

Two studies plant a signpost on the road towards a robust and detailed chromatin interaction map
Chromosome conformation capture technologies are transforming our understanding of genome spatial organization. However, our ability to infer chromatin interactions is limited by two difficult problems: generating high resolution signals, and separating signals from multiple sources of bias. Two new studies address these challenges. The first describes a novel workflow and computational pipeline for high-resolution 4C-seq1, and the second reports a new strategy for simultaneously eliminating multiple sources of bias from Hi-C data2.
While the linear sequence of the human genome was mapped a decade ago, we have only just begun deciphering details of its spatial organization. Recent studies have shown that distant genomic elements (such as enhancers and promoters) can be brought into close proximity by chromatin interactions, transforming our understanding of gene regulation and emphasizing the importance of the 3D chromatin structure. Our ability to map chromatin interactions is being revolutionized by chromosome conformation capture (3C) technology3. All 3C-based methods begin with the digestion of chromatin in fixed cells, followed by the re-ligation of cleaved DNA. In this way, they tie together DNA fragments that are in spatial proximity even if they are remote in sequence4. The characterization of such ligation junctions (for example by sequencing) yields a detailed map of chromatin interactions, averaged over a population of cells. The first 3C technology looked at certain loci, permitting a ‘one-versus-some’ exploration of interactions between preselected regions3. In contrast, 4C5, 6 and Hi-C methods4, 7 profile interactions across the entire genome on a “one-versus-all” and “all-versus-all” basis, respectively. 4C and Hi-C experiments cast a wide net, but these approaches have significant costs: weaker signal at a given sequencing depth, and the multiplication of possible biases with technical or biological sources.
A paper from the de Laat and Tanay groups (van de Werken et al.)1 introduces a 4C-seq method of characterizing chromatin interactions at considerably higher resolution than was previously possible, paving the way for a more accurate and refined description of regulatory interactions at specific functional elements. Their approach begins with the usual steps of cross-linking, digestion and DNA ligation (Figure 1A). The ligated DNA entities, which at this point typically contain multiple restriction fragments, are subjected to an additional round of digestion by a different restriction enzyme. Ligation signals specific to the DNA region of interest (the “viewpoint”) are then amplified using inverse PCR, followed by next-generation sequencing1, 8.
Figure 1.
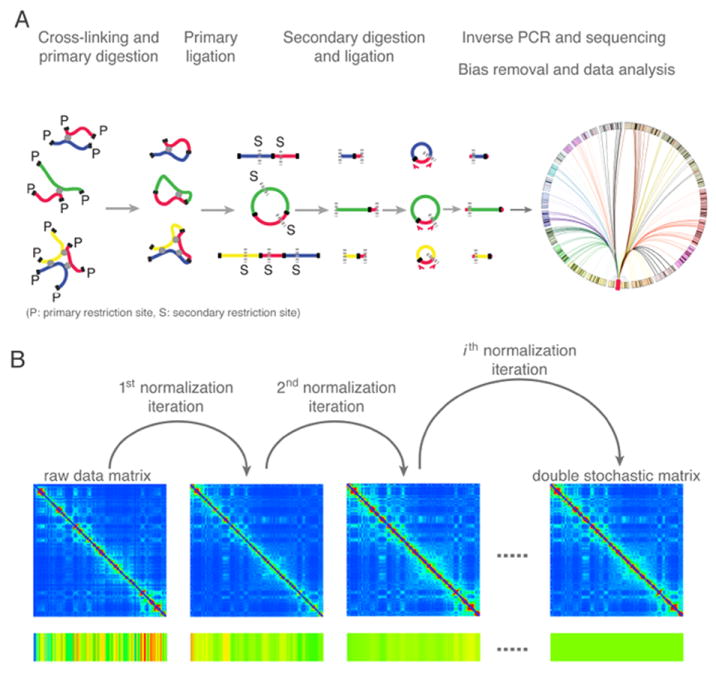
A. Overview of the high-resolution 4C-seq method1, 8. The illustration shows three possible contacts between the viewpoint DNA fragment (red) and other fragments (blue, green, and yellow) mediated by proteins (depicted as grey circles). First, cross-linked chromatin is digested with a 4-base cutting primary restriction enzyme (cutting sites “P” are depicted as solid black lines), followed by proximity ligation. Subsequently, the cross-links are reversed, and DNA fragments are digested with a second, different 4-base cutting restriction enzyme (cutting sites “S” are depicted as dotted grey lines), followed by circularization. Captured fragments containing the viewpoint sequence are then amplified by inverse PCR, followed by high-throughput sequencing. Finally, the “one versus all” contact map is established by computational analysis. B. Illustration of the iterative correction procedure on raw chromosome conformation capture data10. The color bar at the bottom represents the magnitude of the row sums. In an iteration, each entry of the matrix is divided by the product of the visibility biases of the two interacting regions, where the bias of a region is calculated as the normalized sum of the corresponding row. After several iterations, the Hi-C map converges to a normalized matrix where each row and column sums to 1.
