using the intraluminal suture method 19. In brief, the right common carotid artery (CCA) was exposed under an operating microscope. The external carotid artery was ligated close to its distal end. The internal carotid artery (ICA) was isolated and separated from adjacent tissues. A 4‐0 monofilament nylon suture (Ethicon, Somerville, NJ, USA) coated with poly L‐lysine (0.1% [wt/vol]) and heparin (1000 U/mL) was inserted into the MCA lumen located 18–20 mm from the external carotid artery/common carotid artery bifurcation for 2 h to provoke ischemia. As soon as the suture was removed, the desired treatment was administered through a catheter into the ICA in the antegrade direction. In all experiments, body temperature was monitored and maintained at 37°C during ischemia and over the first hour of reperfusion with the use of a feed‐forward temperature controller equipped with a heating lamp and heating pad (Harvard Apparatus; Holliston, MA, USA). Cerebral blood flow was monitored with the use of a PR407–1 straight‐needle laser Doppler flowmeter probe (Perimed, Jäfälla, Stockholm, Sweden) connected to a standard laser Doppler monitor (PF5010 LDPM unit and PF5001 main unit; Perimed, Järfälla). Interruption of blood flow was recorded in the region of ischemic penumbra (2 mm lateral and 2 mm posterior to the bregma).
Determination of the Therapeutic Time Window and Dose Dependence and Effect of Insonation
According to a clinical report, 28.9% of patients arrived at the hospital within 3 h after stroke onset, with a mean hospital arrival time of 5.5 h 20. For the therapeutic time window study, animals were randomly divided into four groups (n = 8 in each group): stroke with no treatment—MCAO only; Xe‐ELIP treatment 2 h after stroke onset; Xe‐ELIP treatment 3 h after stroke onset; Xe‐ELIP treatment 5 h after stroke onset. All rats in each treatment group were given 7 mg/kg (200 μL) of Xe‐ELIP over 5 min into the CCA with modified polyethylene tubing (PE10). The CCA was exposed to 1 MHz continuous wave ultrasound at a peak‐to‐peak pressure amplitude of 0.18 MPa (1 W/cm2 dial setting; Sonitron 2000; Rich‐Mar Corp, Inola, OK, USA) during Xe‐ELIP administration.
For the dose dependence study, each treatment group received either 3.5 mg/kg (100 μL), 7 mg/kg (200 μL), or 14 mg/kg (400 μL) of Xe‐ELIP at 3 h after stroke onset in combination with ultrasound application.
Neurologic Assessment
At days 1, 2, and 3 after surgery, each animal was tested for motor function and neurologic outcomes by recording limb placing, beam walking, and grid walking abilities 19. All behavioral tests were conducted in a quiet and low‐lit room by an observer blinded with respect to the treatment groups. Limb placement was assessed by observing the animal's ability to lift its head and extend its forelimbs toward a table while the animal was suspended over the table by its tail (score 0—no response, score 1—response was sluggish or delayed, score 2—response was rapid and fully executed). The ability to walk across a beam (2.5 × 2.5 × 80 cm) was assessed by monitoring balance maintenance while navigating across the beam. The response scores were assigned as follows: score 0—no foot slip, score 1—traversed with grasping of the lateral side of the beam, score 2—showed difficulty in crawling across the beam but able to traverse, score 3—required more than 10 seconds to traverse the beam due to difficulty in walking, score 4—unable to traverse the beam, score 5—unable to move the body or any limb, score 6—unable to stay on the beam for more than 10 seconds. For the grid‐walking test, the animal was placed on a stainless steel grid floor (40 × 40 × 20 cm with a mesh size of 2 × 2 cm). The total number of steps was counted up to the maximum of 50 steps. The number of foot fault errors misplacing a forelimb or hindlimb and falling through the grid was recorded.
Infarct Volume Measurement
After neurologic assessment on the third day, animals were euthanized and the brains harvested. Using a Jacobowitz brain slicer, 2‐mm thick coronal sections were stained with 2% 2, 3, 5‐triphenyltetrazolium chloride (TTC) 19. Infarct size was normalized with respect to the whole brain volume and presented as normalized infarct volume (%).
Gel Electrophoresis and Immunoblotting
Expression of brain‐derived neurotrophic factor (BDNF), mitogen‐activated protein kinases (MAPK), and Akt were determined. Animals were randomly divided into three groups: (1) sham surgery without ischemic stroke, (2) MCAO without treatment, and (3) MCAO with Xe‐ELIP (7 mg/kg) treatment at 3 h following stroke onset. The fourth 2 mm coronal section (6–8 mm from rostral end) from injury side of the brain was homogenized in 1 mL of RadioImmunoPrecipitation Assay (RIPA) buffer (Cell Signaling Technology, Beverly, MA, USA) containing the protease inhibitor phenylmethlylsulfonyl fluoride (PMSF, 1 mm) and phosphatase inhibitor cocktail (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Whole‐cell protein was extracted, and protein concentrations were measured. Equal amounts of protein (80 μg) were loaded on 12% SDS–polyacrylamide gels with Tris‐glycine running buffer and transferred to a polyvinylidene difluoride membrane (Millipore, Billerica, MA, USA). After blocking in NonMammalian Blocking Reagent (LI‐COR Biosciences, Lincoln, NE, USA) for 1 h, the membranes were incubated with primary antibodies of BDNF (Santa Cruz Biotechnology; diluted 1:250), phospho‐Akt (ser 473; Cell Signaling Technology, diluted 1:250), total Akt (Cell Signaling Technology, diluted 1:500), Phospho‐42/44 MAPK (diluted 1:250, Cell Signaling Technology), and total 42/44 MAPK (diluted 1:500, Cell Signaling Technology) at 4°C overnight. After washing the membranes with Tris‐buffered saline containing 0.1% Tween‐20 (TBS‐T), membranes were incubated with IRDye 800CW Dky anti‐rabbit IgG secondary antibody (H + L; LI‐COR Biosciences) at room temperature for 1 h. After rinsing twice and washing three times for 5 min in TBS‐T. To ensure equivalent protein loading, membranes were reprobed with anti‐β‐actin (Sigma‐Aldrich, St. Louis, MO, USA). The membranes were then scanned with the Odyssey infrared imaging system (LI‐COR Biosciences). The optical densities of all protein bands were analyzed using ImageJ software provided by the National Institutes of health. All target proteins were quantitated by normalizing to β‐actin and calculated as ratio of the corresponding control group.
