

Transformations that entail catalytic generation of reactive entities for in situ use in stereoselective synthesis are in high demand;[1] such processes are more efficient and operationally simpler than when a priori preparation and purification of stoichiometric quantities of sensitive intermediates are required.[2] Successful design of multi-component pathways hinges on high chemoselectivity: A starting material and a reagent must first be catalytically transformed into a new species, which then has to undergo reaction with another substrate. If the catalyst structure is incorporated within the intermediate molecule, reactivity and selectivity can be further controlled. When one or both partners carry several potentially reactive sites, other issues of selectivity must be addressed. Matters of efficiency and site- and/or stereoselectivity need to be resolved at every stage, and conditions must be found to ensure facile catalyst turnover.
Herein, we present a sustainable, three-component, single-vessel catalytic protocol for chemo-, diastereo- and enantioselective conversion of bis(pinacolato)diboron [B2(pin)2], mono-substituted allenes and aldehydes or ketones to 2-B(pin)-substituted homoallylic alkoxides (Scheme 1). Transformations commence by catalytic generation of 2-B(pin)-allylcopper complexes through chemoselective reactions of Cu–B species with allenes, followed by in situ additions to carbonyl substrates.[3] When α,β-unsaturated carbonyls are used, efficient 1,2-allylations remain favored versus boryl or allyl conjugate additions. Catalysts are derived from abundant Cu salts and commercially available precursors of N-heterocyclic carbene (NHC) or phosphine ligands, each offering advantages in chemo- and/or stereoselectivity; allenes are purchased or prepared by high yielding processes.[4] High value products are formed in 18 hours at 4–22 °C with >98% γ-selectivity and 88:12 to >98:2 diastereomeric ratio (d.r.; in 68–92% yield after oxidation); enantioselectivity can be achieved in up to 97:3 enantiomeric ratio (e.r.). To the best of our knowledge, direct catalytic additions of 2-boryl-allyl units to aldehydes are unknown; in cases where 2-boron-substituted allylboron species are prepared separately, alternative modes of site selectivity are observed (more below). There are no catalytic methods for additions of the aforementioned nucleophiles to ketones.[3]
Scheme 1.
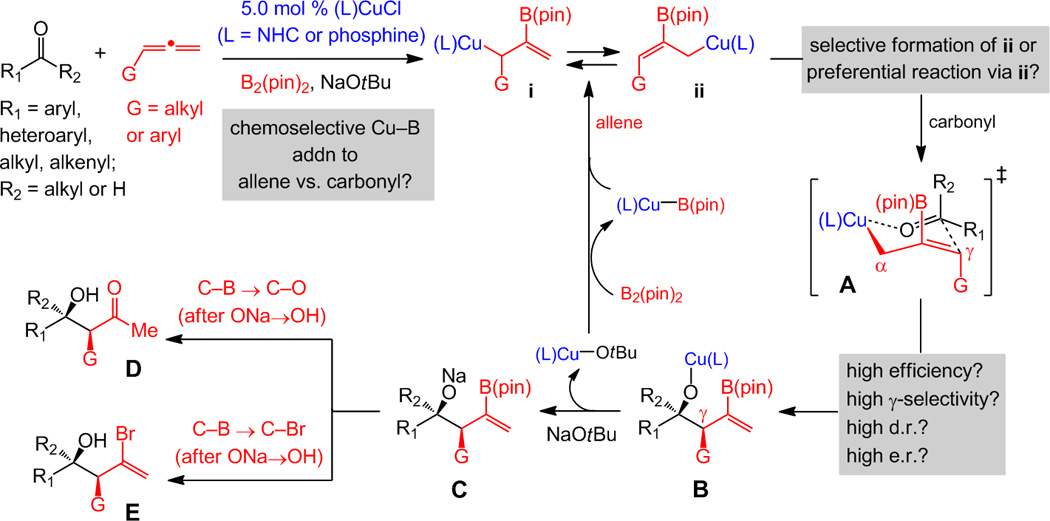
Chemoselective Cu–B addition to an allene, followed by site-, diastereo- and enantioselective addition of the resulting 2-(pinacolato)boron-substituted allylcopper species to aldehydes and ketones would lead to a range of valuable organic molecules by a catalytic multi-component operation; B(pin) = (pinacolato)boron, L = ligand.