Importantly, to maximize the number of interacting fragments, both rounds of digestion employ restriction enzymes with 4-base specificity, which increases the pool of available fragments tenfold in comparison to previously employed 6-base cutters1, 5, 8. One might think that reducing the size of the fragments would suffice to increase resolution. However, increased resolution may also introduce increased bias. For example, by decreasing the average length of the primary restriction fragments, 4C-seq also increases the proportion of primary fragments that do not contain a secondary restriction site. Hence, the final solution contains some fragments that were digested twice and some only once. Because PCR amplification is more efficient for the shorter fragments, a systematic bias can be introduced in the readout. In addition, the experimental coverage is affected by multiple other biases, including the efficiency of the restriction enzymes and the mappability of sequenced fragments to the genome. Improved experimental power goes hand in hand with the need for a more rigorous statistical treatment of data biases.
Van de Werken et al. introduce a computational framework to correct biases by dividing restriction fragments into classes with similar features, such as GC content and fragment length. They apply two complementary strategies to correct the contact intensities. For long-range interactions, they estimate a background probability of coverage for each fragment class, and calculate the enrichment between observed and expected fragment coverage for each class separately. For short-range interactions involving the region surrounding the viewpoint, a different procedure is necessary because contact coverage changes dramatically with sequence distance from the viewpoint. However, because the coverage profiles of different fragment classes represent the same underlying distribution, they can be compared after quantile normalization.
The authors demonstrated the significantly increased resolution of their new 4C-seq method on three different loci: beta-globin, Oct4, and Satb1. They detect ~1000 fragment ends in the 150kb β-globin domain alone, compared to the few dozen reported in previous experiments. They further demonstrate that genome-wide contact profiles are highly reproducible between different viewpoints within the same locus. For instance, in the β-globin locus, 4C-seq robustly detects known DNA interactions and reveals contact details missed by other approaches, which may represent new regulatory interactions. The 4C-seq profile of Oct4 revealed specific contacts between the transcription start site and an unexpectedly distant domain 17 kb upstream. The 4C-seq profile for the Satb1 locus suggests that the large proximal domain acts as a regulatory scaffold, facilitating the high expression of Satb1 in T-cells. Together, these experiments demonstrate that high-resolution 4C-seq is a robust, efficient, and direct method to screen the entire genome for regulatory DNA elements contacting a promoter of interest.
The second paper, contributed by the Mirny and Dekker groups (Imakaev et al.)2, focuses on genome-wide interactions among all regions at lower resolution. As with the 4C-seq method, Hi-C data are affected by multiple technical and biological biases that need to be corrected. This task is difficult, not just because one must imagine all possible sources of bias, but because the magnitude and direction of a given bias will vary between experimental protocols. To address this challenge, Imakaev et al. developed an integrated pipeline that removes many biases from Hi-C data without a priori knowledge of their technical or biological sources. In other words, it removes biases in an unbiased way. The core assumption of Imakaev et al. is that in an unbiased experiment all genomic regions should be detected with the same “visibility”, meaning that they should be seen in the experiment with the same probability. Moreover, the authors assume that the visibility bias of each pairwise contact is factorizable; that is, it can be expressed as the product of two biases that depend only on the respective interacting regions. Based on these assumptions, the matrix of Hi-C raw counts can be iteratively normalized. In an iteration cycle, each entry of the raw Hi-C map is divided by the product of the visibility biases of the two interacting regions, where the bias of a region is calculated as the sum of the corresponding row, normalized over the mean of all row sums. When the biases converge, the Hi-C map is transformed into a normalized relative contact probability matrix whose rows and columns each sum to 1, which is a double stochastic matrix (Figure 1B). Unlike previous attempts to correct Hi-C data using only a single corrective cycle7, this iterative procedure achieves robust and stable normalization and eliminates all factorizable biases. Strikingly, when Imakaev et al.2 apply their method to Hi-C data for a human lymphoblastoid cell line, the biases correlate extremely well with those restriction fragment level biases recently identified using a probabilistic approach9, thereby mutually confirming the power of both methods.
