

Abstract
Protégé was a phase 3, randomized, double-blind, parallel, placebo-controlled 2-year study of three intravenous teplizumab dosing regimens, administered daily for 14 days at baseline and again after 26 weeks, in new-onset type 1 diabetes. We sought to determine efficacy and safety of teplizumab immunotherapy at 2 years and to identify characteristics associated with therapeutic response. Of 516 randomized patients, 513 were treated, and 462 completed 2 years of follow-up. Teplizumab (14-day full-dose) reduced the loss of C-peptide mean area under the curve (AUC), a prespecified secondary end point, at 2 years versus placebo. In analyses of prespecified and post hoc subsets at entry, U.S. residents, patients with C-peptide mean AUC >0.2 nmol/L, those randomized ≤6 weeks after diagnosis, HbA1c <7.5% (58 mmol/mol), insulin use <0.4 units/kg/day, and 8–17 years of age each had greater teplizumab-associated C-peptide preservation than their counterparts. Exogenous insulin needs tended to be reduced versus placebo. Antidrug antibodies developed in some patients, without apparent change in drug efficacy. No new safety or tolerability issues were observed during year 2. In summary, anti-CD3 therapy reduced C-peptide loss 2 years after diagnosis using a tolerable dose.
Immunotherapy that directly inhibits β-cell destruction is an unfulfilled need for treatment of autoimmune type 1 diabetes. Although it may eventually be useful in prediabetes, treatment at clinical onset is an excellent opportunity when patients are easily identified and functional β-cell mass remains (1). Preservation of residual β-cell function, represented by higher levels of C-peptide, facilitates better glycemic control to lessen retinopathy, nephropathy, hypoglycemia, and ketoacidosis (2–4). Immunotherapy given at diagnosis aims to prolong and augment this effect by preventing further β-cell death and possibly also by enabling living β-cells to recover function after resolution of inflammation (5). Clinical trials of different agents have had modest success in this regard, but treatment responses have often waned within 2 years (6–8).
Teplizumab is a nonactivating, Fc-modified, anti-CD3 monoclonal antibody thought to attenuate activated autoreactive T cells mediating β-cell death. These T cells disappear from the peripheral circulation during immunotherapy but return within weeks after stopping treatment (9). Preclinical and clinical studies suggest that the drug may induce regulatory T-cell activity, suggesting augmented immune tolerance (10).
Protégé was a large, randomized, placebo-controlled, double-blinded trial of immunotherapy in type 1 diabetes (11). Recently diagnosed patients (8–35 years of age) were randomized to receive daily infusions of placebo or one of three teplizumab regimens at baseline and at 6 months. The primary outcome, a composite of insulin <0.5 units/kg/day and HbA1c <6.5% (48 mmol/mol) at year 1, had not been validated previously and did not achieve statistical significance. In exploratory analyses, a significant improvement in area under the curve (AUC) mean C-peptide during a 4-h mixed-meal tolerance test (MMTT) was observed in the group treated with a full-dose 14-day course. In certain prespecified subgroups, the AUC mean C-peptide differences versus placebo appeared to be most pronounced in recently diagnosed patients, patients in the U.S., and in younger patients. The drug was generally well tolerated.
A recent study reported that teplizumab treatment reduced β-cell death at 1 year, but the differences versus placebo were not significant earlier, at 6 months (12). The acute (i.e., within 1 year) effects of immunotherapy on β-cell function may not occur through the same mechanisms as longer-term effects that have greater clinical importance. Improvement in C-peptide responses may be seen in type 1 diabetes trials, even with therapies that do not affect immune responses, through mechanisms that may involve recovery of dysfunctional β-cells when inflammation is acutely resolved (5,13). To be of value, a lasting effect on β-cell function and survival is needed.
The objective of this report is to characterize the efficacy and safety of teplizumab over 2 years and identify characteristics associated with response to therapy. Regarding efficacy, we focus on the 14-day full-dose regimen that was administered versus placebo, because at 1 year, efficacy was seen in the 14-day full-dose arm but not in the reduced-dose or curtailed-dose arms (11). Emphasis is given to AUC mean C-peptide because this has become the preferred measure of efficacy in type 1 diabetes immunotherapy (14). To explore the potential implications for dosing in future studies, we also describe the pharmacokinetics and pharmacodynamics of teplizumab, the effect of antidrug antibodies, and the safety profiles of all three dosing regimens.
RESEARCH DESIGN AND METHODS
Details of the trial methodology were published previously (11) and are summarized briefly here and in the Supplementary Data online. Participation was restricted to patients with type 1 diabetes diagnosed according to American Diabetes Association (ADA) criteria (15) within the prior 12 weeks and who required injected insulin therapy. Inclusion also required detectable levels of fasting or stimulated C-peptide and autoantibodies to one or more standard islet autoantigens. Exclusion criteria focused on medical disorders, such as active infections, that might confound results or interfere with safe trial completion. The research protocol was approved by institutional review boards, and all participants or guardians gave written informed consent.
Patients were randomly assigned (2:1:1:1) to one of four parallel treatment groups, with an escalating dose, 14-day course of daily intravenous treatment starting at baseline, and another 14-day course at week 26. For each treatment course, the 14-day full-dose group (n = 209) received a total cumulative teplizumab dose of ∼9,034 µg/m2, the 14-day low-dose group (n = 102) received a total of ∼2,985 µg/m2, the 6-day full-dose group received a total of ∼2,426 µg/m2 over 6 days, followed by 8 days of placebo, and the placebo group (n = 99) received 14 days of placebo infusions. Randomization was stratified by age-group (8–11, 12–17, and 18–35 years) and by country. Dosing was double-blind (patients and study personnel) to conceal allocation. Patients received a nonsteroidal, anti-inflammatory drug (e.g., ibuprofen) and/or antihistamine (e.g., diphenhydramine) to minimize adverse events during each treatment cycle.