In Situ Labeling of DNA Fragmentation by TUNEL Assay
To detect DNA fragmentation in degenerating neurons, animals were sacrificed 24 h after reperfusion, and coronal sections (10 μm) of freshly frozen brain were cut using a cryotome followed by the terminal deoxynucleotidyl transferase dUTP biotin nick end labeling (TUNEL) assay (Roche Diagnostics GmbH, Mannheim, Germany). In brief, the sections were fixed in 4% paraformaldehyde for 20 min at room temperature and permeabilized by incubation with 0.1% Triton X‐100 and 0.1% sodium citrate for 2 min at 4°C. Each slide was incubated with 50 μL of TUNEL reaction mixture at 37°C for 1 h. The slides were also stained with 4′, 6‐diamidino‐2‐phenylindole (DAPI, Invitrogen, Eugene, OR, USA). Images were viewed under a Nikon ECLIPSE Ti fluorescence microscope and photographed with a CoolSNAP photometrics camera. TUNEL‐positive cells were photographed at 100× magnification at three randomly selected fields close to the infarct border. Dead cells were counted using NIS‐Elements BR 3.2 diagnostic software (Nikon Instruments Inc., Melville, NY, USA).
Double Immunofluorescence Staining
At 24 h following MCAO, the brain was removed and fixed in 4% paraformaldehyde solution at 4°C overnight and cryoprotected in 30% sucrose at 4°C overnight. Frozen coronal cryostat sections (20 μm) were stained with antibodies against BDNF (Santa Cruz Biotechnology; diluted 1:50), phospho‐p42/p44 MAPK (Cell Signaling Technology, diluted 1:250), phospho‐Akt (ser 473; Cell Signaling Technology, diluted 1:200), and total Akt (Cell Signaling Technology, diluted 1:200) at 4°C overnight. To counterstain neurons, astrocytes and endothelial cells, the sections were incubated with an antibody against neuron‐specific nuclear protein (NeuN; Millipore, Temecula, CA, USA, diluted 1:200), an antibody against glial fibrillary acidic protein (GFAP; Millipore, diluted 1:2000), or an antibody against rat endothelial cell antigens (RECA, Santa Cruz Biotechnology; diluted 1:50). Alexa Fluor 488 Goat Anti‐Rabbit IgG and Alexa Fluor 555 Goat Anti‐Mouse IgG (Life technologies, Grand Island, NY, USA, diluted 1:1000) were used as secondary antibodies. The slides were examined by fluorescent microscopy (Nikon, Ti‐U, Dusseldorf, Germany).
Statistical Analysis
Nonparametric statistical analyses were performed using the Wilcoxon rank test for two groups or the Kruskal–Wallis analysis of variance for more than two groups. When differences were detected in global comparison, the multiple comparisons of mean ranks for all groups were performed for all pairwise comparisons. Statistical software (Statistica; StatSoft Inc, Tulsa, OK, USA) was used for statistical analyses. Data are presented as mean with standard error. P < 0.05 was considered significant.
Results
Therapeutic Time Window of Xe‐ELIP Treatment
Xenon‐containing echogenic liposomes (7 mg/kg) treatment at 2 h, 3 h, or 5 h after stroke onset was investigated by evaluating normalized infarct size and neurologic disability following transient MCAO. Brain infarctions stained by TTC are shown in Figure 1A–D. In untreated animals, large infarctions developed predominantly involving the cerebral cortex and striatum with a normalized infarct volume of 16 ± 1.8%. Xe‐ELIP administration at 2 h, 3 h, or 5 h after stroke onset reduced the normalized infarct size to 2.9 ± 0.7% (P < 0.001 vs. no treatment), 5.6 ± 1.2% (P < 0.001 vs. no treatment), and 8.5 ± 1.3% (P = 0.004 vs. no treatment), respectively (Figure 1E). Behavioral assessments including limb placement, beam walking, and grid walking ability at days 1 and 3 after surgery are shown in Figure 1F–H, respectively. The group receiving Xe‐ELIP 5 h after stroke onset had marginal improvement in behavioral tasks. Xe‐ELIP administered at 2 or 3 h after stroke onset improved performance in all behavioral tests at day 1, with marked improvement in all tests by day 3.
Figure 1.
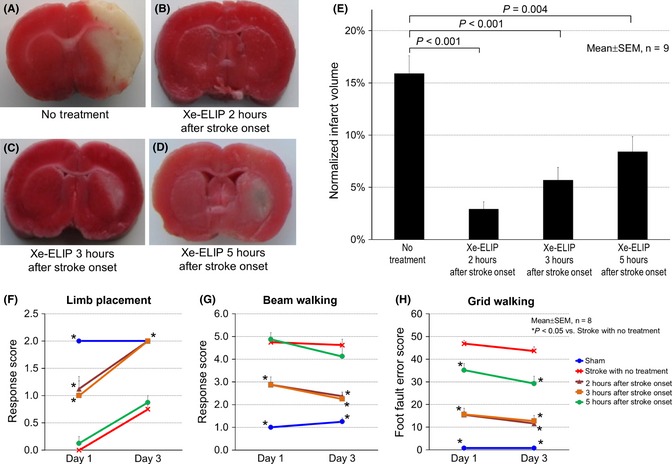
Time window of Xe‐containing echogenic liposomes (Xe‐ELIP) for neuroprotective effect. Representative triphenyltetrazolium chloride (TTC)‐stained coronal brain sections following middle cerebral artery occlusion (MCAO): (A) stroke with no treatment, (B–D) Xe‐ELIP (7 mg/kg) treatment at 2 h (B), 3 h (C), and 5 h (D) after stroke onset. The white areas refer to the infarct regions following MCAO. (E) Quantification of infarct volume demonstrated that Xe‐ELIP administration at 2 h, 3 h, and 5 h after stroke onset reduced infarct volume compared with the no treatment group. Neurologic assessment of (F) limb placement, (G) beam walking, and (H) grid walking corresponded to the TTC‐stained data.