We have shown that 2-B(pin)-substituted allylcopper complexes may be accessed by site-selective Cu–B(pin) addition to allenes (Scheme 1).[5a] Since reactions were performed in the presence of MeOH, in situ protonation of the (pin)B-substituted allylcopper complex resulted in net protoboration and regeneration of a catalytically active Cu–B(pin) complex (via a Cu–OMe intermediate). We wondered if Cu–B additions to allenes could proceed chemoselectively with an aldehyde also present (Scheme 1), and whether, without MeOH, the resulting (pin)B-substituted allylcopper would react with the carbonyl substrate to generate B with high γ- and diastereoselectivity. Such a sequence could proceed via A and involve the less congested Cu center of ii; reaction through i (Scheme 1), which might be in equilibrium with ii,[6] would afford isomeric products with one stereogenic center.[7,8] Another question related to Cu complexes adding efficiently to less reactive ketones and whether d.r. values would be high. Oxidation of the C–B bond (after C→D) would render allylcopper complex ii a metal enolate equivalent, delivering products of value for stereoselective synthesis of biologically active polyketides.[9,10]
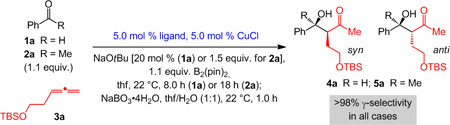
We began by evaluating different Cu-based catalysts (Table 1). Treatment of aldehyde 1a and allene 3a with 5.0 mol % CuCl and salts 6 or 7 affords, after oxidative workup,[11] β-hydroxyketone 4a with complete γ-selectivity and 92:8–94:6 d.r. but in 36% and 41% yield, respectively (entries 1–2).[12] Control experiments show that the moderate efficiency arises from competitive NHC–Cu-catalyzed Cu–B addition to the aldehyde (ca. 90% conv. in 8.0 h without the allene). With the less Lewis basic rac-binap (entry 3), chemoselectivity improves in favor of Cu–B addition to the allene, delivering 4a in 80% yield, >98% γ-selectivity and 95:5 d.r. (<2% B(pin) addition to 1a).[13] In contrast, NHC- or rac-binap-based catalysts promote efficient allylation of ketone 2a (entries 4–6),[14] consistent with a sluggish 1,2-addition of NHC–Cu–B to the carbonyl group.[15] Complete γ-selectivity is observed and diastereoselectivity is high in spite of the diminished size difference between the ketone substituents; unfavorable diaxial interactions in A are likely less severe due to relatively long incipient bonds.[16]
Table 1.
Screening of representative catalyst types.[a]
 | |||||
|---|---|---|---|---|---|
| Entry | R | Ligand | Conv. [%][b] | d.r.[b] | Yield [%][c] |
| 1 | H (1a) | 6 | 47 | 92:8 | 36 |
| 2 | H (1a) | 7 | 58 | 94:6 | 41 |
| 3 | H (1a) | rac-binap | >98 | 95:5 | 80 |
| 4 | Me (2a) | 6 | >98 | 91:9 | 76 |
| 5 | Me (2a) | 7 | >98 | 93:7 | 83 |
| 6 | Me (2a) | rac-binap | >98 | 94:6 | 85 |
Performed under N2 atm.
Determined by analysis of 400 MHz 1H NMR spectra of unpurified mixtures (±2%).
Yields of isolated and purified products (±5%; major isomer for entries 1–3 and both isomers for entries 4–6). See the Supporting Information (SI) for details.

Various aryl-substituted aldehydes can be used (4b-f, Scheme 2); efficiency, γ- and diastereoselectivities are high. Methyl-substituted allene participates are effective substrates (cf. 8a-c, Scheme 2). Aldehydes with aryl groups of diverse electronic and steric attributes are suitable as well (4b-f, 8a-b); those with an electron-deficient substituent (8a-b) require excess allene (5.0 equiv.) since, otherwise, B(pin)-addition predominate. Reactions with alkyl-substituted aldehydes are equally facile and selective (4g and 8c, Scheme 2).
Scheme 2.

Products from sequential 2-B(pin)-substituted allylcopper formation/aldehyde addition/oxidation reactions catalyzed by rac-binap–Cu complex. [a] Reactions performed with 4.0 mol % rac-binap, 4.0 mol % CuCl and 16 mol % NaOtBu under otherwise the same conditions as shown in Table 1, except 5.0 and 2.0 equiv. of allene used for 8a-b and 8c, respectively; >98% conv. in all cases. See the Supporting Information for details.