Intensive diabetes care was provided for all patients. Investigators were instructed to aggressively treat diabetes to a target HbA1c of ≤6.5% and to maintain an insulin dose of ≥0.25 units/kg/day, but insulin adjustment algorithms were not prespecified. Patients used diary cards to record insulin use at screening and for 3 days before each visit at days 91, 140, 273, 364, 448, 546, 616, and 728. Use of agents that might affect islet growth, endogenous insulin secretion, insulin sensitivity, or immune function was not permitted during the study.
HbA1c was measured, and a 4-h MMTT was performed at a screening visit and on days 140, 364, 546, and 728 (HbA1c was also measured on days 273, 448, and 616), and the total AUC mean C-peptide during the MMTT was then calculated (1). After interim analyses determined that the primary end point at 1 year was not met, patients not yet at day 728 continued follow-up, but AUC mean C-peptide, flow cytometry, and anti-drug antibodies were no longer measured to reduce the burden on participants and cost. Anti-cytomegalovirus (CMV) IgG, anti–Epstein-Barr virus (EBV) IgG, and IgM titers were measured at screening and days 28, 91, 140, 210, 273, 364, and 728 to evaluate seropositivity for EBV and CMV; semiquantitative PCR was used to measure viral load for seropositive patients.
Adverse events, including clinically significant hypoglycemia, and abnormal laboratory values were reported by investigators, coded using the Medical Dictionary for Regulatory Activities, and graded using the Common Terminology Criteria for Adverse Events (version 3.0).
Statistical analysis.
Changes from baseline for AUC mean C-peptide, a prespecified secondary end point, HbA1c, and other measures in teplizumab groups were compared with the placebo group using mixed-model repeated-measures analysis (MMRM) models, adjusted for age-group and baseline values. A Mantel-Haenszel test stratified by age-group (8–11, 12–17, and 18–35 years) or Fisher exact test was used for exploratory efficacy analyses of dichotomous outcomes. Two-sided testing was done at an α level of 0.05. Subset analyses compared the 14-day full-dose regimen with placebo; age-groups, regions, and time from diagnosis to randomization were prespecified subsets (11). These analyses were done for hypothesis generation because the primary outcome was not significant at 1 year; therefore, we did not adjust for multiple comparisons. Similar analyses were conducted for the other treatment groups; however, no meaningful findings were observed, so the results are not presented.
The 1-year analysis used a nonparametric analysis and reported median values for AUC mean C-peptide because the distribution was not normal (11); a last observation carried forward analysis was used at the request of regulators, because too few time points existed for a longitudinal analysis at 1 year. For the current report at 2 years, longitudinal analysis (MMRM) was used instead of last observation carried forward. AUC mean C-peptide change from baseline was calculated using [ln(AUC mean C-peptideDay x + 1) − ln(AUC mean C-peptideBaseline + 1)]. The adjusted mean values reported here reflect the logarithm values after adjustment for the covariates (listed above). Consequently, the adjusted means and statistical significance reported here differ from the unadjusted medians and P values reported earlier at 1 year (11). The overall mean of insulin use was calculated for each group using all values after baseline.
Safety and tolerability through year 2 were assessed primarily by summarizing adverse experiences, serious adverse experiences (life-threatening, death, persistent disability, or hospitalization) and adverse experiences of special interest (acute mononucleosis-like illness, infection requiring intravenous antibiotic treatment, demyelinating disease, lymphoma or other malignancy, clinically significant hypoglycemia requiring assistance, grade 3 liver function abnormalities, grade 3 thrombocytopenia, grade 3 neutropenia; and through year 1: rash, grade 4 allergic/hypersensitivity, and grade 4 cytokine-release syndrome).
RESULTS
A high proportion (90% overall) of randomized patients completed 2 years of follow-up (Supplementary Table 1). In the 14-day full-dose group at 2 years, 89% had HbA1c measured and insulin therapy recorded, but only 64% had year 2 AUC mean C-peptide measurements because these were discontinued after final analysis of year 1 data (see Research Design and Methods) (Fig. 1A). Baseline characteristics, including diabetes measures (autoantibodies, C-peptide, insulin dose, and HbA1c), were balanced across treatment groups but not geographic regions. In particular, patients in India had less frequent ICA512 autoantibodies, higher HbA1c, higher insulin use, and lower AUC mean C-peptide, the latter suggesting more advanced disease on average than other regions (11).
FIG. 1.
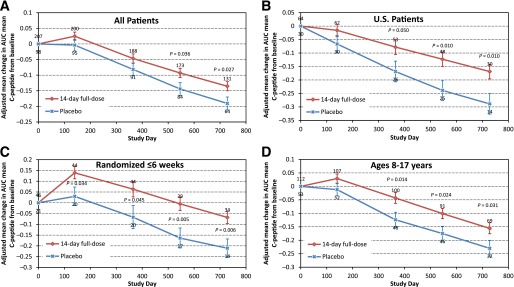
Adjusted mean changes in AUC mean C-peptide from baseline over time in the 14-day full-dose and placebo groups. Bars indicate standard errors; numbers of patients are above (teplizumab) or below (placebo). P values are indicated where significant. Changes in AUC mean C-peptide from baseline were calculated using [ln(AUC mean C-peptideDay x + 1) − ln(AUC mean C-peptideBaseline + 1)]. A: All patients. B: Patients in the U.S. C: Patients randomized ≤6 weeks after diagnosis. D: Subjects 8–17 years of age.
