

Abstract
Although diabetes is the most common cause of end-stage renal disease (ESRD) worldwide, most people with diabetic nephropathy will never develop ESRD but will instead die of cardiovascular (CV) disease (CVD). The first evidence of kidney injury in diabetes is often microalbuminuria, itself also an independent risk marker for CVD. Although the two processes are closely associated, the recent failure of antialbuminuric therapies to affect CV outcomes has encouraged a reconsideration of how albuminuria may occur in diabetes and how increased urinary albumin excretion may be indicative of CV risk. The relationship between CVD and urinary albumin content (even within the normal range) is widely considered to reflect the common underlying pathology of endothelial dysfunction. At the same time, recent years have witnessed a growing appreciation that diabetic albuminuria commonly arises from damage to glomerular podocytes, specialized epithelial cells acting as the final barrier to macromolecular flow into the urinary filtrate. These superficially discordant paradigms can be assimilated by the emerging concept of endothelial-podocyte crosstalk across the glomerular filtration barrier, whereby the actions of one type of cell may profoundly influence the function of the other. The bidirectional nature of this paracrine network is illustrated by the actions of the vascular endothelial growth factor-A (VEGF-A)/VEGF receptor-2 and activated protein C systems, among others. Identification of novel mediators of endothelial-podocyte crosstalk may lead to the development of more effective treatments for diabetic nephropathy and its sequelae.
Even though it is the most common cause of end-stage renal disease (ESRD) worldwide, most people with diabetic nephropathy will never reach the stage of requiring dialysis or a kidney transplant but will instead die of cardiovascular (CV) disease (CVD). Often, the earliest indicator of kidney damage in diabetes is the abnormal passage of the protein albumin into the urinary filtrate, which is widely considered to reflect underlying endothelial dysfunction and is itself an independent predictor of CVD. Despite their well-recognized association, the manner in which endothelial dysfunction may result in albuminuria is incompletely understood. The passage of albumin into the urine is normally impeded by a three-layered glomerular filtration barrier composed of fenestrated glomerular endothelial cells lining the capillary loops, interdigitating podocytes lining the urinary space, and an interpositioned glomerular basement membrane (Fig. 1A–C). Recent evidence indicates that paracrine communication between the fenestrated endothelium and podocytes plays a critical role in maintaining filtration barrier integrity and that this communication may be disrupted by hyperglycemia and its associated hemodynamic and metabolic perturbations leading to albumin leakage. Here, we consider the roles of the superficially competing paradigms of endothelial dysfunction and podocyte injury in diabetic nephropathy and how these phenomena can be reconciled by the emerging concept of endothelial-podocyte crosstalk.
FIG. 1.

A: Scanning electron micrograph illustrating the fenestrated glomerular endothelium. B: Scanning electron micrograph illustrating interdigitating podocyte foot processes wrapped around the glomerular capillary wall. C: Transmission electron micrograph showing the structure of the glomerular filtration barrier composed of fenestrated endothelium, glomerular basement membrane, and interdigitating podocytes bridged by a molecular slit diaphragm. D: Transmission electron micrograph of the glomerular filtration barrier from a diabetic endothelial NO synthase–deficient mouse showing podocyte foot process effacement in the presence of normal glomerular endothelial fenestrations and ultrastructure.
MICROALBUMINURIA AS A MARKER OF CV RISK
Microalbuminuria, the persistent detectable excretion of albumin into the urine below conventional dipstick thresholds, may affect up to one-third of individuals with diabetes and is an independent risk marker for CVD. In the Prevention of Renal and Vascular End Stage Disease (PREVEND) study of more than 40,000 individuals, for example, a doubling of morning urine albumin concentration over ∼3 years was associated with an almost 30% increase in the relative risk of CV mortality, with this relationship being apparent even at levels of albuminuria below the typical cutoff for defining microalbuminuria (1). Similarly, in a post hoc analysis of the Heart Outcomes Prevention Evaluation (HOPE) trial of individuals already at high CV risk, baseline microalbuminuria in patients with diabetes was associated with an almost doubling in CV risk; again, with this association exhibiting a continuous pattern even within the normoalbuminuric range (2). During the past decade, this well-established risk association has provided the foundation for a number of clinical trials designed to investigate whether therapies that reduce the magnitude of urinary albumin may simultaneously reduce the incidence and prevalence of CVD.
DOES TARGETING ALBUMINURIA REDUCE CV EVENTS?
For almost all individuals with albuminuria in diabetes, the mainstay of treatment is control of blood pressure, particularly with agents that block the renin-angiotensin system (RAS). For instance, in the Reduction of Endpoints in NIDDM with the Angiotensin II Antagonist Losartan (RENAAL) study of albuminuric individuals with type 2 diabetes, treatment with losartan, the angiotensin II receptor blocker (ARB), resulted in a risk reduction of 28% for developing ESRD and 25% for doubling of serum creatinine, effectively delaying the need for hemodialysis by ∼2 years (3). Intriguingly, subsequent post hoc analyses of the RENAAL study cohort revealed that albuminuria reduction during the initial 6 months of therapy was the only predictor of CV outcome, with a 50% reduction in albuminuria being accompanied by an 18% reduction in CV events (4).
Beyond conventional RAS blockade, however, the effects of additional antialbuminuric therapies on CV events have been generally disappointing. In the Ongoing Telmisartan Alone and in Combination with Ramipril Global Endpoint Trial (ONTARGET) study, dual RAS blockade with an ACE inhibitor and an ARB conferred no significant benefit on the primary outcome of death or CV events compared with either agent in isolation, despite a reduction in preexisting proteinuria and in the rate of new-onset microalbuminuria (5,6). Similarly, although the antialbuminuric properties of the direct renin inhibitor, aliskiren, were effectively demonstrated in the Aliskiren in the Evaluation of Proteinuria in Diabetes (AVOID) trial (7), the CV outcome study for aliskiren, Aliskiren Trial in Type 2 Diabetes Using Cardio-Renal Endpoints (ALTITUDE), was prematurely terminated due to futility as well as safety concerns (8).
