

At 30,000 feet, ATP-sensitive potassium (KATP) channels are seen coupling the metabolic status of the cell to its electrical excitability. But down at the molecular level of the β-cell, there is a lot more coupling going on in exactly how the KATP channel does this. Coupling is developed in the conceptual framework of linkage (1), which thermodynamically quantifies how conformational states of high and low affinity in regulatory ligand binding sites reciprocally link with additional sites, and active and inactive conformations of proteins. It is also at the heart of a molecular medicine puzzler with a satisfying solution originating from a study by Proks et al. (2) reported in this issue.
The main gate of the KATP channel opens and shuts to regulate β-cell electrical excitability and insulin secretion. Only a small number of the channels need be open for outflow of potassium ions to keep the β-cell electrically at rest, thus preventing insulin secretion. In a high glucose setting, however, β-cell metabolism generates ATP at the expense of ADP, which effectively shuts all KATP channels to stop potassium ion outflow. This leads to activation of voltage-dependent calcium channels and calcium influx, which triggers secretory granule exocytosis mediating insulin secretion.
Regulatory ligand binding sites link the main gate of the KATP channel to glucose metabolism via ATP, MgATP, and MgADP (3). The β-cell KATP channel is a hetero-octameric protein of four inner pore-forming Kir6.2 and four outer sulfonylurea receptor 1 (SUR1) subunits (Fig. 1). The gate is formed by concerted action of all four Kir6.2 subunits (4). Each Kir6.2 subunit also has an independent ATP site whose occupancy effectively can shut the gate and can keep it shut (5), referred to as inhibition. Pharmacological ligands also inhibit the KATP channel. Each SUR1 subunit has an independent sulfonylurea site (6) whose occupancy not so effectively shuts the same gate. Partial linkage of sulfonylurea site occupancy and gate closure is one of two keys to solving the diabetes puzzler—even when all SUR1 sulfonylurea sites are saturated KATP channels exhibit 50–80% inhibition (2,7–9). On its own, this is not enough inhibition to trigger secretion. Each SUR1 also has binding sites for MgATP and MgADP (MgAXPs) whose occupancy can open the same gate and can keep it open (10,11), referred to as activation. MgATP can be hydrolyzed with bound MgADP markedly prolonging activation (12). KATP channels activated by MgAXPs bound at SUR1 prevent insulin secretion.
FIG. 1.
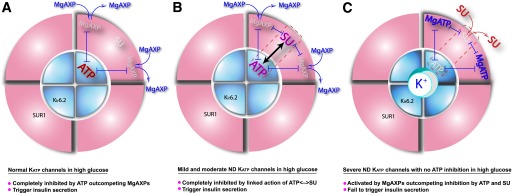
Linkage relationships define how metabolic nucleotides interact with antidiabetic sulfonylureas and inform the molecular medicine of ND. The KATP channel is an octameric protein with four outer regulatory SUR1 subunits and four inner pore-forming Kir6.2 subunits. Cytosolic metabolic nucleotides and sulfonylurea (SU) ligands bind all of these subunits but are shown above for one SUR1-Kir6.2 subunit pair for clarity. Regulation is integrated mainly through a series of reciprocal heterotropic ligand–ligand interactions indicated by double arrows when enhancing each other and double perpendicular stops when opposing each other. Proks et al. (2) show convincing evidence for SU at SUR1 and ATP at Kir6.2 in positive linkage enhancing one another’s inhibition of the KATP channel, which has critical consequences for understanding ND treatment as follows. A: Normal KATP channels in high glucose. ATP/MgATP ratio is high favoring ATP in ATP |––| MgAXP negative linkages, which completely inhibits the KATP channels, stimulates insulin secretion, and normalizes high blood glucose. B: Mild and moderate ND KATP channels, which have mild or moderate effects on ATP inhibition, in high glucose. ATP/MgATP ratio is high and enough ATP inhibition remains to combine and positively link with SU inhibition, ATP<–>SU, each of which negatively links with MgAXP, ATP |––| MgAXP, and SU |––| MgAXP to outcompete MgAXP activation. This results in full inhibition of the channels, triggering insulin secretion and normalizing blood glucose. C: Severe ND KATP channels with severely reduced or absent ATP inhibition in high glucose. The ATP/MgATP ratio is high but severe disease mutations (e.g., Kir6.2-G334D, -L164P, and -I296L) leave insufficient ATP inhibition, be it wiping out the ATP site or the gate, for enhanced linkage effects, so that current sulfonylureas cannot compete alone, and MgAXP activation is favored, resulting in activated KATP channels hyperpolarizing the β-cell and turning off insulin secretion, so blood glucose remains high.