Aryl ketones are converted to products bearing tertiary hydroxyl groups in ≥79% yield, >98% γ-selectivity, and ≥91:9 d.r. with NHC– or rac-binap–Cu complexes (Scheme 3). Sterically congested ketones (cf. 5b-d), those that contain electron-withdrawing (cf. 5b, 5e-f) or donating substituents (cf. 5c) react with high efficiency and selectivity. The transformation with an ethyl ketone (cf. 5g) is facile but slightly less diastereoselective, presumably due to the reduced size difference between the carbonyl substituents (cf. A, Scheme 1). Heterocyclic substituents are tolerated (e.g., 5h in ≥93:7 d.r.)[17] Cyclic ketones are effective: 5i is isolated in 82–88% yield and up to 94:6 d.r. (Scheme 3). Efficient and selective formation of 9 (83–87% yield, >98:2 d.r.) is notable: it offers an attractive alternative to a propionate ketone-aldol process, where, as would be true in all cases, access to the trisubstituted enolate in high selectivity would be difficult. Unlike reactions with aldehydes, however, use of alkyl-substituted ketones leads to a preponderance of side reactions; it is plausible that the lower electrophilicity of aliphatic ketones renders enolization by NaOtBu and the ensuing undesired reactions more competitive. Products expected from additions to aliphatic ketones could be synthesized by catalytic hydrogenation of the corresponding tertiary allylic alcohols derived from transformations with α,β-unsaturated carbonyls.
Scheme 3.
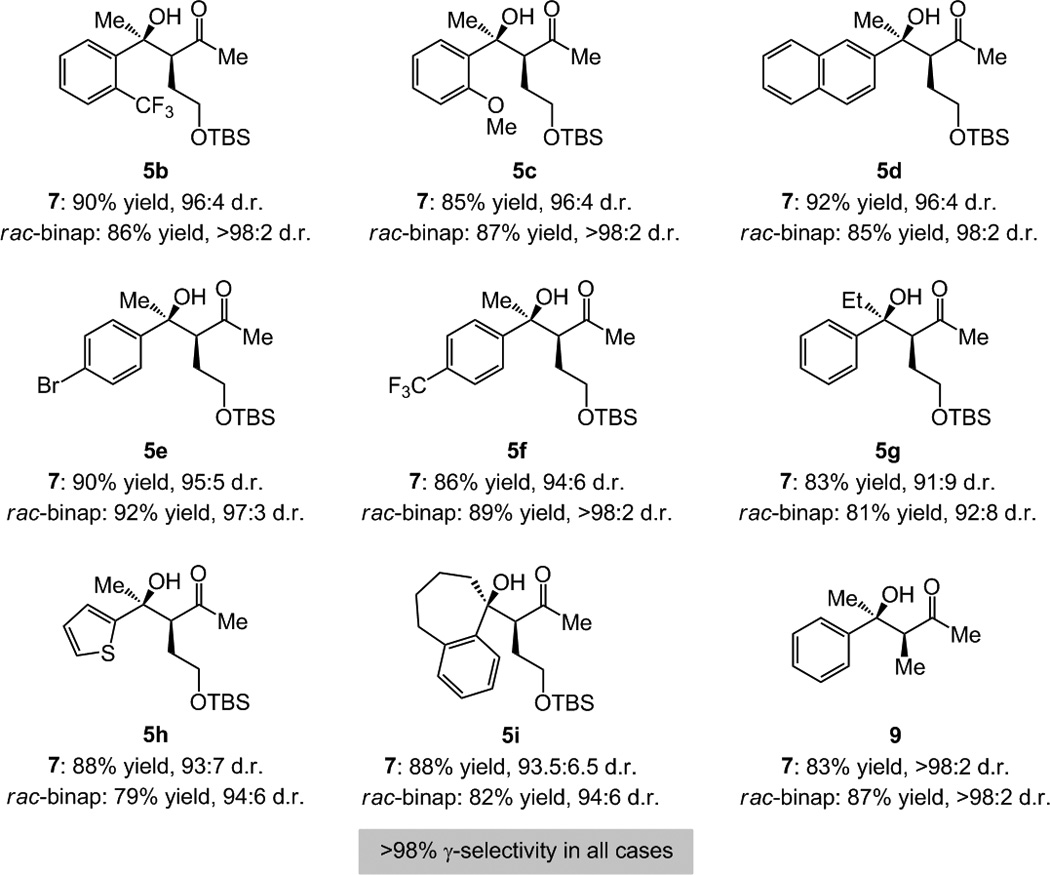
Products from sequential catalytic allylcopper formation/addition to aryl ketones followed by C–B oxidation; >98% conv. in all cases. See Table 1 for conditions and the Supporting Information for experimental and analytical details.