HbA1c.
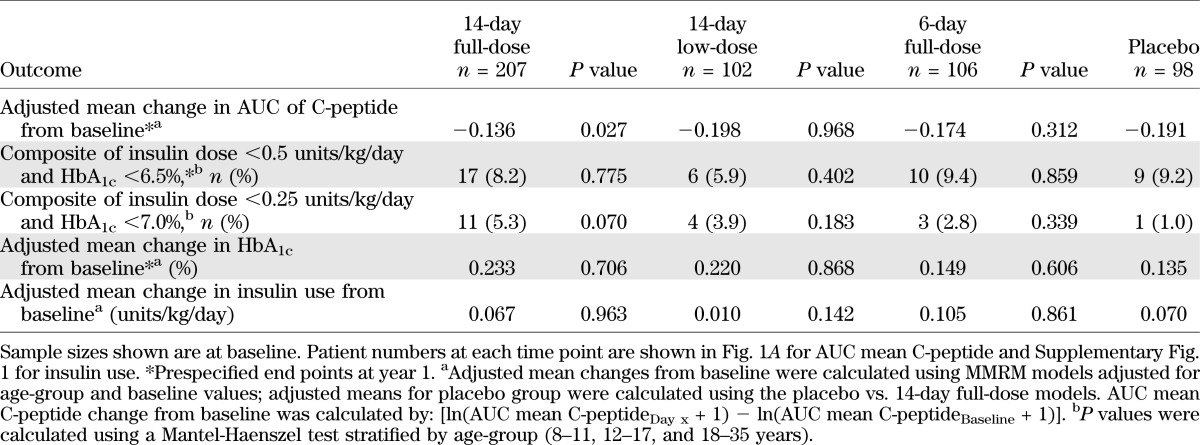
Intensive diabetes care with insulin was provided for all patients. There was no significant difference in HbA1c change from baseline comparing the teplizumab and placebo groups over the 2-year study at any time point (see Table 1), suggesting that glycemic control was maintained to a comparable extent across treatment groups.
TABLE 1.
Outcomes at year 2
Efficacy measures during 2 years of follow-up
AUC mean C-peptide.
Teplizumab treatment (14-day full-dose) reduced the loss of AUC mean C-peptide at 2 years versus placebo (P = 0.027; Fig. 1A and Table 1). The adjusted mean differences in AUC mean C-peptide change from baseline at 2 years favored the 14-day full-dose regimen versus placebo in analyses of all patients, patients in the U.S., and patients randomized ≤6 weeks after diagnosis (Figs. 1B and C and 2 and Table 2). The results in these prespecified subsets suggested larger treatment effects in patients with characteristics consistent with less advanced disease. Therefore, additional analyses were conducted to explore the treatment effects in other patient subsets at entry defined by 1) HbA1c<7.5%, the ADA recommendation for type 1 diabetes control in children 13–19 years of age (16); 2) insulin use <0.4 units/kg/day, the lower limit of typical type 1 diabetes insulin needs (17); 3) AUC mean C-peptide >0.65 nmol/L, the mean at baseline (11); and 4) AUC mean C-peptide >0.2 nmol/L, a value including ≥90% of newly diagnosed patients and comparable (18) to an amount of insulin reserve thought to be clinically beneficial (19).
FIG. 2.

Adjusted mean difference is shown in AUC mean C-peptide change from baseline at 2 years among subsets at study entry in the 4-day full-dose group vs. placebo. The ♦ indicate least squares means; bars indicate 95% CIs. AUC mean C-peptide change from baseline was calculated using [ln(AUC mean C-peptideDay x + 1) − ln(AUC mean C-peptideBaseline + 1)].
TABLE 2.