Beyond agents that act at the level of the RAS, a similar disconnect between albuminuria reduction and CV events has been observed. In a phase III study, treatment with avosentan, the predominant endothelin type A receptor antagonist, for example, resulted in a >40% reduction in the albumin-to-creatinine ratio (ACR) yet had no effect on the primary composite outcome of death, ESRD, or doubling of serum creatinine while increasing the risk of heart failure (9). Thus, whereas there is a continuous association between albumin excretion and CV risk, strategies that reduce the former appear to be largely ineffective at affecting the latter. This apparent disconnect demands a reconsideration of how albuminuria may occur in diabetes and how this may relate to CV risk.
THE RELATIONSHIP BETWEEN ENDOTHELIAL DYSFUNCTION AND ALBUMINURIA IN DIABETES
Although the abnormal passage of large amounts of albumin into the renal tubule may plausibly contribute to the pathogenesis of renal decline, the increased CV risk among individuals with microalbuminuria cannot be attributed to a pathogenetic effect of the protein albumin itself, but rather implies a common underlying etiology. For the past two decades, this relationship has been widely attributed to vascular dysfunction, a notion first espoused by Deckert et al. (10) and commonly termed the Steno hypothesis.
An extensive body of evidence implicates injury to the vascular endothelium as the root cause of diabetes complications, where endothelial injury may arise after the overproduction of oxygen free radicals and the activation of a number of interrelated pathogenetic pathways, elegantly reviewed and assimilated by the seminal work of Brownlee (11). Although such a unifying hypothesis is viewed as a paradigm-shifting advance, biological processes other than glucose-induced oxidative stress also play an important auxiliary role in diabetes-associated endothelial dysfunction. For instance, hyperglycemia can independently lead to a reduction in the bioavailability and responsiveness of endothelial-derived vasoactive mediators, such as nitric oxide (NO) (12), which ordinarily promotes vascular smooth muscle relaxation and inhibits platelet aggregation and leukocyte adhesion at the vascular endothelium. Accelerating factors, such as comorbid hypertension, may further compound vascular damage induced by high ambient glucose concentrations acting through enhanced physical shear stress and flow-related forces.
Endothelial dysfunction likely plays a central role in the development of vascular disease in diabetes, but clinical studies have also directly linked the same process to renal injury. For instance, observations using the measurement of plasma levels of von Willebrand factor as a marker of endothelial injury have revealed an association between the degree of nephropathy and the extent of endothelial damage in type 1 (13) and type 2 diabetes (14), with increased albumin excretion and von Willebrand factor levels conferring a greater than threefold increased relative risk of CV events (14). However, although the importance of endothelial damage induced by hyperglycemia and its associated perturbations in diabetes is without question, the manner in which endothelial injury may result in impaired glomerular permselectivity to macromolecules in diabetes is less clear.
One plausible mechanism by which endothelial dysfunction may lead to increased passage of albumin into the urinary filtrate is through a direct effect of hyperglycemia on the barrier function of the fenestrated glomerular endothelium. However, although appealing in its simplicity, this mechanism fails to account for the complex structural and molecular interplay of the multiple layers of the glomerular filtration barrier. All three layers of the glomerular filtration barrier likely function to regulate permselectivity to macromolecules, although the relative contribution of each layer remains controversial. In the case of the glomerular endothelium, the presence of large fenestrations (Fig. 1A) in the absence of a bridging fenestral diaphragm was initially interpreted as indicating a minimal contribution of this layer to albumin retention.
More recently, however, a barrier function has been inferred from the subsequent identification of a complex glycocalyx network of proteoglycans and glycoproteins covering the fenestral regions. For instance, investigators recently reported that exposure of cultured glomerular endothelial cells to oxidative stress caused loss of the glycocalyx and shedding of glycosaminoglycans that ultimately resulted in an increase in transendothelial albumin passage (15). Nevertheless, it is possible for glomerular endothelial abnormalities to exist in the absence of significant proteinuria. In preeclampsia of pregnancy, for example, the characteristic glomerular lesion is endotheliosis, recognized by the presence of swollen glomerular endothelial cells encroaching on the capillary lumen. Although pathologically implicated in the proteinuria of preeclampsia, endotheliosis may also be observed in a significant proportion of women with healthy pregnancies and no proteinuria (16). Similarly, in the case of Shiga toxin–associated hemolytic uremic syndrome, albuminuria may be mild or even absent despite the endothelial cell being a primary site of injurious insult (17).
PODOCYTE INJURY AT THE CENTER OF DIABETIC ALBUMINURIA
An expanding body of literature has evolved implicating endothelial dysfunction as a major contributor to the pathogenesis of diabetes complications, but a comparable body of literature has simultaneously evolved implicating injury to glomerular podocytes as the major cause of albuminuria in diabetes. Podocytes are epithelial cells, exclusive to the renal glomerulus, lying along the basal lamina in intimate apposition to the glomerular capillary wall. Their long interdigitating foot processes bridged by a molecular slit diaphragm represent the last obstacle to macromolecular leakage into the urinary filtrate (Fig. 1B and C). This formidable barrier function is facilitated by a foot process actin cytoskeleton interacting with proteins of the slit diaphragm, an adherens junction-like structure covering the filtration slits. The important barrier function of the podocyte is readily illustrated by the heavy albuminuria affecting individuals with congenital nephrotic syndrome of the Finnish subtype, a disease caused by an inherited mutation in the gene encoding nephrin, the podocyte-specific slit diaphragm protein (18). Since this initial discovery, mutations in the genes for a number of other podocyte cytoskeletal or signaling proteins have been linked to proteinuria, reinforcing the critical role that these cells play in glomerular permselectivity.