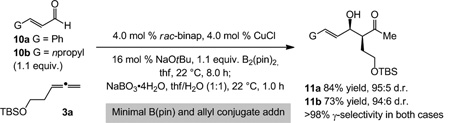
Reactions with unsaturated carbonyls bear the extra complication of possible competitive B(pin) or allyl conjugate addition.[18] Nonetheless, we find that subjection of enals 10a-b to the reaction conditions with 3a and 4.0 mol % rac-binap–Cu complex leads to complete allene consumption, furnishing allylic alcohols 11a-b in ≥73% yield and up to 95:5 d.r. [Eq. (1)]. The challenge of such catalytic transformations is underlined by the finding that without allene 3a, under otherwise identical conditions, there is complete consumption of 10a in only 4.0 hours (>98% conv. for synthesis of 11a-b in 8.0 h).[19]
 |
(1) |
High diastereoselectivities are observed with α,β-unsaturated ketones in spite of the reduced size difference between the carbonyl substituents (vs. aryl ketones; Table 2). With either 5.0 mol % 7 or rac-binap, enones 12a-b are converted to tertiary allylic alcohols in 53–77% yield (entries 1–4, Table 2); the remainder of the material is consumed by adventitious B(pin) conjugate addition (1H NMR analysis).[20] Dienones are appropriate substrates (entries 5–6, Table 2); the increased efficiency in the formation of 13c (81–86% yield) is likely because the hindered cyclic moiety discourages conjugate addition. Diastereoselectivities are higher with the NHC-based catalysts; the larger size of the P-based ligand might destabilize the chair-type transition state for the carbonyl addition (cf. A, Scheme 1). Reactions shown in Scheme 3 do not exhibit such a trend, as the more sizeable aryl units of the ketone better enforce a chair-like transition structure.
Table 2.
Sequential catalytic reactions with α,β-unsaturated ketones.[a]
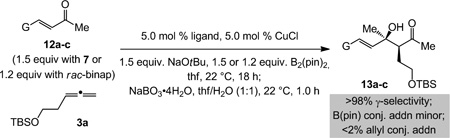 | ||||
|---|---|---|---|---|
| Entry | Substrate; G | Ligand | d.r.[b] | Yield [%][c] |
| 1 | 12a; Ph | 7 | >98:2 | 64 |
| 2 | 12a; Ph | rac-binap | 91:9 | 68 |
| 3 | 12b; npent | 7 | >98:2 | 53 |
| 4 | 12b; npent | rac-binap | 91.5:8.5 | 77 |
| 5 | 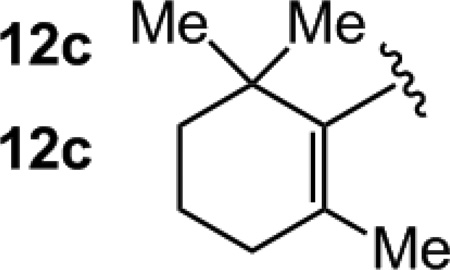 |
7 | 90:10 | 86 |
| 6 | rac-binap | 87:13 | 81 | |
Performed under N2 atm; 1.5 and 1.2 equiv. B2(pin)2 used with 7 and rac-binap, respectively.
Determined by analysis of 400 MHz 1H NMR spectra of unpurified mixtures (±2%).
Yields of isolated and purified products (±5%; both isomers). See the Supporting Information (SI) for details.