Adjusted mean change from baseline at year 2 for AUC mean C-peptide in the 14-day full-dose and placebo groups by characteristics at study entry
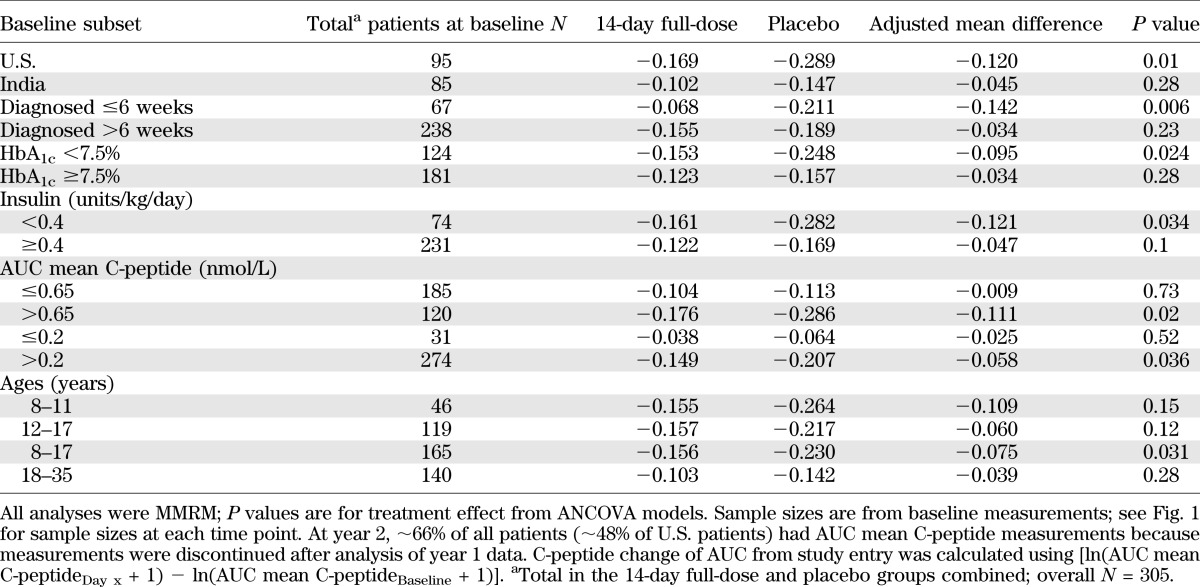
Importantly, patients randomized ≤6 weeks after diagnosis had the largest treatment difference versus placebo among the baseline subsets examined (Fig. 2 and Table 2). Subsets with U.S. residence, HbA1c <7.5%, insulin use <0.4 units/kg/day, AUC mean C-peptide >0.65 nmol/L at entry, or AUC mean C-peptide >0.2 nmol/L also had much larger differences versus placebo compared with subsets of patients from India, higher HbA1c, higher insulin use, and lower C-peptide, respectively. Of note, for patients in the U.S. and India, mean baseline HbA1c was 7.6% (60 mmol/mol) and 9.7% (83 mmol/mol), insulin use was 0.47 and 0.98 units/kg/day, and AUC mean C-peptide was 0.77 and 0.53 nmol/L, respectively (11).
Age-groups were also prespecified for analyses, served as an enrollment stratification criterion, and served as an adjustment covariate in analyses. Although the difference versus placebo was small and not statistically significant in 18- to 35-year-olds, treatment effects were larger in the 8- to 11- and 12- to 17-year-olds, and these groups were combined (Table 2). In the combined 8- to 17-year-old subset, differences in AUC mean C-peptide change from baseline favored the 14-day full-dose versus placebo group (P < 0.05 at 1 year and all subsequent time points; Figs. 1D and 2 and Table 2). Among age subsets, a large difference versus placebo was seen in 8–11 years of age, but the P value was not significant until combined with ages 12–17 years, perhaps due to the smaller number of patients in the youngest group.
Insulin use.
After an initial decline from baseline, adjusted mean insulin use increased progressively over time (Supplementary Fig. 1 and Table 1). It was prespecified to look at the largest countries in the trial (U.S. and India), and there were important regional differences in insulin use, HbA1c, and C-peptide at study entry, as described above. The overall adjusted mean insulin use (units/kg/day) at all times after baseline in the 14-day full-dose versus placebo groups was 0.59 versus 0.62 for all patients (not significant) and 0.44 vs. 0.50 (P = 0.02) for U.S. patients (data not shown). For individual time points, the difference versus placebo was statistically significant at day 448 in U.S. patients (Supplementary Fig. 1). Compared with placebo, a greater proportion of patients in the 14-day full-dose group met the modified composite end point of HbA1c <7% (53 mmol/mol) and insulin use <0.25 units/kg/day, and the differences were statistically significant at days 91, 273, 364, and 616 (Supplementary Fig. 2). Despite being blind to treatment, at 1 year, 5.3% (11/207) of patients in the 14-day full-dose group were not taking insulin, compared with 0% (0/98) in the placebo group (P = 0.02). At year 2, 3 of these 11 patients remained off insulin, whereas all placebo patients were still taking insulin (P > 0.05).
Teplizumab pharmacokinetics, immunogenicity, and effects on T cells.
Higher levels of anti-teplizumab (anti-drug) antibodies were seen in cycle 2 than cycle 1 for all three teplizumab regimens (Supplementary Table 2). For typical patients in the 14-day full-dose group who did not make anti-drug antibodies, teplizumab levels peaked on day 14 with concentration minimum and maximums (mean ± SD) of 418 ± 225 and 826 ± 391 ng/mL, respectively. However, teplizumab clearance increased with maximum observed anti-drug antibody concentrations, and some patients demonstrated a strong anti-drug antibody response after ∼10 days of cycle 2 dosing, with an abrupt reduction of bioavailability and increase in drug clearance (Supplementary Fig. 3). There did not appear to be a meaningful correlation between anti-drug antibody levels and AUC mean C-peptide changes from baseline (Supplementary Table 3), nor with response using the modified composite HbA1c plus insulin usage end point (data not shown). Additional details are in the Supplementary Data online.
Circulating levels of CD4+ and CD8+ T cells were transiently reduced during each cycle of treatment but not in the placebo group (Fig. 3A). The effects of teplizumab on CD4+ T cells appeared to diminish at anti-drug antibody levels >5,000 ng/mL (Supplementary Fig. 4). This level was observed in 19% of patients in the 14-day full-dose group after the second course of drug (Supplementary Table 2). During treatment, teplizumab was transiently bound to CD3 molecules on surfaces of CD4+ and CD8+ T cells (Fig. 3B). There was some evidence of down-modulation (Fig. 3C) and an increase in the percentage of circulating forkhead box P3 (Foxp3)+ CD8+ (but not CD4+) T cells during teplizumab dosing (Fig. 3D).