The central role that podocyte injury plays in the pathogenesis of diabetic albuminuria has been inferred from clinical studies and experimental models. Numerous studies of human biopsy tissue have demonstrated a relationship between podocyte loss or pathological widening of podocyte foot processes and the albumin excretion rate (AER). For instance, serial biopsy specimens taken from patients with type 1 diabetes during a 3-year period showed a significant correlation between a decrease in podocyte number and a change in AER over time (19). Similarly, when glomerular morphological characteristics were evaluated in biopsy specimens obtained from Pima Indians with type 2 diabetes, the strongest predictor of albuminuria progression was the number of podocytes per glomerulus, with fewer podocytes predicting more rapid progression (20). With respect to podocyte foot processes, a study of individuals with type 2 diabetes revealed that the AER correlated inversely with filtration slit length density and directly with foot process width (21). Other studies have described similar findings when assessing the expression of podocyte-specific proteins on a molecular level, most notably reporting a reduction in the expression of nephrin, the slit diaphragm protein, in human diabetic nephropathy (22).
Further evidence for a direct effect of podocyte injury in the pathogenesis of diabetic albuminuria comes from the evaluation of the therapeutic effects of RAS blockade. The antialbuminuric effect of RAS inhibition in diabetic nephropathy was originally attributed to its actions in lowering systemic and intraglomerular pressure, the latter achieved through the well-described preferential vasodilatory effects of these agents at the level of the efferent arteriole. Although these actions are undisputed, more recent evidence has emerged supporting a local podocyte-specific RAS, most convincingly demonstrated by the development of proteinuria in rats overexpressing the angiotensin II type 1 receptor in podocytes (23). Thus, whereas the beneficial effects of RAS blockade in reducing CV events may be attributable to a global improvement in endothelial function, actions at the level of the podocyte may attenuate albuminuria without affecting the systemic vasculature. This may be one mechanism underlying the persistence of CV events despite further lowering of albuminuria in clinical studies of dual-RAS blockade or direct renin inhibition.
ENDOTHELIAL-PODOCYTE CROSSTALK: THE MISSING LINK BETWEEN ENDOTHELIAL DYSFUNCTION AND ALBUMINURIA IN DIABETES?
In diabetes, endothelial insult predisposes to albuminuria and CVD, yet at the same time, the primary renal cell exhibiting evidence of damage and predisposing to albuminuria is often the podocyte. How can these two superficially competing phenomena be reconciled? With the aid of modern technological advances in the inducible and cell-specific manipulation of gene dosage, it is now apparent that the glomerular filtration barrier is not simply an inert sieve-like obstacle impeding macromolecular flow, but rather, it is a complex dynamic structure in which the actions of one of its components can profoundly affect the function of another. Viewed in this light, one way in which endothelial dysfunction may result in podocyte injury and albuminuria in diabetes is through altered paracrine communication, or crosstalk, between the two cell types across the filtration barrier. In this scenario, endothelial dysfunction may predispose to vascular disease in multiple tissue beds through altered crosstalk with resident cells. Altered communication within the large vessels between endothelial cells and vascular smooth muscle cells may predispose to atherosclerosis (24), and altered endothelial-podocyte crosstalk in the kidney may manifest as albuminuria.
We recently demonstrated the in vivo importance of endothelial-podocyte crosstalk in the development of diabetic albuminuria, showing that diabetic mice with endothelial dysfunction induced by genetic deficiency of the ostensibly endothelial specific gene, endothelial NO synthase, develop a podocyte-specific injury and heavy albuminuria (Fig. 1D) (25). Moreover, this podocyte injury was responsive to RAS blockade (25). Although NO evidently has some regulatory role in filtration barrier integrity, the heavy albuminuria repeatedly demonstrated in diabetic endothelial NO synthase–deficient mice in multiple studies is unlikely solely a consequence of decreased NO production, because NO itself may increase glomerular albumin permeability (26). Indeed, further evidence for the importance of alternative endothelial-derived secreted factor(s) in mediating podocyte barrier function comes from the observation that culture medium conditioned by glomerular endothelial cells exposed to shear stress impairs podocyte transepithelial resistance in vitro (27).
EVIDENCE FOR A PARACRINE COMMUNICATION NETWORK WITHIN THE RENAL GLOMERULUS
The complex role of a dynamic signaling network across the filtration barrier is most readily illustrated by the example of the vascular endothelial growth factor-A (VEGF-A)/VEGF receptor 2 (VEGFR-2) system, which is essential for normal glomerular development and adult renal homeostasis and which is exquisitely sensitive to the shifting hemodynamic and metabolic stresses imposed by the diabetic milieu. VEGF-A is a pleiotropic growth factor that plays a critical role in endothelial cell survival, differentiation, proliferation, and migration. These actions are mediated by two transmembrane tyrosine kinase receptors: VEGFR-1 and VEGFR-2, with signaling via VEGFR-2 appearing both necessary and sufficient for the normal biological response. VEGF-A is abundantly expressed by podocytes as the isoforms VEGF121, VEGF165 (most abundant), and VEGF189 generated by alternative messenger RNA splicing. Within the glomerulus, VEGFR-2 is expressed primarily (or possibly exclusively) on endothelial cells, with controversy existing about the in vivo presence of an active VEGFR-2 signaling system within glomerular podocytes (28). Whereas podocyte-specific VEGFR-2 deletion is accompanied by an unremarkable phenotype, endothelial VEGFR-2 deletion results in global defects in the glomerular microvasculature (28). In this context, our earlier observation of podocyte injury after VEGFR-2 inhibition in rodents with varying degrees of hypertension (29) suggests that endothelial injury induced by VEGFR-2 blockade may, itself, ultimately affect podocyte homeostasis (Fig. 2). Collectively, however, these observations emphasize the predominance of a paracrine VEGF-A/VEGFR-2 system within the renal glomerulus acting across the glomerular basement membrane and contrary to the flow of urine.