Several matters of efficiency and selectivity underscore the distinguishing advantages of the present approach. Pd-catalyzed diboron additions to allenes deliver 2-B(pin)-substituted allylborons, which, unlike the reactions described above, react with aldehydes to generate acetate aldol products (after oxidation; Scheme 4, part 1);[21] related additions to ketones would require an additional catalyst (not reported). Our attempts to utilize the same B(pin)-substituted set of allylboron species as precursors to allylcopper intermediates, as summarized in Scheme 4 (part 2), led to predominance of the same mode of site selectivity as mentioned above (cf. part 1); this is largely because the requisite 2-B(pin)-allylcopper species cannot be generated efficiently through such a pathway.[22] A different type of Pd-catalyzed diboron addition to allenes furnish achiral B(pin)-substituted allyboron isomers bearing a trisubstituted vinylboron moiety[23] (Scheme 4, part 3 vs. part 1). These latter entities, similar to allylcopper ii in Scheme 1, can react with aldehydes to yield product isomers represented by B (R2 = H, Scheme 1); however, enantioselective reactions with aldehydes as well as any additions to ketones, would require the use of a second catalyst. It is unlikely that allylcopper species are converted to allylboron intermediates that then undergo addition; otherwise, two equivalents of B2(pin)2 would be required for complete conversion (vs. 1.1–1.5 equiv. used), additions to aldehydes would likely not be subject to enantioselective catalysis (see below), and there would be minimal reaction with ketones.
Scheme 4.
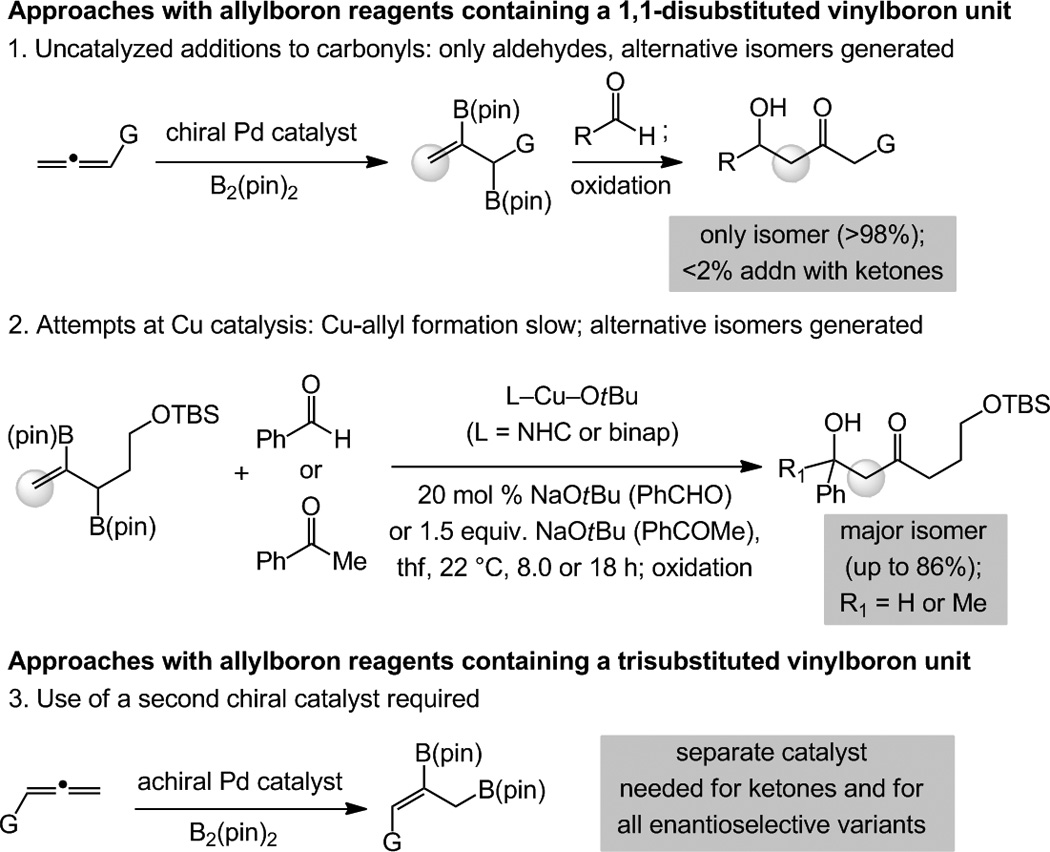
Strategies involving initial preparation and use of 2-B(pin)-substituted allylboron reagents leads to alternative product isomers or demand the identification and use of a second catalyst, in contrast to the present Cu-catalyzed approach.
Vinyl bromides (Scheme 5) are another set of useful isolable derivatives that can be accessed.[11] By subjection of the product mixture to CuBr214a-b, originating from reaction of phenyl-substituted allene 3b, and 15a-b[24] are obtained in 77–89% yield and 88:12 to >98:2 d.r., respectively. As far as we are aware, methods for stereoselective synthesis of this type of vinyl halide-containing tertiary alcohols by direct allyl additions to ketones are unavailable.[25,26]
Scheme 5.