FIG. 3.

Flow cytometry for CD4+ (left) and CD8+ T cells (right). A: Cell counts. B: CD3 occupancy/cell. C: CD3/TCR (T-cell receptor) modulation on cells. D: Percentage of cells positive for Foxp3 marker. Symbols indicate means, and bars indicate standard errors. A and C: Number of patients is above (placebo) or below (teplizumab). B and D: Number of patients is above (teplizumab) or below (placebo).
Safety and tolerability.
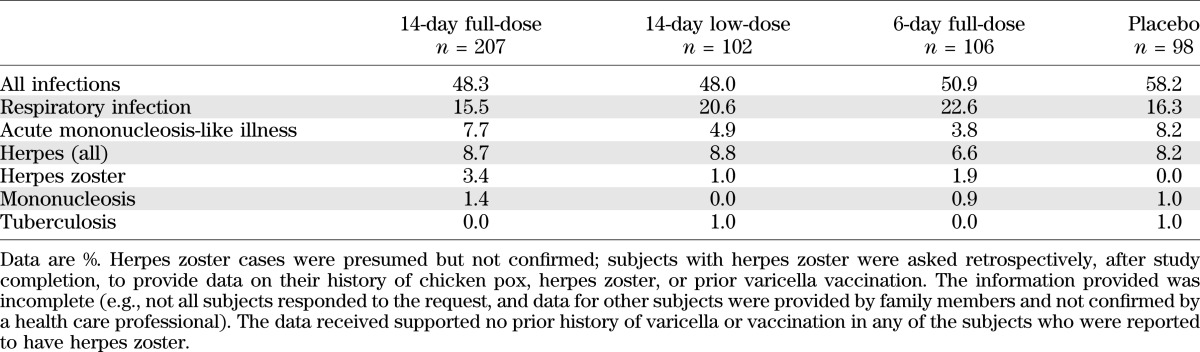
There were no differences in adverse events or serious adverse events among groups at year 2 (Table 3). Grade 3 adverse events were increased in teplizumab groups, but this difference versus placebo was primarily due to lymphopenia, an expected consequence of the mechanism of action. In particular, no differences were apparent between groups in the incidence of infections overall, or by specific types, with the possible exception of herpes zoster (10 teplizumab patients vs. no placebo patients, all nonserious; Table 4). Information on history was incomplete, but there was no convincing evidence of a history of varicella or varicella vaccination in any of the patients who reported herpes zoster (12–34 years of age). Three events occurred within 28 days of starting cycle 1 of treatment, whereas five occurred after 270 days when drug is no longer detectable in the circulation. Other herpes virus infections, including CMV and EBV, did not appear to increase in frequency during the 2 years of the trial. The most common infection was upper respiratory infection (16.3% of placebo vs. 15.5% of 14-day full-dose patients), which did not differ appreciably between teplizumab groups and placebo (Table 4). As expected, no rashes or cytokine release events occurred during the second year because the drug was not administered during this period.
TABLE 3.
Adverse events in the safety population in the complete 2-year study
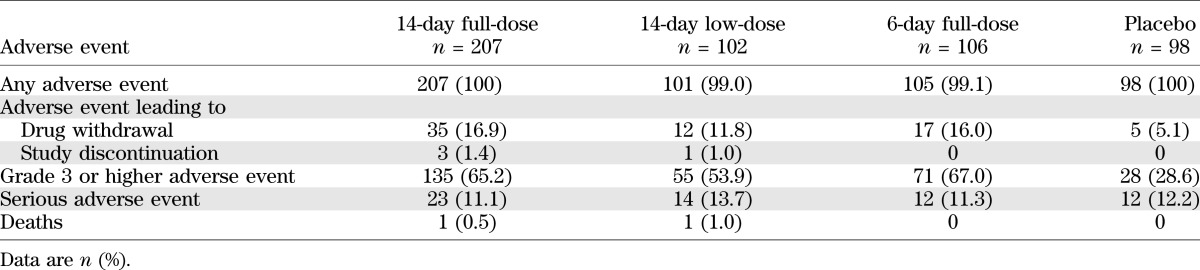
TABLE 4.
Incidence of infections in the complete 2-year study
DISCUSSION
In this report of data from the complete Protégé trial, patients with new-onset type 1 diabetes who received a full course of teplizumab (14-day full-dose) had significant improvement in stimulated C-peptide responses compared with placebo-treated subjects (P = 0.027). This effect was strongest in particular subsets, including children, those randomized ≤6 weeks after diagnosis, and in U.S. participants. As reported earlier, no significant differences were observed between the teplizumab and placebo treatment groups using a previously unvalidated primary composite end point (insulin <0.5 units/kg/day and HbA1c <6.5% at year 1). Although investigators were instructed to treat aggressively to HbA1c <6.5% and to maintain insulin >0.25 units/kg/day, this may have been unrealistic given that, after a nadir of ∼6.9% (52 mmol/mol) at 90 days, mean HbA1c increased to ∼7.9% (63 mmol/mol) at year 1 (11) and to 8.4% (68 mmol/mol) at year 2. Further, C-peptide may be a more objective and reliable outcome than insulin use and HbA1c because it is a more direct indicator of endogenous insulin secretion and cannot be easily measured or manipulated by patients or their physicians. AUC mean C-peptide is now the most widely used end point for type 1 diabetes interventions and is accepted by the U.S. Food and Drug Administration as a primary end point for these trials (6,8,20–22).
