

Abstract
The epidemic of obesity imposes unprecedented challenges on human adipose tissue (WAT) storage capacity that may benefit from adaptive mechanisms to maintain adipocyte functionality. Here, we demonstrate that changes in the regulatory feedback set point control of Insig1/SREBP1 represent an adaptive response that preserves WAT lipid homeostasis in obese and insulin-resistant states. In our experiments, we show that Insig1 mRNA expression decreases in WAT from mice with obesity-associated insulin resistance and from morbidly obese humans and in in vitro models of adipocyte insulin resistance. Insig1 downregulation is part of an adaptive response that promotes the maintenance of SREBP1 maturation and facilitates lipogenesis and availability of appropriate levels of fatty acid unsaturation, partially compensating the antilipogenic effect associated with insulin resistance. We describe for the first time the existence of this adaptive mechanism in WAT, which involves Insig1/SREBP1 and preserves the degree of lipid unsaturation under conditions of obesity-induced insulin resistance. These adaptive mechanisms contribute to maintain lipid desaturation through preferential SCD1 regulation and facilitate fat storage in WAT, despite on-going metabolic stress.
The epidemic of obesity is testing the capacity of white adipose tissue (WAT) to cope with an unprecedented nutritional pressure and demand to expand. We and others have proposed that the expansion and dysfunction of WAT may be an important pathogenic contributor to obesity-associated metabolic complications (1). However, a recurrent observation from clinical studies is that obese patients maintain a remarkable capacity to store fat before adverse metabolic effects occur. Moreover, we have recently demonstrated, using a monozygotic twin cohort discordant for obesity, that while expanding, the WAT adapts its biochemical features to maintain its biophysical characteristics and preserves its functionality by increasing the degree of fatty acid (FA) unsaturation and chain length in adipocyte membrane phospholipids (PLs). This implies the existence of adaptive homeostatic mechanisms that preserve WAT functionality (2).
Intrigued by these and other observations showing that improving WAT expandability and storage capacity ameliorates insulin sensitivity (3), we hypothesized the existence of adaptive responses that optimize fat storage and preserve WAT homeostasis. These adaptations may help WAT to cope with the added storage demands imposed by a positive energy balance through changes in transcriptional and biochemical cycles. The interest in identifying these adaptive mechanisms is fueled by their potential as biomarkers of metabolic stress (4), with diagnostic, prognostic, and therapeutic value in human obesity.
SREBPs are transcription factors that control genes involved in FA and triglyceride (TG) synthesis via SREBP1 or cholesterol biosynthesis through SREBP2. Owing to alternative splicing, two isoforms of SREBP1 exist. The highly active Srebp1a isoform is predominantly expressed in vitro, whereas Srebp1c, a less active form, is predominantly expressed in vivo. Demonstrated SREBP1 targets include genes involved in rate-limiting de novo lipogenesis steps (e.g., acetyl-CoA carboxylase [Acc] and FA synthase [Fas]) (5–7), FA desaturation (e.g., stearoyl-CoA desaturase 1 [Scd1]) (6,8) and elongation (e.g., elongase 6 [Elovl 6]) (9,10), FA re-esterification (e.g., mitochondrial glycerol-3-phosphate acyltransferase [Gpat1]) (11), and PL synthesis (e.g., CTP: phosphocholine cytidylyltransferase α [Cctα]) (12,13). Given these pleiotropic effects of SREBP1 on lipid metabolism, we hypothesized that SREBP1 may play a leading role in adaptive responses in WAT. Moreover, because a key event in this response is the post-translational maturation and activation of SREBP1 and this process is highly dependent on the activity of insulin-induced genes 1 and 2 (Insig1 and Insig2), we hypothesized that the set point linking SREBP1 and Insig1 may be important for the process of WAT adaptation to excess lipid storage.
It is well established that under conditions characterized by high sterol levels or elevated polyunsaturated/unsaturated FAs, SREBP1 is physiologically inactivated. Under these conditions, SREBP1 remains anchored in the endoplasmic reticulum (ER) as part of a tripartite complex with SREBP cleavage-activating protein (SCAP) and Insig1 or Insig2 (14,15), which exert a permissive retaining effect on SREBP1, preventing its maturation and activation. This regulatory system is highly sensitive and rapidly activated in response to conditions requiring SREBP1 activation (e.g., decreased polyunsaturated FA, anabolic effects of insulin, or excess saturated FA). In these situations, the ER–anchoring activity of Insigs is diminished, allowing SCAP to escort SREBP1 from the ER to the Golgi to be cleaved, producing a soluble and transcriptionally active SREBP1 mature protein.