There was no difference in core body temperature between the groups during MCAO and the initial hours of reperfusion. Regional cerebral blood perfusion was measured in the region of ischemic penumbra (2 mm lateral and 2 mm posterior to the bregma) before and after MCA occlusion, after reperfusion, just before Xe delivery, and after Xe delivery. There was no change of flow detected in the ischemic penumbra region due to Xe‐ELIP delivery at a dosage range of 7–14 mg/kg.
Xe‐ELIP Dose Dependence for Neuroprotection
We investigated the dose dependence of Xe‐ELIP administrated 3 h after stroke onset. Maximal reduction in infarct volume was achieved at a dose of 14 mg/kg (normalized infarct volume of 3.3 ± 0.7%, P < 0.001 vs. no treatment; Figure 2A–E). Although 7 mg/kg was less effective than 14 mg/kg, 7 mg/kg also reduced infarct size (5.6 ± 1.2%, P < 0.001 vs. no treatment). The lowest dose of Xe‐ELIP (3.5 mg/kg) treatment showed little improvement in infarct size reduction (12 ± 3.0%, P > 0.05 vs. no treatment). Behavioral tests demonstrated improved neurologic function for both the 14 mg/kg and the 7 mg/kg treatment groups (Figure 2F–H).
Figure 2.
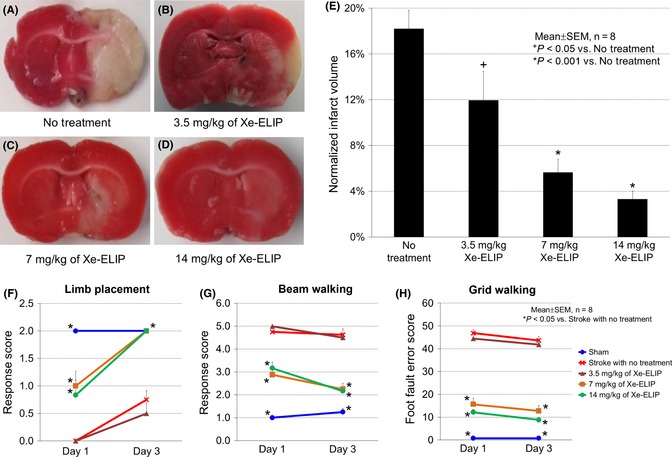
Dose response of Xe‐containing echogenic liposomes (Xe‐ELIP) for neuroprotective effect. Coronal brain sections (stained with triphenyltetrazolium chloride) following middle cerebral artery occlusion: (A) stroke with no treatment, (B–D) Xe‐ELIP treatment with 3.5 mg/kg (B), 7 mg/kg (C), and 14 mg/kg (D) at 3 h after stroke onset. (E) Quantification of the infarct volumes. Neurologic assessment of (F) limb placement, (G) beam walking, and (H) grid walking.
Ultrasound‐Enhanced Xe Delivery to Cerebral Ischemic Injury In Vivo
Ultrasound application over the CCA during Xe‐ELIP delivery to the cerebral circulation demonstrated a further decrease in infarct volume (Figure 3). The normalized infarct volume with no treatment was 21 ± 2.5%. Air‐ELIP (5 mg/kg) treatment demonstrated no difference in infarct size (P > 0.05 vs. no treatment). Xe‐ELIP treatment (7 mg/kg) decreased the normalized infarct volume to 6.1 ± 0.5% (P < 0.001 vs. no treatment). Ultrasound triggered additional Xe release from Xe‐ELIP resulting in a further reduction in the normalized infarct size to 2.9 ± 0.7% (P < 0.001 vs. no treatment, P < 0.05 vs. no ultrasound).
Figure 3.
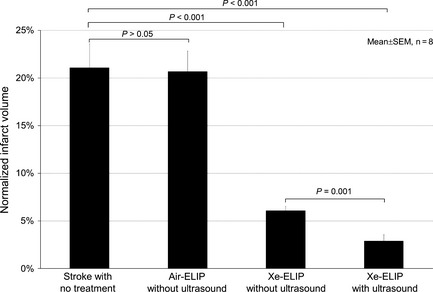
Ultrasound‐enhanced neuroprotective effect of local delivery of Xe‐containing echogenic liposomes (Xe‐ELIP). Normalized infarct volume comparison demonstrates decreased infarct volume with Xe‐ELIP alone and further reduction with Xe‐ELIP combined with ultrasound activation. No neuroprotection was found with air‐ELIP treatment.
Cell Death Evaluation
Cell death evaluation 24 h after ischemic stroke demonstrated a decrease in the number of TUNEL‐labeled cells (used as index of cell death, 39 ± 9.0 cells/mm2) in the penumbra with Xe‐ELIP treatment at 3 h following stroke onset compared with 113 ± 10 cells/mm2 (P = 0.002) in the untreated stroke. DAPI, neuron‐specific nuclear protein (NeuN; Figure 4A), and GFAP (Figure 4B) were counterstained to address a possible colocalization of TUNEL‐positive cells with neurons, astrocytes/glia cells, and endothelial cells. TUNEL‐positive cells were colocalized within the neuronal cells (Figure 4A) but not astrocytes (Figure 4B).
Figure 4.