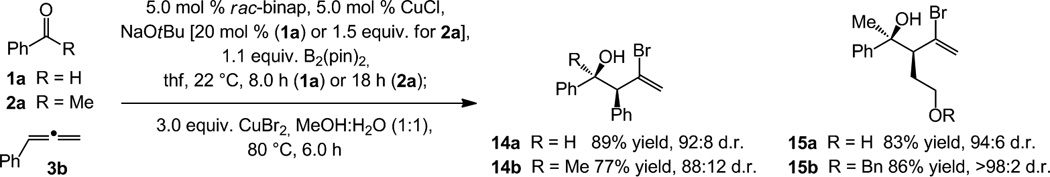
The Cu-catalyzed protocol can be used to obtain valuable vinylbromides with high efficiency.
Phosphine-Cu complexes promote the catalytic process in up to 97:3 enantiomeric ratio (e.r.);[27] reactions with aryl-, alkenyl- or alkyl-substituted carbonyls afford products that are not readily accessible in high selectivity by catalytic enantioselective aldol additions (Scheme 6).[28] With various chiral NHC-based complexes, enantioselectivities do not exceed 60:40 e.r.[4] Two purchasable chiral bis-phosphines (16 and 17) provide optimal enantioselectivities for additions to aldehydes and ketones;[29] the precise origin of such variations is the subject of current studies.
Scheme 6.
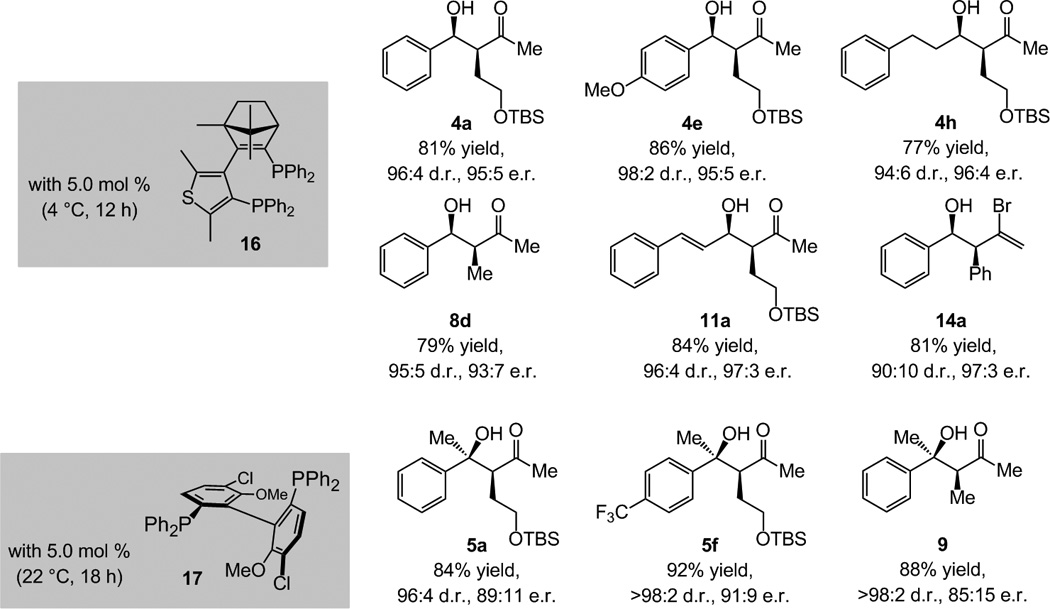
Cu-catalyzed γ- and diastereoselective coupling of allenes with carbonyls can be performed enantioselectively through the use of chiral bis-phosphine ligands; >98% conv. in all cases. For exact conditions, see Table 1 and Scheme 5.
Design of more effective chiral catalysts for reactions with ketones as well as applications to natural product synthesis are in progress.
Supplementary Material
Footnotes
Financial support was provided by the NSF (CHE-1111074) and the NIH (GM-57212). We thank Dr. F. Haeffner and E. M. Vieira for valuable discussions.
References & Footnotes
- 1.For a review on multi-component catalytic reactions, see: Touré BB, Hall DG. Chem. Rev. 2009;109:4439. doi: 10.1021/cr800296p.