The AUC mean C-peptide treatment difference versus placebo did not appear to change markedly during the second year of follow-up (Fig. 1A), although the study was not designed to test hypotheses regarding time-trends. A particularly strong treatment effect was found in patient subsets that shared characteristics of early type 1 diabetes, including treatment sooner after diagnosis, lower baseline insulin use, greater C-peptide, and lower HbA1c at baseline. The higher baseline HbA1c and lower C-peptide levels suggest the patients in India had more advanced disease. Larger treatment effects on AUC mean C-peptide were also observed in subjects 8–17 years of age, who have a more rapid C-peptide decline, on average, than adults.
The modified composite end point (insulin <0.25 units/kg/day and HbA1c <7.0%) and insulin use also suggested a treatment benefit on insulin use. In the U.S., the overall insulin use was less in the teplizumab-treated subjects compared with those receiving placebo. This trend was not seen when subjects outside the U.S. were included, perhaps reflecting different patterns of insulin use in other countries. Together, the results suggest teplizumab treatment preserves endogenous insulin, thereby reducing needs for exogenous insulin to maintain glycemic control.
Immunotherapy must meet a high safety standard because clinical type 1 diabetes can be managed using insulin. However, good metabolic control is often difficult to achieve safely with insulin: a recent study of 25,833 type 1 diabetic patients revealed that 7% reported severe hypoglycemic events (seizure or coma) and 8% reported diabetic ketoacidosis in the prior 12 months (23). High doses of anti-CD3 immunotherapy are associated with tolerability/toxicity issues (9,24), whereas low doses appear to be ineffective (11,25). One phase 2 trial of teplizumab (9) used a high dose (37 mg total per course per 1.9-m2 subject) and observed a high (28%) incidence of grade 2 or greater adverse events associated with infusion (primarily fever, nausea, vomiting, and rigors), whereas the incidence was only 6% in an earlier trial (6). The Protégé trial used a dose of 17 mg total per course (for a 1.9-m2 patient), comparable with that used in the earlier trial (6), and experienced similar excellent tolerability. Conversely, very low-dose otelixizumab (another nonactivating Fc-modified anti-CD3 monoclonal antibody), dosed at 3.1 mg over 8 days, did not preserve β-cell function in a double-blinded phase 3 study (25), and the Protégé treatment arms with lower cumulative dose were also ineffective (11). Overall, the 14-day full-dose regimen of Protégé appears to provide sufficient drug to influence efficacy measures, with acceptable tolerability and safety.
Treatment-related adverse experiences were mostly limited to the dosing period and generally resolved within 14 days (11). Most (transient cytopenias, transient mild laboratory or clinical manifestations of cytokine release such as rash, headache, nausea, and vomiting) were moderate, manageable, and expected as a manifestation of the intended mechanism of action. Along with transient small increases in aminotransferases, these also represented the main differences versus placebo in year 1 safety analyses (11). Use of effective stopping rules (based on liver function tests to delimit cytokine-release syndrome) served to lessen adverse events compared with earlier studies, allowing 90.6% of treated patients to complete a full course of drug (11).
The observed reduction in circulating CD4+ and CD8+ cells likely reflects transient margination of the T-cell compartment and apoptosis of some activated T-cell subsets. Both may be relevant mechanisms of action of teplizumab, wherein the T-effector cells, which are maintaining an inflammatory environment in the pancreas, are preferentially depleted while regulatory T cells are favored. Flow cytometry analysis of peripheral blood in the treated Protégé patients suggested that Foxp3 expression might be increased in CD8+ but not CD4+ T cells during periods of maximum drug binding to T cells. Previous studies have shown that teplizumab induces activation of CD8+ T cells with regulatory function (26,27). In addition, CD4+ and CD8+ cells are directed to the lamina propria, where they appear to acquire regulatory function, although cell deletion may also be involved in the drug action.
Longer-term changes in patient immune function, such as persistent low CD4 counts (9) and reactivation of EBV infection, were reported from previous studies that used much higher anti-CD3 doses (28). At the lower doses used in Protégé, EBV reactivation was rare, and acute mononucleosis syndrome was not increased versus placebo. A possible dose-related increase in herpes zoster was seen, with 10 cases (that could not be subsequently confirmed) reported among teplizumab patients; no cases occurred in the placebo group. Of note, in a subsequent phase 3, double-blind, randomized study (n = 254) with identical teplizumab dosing (NCT00920582), after 2 years of follow-up, the only patient with herpes zoster was a placebo patient (data on file).
To be meaningful, treatment effects must be maintained for multiple years. Repeated dosing might be advantageous if it increases durability without causing new or cumulative side effects. Protégé did not include an arm with a single drug cycle and cannot answer whether two drug cycles confer greater benefit or duration than a single cycle. Nonetheless, Protégé did not identify any cumulative, persistent, or unexpected safety or tolerability issues. Although high levels of anti-drug antibodies occurred late in the second cycle in about one-sixth of all patients and appeared to accelerate drug clearance, this did not appear to affect efficacy end points.
The large number and diverse characteristics of Protégé patients enables more precise estimates of treatment effects than smaller trials, increases generalizability, and allows for meaningful subset analyses. The 2-year follow-up provides evaluation of efficacy and safety during placebo-controlled double-blind conditions for a longer period than previous trials. The double-blind design reduces the potential for bias. Limitations of the study include the heterogeneous baseline patient disease status, post hoc analyses without adjustment for multiple comparisons, and elimination of AUC mean C-peptide measurements in some patients after the primary analysis at year 1, which may have reduced statistical power. Another limitation is the lack of information on HLA-DQ/DR or other genotypes that might identify patient subsets with greater response.