FIG. 2.

Podocyte injury resulting from VEGFR-2 inhibition in vivo. Reprinted with permission from Advani et al. (29), copyright (2007) National Academy of Sciences, U.S.A. Transmission electron micrographs of representative podocytes from vehicle-treated animals. Sprague-Dawley (SD) rat (A), spontaneously hypertensive rat (SHR) (B), and transgenic TGR(mRen-2)27 rat (C). After VEGFR-2 inhibition with the small molecule vandetanib, SD rat (D), SHR (E), and TGR(mRen-2)27 rat (F). Among vandetanib-treated groups, SD rats had occasional podocyte pseudocysts (*); SHRs showed a predominance of proteinaceous adsorption droplets (arrow), with occasional foot process fusion (arrowhead); and TGR(mRen-2)27 rats demonstrated severe podocyte injury with abundant adsorption droplet accumulation and some foot process fusion (arrowhead). Original magnification ×6,600.
VEGF-A is essential for glomerular development and renal homeostasis. Podocyte-derived VEGF-A is required during glomerular development for the formation of normal capillaries and endothelial cell fenestrations. Similarly, inducible podocyte-specific VEGF-A deletion in the adult kidney may result in endotheliosis and a thrombotic microangiopathy-like picture analogous to the renal injury that occurs in patients receiving VEGF-A antagonists in the oncology setting (30). Illustrating the importance of a “dosage-sensitivity” of the VEGF-A/VEGFR-2 system, gain-of-function models have shown that glomerular VEGF-A overexpression also causes podocytopathy, together with thickening of the glomerular basement membrane and proteinuria, despite surprisingly little endothelial cell injury (31,32).
The role that the VEGF-A/VEGFR-2 system plays in diabetic nephropathy is complex and is sometimes termed the VEGF-A paradox. A number of experimental studies have described an increase in the expression of VEGF-A/VEGFR-2 in the kidneys of rodents with diabetes (33), whereas the opposite effect has been observed in biopsy specimens of patients with diabetic nephropathy (34). Blockade of VEGF-A or VEGFR-2 may attenuate albuminuria development in diabetic wild-type rodents (35,36). Similarly, inducible overexpression of an endogenous VEGF-A inhibitor, soluble fms-like tyrosine kinase-1, attenuates albuminuria development in diabetic mice (37). In contrast to these results, inducible podocyte-specific VEGF-A deletion has also been reported to actually accelerate renal injury in the context of experimental diabetes (38). Collectively, this growing body of work highlights the intricacies of an intraglomerular crosstalk system where subtle effects of disease microenvironment, gene dosage, or manner of VEGF-A blockade may have profound effects on the glomerular phenotype.
OTHER MEDIATORS OF ENDOTHELIAL-PODOCYTE CROSSTALK
Although most extensively investigated, the VEGF-A/VEGFR-2 system is not the sole mediator of endothelial-podocyte crosstalk (Table 1). Other identified pathways include the stromal cell–derived factor-1 (SDF-1)/C-X-C chemokine receptor type 4 (CXCR4) axis, the angiopoietins, and the semaphorins.
TABLE 1.
Currently identified mediators of endothelial-podocyte communication in renal development, health, and disease
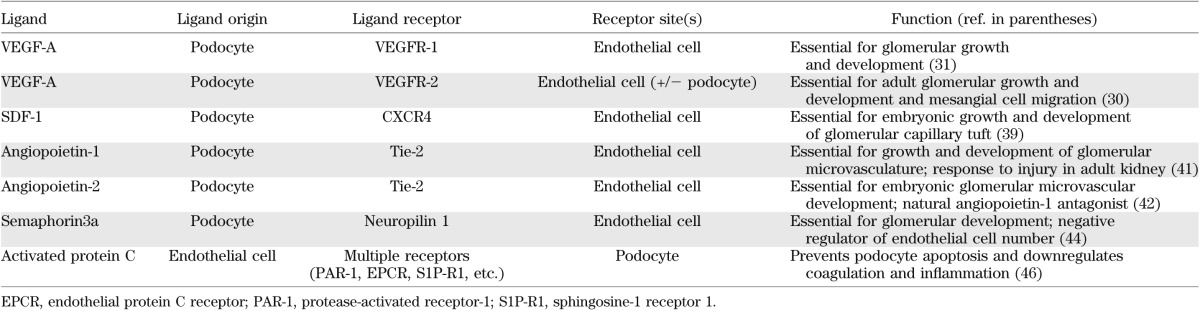
The chemokine SDF-1 is the primary ligand for the G-protein coupled receptor CXCR4, playing a pivotal role in the trafficking of stem cells under physiologic and malignant conditions. Interestingly, SDF-1/CXCR4 interaction is also essential for the development of the renal vasculature. SDF-1–producing podocytes during embryonic development appear to reside close to CXCR4 receptors present on glomerular endothelial cells, and selective endothelial CXCR4 inactivation will result in abnormal renal vessel formation (39). Compared with the analogously angiogenic VEGF-A/VEGFR-2 system, however, relatively little is known about paracrine intraglomerular SDF-1/CXCR4 signaling in the disease state, with an initial report describing an antialbuminuric effect of a short RNA oligonucleotide sequence directed against SDF-1 when administered to diabetic mice (40).