A gold mine of linkages. These regulatory ligand sites not only link to the gate but also to each other, enhancing and opposing one another in an exquisite regulatory mechanism orchestrating changes in glucose metabolites into changes in KATP channel gating governing insulin secretion (Fig. 1). There is positive linkage where ATP inhibition at Kir6.2 and sulfonylurea inhibition at SUR1 enhance each other (2), which turns out to be the second key to the puzzler. In addition, ATP inhibition at Kir6.2 and MgAXP activation at SUR1 show negative linkage, opposing each other. Sulfonylurea inhibition and MgAXP activation at SUR1 also show negative linkage.
The diabetes puzzler arose from initial success transferring patients with neonatal diabetes (ND) caused by ATP site mutations from insulin injection to oral sulfonylurea (13,14). The pharmacogenetic heuristic was that sulfonylurea sites at SUR1 provide an alternate route to gate closure to stimulate insulin secretion when ATP sites at Kir6.2 are mutated. However, Masia et al. (15) established a striking exception to the rule of thumb. A patient with ND caused by Kir6.2-G334D was unresponsive to sulfonylurea. The mutant KATP channel essentially exhibited no inhibition by ATP, as expected from earlier characterization of the mouse mutant channel that positioned G334 in the ATP site (16–18). Thus, the puzzler—how could a mutation in the inhibitory ATP site of Kir6.2 render the KATP channel unresponsive to sulfonylurea in vivo? A cellular explanation preferred by Masia et al. (15) is that G334D disruption of insulin secretion leads to progressive loss of β-cell mass until sulfonylurea is ineffective. Mouse ND models do show coordinate losses in β-cell mass and responsiveness to sulfonylurea, but the losses require uncompensated diabetes (19), unlike clinical cases compensated by insulin replacement. They concluded that the moderate loss in sulfonylurea sensitivity observed in vitro is unlikely to account for sulfonylurea unresponsiveness in the G334D patient.
Not necessarily so fast. Proks et al. (2) provide convincing evidence for an alternate, molecular mechanism: Unresponsiveness to sulfonylurea drugs in the G334 patient and others with severely reduced ATP inhibition can be explained by loss of the positive linkage interaction between ATP sites on Kir6.2 and sulfonylurea sites on SUR1. Uncommon command of heterotropic ligand linkage interactions in the KATP channel enabled Proks et al. to design incisive experimental and theoretical tests. Regardless of whether the ND mutation is in gating (I296L) or ATP site (G334D) domains of Kir6.2, in the absence of ATP inhibition, the incomplete sulfonylurea inhibition cannot out battle physiological MgAXP activation, with consequent hyperpolarization of the β-cell preventing adequate insulin secretion. Absent any ATP inhibition, the best sulfonylureas are unable to completely inhibit KATP channels for insulin secretion at the molecular level, with or without loss of β-cell mass at the cellular level in ND patients.
How else do SUR1 and Kir6.2 talk to each other and to themselves? Additional mutations including in SUR1 (F132V, distant from the sulfonylurea binding site and not altering nucleotide-independent gating) also cause ND unresponsive to sulfonylurea (20). Finding out how such ND SUR1 mutations might lesion the linkage with sulfonylurea inhibition will be exciting. Also, how might four independent ATP sites that show no cooperativity (5) link with a concerted inhibition gate (4)? A sequential ligand-dependent, conditional homotropic ligand linkage model has been proposed where only ATP-bound sites engage the linkage to the inhibited conformation of the gate (17), consistent with sequential interaction Adair-Koshland-Nemethy-Filmer and Hammes-Wu models (21).
The findings of Proks et al. (2) raise additional clinical questions: Are “full agonist” sulfonylureas possible, which would more strongly displace the gating equilibria of severe ND mutant KATP channels to complete inhibition, and so provide a sulfonylurea option for ND patients with severe reductions in ATP inhibition? Also, let us not lose sight of the loss in β-cell mass model in ND (15,19). In some ND patients, a loss in ATP<–>sulfonylurea linkage might have to combine with loss of β-cell mass to effect unresponsiveness to sulfonylurea. We actually do not have the measurements needed in the ND cases to know! Clinical measures of changes in functional β-cell mass including plasma C-peptide levels might correlate with changes in glycemic control over time in ND. Could improvements in functional β-cell mass underlie the reduction over time in sulfonylurea dose for normoglycemia that typically occurs after transfer from insulin therapy? Given dependence of loss in β-cell mass on genetic predisposition and lifestyle, can patients with ND caused by certain KATP channel mutations go from sulfonylurea responsive to unresponsive, as a function of progressive loss in β-cell mass, as often occurs in later stages of type 2 diabetes?
ACKNOWLEDGMENTS
This work was supported by an American Diabetes Association Basic Science Award (RA-1-06-39) and the Pittsburgh Foundation Medical Research Fund (M2009-0041).
No potential conflicts of interest relevant to this article were reported.
Footnotes
See accompanying original article, p. 3909.