Rodent studies reported full reversal of diabetes using anti-CD3 immunotherapy, but only when given immediately at disease onset (29,30). In clinical trials, delays due to required screening and enrollment procedures may lead to lower drug efficacy. In actual clinical settings, immunotherapy could be initiated promptly at the time of diagnosis. Further, the peak incidence of diabetes occurs in 8- to 11-year-olds, and subjects 8–17 years of age appeared to have a greater drug response than older patients.
In summary, continued follow-up for a second year demonstrated a benefit of teplizumab treatment on AUC mean C-peptide and a possible benefit on insulin needs. Most importantly, these analyses identified baseline characteristics associated with greater treatment efficacy. No new safety or tolerability issues emerged. These post hoc findings are hypothesis-generating, and confirmation is needed; nevertheless, they suggest that future studies of CD3 immunotherapy should consider recruiting young patients with better glucose control and greater remaining endogenous insulin secretion and initiating treatment immediately upon diagnosis.
ACKNOWLEDGMENTS
This research was supported by MacroGenics, Inc. (Rockville, MD). D.C., E.B., S.J., S.K., and A.G.D. are full-time employees and K.E.S. is a part-time employee of MacroGenics, Inc. and have stock options as part of an employee-offering program. W.H., R.J.F., N.S., and J.L. received research support for this study from MacroGenics. J.L. has received advisory board support from Johnson & Johnson. R.J.F. has received research support within the past 3 years from Tolerx (to study otelixizumab for recent-onset type 1 diabetes), unrelated research support from the U.S. National Institutes of Health (grants R21-HD-059292, T35-DK-007405, and U01-DK-085465), JDRF (grant 2011-597), Gabrielle’s Angel Foundation, Eli Lilly, Diamyd, Pfizer, Bristol-Myers Squibb, Takeda, and Novo Nordisk, and unrestricted research support from Le Bonheur Foundation (Memphis, TN). W.H. chaired the data safety and monitoring board for the BHT-3021-01 insulin plasmid trial (Bayhill Pharmaceuticals) and received research support from Novo Nordisk. K.C.H. has received research support from MacroGenics and support from the JDRF (grant 2008-502) for laboratory studies on patients’ samples from Protégé. No other potential conflicts of interest relevant to this article were reported.
W.H., R.J.F., N.S., D.C., A.G.D., and K.C.H. contributed to planning of protocol-stated analyses and post hoc analyses. All authors contributed to study implementation and supervision of data collection at the sites. D.C. designed and did the statistical analysis and verified its accuracy. D.C. and A.G.D. contributed to compiling the official clinical study report. E.B., S.J., K.E.S., S.K., K.C.H., and J.L. contributed to the study design. All authors had full access to the data, helped draft the report or critically revise the draft, contributed to data interpretation, and reviewed and approved the final version of the report. W.H. and J.L. are the guarantors of this work, and as such, had full access to all the data, and take full responsibility for the integrity of the data and the accuracy of the data analysis.
Parts of this study were presented in abstract form at the 71st Scientific Sessions of the American Diabetes Association, San Diego, California, 24–28 June 2011; at the 47th European Association for the Study of Diabetes, Lisbon, Portugal, 12–16 September 2011; and at the 48th European Association for the Study of Diabetes, Berlin, Germany, 1–5 October 2012.
The authors thank Philip Ross, MedStrat Communications, for editorial assistance in preparing the manuscript, with support from MacroGenics, Inc.
Footnotes
Clinical trial reg. no. NCT00385697, clinicaltrials.gov.
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db13-0236/-/DC1.
A list of all the Protégé trial investigators can be found in the Supplementary Data online.
See accompanying commentary, p. 3669.
REFERENCES
- 1.Steele C, Hagopian WA, Gitelman S, et al. Insulin secretion in type 1 diabetes. Diabetes 2004;53:426–433 [DOI] [PubMed] [Google Scholar]
- 2.Binder C, Faber OK. Residual beta-cell function and its metabolic consequences. Diabetes 1978;27(Suppl. 1):226–229 [DOI] [PubMed] [Google Scholar]
- 3.Steffes MW, Sibley S, Jackson M, Thomas W. Beta-cell function and the development of diabetes-related complications in the diabetes control and complications trial. Diabetes Care 2003;26:832–836 [DOI] [PubMed] [Google Scholar]
- 4.Madsbad S, Alberti KG, Binder C, et al. Role of residual insulin secretion in protecting against ketoacidosis in insulin-dependent diabetes. BMJ 1979;2:1257–1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sherry NA, Kushner JA, Glandt M, Kitamura T, Brillantes AM, Herold KC. Effects of autoimmunity and immune therapy on beta-cell turnover in type 1 diabetes. Diabetes 2006;55:3238–3245 [DOI] [PubMed] [Google Scholar]
- 6.Herold KC, Hagopian WA, Auger JA, et al. Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. N Engl J Med 2002;346:1692–1698 [DOI] [PubMed] [Google Scholar]