The research of Imakaev et al.2 is not limited to error analysis. They perform an eigenvector decomposition of the corrected probability map, and discover interesting features of the multi-level chromatin organization that are not readily detectable in standard heat maps. In particular, they find that contact probabilities depend mainly upon two factors: one related to the genomic sequence and local epigenetic chromatin states, and a second related to the region’s position along the chromosome arm. They also apply their pipeline to the Hi-C data of human and mouse, demonstrating remarkable evolutionary conservation of genome-wide chromosome organization in syntenic regions at the megabase level.
As demonstrated by these two papers, we are seeing dramatic improvements in experimental technology and analysis methods to determine chromatin contact maps, a highly promising path towards understanding 3D genome structures. Such knowledge would drastically change our view of the human genome landscape. Combined with diverse genomic, epigenomic, and imaging data, the community will soon be ready to establish detailed structure-function maps of the genome.
This is a commentary on article van de Werken HJ, Landan G, Holwerda SJ, Hoichman M, Klous P, Chachik R, Splinter E, Valdes-Quezada C, Oz Y, Bouwman BA, Verstegen MJ, de Wit E, Tanay A, de Laat W. Robust 4C-seq data analysis to screen for regulatory DNA interactions. Nat Methods. 2012;9(10):969-72.
References
- 1.van de Werken HJG, et al. 4C-seq for robust identification of physical interactions between genes and regulatory elements. Nature Methods. 2012 [Google Scholar]
- 2.Imakaev M, et al. Iterative correction of Hi-C data reveals new features of chromosome organization. Nature Methods. 2012 doi: 10.1038/nmeth.2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dekker J, Rippe K, Dekker M, Kleckner N. Capturing chromosome conformation. Science. 2002;295:1306–1311. doi: 10.1126/science.1067799. [DOI] [PubMed] [Google Scholar]
- 4.Hakim O, Misteli T. SnapShot: Chromosome confirmation capture. Cell. 2012;148:1068.e1061–1062. doi: 10.1016/j.cell.2012.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Simonis M, et al. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C) Nat Genet. 2006;38:1348–1354. doi: 10.1038/ng1896. [DOI] [PubMed] [Google Scholar]
- 6.Zhao Z, et al. Circular chromosome conformation capture (4C) uncovers extensive networks of epigenetically regulated intra- and interchromosomal interactions. Nat Genet. 2006;38:1341–1347. doi: 10.1038/ng1891. [DOI] [PubMed] [Google Scholar]
- 7.Lieberman-Aiden E, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Splinter E, de Wit E, van de Werken HJG, Klous P, de Laat W. Determining long-range chromatin interactions for selected genomic sites using 4C-seq technology: From fixation to computation. Methods (San Diego, Calif) 2012 doi: 10.1016/j.ymeth.2012.04.009. [DOI] [PubMed] [Google Scholar]
- 9.Yaffe E, Tanay A. Probabilistic modeling of Hi-C contact maps eliminates systematic biases to characterize global chromosomal architecture. Nat Genet. 2011;43:1059–1065. doi: 10.1038/ng.947. [DOI] [PubMed] [Google Scholar]
- 10.Kalhor R, Tjong H, Jayathilaka N, Alber F, Chen L. Genome architectures revealed by tethered chromosome conformation capture and population-based modeling. Nature biotechnology. 2012;30:90–98. doi: 10.1038/nbt.2057. [DOI] [PMC free article] [PubMed] [Google Scholar]