The importance and dependence on Insig1 for the regulation of SREBP1 activity and lipogenic capacity has been demonstrated in vitro and in vivo. Overexpressing Insig1 in 3T3-L1 adipocytes inhibits both adipogenesis and lipid accumulation (16). Similarly, peroxisome proliferator–activated receptor-γ agonists can induce Insig1 expression in vitro and in vivo in WAT (17,18) while promoting adipocyte differentiation, suggesting that Insig1 acts as a control set point to limit the amount of fat that can be stored under anabolic conditions. Coordinating this regulation, insulin itself can activate SREBP1 and promote lipogenesis, while, simultaneously, Insig1 expression is induced and restricts further SREBP1 activation. This has been demonstrated in vitro (17,19,20) and in vivo (21). These findings indicate a physiological negative feedback regulatory loop consisting in the regulation of Insig1 expression by SREBP1 in order to finely control its own cleavage. Akin to that, this type of autoregulatory feedback loop has already been suggested for the physiological control of cholesterol synthesis in liver via Insig1/SREBP (21,22). However, pathophysiological dysregulation of this loop in WAT in the context of obesity has never been considered.
In addition to genes involved in de novo synthesis of FAs and TG storage, SREBP1-target genes are also involved in the supply of specialized FAs used for critical cellular roles, including maintenance of membrane lipid homeostasis, lipid modification of proteins, and effectors of intracellular signaling (23). On the basis of previous knowledge, we hypothesized that in an insulin-resistance (IR) state, the downregulation of Insig1 will lead to a resetting of the physiological negative Insig1/SREBP1 regulatory feedback loop to optimize active SREBP1 levels in an attempt to maintain adipose lipid homeostasis and preserve the synthesis of specific lipid species essential for cell functionality. A similar concept has been described in a recent report that showed that in Caenorhabditis elegans and mammalian cells, genetic limitation in the biosynthesis of phosphatidylcholine (PC) PLs led to changes in membrane composition and functionality, promoted increased levels of nuclear mature SREBP1, and elevated lipogenesis. This study proposes a conserved feedback loop responding to PC levels and ensuring adequate levels of PC production (24). In a different experimental model, perturbation of phosphatidylethanolamine (PE) synthesis in the Drosophila heart leads to alteration in PE distribution in cardiac sarcolemmal membranes. Attempts to compensate this specific lipid perturbation involved adaptive elevation in SREBP1 mature protein levels and inappropriately increased lipogenesis (25) and associated pathology.
Here, we investigate whether coordinated regulation of Insig1/SREBP1 could work as an adaptive mechanism facilitating lipid biosynthesis to maintain WAT lipid homeostasis, defying the recognized antilipogenic effect determined by IR. In agreement with this model, our results indicate that decreased Insig1 expression in WAT is a common feature of murine models of diet- and genetic-induced obesity exposed to excessive nutrient pressure and is operative in human obesity and IR. In support of this concept, we have in vitro and in vivo evidence that decreased Insig1, as observed in obese individuals, facilitates adipocyte differentiation and fat deposition. Finally, we discuss whether this adaptive response may contribute to preserve lipid generation and storage under adverse metabolic conditions.
RESEARCH DESIGN AND METHODS
Human samples.
All subjects from the five cohorts studied gave their consent after being informed of the nature, purpose, and possible risks of the different studies. All ethical committees gave their approval.
Animal care.
Animals were housed in a temperature-controlled room with a 12-h light/dark cycle. Food and water were available ad libitum. Mice were fed a standard chow diet or a 45% high-fat diet (HFD; Research diet D12451) according to the experiment. All animal protocols were approved by the U.K. Home Office and the University of Cambridge, and mice were cared for according to the Guiding Principles for Research Involving Animals and Human Beings. The ob/ob and lean control mice (mix 129Sv-C57BL6) were generated as described in Medina-Gomez et al. (26).
Insig1 knockout mouse.
This mouse was generated by AstraZeneca Transgenics and Comparative Genomics (ATCG), AstraZeneca R&D MoIndal (Sweden) (Supplementary Fig. 11). For this study, tissue samples were collected from 11- to 12-week-old male mice. Dual-energy X-ray absorptiometry (Lunar Corporation) was used to measure body composition.
Cell culture and in vitro insulin resistance studies.
3T3-L1 preadipocytes were cultured as previously reported (27). IR was induced at day 9 of differentiation by incubating the cells in Dulbecco’s modified Eagle’s medium (25 mmol/L glucose) containing 100 nmol/L insulin for 16 h.
Stable Insig1 knockdown 3T3-L1 cell line.
Insig1 stable knockdown and control cell line were generated as previously described (28). Cells were cultured, differentiated into adipocytes, and stained for Oil-Red-O, as described previously (29). TG content was determined as previously reported (30).