A further family of angiogenic proteins implicated in endothelial-podocyte crosstalk is the angiopoietin family. During development, podocyte-derived angiopoietin-1 binds its tyrosine kinase receptor, Tie-2, to promote glomerular microvascular growth (41). In contrast to the VEGF-A/VEGFR-2 system, the angiopoietin-1/Tie-2 network appears to be dispensable in the adult kidney, although it acts to preserve microvascular integrity when challenged by experimental diabetes (41). Angiopoietin-2 is commonly considered as a natural antagonist of angiopoietin-1, competitively binding to the Tie-2 receptor and disrupting angiogenesis. Significantly, podocyte-specific overexpression of angiopoietin-2 induces endothelial apoptosis and albuminuria (42), whereas the growth factor itself is upregulated in diabetic nephropathy (43).
Semaphorins are a group of secreted and membrane proteins originally identified for their inhibitory actions on axonal growth but more recently recognized as playing an important role in renal development and in endothelial-podocyte crosstalk. The semaphorin, Sema3a, is essential for normal glomerular development and is continually expressed by mature podocytes, whereas overexpression of podocyte Sema3a results in endothelial apoptosis and Sema3a deletion is accompanied by endothelial overgrowth (44,45), effects that have again been attributed to altered VEGF-A/VEGFR-2 signaling.
ENDOTHELIAL-DERIVED FACTORS SIGNALING TO THE GLOMERULAR PODOCYTE
Close examination of the endothelial-podocyte crosstalk mediators reviewed thus far will reveal that all of these factors are podocyte-derived proteins signaling to glomerular endothelial cells. This may reflect a relative protein abundance needed when acting contrary to urine flow. In contrast, endothelial-derived factors that signal to closely apposed podocytes, and thus traveling in the direction of the urinary filtrate, may be present at comparatively lower levels, impeding their identification by current experimental methodologies. Perhaps the best example of the reciprocal nature of endothelial-podocyte communication is the activated protein C pathway. Protein C is activated by the thrombin/thrombomodulin complex on the surface of endothelial cells, where it serves to downregulate coagulation, enhance anti-inflammatory effects, and prevent apoptosis. In diabetes, reduced formation of activated protein C results in increased albuminuria and podocyte apoptosis (46), recently discovered to be mediated through epigenetic regulation of oxidative stress in podocytes (47) (Fig. 3). Intriguingly, decreased activated protein C is also associated with an increased risk of myocardial infarction and the severity of lesions within the coronary vessels (48).
FIG. 3.

Effects of activated protein C on mesangial matrix accumulation and oxidative stress in podocytes. Reprinted with permission from Bock et al. (47). Glomerular profiles from kidney sections stained with periodic acid Schiff (PAS) or for peroxynitrite (ONOO−), which is an indicator of reactive oxygen species formation, or the redox enzyme p66Shc. In diabetic mice (DM) with impaired thrombomodulin-dependent protein C activation (TMP/P DM), there is enhanced accumulation of mesangial matrix accompanied by increased ONOO− and p66Shc expression in peripherally arranged podocytes. These effects are negated by crossing TMP/P DM mice with transgenic mice with increased plasma activated protein C (TMP/P × APChigh DM) (47). WT, wild-type.
FUTURE CONSIDERATIONS IN THE IDENTIFICATION OF MEDIATORS OF ENDOTHELIAL-PODOCYTE CROSSTALK AS THERAPEUTIC TARGETS
Endothelial-podocyte crosstalk is emerging as a complex paracrine-signaling network that is 1) essential for renal development and glomerular homeostasis and 2) altered in the disease setting, manifesting most commonly as abnormal albumin excretion. Elucidation of novel mediators of this crosstalk system may facilitate future therapeutic strategies that not only delay renal decline but, should the same processes prove to be recapitulated in other vascular beds, may also simultaneously attenuate other diabetes complications, including CVD (Fig. 4). Impeding the search for these communicants, however, are a number of questions that require addressing and a number of obstacles that need to be circumvented.
FIG. 4.

Illustrating the manner by which endothelial dysfunction, induced by hyperglycemia and associated accelerating factors, may predispose to CVD and albuminuria, the latter occurring through altered communication between glomerular endothelial cells and closely apposed podocytes.
To begin with, how do paracrine communicators cross the glomerular basement membrane to exert their effects? In this regard, new insights have been obtained through the study of heparan sulfate proteoglycans. In elegant studies, investigators recently demonstrated that the podocyte-specific transcription factor, WT1, regulates the expression of heparan sulfate 6-O-endosulfatases that function to modulate the bioavailability of signaling molecules, including VEGF-A, within the glomerular filtration barrier (49). These observations add a further layer of complexity to our understanding of the crosstalk network, with the biological effects of proteins dependent not only on their abundance but also on their relative bioavailability. Similarly highlighting the importance of bioavailability, a previous study of human biopsy tissue reported that whereas VEGF-A expression was increased in diabetic glomeruli, VEGF-A receptor activation was more tightly regulated, being decreased in more severely injured glomeruli (50).
Second, whereas the crosstalk mediators identified to date are all protein-based molecules, there is no particular reason to assume that all of the important protagonists are proteins. For instance, lipid mediators, most notably the arachidonic acid–derived prostanoids, are important autocrine and paracrine signaling molecules. Cyclooxygenase-derived prostanoids play an established yet complex role in renal (patho)physiology, and several prostanoid receptors have been identified on the surface of glomerular podocytes (51). The microRNAs are a group of highly conserved, short noncoding RNA sequences that posttranscriptionally regulate gene expression by binding to specific messenger RNA transcripts and functionally inhibiting protein translation or promoting mRNA degradation. A number of studies have shown that microRNAs may play a role in the pathogenesis of diabetic nephropathy (52), while paracrine microRNA-regulated communication between endothelial cells and their neighbors has recently been reported (53).