- 2.For a relevant discussion, see: Bower JF, Kim IS, Patman RL, Krische MJ. Angew. Chem. Int. Ed. 2009;48:34. doi: 10.1002/anie.200802938. 2-Substituted allylmetal species cannot be accessed by Ir- and Ru-catalyzed reductive couplings with allenes, and there are no related processes involving ketones
- 3.Yus M, González-Gómez JC, Foubelo F. Chem. Rev. 2011;111:7774. doi: 10.1021/cr1004474. [DOI] [PubMed] [Google Scholar]
- 4.See the Supporting Information for details.
- 5. Jung B, Hoveyda AH. J. Am. Chem. Soc. 2012;134:1490. doi: 10.1021/ja211269w. For a related study, see: Yuan W, Ma S. Adv. Synth. Catal. 2012;354:1867.
- 6.The allylcopper species are represented as η1 (vs. π-allyl) complexes, based on computational studies and a report regarding the electronically related allylzinc systems by Okuda. See: Lichtenberg C, Engel J, Spaniol TP, Englert U, Raabe G, Okuda J. J. Am. Chem. Soc. 2012;134:9805. doi: 10.1021/ja303480a.
- 7.Catalytic coupling reactions of allenes and aldehydes, involving in situ-generated allylmetal complexes, have been reported; none utilize abundant metal salts (e.g., Cu) and allyl units are not 2-B(pin)-substituted. For Ircatalyzed versions, see: Skucas E, Bower JF, Krische MJ. J. Am. Chem. Soc. 2007;129:12678. doi: 10.1021/ja075971u. For Ru-catalyzed diastereoselective variants, see: Zbieg JR, McInturff EL, Leung JC, Krische MJ. J. Am. Chem. Soc. 2011;133:1141. doi: 10.1021/ja1104156.
- 8.For an overview of (largely Pd-catalyzed) three-component coupling reactions with allenes, affording reagents that can be used in allyl addition processes, see: Jeganmohan M, Cheng C-H. Chem. Commun. 2008:3101. doi: 10.1039/b800440d.
- 9.For three-component catalytic non-enantioselective access to ketone aldol-type products, see: Tanabe Y, Matsumoto N, Higashi T, Misaki T, Itoh T, Yamamoto M, Mitarai K, Nishii Y. Tetrahedron. 2002;58:8269.
- 10.For a review on polyketide natural products, see: O’Hagan D. The Polyketide Metabolites. Ellis Horwood: Chichester, U.K.; 1991.
- 11.The vinylboron products are unstable to silica gel; such instability might originate from boron chelation with the neighboring alcohol.
- 12.Similar results were obtained with aryl-substituted imidazolium or alkyl-substituted imidazolinium salts; see the Supporting Information.
- 13.For direct Ti-catalyzed aldol addition involving ketones and aryl aldehydes, see: Mahrwald R, Schetter B. Org. Lett. 2006;8:281. doi: 10.1021/ol052637z.
- 14.Excess NaOtBu is needed for reactions with ketones, presumably because L–Cu–B(pin) regeneration by reaction of the more sterically congested Cualkoxide B with B2(pin)2 is slower via σ-bond metathesis and must proceed by alkoxide-assisted ligand exchange (vs. 16 mol % base needed with an aldehyde). See: Gao F, Carr JL, Hoveyda AH. Angew. Chem. Int. Ed. 2012;51:6613. doi: 10.1002/anie.201202856.
- 15.Treatment of ketone 2a to the conditions in entry 5 of Table 1 but in the absence of allene 3a results in <2% conversion after 18 hours.
- 16.Further Cu–B additions to the vinylboron products do not occur; see: Lee Y, Jang H, Hoveyda AH. J. Am. Chem. Soc. 2009;131:18234. doi: 10.1021/ja9089928.
- 17.Reactions with pyridyl aldehydes or ketones lead to <2% conversion.
- 18.For Cu-catalyzed B(pin) conjugate additions to unsaturated carbonyls, see: Hartmann E, Vyas DJ, Oestreich M. Chem. Commun. 2011;47:7917. doi: 10.1039/c1cc10528k.
- 19.High chemoselectivity in favor of reaction initiating with addition to the allene in spite of a slower rate of transformation with an enal might be due to a substantially faster rate of Cu–allene coordination.
- 20.As with aldehydes (Table 1), the initial Cu–B(pin) additions are more chemoselective with the P-based complex. Since 1,2-addition of B(pin) to ketones is slow, excess enone is used with 7 to maximize efficiency.