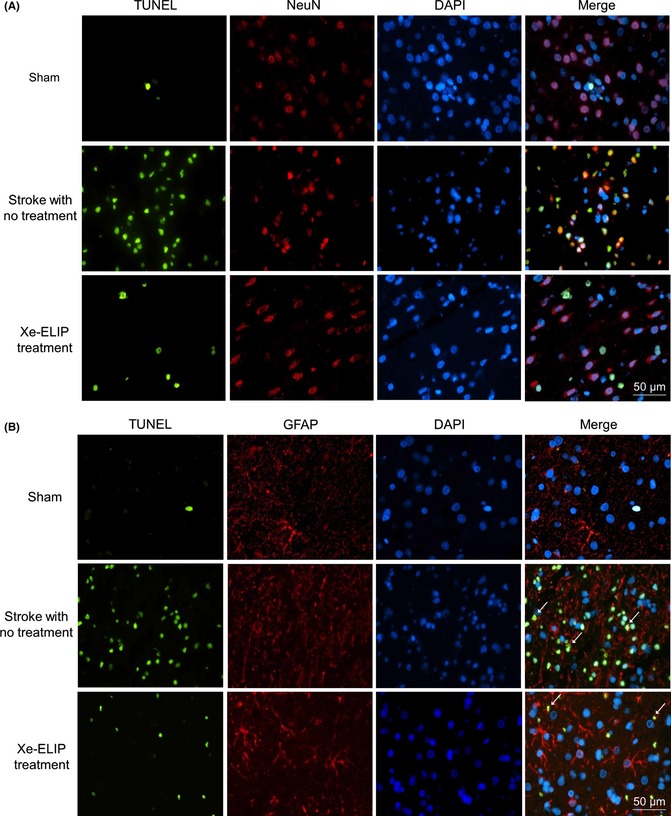
Cell death detected by TUNEL staining and colocalization with neuronal cells (A) and astrocytes (B). TUNEL staining in the penumbral region of the brain; sham, stroke with no treatment, and stroke with Xe‐containing echogenic liposomes (Xe‐ELIP) treatment. Xe‐ELIP treatment demonstrated reduction in cell death. The TUNEL‐positive staining is colocalized within the neuronal cells but not astrocytes. Green—TUNEL‐positive staining; Blue—DAPI counterstaining of nuclei; Red—neuron‐specific nuclear protein (NeuN) or glial fibrillary acidic protein (GFAP).
BDNF Expression and Immunoreactivity
Western blot was performed to examine BDNF expression in stroke with and without Xe‐ELIP treatment. BDNF levels decreased from 1.1 ± 0.1 (sham) to 0.7 ± 0.1 (P < 0.05) at 24 h after stroke with no treatment. Conversely, Xe‐ELIP administration increased BDNF expression to 1.5 ± 0.1 (P < 0.01 vs. no treatment; Figure 5A,B).
Figure 5.
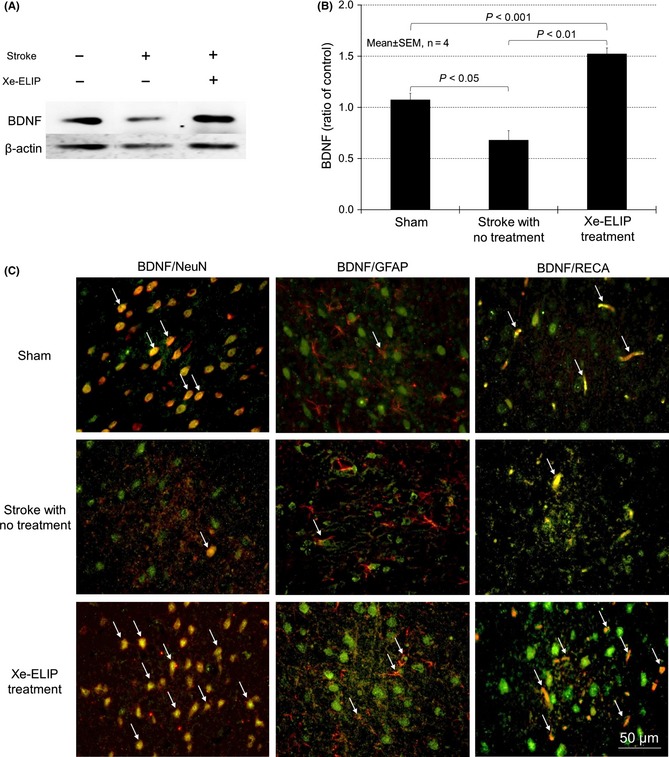
Brain‐derived neurotrophic factor (BDNF) expression and colocalization with neurons, astrocytes, and endothelial cells. Western blot analysis of BDNF expression (A) and quantitation (B). Double immunofluorescence staining for BDNF (green) and NeuN (red), glial fibrillary acidic protein (GFAP) (red), or rat endothelial cell antigen (RECA) (red) in the penumbral region of the brain tissue at 24 h poststroke onset (C). BDNF expression was found in the neurons, astrocytes, and endothelial cells in normal animals. At 24 h after stroke, BDNF expression was reduced, but enhanced BDNF immunoreactivity was found following Xe‐containing echogenic liposomes treatment. This was colocalized with NeuN, GFAP, and RECA (white arrows in merged figures). For western blot, BDNF proteins were quantitated by normalizing to β‐actin and calculated as ratio of the corresponding control group.
To address a possible colocalization of BDNF with neurons, astrocytes/glia cells, and endothelial cells, double immunofluorescent staining was performed. BDNF immunoreactivity was enhanced in Xe‐ELIP‐treated group (Figure 5C). Most of the BDNF immunoreactivity is colocalized with neuronal cells and endothelial cells. There was little colocalization between BDNF immunoreactivity and astrocytes.