Third, a number of pathways and processes recognized for their importance in the development and progression of diabetic nephropathy may play a specific role in regulating endothelial-podocyte crosstalk. For instance, podocytes express the receptor for advanced glycation end product (54), raising the intriguing possibility that the advanced glycation end product/receptor for advanced glycation end product system may mediate its effects, at least partly, through paracrine actions across the filtration barrier. Similarly, proinflammatory chemokines, such as monocyte chemotactic protein-1, produced by the endothelium, may contribute to glomerular injury in diabetes by virtue of expression of their receptors on podocytes (55). Alternatively, intracellular signaling cascades that may be detrimental (e.g., protein kinase C isoforms (11), NADPH oxidases (56), and the Janus kinase/signal transducers and activators of transcription pathway (57)) or protective (e.g., AMP-activated protein kinase (58)) may exert their effects by altering the secretion of paracrine communicants or by regulating the cellular response to crosstalk mediators.
Fourth, although much attention has recently been focused on the role of the podocyte in preventing albuminuria, the pivotal action of the mesangial cell in glomerular homeostasis should not be forgotten. Mesangial cells ordinarily play a vital role in the normal formation of the glomerular capillary tuft, whereas podocyte-specific VEGF-A deletion is accompanied not only by defects in glomerular endothelial development but also by an inability of mesangial cells to migrate into the developing glomerulus (31). Thus, a third dimension may need to be considered where the three types of cells—endothelial, podocyte, and mesangial—act in concert to influence their global function.
Finally, even though our understanding of paracrine communication within the glomerulus has been aided immensely by the evolution of modern genetic tools, the limitations of current technology still impede progress. Compared with the relative ease in which gene dosage may now be altered in podocytes, endothelial-specific manipulations are limited to the global endothelium, rather than the fenestrated glomerular endothelial cell, and as such, discrimination of intraglomerular effects from hemodynamic actions at the level of the afferent and efferent arterioles or generalized systemic effects cannot currently be achieved.
SUMMARY
In attempting to understand the manner by which high ambient glucose concentrations may lead to micro- and macrovascular complications, two paradigms have emerged in tandem: 1) endothelial dysfunction is commonly the initiating insult predisposing to tissue injury in diabetes, and 2) albuminuria commonly reflects underlying damage to glomerular podocytes. With the advancement in genetic manipulation technologies, these paradigms are beginning to be assimilated through the emerging appreciation of a complex crosstalk system between endothelial cells and podocytes within the glomerular filtration barrier. Understanding the mediators of this communication may lead to the development of novel therapies that not only function to prevent renal decline but may also simultaneously attenuate CV events, the major cause of morbidity and mortality in patients with diabetic nephropathy.
ACKNOWLEDGMENTS
This work was supported by a Diabetes Innovation Award from Novo Nordisk to A.A. F.S.S. is supported by a postdoctoral fellowship from the Banting and Best Diabetes Centre, and A.A. is supported by a Canadian Diabetes Association Clinician Scientist Award. No other potential conflicts of interest relevant to this article were reported.
F.S.S. and A.A. wrote and edited the manuscript.
The authors thank Dr. Kathryn E. White (EM Research Services, Newcastle University, Newcastle, U.K.) for provision of the electron micrographs and Kryski Biomedia for the elegant artwork.
REFERENCES
- 1.Hillege HL, Fidler V, Diercks GF, et al. Prevention of Renal and Vascular End Stage Disease (PREVEND) Study Group Urinary albumin excretion predicts cardiovascular and noncardiovascular mortality in general population. Circulation 2002;106:1777–1782 [DOI] [PubMed] [Google Scholar]
- 2.Gerstein HC, Mann JF, Yi Q, et al. HOPE Study Investigators Albuminuria and risk of cardiovascular events, death, and heart failure in diabetic and nondiabetic individuals. JAMA 2001;286:421–426 [DOI] [PubMed] [Google Scholar]
- 3.Brenner BM, Cooper ME, de Zeeuw D, et al. RENAAL Study Investigators Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med 2001;345:861–869 [DOI] [PubMed] [Google Scholar]
- 4.de Zeeuw D, Remuzzi G, Parving HH, et al. Proteinuria, a target for renoprotection in patients with type 2 diabetic nephropathy: lessons from RENAAL. Kidney Int 2004;65:2309–2320 [DOI] [PubMed] [Google Scholar]
- 5.Mann JF, Schmieder RE, McQueen M, et al. ONTARGET investigators Renal outcomes with telmisartan, ramipril, or both, in people at high vascular risk (the ONTARGET study): a multicentre, randomised, double-blind, controlled trial. Lancet 2008;372:547–553 [DOI] [PubMed] [Google Scholar]
- 6.Yusuf S, Teo KK, Pogue J, et al. ONTARGET Investigators Telmisartan, ramipril, or both in patients at high risk for vascular events. N Engl J Med 2008;358:1547–1559 [DOI] [PubMed] [Google Scholar]
- 7.Parving HH, Persson F, Lewis JB, Lewis EJ, Hollenberg NK, AVOID Study Investigators Aliskiren combined with losartan in type 2 diabetes and nephropathy. N Engl J Med 2008;358:2433–2446 [DOI] [PubMed] [Google Scholar]