- 21.Woodward AR, Burks HE, Chan LM, Morken JP. Org. Lett. 2005;7:5505. doi: 10.1021/ol052312i. [DOI] [PubMed] [Google Scholar]
- 22.Because C–B to C–Cu transmetallation is inefficient, uncatalyzed allylboration of aldehydes and metal-alkoxide-catalyzed addition of the same reagents to ketones predominate (NaOtBu is needed for C–Cu formation).
- 23. Yang F-Y, Cheng C-H. J. Am. Chem. Soc. 2001;123:761. doi: 10.1021/ja005589g. For reactions with (pin)B–SiMe2Ph and diastereoselective addition of 2-silyl-substituted allylborons to aldehydes, see: Chang K-J, Rayabarapu DK, Yang F-Y, Cheng C-H. J. Am. Chem. Soc. 2005;127:126. doi: 10.1021/ja044662q. For related studies, see: Suginome M, Nakamura H, Matsuda T, Ito Y. J. Am. Chem. Soc. 1998;120:4248. Zbieg JR, Moran J, Krische MJ. J. Am. Chem. Soc. 2011;133:10582. doi: 10.1021/ja2046028.
- 24.Alcohol 15a is derived from reaction of 3a. For references regarding synthesis of 2-halogen-substituted products see the Supporting Information.
- 25.For catalytic diastereoselective allyl additions to ketones (not 2-B(pin)-substituted), see: Ren H, Dunet G, Mayer P, Knochel P. J. Am. Chem. Soc. 2007;129:5376. doi: 10.1021/ja071380s. Peng Z, Blümke TD, Mayer P, Knochel P. Angew. Chem. Int. Ed. 2010;49:8516. doi: 10.1002/anie.201003813. Takeda T, Yamamoto M, Yoshida S, Tsubouchi A. Angew. Chem. Int. Ed. 2012;51:7263. doi: 10.1002/anie.201202808. For reactions of ketones and stoichiometric amounts of allylboronic acids (not 2-B-substituted), isolated from allylic alcohols by a Pd-catalyzed process, see: Raducan M, Alam R, Szabó KJ. Angew. Chem. Int. Ed. 2012;51:13050. doi: 10.1002/anie.201207951. See the Supporting Information for additional references.
- 26.For use of enantiomerically pure reagents for additions of substituted allyl units to carbonyls (no 2-B(pin) group), see the Supporting Information.
- 27.For catalytic enantioselective allyl additions to ketones (not 2-B(pin)- substituted), see: Wada R, Oisaki K, Kanai M, Shibasaki M. J. Am. Chem. Soc. 2004;126:8910. doi: 10.1021/ja047200l. Wadamoto M, Yamamoto H. J. Am. Chem. Soc. 2005;127:14556. doi: 10.1021/ja0553351. Lou S, Moquist PN, Schaus SE. J. Am. Chem. Soc. 2006;128:12660. doi: 10.1021/ja0651308. Jiang X, Cao Y, Wang Y, Liu L, Shen F, Wang R. J. Am. Chem. Soc. 2010;132:15328. doi: 10.1021/ja106349m.
- 28.Direct enantioselective catalytic aldol additions with aldehydes have been reported (none with ketones). Reactions with cyclic ketones (enol precursors) are more common and the large majority of acyclic cases involve reactions with strongly electron-deficient aryl aldehydes and generate products that a bear a Me or an alkoxy unit adjacent to the carbonyl (higher reactivity of the enol); low to moderate regioselectivity is typically observed (propionate vs. acetate aldol). For example, see: Luo S, Xu H, Li J, Zhang L, Cheng J-P. J. Am. Chem. Soc. 2007;129:3074. doi: 10.1021/ja069372j. Aratake S, Itoh T, Okano T, Nagae N, Sumiya T, Shoji M, Hayashi Y. Chem. Eur. J. 2007;13:10246. doi: 10.1002/chem.200700363. Reactions with aliphatic aldehydes are scarce (none with enals); one reported example requires >140 h; see: Ma G, Bartoszewicz A, Ibrahem I, Córdova A. Adv. Synth. Catal. 2011;353:3114.
- 29.For example, 4a is generated in 95:5 d.r. and 90:10 e.r. (87% yield) when 17 is used, whereas 5a is formed in 92:8 d.r. and 76:24 e.r. (82% yield) with 16.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.