Phosphorylated Akt Expression and Immunoreactivity
As the Akt pathway is related to BDNF/TrkB (neurotrophic tyrosine kinase receptor, type 2)‐induced neuroprotection 21. We investigated the phosphorylated Akt expression and its colocalization with neuronal cells, astrocytes, and endothelial cells. Western blot was performed to examine Akt expression in stroke with and without Xe‐ELIP treatment. The expression levels of phospho‐Akt and total Akt were 0.8 ± 0.1 and 0.9 ± 0.01 without Xe‐ELIP treatment. Expression levels were upregulated by Xe‐ELIP to 1.7 ± 0.4 (P = 0.28) and 1.4 ± 0.1 (P = 0.03), respectively (Figure 6A,B). Phospho‐Akt immunoreactivity increased in both neuronal cells and astrocytes/glia cells at 24 h after Xe‐ELIP treatment, but not in the endothelial cells (Figure 6C). Akt immunoreactivity was mostly located in the neuronal cells.
Figure 6.
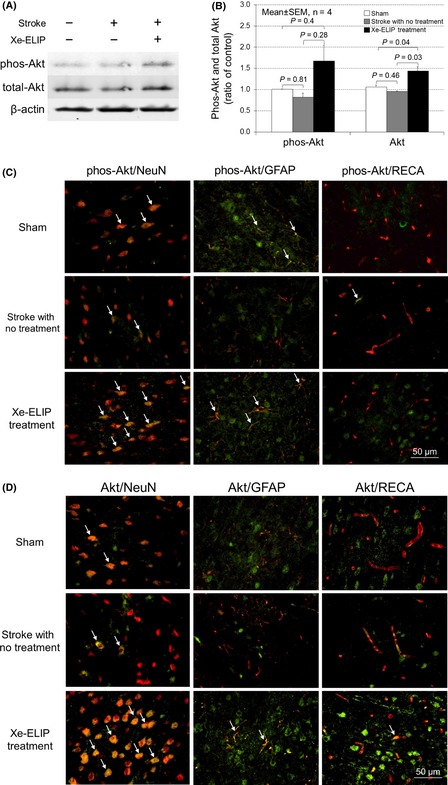
Phosphor‐Akt and total Akt expression and colocalization with neurons, astrocytes, and endothelial cells. Western blot analysis of Akt expression (A) and quantitation (B). Double immunofluorescence staining for colocalization of phosphor‐Akt (C) and total Akt (D) (green) with NeuN (red), glial fibrillary acidic protein (red), or rat endothelial cell antigens (red) in the penumbral region of the brain tissue at 24 h poststroke onset. Phospho‐Akt immunoreactivity increased in both neuronal cells and astrocytes/glia cells at 24 h after Xe‐containing echogenic liposomes treatment, but not in the endothelial cells.
Colocalization of Phosphorylated 42/44 MAPK Immunoreactivity
The MAPK signaling pathway is also related to BDNF‐induced neuroprotection. The concomitant activation of the MAPK and Akt signaling pathways is required to develop an integrated network to regulate apoptotic cell death. We investigated the phosphorylation of p42/44 MAPK and its colocalization with neurons, astrocytes, and endothelial cells. Western blot was performed to examine phospho‐42/44 MAPK expression in stroke with and without Xe‐ELIP treatment. Phospho‐42 MAPK and phospho‐44 MAPK expressions increased to 2.7 ± 0.3 (P < 0.001 vs. no treatment) and 1.9 ± 0.4 (P = 0.07 vs. no treatment), respectively, at 24 h after Xe‐ELIP administration (Figure 7A,B). The phospho‐42/44 MAPK immunoreactivity demonstrated colocalization with the neuronal cells, astrocyte/glia cells, and endothelial cells (Figure 7).
Figure 7.
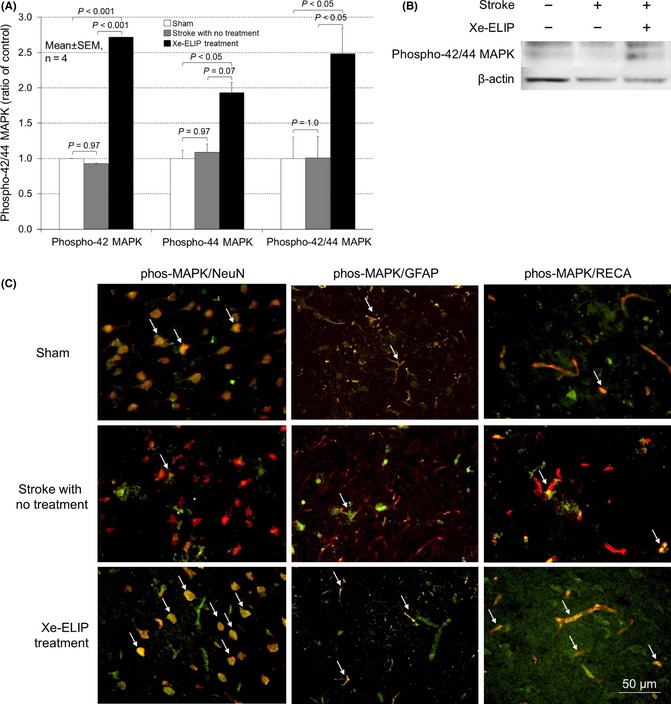
Phosphor‐42/44 mitogen‐activated protein kinases (MAPK) expression and colocalization with neurons, astrocytes, and endothelial cells. Western blot analysis of Phosphor‐42/44 MAPK expression and quantitation (A, B). Double immunofluorescence staining for colocalization of Phosphor‐42/44 MAPK (green) with NeuN (red), glial fibrillary acidic protein (red), or rat endothelial cell antigens (red) in the penumbral region of the brain tissue at 24 h poststroke onset (C). The phospho‐42/44 MAPK immunoreactivity demonstrated colocalization with the neuronal cells, astrocyte/glia cells, and endothelial cells.
Discussion
Continuing upon our previous work in developing Xe‐ELIP as a treatment strategy to reduce infarct volume and improve neurologic function in ischemia 19, the present study addresses the infusion time window for efficacy, the optimal Xe‐ELIP dose, and addresses molecular mechanisms for this neuroprotective effect. We have found that Xe‐ELIP administered within 5 h after stroke onset at a dosage range of 7–14 mg/kg provides the most efficacious neuroprotection. Mechanisms include the activation of BDNF, Akt, and MAPK. Injection of Xe‐ELIP within 5 h following stroke onset demonstrated efficacy, but to a lesser extent at 24 h postocclusion. These findings indicate that Xe‐ELIP have a clinically applicable therapeutic time window for treatment of stroke when administered to patients in the field.