- 8.Parving HH, Brenner BM, McMurray JJ, et al. ALTITUDE Investigators Cardiorenal end points in a trial of aliskiren for type 2 diabetes. N Engl J Med 2012;367:2204–2213 [DOI] [PubMed] [Google Scholar]
- 9.Mann JF, Green D, Jamerson K, et al. ASCEND Study Group Avosentan for overt diabetic nephropathy. J Am Soc Nephrol 2010;21:527–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deckert T, Feldt-Rasmussen B, Borch-Johnsen K, Jensen T, Kofoed-Enevoldsen A. Albuminuria reflects widespread vascular damage. The Steno hypothesis. Diabetologia 1989;32:219–226 [DOI] [PubMed] [Google Scholar]
- 11.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001;414:813–820 [DOI] [PubMed] [Google Scholar]
- 12.Brodsky SV, Morrishow AM, Dharia N, Gross SS, Goligorsky MS. Glucose scavenging of nitric oxide. Am J Physiol Renal Physiol 2001;280:F480–F486 [DOI] [PubMed] [Google Scholar]
- 13.Stehouwer CD, Stroes ES, Hackeng WH, Mulder PG, Den Ottolander GJ. von Willebrand factor and development of diabetic nephropathy in IDDM. Diabetes 1991;40:971–976 [DOI] [PubMed] [Google Scholar]
- 14.Stehouwer CD, Nauta JJ, Zeldenrust GC, Hackeng WH, Donker AJ, den Ottolander GJ. Urinary albumin excretion, cardiovascular disease, and endothelial dysfunction in non-insulin-dependent diabetes mellitus. Lancet 1992;340:319–323 [DOI] [PubMed] [Google Scholar]
- 15.Singh A, Ramnath RD, Foster RR, et al. Reactive oxygen species modulate the barrier function of the human glomerular endothelial glycocalyx. PLoS ONE 2013;8:e55852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Strevens H, Wide-Swensson D, Hansen A, et al. Glomerular endotheliosis in normal pregnancy and pre-eclampsia. BJOG 2003;110:831–836 [PubMed] [Google Scholar]
- 17.Petruzziello-Pellegrini TN, Yuen DA, Page AV, et al. The CXCR4/CXCR7/SDF-1 pathway contributes to the pathogenesis of Shiga toxin-associated hemolytic uremic syndrome in humans and mice. J Clin Invest 2012;122:759–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lenkkeri U, Männikkö M, McCready P, et al. Structure of the gene for congenital nephrotic syndrome of the finnish type (NPHS1) and characterization of mutations. Am J Hum Genet 1999;64:51–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.White KE, Bilous RW, Marshall SM, et al. Podocyte number in normotensive type 1 diabetic patients with albuminuria. Diabetes 2002;51:3083–3089 [DOI] [PubMed] [Google Scholar]
- 20.Meyer TW, Bennett PH, Nelson RG. Podocyte number predicts long-term urinary albumin excretion in Pima Indians with type II diabetes and microalbuminuria. Diabetologia 1999;42:1341–1344 [DOI] [PubMed] [Google Scholar]
- 21.Dalla Vestra M, Masiero A, Roiter AM, Saller A, Crepaldi G, Fioretto P. Is podocyte injury relevant in diabetic nephropathy? Studies in patients with type 2 diabetes. Diabetes 2003;52:1031–1035 [DOI] [PubMed] [Google Scholar]
- 22.Langham RG, Kelly DJ, Cox AJ, et al. Proteinuria and the expression of the podocyte slit diaphragm protein, nephrin, in diabetic nephropathy: effects of angiotensin converting enzyme inhibition. Diabetologia 2002;45:1572–1576 [DOI] [PubMed] [Google Scholar]
- 23.Hoffmann S, Podlich D, Hähnel B, Kriz W, Gretz N. Angiotensin II type 1 receptor overexpression in podocytes induces glomerulosclerosis in transgenic rats. J Am Soc Nephrol 2004;15:1475–1487 [DOI] [PubMed] [Google Scholar]
- 24.Zhang D, Chen Y, Xie X, et al. Homocysteine activates vascular smooth muscle cells by DNA demethylation of platelet-derived growth factor in endothelial cells. J Mol Cell Cardiol 2012;53:487–496 [DOI] [PubMed] [Google Scholar]
- 25.Yuen DA, Stead BE, Zhang Y, et al. eNOS deficiency predisposes podocytes to injury in diabetes. J Am Soc Nephrol 2012;23:1810–1823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li B, Yao J, Morioka T, Oite T. Nitric oxide increases albumin permeability of isolated rat glomeruli via a phosphorylation-dependent mechanism. J Am Soc Nephrol 2001;12:2616–2624 [DOI] [PubMed] [Google Scholar]
- 27.Slater SC, Ramnath RD, Uttridge K, et al. Chronic exposure to laminar shear stress induces Kruppel-like factor 2 in glomerular endothelial cells and modulates interactions with co-cultured podocytes. Int J Biochem Cell Biol 2012;44:1482–1490 [DOI] [PubMed] [Google Scholar]
- 28.Sison K, Eremina V, Baelde H, et al. Glomerular structure and function require paracrine, not autocrine, VEGF-VEGFR-2 signaling. J Am Soc Nephrol 2010;21:1691–1701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Advani A, Kelly DJ, Advani SL, et al. Role of VEGF in maintaining renal structure and function under normotensive and hypertensive conditions. Proc Natl Acad Sci U S A 2007;104:14448–14453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eremina V, Jefferson JA, Kowalewska J, et al. VEGF inhibition and renal thrombotic microangiopathy. N Engl J Med 2008;358:1129–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eremina V, Sood M, Haigh J, et al. Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal diseases. J Clin Invest 2003;111:707–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Veron D, Reidy KJ, Bertuccio C, et al. Overexpression of VEGF-A in podocytes of adult mice causes glomerular disease. Kidney Int 2010;77:989–999 [DOI] [PubMed] [Google Scholar]
- 33.Cooper ME, Vranes D, Youssef S, et al. Increased renal expression of vascular endothelial growth factor (VEGF) and its receptor VEGFR-2 in experimental diabetes. Diabetes 1999;48:2229–2239 [DOI] [PubMed] [Google Scholar]