REFERENCES
- 1.Wyman J., Jr Linked functions and reciprocal effects in hemoglobin: a second look. Adv Protein Chem 1964;19:223–286 [DOI] [PubMed] [Google Scholar]
- 2.Proks P, de Wet H, Ashcroft FM. Molecular mechanism of sulphonylurea block of KATP channels carrying mutations that impair ATP inhibition and cause neonatal diabetes. Diabetes 2013;62:3909–3919 [DOI] [PMC free article] [PubMed]
- 3.Proks P, Ashcroft FM. Modeling K(ATP) channel gating and its regulation. Prog Biophys Mol Biol 2009;99:7–19 [DOI] [PubMed] [Google Scholar]
- 4.Drain P, Geng X, Li L. Concerted gating mechanism underlying KATP channel inhibition by ATP. Biophys J 2004;86:2101–2111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Markworth E, Schwanstecher C, Schwanstecher M. ATP4- mediates closure of pancreatic β-cell ATP-sensitive potassium channels by interaction with 1 of 4 identical sites. Diabetes 2000;49:1413–1418 [DOI] [PubMed] [Google Scholar]
- 6.Dörschner H, Brekardin E, Uhde I, Schwanstecher C, Schwanstecher M. Stoichiometry of sulfonylurea-induced ATP-sensitive potassium channel closure. Mol Pharmacol 1999;55:1060–1066 [DOI] [PubMed] [Google Scholar]
- 7.Gribble FM, Tucker SJ, Ashcroft FM. The interaction of nucleotides with the tolbutamide block of cloned ATP-sensitive K+ channel currents expressed in Xenopus oocytes: a reinterpretation. J Physiol 1997;504:35–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Proks P, Reimann F, Green N, Gribble F, Ashcroft F. Sulfonylurea stimulation of insulin secretion. Diabetes 2002;51(Suppl. 3):S368–S376 [DOI] [PubMed] [Google Scholar]
- 9.Gribble FM, Reimann F. Sulphonylurea action revisited: the post-cloning era. Diabetologia 2003;46:875–891 [DOI] [PubMed] [Google Scholar]
- 10.Kakei M, Kelly RP, Ashcroft SJ, Ashcroft FM. The ATP-sensitivity of K+ channels in rat pancreatic B-cells is modulated by ADP. FEBS Lett 1986;208:63–66 [DOI] [PubMed] [Google Scholar]
- 11.Misler S, Falke LC, Gillis K, McDaniel ML. A metabolite-regulated potassium channel in rat pancreatic B cells. Proc Natl Acad Sci USA 1986;83:7119–7123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zingman LV, Alekseev AE, Bienengraeber M, et al. Signaling in channel/enzyme multimers: ATPase transitions in SUR module gate ATP-sensitive K+ conductance. Neuron 2001;31:233–245 [DOI] [PubMed] [Google Scholar]
- 13.Sagen JV, Raeder H, Hathout E, et al. Permanent neonatal diabetes due to mutations in KCNJ11 encoding Kir6.2: patient characteristics and initial response to sulfonylurea therapy. Diabetes 2004;53:2713–2718 [DOI] [PubMed] [Google Scholar]
- 14.Pearson ER, Flechtner I, Njølstad PR, et al. Neonatal Diabetes International Collaborative Group Switching from insulin to oral sulfonylureas in patients with diabetes due to Kir6.2 mutations. N Engl J Med 2006;355:467–477 [DOI] [PubMed] [Google Scholar]
- 15.Masia R, Koster JC, Tumini S, et al. An ATP-binding mutation (G334D) in KCNJ11 is associated with a sulfonylurea-insensitive form of developmental delay, epilepsy, and neonatal diabetes. Diabetes 2007;56:328–336 [DOI] [PubMed] [Google Scholar]
- 16.Drain P, Li LH, Wang J. KATP channel inhibition by ATP requires distinct functional domains of the cytoplasmic C terminus of the pore-forming subunit. Proc Natl Acad Sci USA 1998;95:13953–13958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li L, Geng X, Yonkunas M, et al. Ligand-dependent linkage of the ATP site to inhibition gate closure in the KATP channel. J Gen Physiol 2005;126:285–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Antcliff JF, Haider S, Proks P, Sansom MS, Ashcroft FM. Functional analysis of a structural model of the ATP-binding site of the KATP channel Kir6.2 subunit. EMBO J 2005;24:229–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Remedi MS, Kurata HT, Scott A, et al. Secondary consequences of beta cell inexcitability: identification and prevention in a murine model of K(ATP)-induced neonatal diabetes mellitus. Cell Metab 2009;9:140–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rafiq M, Flanagan SE, Patch AM, Shields BM, Ellard S, Hattersley AT, Neonatal Diabetes International Collaborative Group Effective treatment with oral sulfonylureas in patients with diabetes due to sulfonylurea receptor 1 (SUR1) mutations. Diabetes Care 2008;31:204–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hammes GG, Wu CW. Kinetics of allosteric enzymes. Annu Rev Biophys Bioeng 1974;3:1–33 [DOI] [PubMed] [Google Scholar]