We delivered Xe‐ELIP at 2 h after stroke onset and have found that Xe‐ELIP has a therapeutic time window of at least 5 h after stroke onset. The key pathology of early brain ischemia involves the overstimulation of excitatory NMDA receptors by increased extracellular glutamate, resulting in a massive calcium influx into the neurons 22. The increase in intracellular calcium occurs within minutes after the onset ischemia followed by initiation of “cytotoxic cascades” involved in activation of a series of downstream signal pathways 23. Varying degrees of NMDA excitotoxicity then lead to neuronal apoptosis/necrosis 24. In general, the therapeutic time window for NMDA receptor antagonists in focal ischemia is short, approximately 1–2 h after stroke onset 25. However, several NMDA subunits such as NR2A and NR2B receptor mRNAs are upregulated in the cortex at 3 h after reperfusion following MCAO. This effect is accompanied by an increase in NR2A and NR2B immunoreactivity in the penumbra at 6–24 h after reperfusion 26. Late increase in NMDA receptor expression can make neuronal cells more vulnerable to injury. Therefore, blocking of NMDA receptors 2 h after stroke onset by Xe delivery may still provide some beneficial effects to neuronal cells.
Our data suggest that Xe‐ELIP provides neuroprotection through other molecular mechanisms in addition to NMDA inhibition. Xe also has neuroprotective and cardioprotective effects via preconditioning. Mechanisms related to Xe preconditioning involve preservation of mitochondrial function 27, natural induction of hypoxia‐inducible factor 28, and increase in neurotrophic factors 29. Neurotrophic factors such as BDNF and nerve growth factor (NGF) play important roles in neuronal survival 30. Treatment with BDNF has shown neuroprotective effects in ischemic stroke 31. Xe upregulates pCREB, resulting in enhanced expression of BDNF 32. Our data demonstrated that Xe‐ELIP treatment stabilized BDNF expression associated with ischemic injury in neuronal cells.
Western blotting is a useful tool to quantity molecular changes in the signaling pathway. Our Western blot data demonstrated that Xe‐ELIP administration attenuated the reduction in phosphorylation of both Akt (Figure 6) and p42/44 MAPK (Figure 7) 24 h after stroke onset. However, the brain is composed of various types of cells, which may respond differently to ischemic injury and Xe‐ELIP treatment. Our double immunofluorescent staining revealed that increased phosphor‐Akt and phosphor‐42/44 MAPK expressions are strongly colocalized within the neurons (Figures 6C and 7C). These are key proteins in the signaling pathways that contribute to regulation of cell survival 33. Activation of a BDNF receptor, known as TrkB, results in activation of the MAP kinase or PI3K/Akt cascade via several intermediate steps. However, the roles of these pathways are distinct in each cell type in the neurovascular unit. Strong colocalization of BDNF, p42/44 MAPK, and p‐Akt indicates that the MAPK and Akt pathways are important in Xe‐induced neuronal cell survival. The MAPK signaling pathway promotes cell survival through a dual mechanism including inactivation of proapoptotic proteins and upregulation of prosurvival protein, and cAMP‐responsive element‐binding (CREB) proteins. The MAPK and Akt signaling pathways interact to develop an integrated network, and the regulation of apoptosis requires coordinated changes in both signaling pathways 34. Our data demonstrated upregulation of both Akt and MAPK concomitantly with inhibition of cell death. TUNEL staining 24 h following ischemia showed that Xe‐ELIP treatment markedly reduced neuronal death (Figure 4A). The reduced infarct volume accompanied by a decrease in TUNEL‐labeled cells following Xe‐ELIP treatment further supports the neuroprotective effect of Xe delivered via Xe‐ELIP. This may account for a longer neuroprotective effect of Xe‐ELIP (best within 5 h but efficacious up to 24 h).
Brain‐derived neurotrophic factor immunoreactivity is colocalized within the endothelial cells (Figure 5). The endothelial cells are known to secrete a potent amount of BDNF for neuroprotection. It has shown that BDNF containing endothelial‐conditioned media upregulates prosurvival Akt signaling and protects neurons against hypoxic injury 35. A homeostatic coupling from the cerebral endothelial cells to the neurons may play an important role in maintaining neurovascular function 21, 35. Our in vivo data demonstrated a strong colocalization of BDNF within the endothelial cells accompanied by activation of Akt in both the neurons and the endothelial cells. In the human umbilical vein endothelial cells (HUVEC) culture system, Xe preconditioning prevents inflammatory factor expression induced by tumor necrosis factor‐α (TNF‐α) including intracellular cell adhesion molecule 1 (ICAM‐1), vascular cell adhesion molecule 1 (VCAM‐1), and nuclear factor κB (NF‐κB) 36. This indicates that, in addition to targeting to neurons, Xe is likely to protect the neurovascular unit function by affecting the cerebral vascular endothelium.
There is a weak colocalization of TUNEL‐positive cells with astrocytes. Astrocytes are more resistant than neurons to oxygen‐glucose deprivation 37. For example, most neurons will die after 60–90 min of oxygen–glucose deprivation (OGD), while astrocytes only suffer a similar extent of injury after 4–6 h OGD 38, 39. Other in vivo studies also suggest that astrocytes are better preserved than neurons following ischemia 40, 41. Our data demonstrated that ischemic damage of 2 h resulted in little astrocyte death (Figure 4B).
Astrocytes play an important role in pro‐inflammation and in protecting neurons from antioxidant damage 37. Xe has both antioxidant and antiinflammatory effects 7. There was a colocalization of phos‐Akt with astrocytes (Figure 6C). To determine whether the activation of Akt is related to astrocytes function, further experiments are required.