- 34.Lindenmeyer MT, Kretzler M, Boucherot A, et al. Interstitial vascular rarefaction and reduced VEGF-A expression in human diabetic nephropathy. J Am Soc Nephrol 2007;18:1765–1776 [DOI] [PubMed] [Google Scholar]
- 35.de Vriese AS, Tilton RG, Elger M, Stephan CC, Kriz W, Lameire NH. Antibodies against vascular endothelial growth factor improve early renal dysfunction in experimental diabetes. J Am Soc Nephrol 2001;12:993–1000 [DOI] [PubMed] [Google Scholar]
- 36.Sung SH, Ziyadeh FN, Wang A, Pyagay PE, Kanwar YS, Chen S. Blockade of vascular endothelial growth factor signaling ameliorates diabetic albuminuria in mice. J Am Soc Nephrol 2006;17:3093–3104 [DOI] [PubMed] [Google Scholar]
- 37.Ku CH, White KE, Dei Cas A, et al. Inducible overexpression of sFlt-1 in podocytes ameliorates glomerulopathy in diabetic mice. Diabetes 2008;57:2824–2833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sivaskandarajah GA, Jeansson M, Maezawa Y, Eremina V, Baelde HJ, Quaggin SE. Vegfa protects the glomerular microvasculature in diabetes. Diabetes 2012;61:2958–2966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Takabatake Y, Sugiyama T, Kohara H, et al. The CXCL12 (SDF-1)/CXCR4 axis is essential for the development of renal vasculature. J Am Soc Nephrol 2009;20:1714–1723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sayyed SG, Hägele H, Kulkarni OP, et al. Podocytes produce homeostatic chemokine stromal cell-derived factor-1/CXCL12, which contributes to glomerulosclerosis, podocyte loss and albuminuria in a mouse model of type 2 diabetes. Diabetologia 2009;52:2445–2454 [DOI] [PubMed] [Google Scholar]
- 41.Jeansson M, Gawlik A, Anderson G, et al. Angiopoietin-1 is essential in mouse vasculature during development and in response to injury. J Clin Invest 2011;121:2278–2289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Davis B, Dei Cas A, Long DA, et al. Podocyte-specific expression of angiopoietin-2 causes proteinuria and apoptosis of glomerular endothelia. J Am Soc Nephrol 2007;18:2320–2329 [DOI] [PubMed] [Google Scholar]
- 43.Sun H, Zheng JM, Chen S, Zeng CH, Liu ZH, Li LS. Enhanced expression of ANGPTL2 in the microvascular lesions of diabetic glomerulopathy. Nephron, Exp Nephrol 2007;105:e117–e123 [DOI] [PubMed] [Google Scholar]
- 44.Reidy KJ, Villegas G, Teichman J, et al. Semaphorin3a regulates endothelial cell number and podocyte differentiation during glomerular development. Development 2009;136:3979–3989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Villegas G, Tufro A. Ontogeny of semaphorins 3A and 3F and their receptors neuropilins 1 and 2 in the kidney. Gene Expr Patterns 2002;2:151–155 [DOI] [PubMed] [Google Scholar]
- 46.Isermann B, Vinnikov IA, Madhusudhan T, et al. Activated protein C protects against diabetic nephropathy by inhibiting endothelial and podocyte apoptosis. Nat Med 2007;13:1349–1358 [DOI] [PubMed] [Google Scholar]
- 47.Bock F, Shahzad K, Wang H, et al. Activated protein C ameliorates diabetic nephropathy by epigenetically inhibiting the redox enzyme p66Shc. Proc Natl Acad Sci U S A 2013;110:648–653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zorio E, Navarro S, Medina P, et al. Circulating activated protein C is reduced in young survivors of myocardial infarction and inversely correlates with the severity of coronary lesions. J Thromb Haemost 2006;4:1530–1536 [DOI] [PubMed] [Google Scholar]
- 49.Schumacher VA, Schlötzer-Schrehardt U, Karumanchi SA, et al. WT1-dependent sulfatase expression maintains the normal glomerular filtration barrier. J Am Soc Nephrol 2011;22:1286–1296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hohenstein B, Hausknecht B, Boehmer K, Riess R, Brekken RA, Hugo CP. Local VEGF activity but not VEGF expression is tightly regulated during diabetic nephropathy in man. Kidney Int 2006;69:1654–1661 [DOI] [PubMed] [Google Scholar]
- 51.Bek M, Nüsing R, Kowark P, Henger A, Mundel P, Pavenstädt H. Characterization of prostanoid receptors in podocytes. J Am Soc Nephrol 1999;10:2084–2093 [DOI] [PubMed] [Google Scholar]
- 52.Kato M, Zhang J, Wang M, et al. MicroRNA-192 in diabetic kidney glomeruli and its function in TGF-beta-induced collagen expression via inhibition of E-box repressors. Proc Natl Acad Sci U S A 2007;104:3432–3437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Robinson HC, Baker AH. How do microRNAs affect vascular smooth muscle cell biology? Curr Opin Lipidol 2012;23:405–411 [DOI] [PubMed] [Google Scholar]
- 54.Tanji N, Markowitz GS, Fu C, et al. Expression of advanced glycation end products and their cellular receptor RAGE in diabetic nephropathy and nondiabetic renal disease. J Am Soc Nephrol 2000;11:1656–1666 [DOI] [PubMed] [Google Scholar]
- 55.Tarabra E, Giunti S, Barutta F, et al. Effect of the monocyte chemoattractant protein-1/CC chemokine receptor 2 system on nephrin expression in streptozotocin-treated mice and human cultured podocytes. Diabetes 2009;58:2109–2118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Eid AA, Gorin Y, Fagg BM, et al. Mechanisms of podocyte injury in diabetes: role of cytochrome P450 and NADPH oxidases. Diabetes 2009;58:1201–1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hodgin JB, Nair V, Zhang H, et al. Identification of cross-species shared transcriptional networks of diabetic nephropathy in human and mouse glomeruli. Diabetes 2013;62:299–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sharma K, Ramachandrarao S, Qiu G, et al. Adiponectin regulates albuminuria and podocyte function in mice. J Clin Invest 2008;118:1645–1656 [DOI] [PMC free article] [PubMed] [Google Scholar]