- 7.Pescovitz MD, Greenbaum CJ, Krause-Steinrauf H, et al. Type 1 Diabetes TrialNet Anti-CD20 Study Group Rituximab, B-lymphocyte depletion, and preservation of beta-cell function. N Engl J Med 2009;361:2143–2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buzzetti R, Cernea S, Petrone A, et al. DiaPep Trialists Group C-peptide response and HLA genotypes in subjects with recent-onset type 1 diabetes after immunotherapy with DiaPep277: an exploratory study. Diabetes 2011;60:3067–3072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Herold KC, Gitelman S, Greenbaum C, et al. Immune Tolerance Network ITN007AI Study Group Treatment of patients with new onset Type 1 diabetes with a single course of anti-CD3 mAb Teplizumab preserves insulin production for up to 5 years. Clin Immunol 2009;132:166–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Herold KC, Gitelman SE, Masharani U, et al. A single course of anti-CD3 monoclonal antibody hOKT3gamma1(Ala-Ala) results in improvement in C-peptide responses and clinical parameters for at least 2 years after onset of type 1 diabetes. Diabetes 2005;54:1763–1769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sherry N, Hagopian W, Ludvigsson J, et al. Protégé Trial Investigators Teplizumab for treatment of type 1 diabetes (Protégé study): 1-year results from a randomised, placebo-controlled trial. Lancet 2011;378:487–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lebastchi J, Deng S, Lebastchi AH, et al. Immune therapy and β-cell death in type 1 diabetes. Diabetes 2013;62:1676–1680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ablamunits V, Sherry NA, Kushner JA, Herold KC. Autoimmunity and beta cell regeneration in mouse and human type 1 diabetes: the peace is not enough. Ann N Y Acad Sci 2007;1103:19–32 [DOI] [PubMed] [Google Scholar]
- 14.Greenbaum CJ, Beam CA, Boulware D, et al. Type 1 Diabetes TrialNet Study Group Fall in C-peptide during first 2 years from diagnosis: evidence of at least two distinct phases from composite Type 1 Diabetes TrialNet data. Diabetes 2012;61:2066–2073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.American Diabetes Association Diagnosis and classification of diabetes mellitus. Diabetes Care 2006;29(Suppl. 1):S43–S48 [PubMed] [Google Scholar]
- 16.American Diabetes Association Standards of medical care in diabetes—2011. Diabetes Care 2011;34(Suppl. 1):S11–S61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hirsch IB, Skyler JS. Chapter 17 – The Management of Type 1 Diabetes. Updated 2009. Available from: http://www.endotext.org/diabetes/diabetes17/diabetesframe17.htm Accessed 5 November 2012
- 18.Besser RE, Shields BM, Casas R, Hattersley AT, Ludvigsson J. Lessons from the mixed-meal tolerance test: use of 90-minute and fasting C-peptide in pediatric diabetes. Diabetes Care 2013;36:195–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.The Diabetes Control and Complications Trial Research Group Effect of intensive therapy on residual beta-cell function in patients with type 1 diabetes in the diabetes control and complications trial. A randomized, controlled trial. Ann Intern Med 1998;128:517–523 [DOI] [PubMed] [Google Scholar]
- 20.Ludvigsson J, Faresjö M, Hjorth M, et al. GAD treatment and insulin secretion in recent-onset type 1 diabetes. N Engl J Med 2008;359:1909–1920 [DOI] [PubMed] [Google Scholar]
- 21.Mandrup-Poulsen T. IAPP boosts islet macrophage IL-1 in type 2 diabetes. Nat Immunol 2010;11:881–883 [DOI] [PubMed] [Google Scholar]
- 22.Ludvigsson J, Krisky D, Casas R, et al. GAD65 antigen therapy in recently diagnosed type 1 diabetes mellitus. N Engl J Med 2012;366:433–442 [DOI] [PubMed] [Google Scholar]
- 23.Beck RW, Tamborlane WV, Bergenstal RM, Miller KM, DuBose SN, Hall CA, T1D Exchange Clinic Network The T1D Exchange clinic registry. J Clin Endocrinol Metab 2012;97:4383–4389 [DOI] [PubMed] [Google Scholar]
- 24.Keymeulen B, Vandemeulebroucke E, Ziegler AG, et al. Insulin needs after CD3-antibody therapy in new-onset type 1 diabetes. N Engl J Med 2005;352:2598–2608 [DOI] [PubMed] [Google Scholar]
- 25.Sprangers B, Van der Schueren B, Gillard P, Mathieu C. Otelixizumab in the treatment of type 1 diabetes mellitus. Immunotherapy 2011;3:1303–1316 [DOI] [PubMed] [Google Scholar]
- 26.Bisikirska B, Colgan J, Luban J, Bluestone JA, Herold KC. TCR stimulation with modified anti-CD3 mAb expands CD8+ T cell population and induces CD8+CD25+ Tregs. J Clin Invest 2005;115:2904–2913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ablamunits V, Bisikirska B, Herold KC. Acquisition of regulatory function by human CD8(+) T cells treated with anti-CD3 antibody requires TNF. Eur J Immunol 2010;40:2891–2901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keymeulen B, Candon S, Fafi-Kremer S, et al. Transient Epstein-Barr virus reactivation in CD3 monoclonal antibody-treated patients. Blood 2010;115:1145–1155 [DOI] [PubMed] [Google Scholar]
- 29.Chatenoud L, Thervet E, Primo J, Bach JF. Anti-CD3 antibody induces long-term remission of overt autoimmunity in nonobese diabetic mice. Proc Natl Acad Sci U S A 1994;91:123–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chatenoud L, Primo J, Bach J-F. CD3 antibody-induced dominant self tolerance in overtly diabetic NOD mice. J Immunol 1997;158:2947–2954 [PubMed] [Google Scholar]