Our dose response data indicate that it is critical to provide an adequate dose of Xe via Xe‐ELIP to achieve neuroprotection. Xe as a gas has a fast “wash out” rate in the brain. These data demonstrate that a dose of 7–14 mg/kg of Xe‐ELIP provides the best therapeutic effect. To translate this delivery strategy from animals into clinical studies, we must account for the differences in the brain structure between rats and human. The human brain has a larger proportion of white matter relative to the rodent brain. To translate to humans, we must first investigate the therapeutic time window and dose dependence of Xe‐ELIP neuroprotection using a larger animal model. Differences in transit time in humans, the pulmonary circulation and other hemodynamic effects, also emphasize the necessity of optimizing this therapeutic strategy in a large animal model.
According to clinical report, the average time interval between the onset of stroke symptoms and hospital arrival was 330 min (5.5 h), and 28.9% of the patients arrives at the hospital within 3 h 20. We chose the 3‐h time point for dose dependence study as it is the closest time window in the clinical setting. A follow‐up dose dependence study with an extended therapeutic window up to 5 h may allow to determine complete potential therapeutic benefits of Xe‐ELIP.
To investigate the therapeutic time window and dose dependence of Xe‐ELIP, we evaluated histologic changes and improvement of neurologic behavior for 3 days following stroke. Stroke‐induced functional impairment can be divided into acute and long‐term phases. Further studies to address the effect of Xe‐ELIP on long‐term behavioral improvement will provide more comprehensive therapeutic values in restoring neurologic functions and allowing for improved daily patient lives following stroke.
Conclusions
We have demonstrated that Xe‐ELIP can reduce infarct volume and improve neurologic function in an animal model of ischemia. These data help to determine the time window of injection for therapeutic efficacy and the optimal efficacious dose. Our data indicate that Xe‐ELIP administered within 5 h after stroke onset at a dose of 7–14 mg/kg provides considerable neuroprotection. This novel liposomal strategy to deliver Xe has the potential for neuroprotection in patients following acute stroke.
Conflict of Interest
Drs Hyunggun Kim, David D. McPherson, Shao‐Ling Huang and The University of Texas Health Science Center at Houston have research‐related financial interests in Zymo Pharmaceuticals, LLC.
Acknowledgments
This work was supported in part by the National Institutes of Health (NS067454, Dr Huang; HL074002, HL059586, Dr McPherson) and the Center for Clinical and Translational Sciences at The University of Texas Health Science Center, which is funded by the National Institutes of Health Clinical and Translational Award UL1 TR000371 from the National Center for Advancing Translational Science. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Advancing Translational Science or the National Institutes of Health.
References
- 1. Towfighi A, Saver JL. Stroke declines from third to fourth leading cause of death in the United States: Historical perspective and challenges ahead. Stroke 2011;42:2351–2355. [DOI] [PubMed] [Google Scholar]
- 2. Suwanwela N, Koroshetz WJ. Acute ischemic stroke: Overview of recent therapeutic developments. Annu Rev Med 2007;58:89–106. [DOI] [PubMed] [Google Scholar]
- 3. Kaufmann AM, Firlik AD, Fukui MB, Wechsler LR, Jungries CA, Yonas H. Ischemic core and penumbra in human stroke. Stroke 1999;30:93–99. [DOI] [PubMed] [Google Scholar]
- 4. Fan X, Kavelaars A, Heijnen CJ, Groenendaal F, van Bel F. Pharmacological neuroprotection after perinatal hypoxic‐ischemic brain injury. Curr Neuropharmacol 2010;8:324–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fisher M. New approaches to neuroprotective drug development. Stroke 2011;42:S24–S27. [DOI] [PubMed] [Google Scholar]
- 6. Irani Y, Pype JL, Martin AR, et al. Noble gas (argon and xenon)‐saturated cold storage solutions reduce ischemia‐reperfusion injury in a rat model of renal transplantation. Nephron Extra 2011;1:272–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nakao A, Sugimoto R, Billiar TR, McCurry KR. Therapeutic antioxidant medical gas. J Clin Biochem Nutr 2009;44:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Qipshidze N, Metreveli N, Mishra PK, Lominadze D, Tyagi SC. Hydrogen sulfide mitigates cardiac remodeling during myocardial infarction via improvement of angiogenesis. Int J Biol Sci 2012;8:430–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Banks P, Franks NP, Dickinson R. Competitive inhibition at the glycine site of the N‐methyl‐D‐aspartate receptor mediates xenon neuroprotection against hypoxia‐ischemia. Anesthesiology 2010;112:614–622. [DOI] [PubMed] [Google Scholar]
- 10. Dickinson R, Franks NP. Bench‐to‐bedside review: Molecular pharmacology and clinical use of inert gases in anesthesia and neuroprotection. Crit Care 2010;14:229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dworschak M. Pharmacologic neuroprotection–is xenon the light at the end of the tunnel? Crit Care Med 2008;36:2477–2479. [DOI] [PubMed] [Google Scholar]
- 12. Sanders RD, Maze M. Xenon: From stranger to guardian. Curr Opin Anaesthesiol 2005;18:405–411. [DOI] [PubMed] [Google Scholar]
- 13. Buchanan KD, Huang S, Kim H, Macdonald RC, McPherson DD. Echogenic liposome compositions for increased retention of ultrasound reflectivity at physiologic temperature. J Pharm Sci 2008;97:2242–2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Coussios CC, Holland CK, Jakubowska L, et al. In vitro characterization of liposomes and Optison by acoustic scattering at 3.5 MHz. Ultrasound Med Biol 2004;30:181–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hamilton A, Rabbat M, Jain P, et al. A physiologic flow chamber model to define intravascular ultrasound enhancement of fibrin using echogenic liposomes. Invest Radiol 2002;37:215–221. [DOI] [PubMed] [Google Scholar]
- 16. Huang S, Hamilton AJ, Tiukinhoy SD, et al. Liposomes as ultrasound imaging contrast agents and as ultrasound‐sensitive drug delivery agents. Cell Mol Biol Lett 2002;7:233–235. [PubMed] [Google Scholar]
- 17. Huang SL, Hamilton AJ, Nagaraj A, et al. Improving ultrasound reflectivity and stability of echogenic liposomal dispersions for use as targeted ultrasound contrast agents. J Pharm Sci 2001;90:1917–1926. [DOI] [PubMed] [Google Scholar]
- 18. Smith DA, Porter TM, Martinez J, et al. Destruction thresholds of echogenic liposomes with clinical diagnostic ultrasound. Ultrasound Med Biol 2007;33:797–809. [DOI] [PubMed] [Google Scholar]
- 19. Britton GL, Kim H, Kee PH, et al. In vivo therapeutic gas delivery for neuroprotection with echogenic liposomes. Circulation 2010;122:1578–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Maze LM, Bakas T. Factors associated with hospital arrival time for stroke patients. J Neurosci Nurs 2004;36:136–141, 55. [PubMed] [Google Scholar]
- 21. Guo S, Som AT, Waeber C, Lo EH. Vascular neuroprotection via TrkB‐ and Akt‐dependent cell survival signaling. J Neurochem 2012;123(Suppl 2):58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: An integrated view. Trends Neurosci 1999;22:391–397. [DOI] [PubMed] [Google Scholar]
- 23. Stankowski JN, Gupta R. Therapeutic targets for neuroprotection in acute ischemic stroke: Lost in translation? Antioxid Redox Signal 2011;14:1841–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Broughton BR, Reutens DC, Sobey CG. Apoptotic mechanisms after cerebral ischemia. Stroke 2009;40:e331–e339. [DOI] [PubMed] [Google Scholar]
- 25. Hsu CY, Ahmed SH, Lees KR. The therapeutic time window–theoretical and practical considerations. J Stroke Cerebrovasc Dis 2000;9:24–31. [DOI] [PubMed] [Google Scholar]
- 26. Gappoeva MU, Izykenova GA, Granstrem OK, Dambinova SA. Expression of NMDA neuroreceptors in experimental ischemia. Biochemistry (Mosc) 2003;68:696–702. [DOI] [PubMed] [Google Scholar]
- 27. Mio Y, Shim YH, Richards E, Bosnjak ZJ, Pagel PS, Bienengraeber M. Xenon preconditioning: The role of prosurvival signaling, mitochondrial permeability transition and bioenergetics in rats. Anesth Analg 2009;108:858–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ma D, Lim T, Xu J, et al. Xenon preconditioning protects against renal ischemic‐reperfusion injury via HIF‐1alpha activation. J Am Soc Nephrol 2009;20:713–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Valleggi S, Cavazzana AO, Bernardi R, et al. Xenon up‐regulates several genes that are not up‐regulated by nitrous oxide. J Neurosurg Anesthesiol 2008;20:226–232. [DOI] [PubMed] [Google Scholar]
- 30. Mattson MP, Duan W, Chan SL, et al. Neuroprotective and neurorestorative signal transduction mechanisms in brain aging: Modification by genes, diet and behavior. Neurobiol Aging 2002;23:695–705. [DOI] [PubMed] [Google Scholar]
- 31. Zhang Y, Pardridge WM. Neuroprotection in transient focal brain ischemia after delayed intravenous administration of brain‐derived neurotrophic factor conjugated to a blood‐brain barrier drug targeting system. Stroke 2001;32:1378–1384. [DOI] [PubMed] [Google Scholar]
- 32. Ma D, Hossain M, Pettet GK, et al. Xenon preconditioning reduces brain damage from neonatal asphyxia in rats. J Cereb Blood Flow Metab 2006;26:199–208. [DOI] [PubMed] [Google Scholar]
- 33. Hetman M, Kanning K, Cavanaugh JE, Xia Z. Neuroprotection by brain‐derived neurotrophic factor is mediated by extracellular signal‐regulated kinase and phosphatidylinositol 3‐kinase. J Biol Chem 1999;274:22569–22580. [DOI] [PubMed] [Google Scholar]
- 34. Marushige K, Marushige Y. Changes in the mitogen‐activated protein kinase and phosphatidylinositol 3‐kinase/Akt signaling associated with the induction of apoptosis. Anticancer Res 1999;19:3865–3871. [PubMed] [Google Scholar]
- 35. Guo S, Kim WJ, Lok J, et al. Neuroprotection via matrix‐trophic coupling between cerebral endothelial cells and neurons. Proc Natl Acad Sci U S A 2008;105:7582–7587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liu W, Liu Y, Chen H, Liu K, Tao H, Sun X. Xenon preconditioning: Molecular mechanisms and biological effects. Med Gas Res 2013;3:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Barreto G, White RE, Ouyang Y, Xu L, Giffard RG. Astrocytes: Targets for neuroprotection in stroke. Cent Nerv Syst Agents Med Chem 2011;11:164–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Almeida A, Delgado‐Esteban M, Bolanos JP, Medina JM. Oxygen and glucose deprivation induces mitochondrial dysfunction and oxidative stress in neurones but not in astrocytes in primary culture. J Neurochem 2002;81:207–217. [DOI] [PubMed] [Google Scholar]
- 39. Giffard RG, Swanson RA. Ischemia‐induced programmed cell death in astrocytes. Glia 2005;50:299–306. [DOI] [PubMed] [Google Scholar]
- 40. Chen Y, Swanson RA. Astrocytes and brain injury. J Cereb Blood Flow Metab 2003;23:137–149. [DOI] [PubMed] [Google Scholar]
- 41. Li Y, Chopp M, Zhang ZG, Zhang RL. Expression of glial fibrillary acidic protein in areas of focal cerebral ischemia accompanies neuronal expression of 72‐kDa heat shock protein. J Neurol Sci 1995;128:134–142. [DOI] [PubMed] [Google Scholar]