RNA preparation and qRT-PCR.
RNA extraction quantitative (q)RT-PCR were performed as previously reported (26,28). The input value of the gene of interest was standardized to 18S.
Western blot analysis.
Protein isolation from cells and murine and human adipose tissue was carried out as reported (28,31). Proteins were electrotransfered on polyvinylidene fluoride membranes (Millipore) and probed with phosphorylated (p)AKT (Thr308), total AKT, α-tubulin (Cell Signaling), SREBP1 (Santa Cruz Biotechnology), INSIG1, and β-actin (Abcam) antibodies.
Total FA profiling-gas chromatography–mass spectrometry experiments.
Lipid profiling of adipose tissue (human and murine) was assessed by gas chromatography–mass spectrometry as described (32). Subfractionation of adipose tissue was performed as reported (33). Shotgun lipidomics were performed as described previously (26,34).
De novo FA synthesis assay.
Insig1 KD and control cells were differentiated in 24-well plates. After overnight serum starvation, both cell lines were treated with or without 200 nmol/L C75 (a Fas inhibitor) and with or without insulin for 30 min before adding 13C-labeled sodium acetate into the medium. Incorporation of 13C-sodium acetate into lipids was assessed during 60 min. Cells were then lysed with 1 volume of 1N KOH, and the supernatant was transferred to a screwtop vial. Subsequently, 1 volume of 5 mol/L KOH in 15% methanol was added and the lysate heated for 45 min at 65°C to saponify. Then 1 volume of cold 5 mol/L HCL was added to acidify the reaction in a fume cupboard, followed by the addition of 1 volume of cold absolute ethanol. Saponified lipids were extracted twice by adding 7.5 volume of n-hexane. Hexane containing the radioactive lipid mixture was mixed with the compatible organic solvent scintillate, and radioactivity was measured using liquid scintillation counter.
Statistical analysis.
The data presented here were analyzed using SPSS 19 software (IBM) using ANOVA, a general-linear model for ANCOVA, and the Student t test (unpaired). The significance level was set at P < 0.05. Principle component analysis was carried out using Simca 12 software (Umetrics).
RESULTS
Downregulation of Insig1 and Srebp1 mRNA and maintenance of SREBP1 mature protein in WAT of murine models of diet- and genetic-induced obesity.
We examined whether Srebp1 and Insig1 mRNA levels were altered in hypertrophic white adipocytes from mice undergoing nutritional excess, obesity, and IR. Our data show that 6 months on the HFD caused obesity and IR, as indicated by decreased levels of p-AKT in WAT (Supplementary Fig. 1A). At the transcriptional level, Srebp1c and Insig1 mRNA expression was decreased (Fig. 1A). These changes were not observed after 15 days on the HFD (Fig. 1B), when carbohydrate metabolism was still unaffected and the animals were healthy and actively expanding their WAT (Supplementary Fig. 1B). Interestingly, despite a reduction in Srebp1c mRNA expression, 6 months on the HFD was associated with increased levels of transcriptionally active mature SREBP1 protein in WAT compared with chow-fed animals (Fig. 1C and Supplementary Fig. 1C). Maintenance of SREBP1 maturation may result from reduced Insig1 mRNA expression. Of note, the level of SREBP1 protein was similar in WAT from 15 days’ HFD and chow-fed animals (Fig. 1D and Supplementary Fig. 1D).
FIG. 1.

Analysis of Insigs and Srebp1 mRNA expression and of SREBP1 mature protein levels in murine models of diet- and genetic-induced obesity. RT-PCR analysis was performed on RNA obtained from gonadal WAT of male WT C57BL6 mice fed an HFD for 6 months (n = 7 HFD; n = 9 chow) (A) or 15 days (n = 8 each group) (B) vs. their respective controls fed a standard chow diet and from gonadal WAT of males ob/ob and WT littermates when mice were 4 months old (n = 9 WT n = 8 ob/ob) (E) or 1 month old (n = 8 WT, n = 6 ob/ob) (F). Western blot analysis of SREBP1 mature protein levels was performed on gonadal WAT of the same groups of mice: WT C57BL6 mice fed an HFD for 6 months (n = 7 HFD; n = 7 chow) (C) or 15 days (n = 7 each group) (D) vs. their respective controls fed a standard chow diet and on gonadal WAT of male ob/ob and WT littermates when mice were 4 months old (n = 4 for both WT and ob/ob) (G) or 1 month old (n = 4 for both WT and ob/ob) (H). SREBP1 mature protein was normalized to α-tubulin as the loading control. Blots were analyzed by densitometry. WT C57BL6 fed 6 months on the HFD vs. chow diet: *P < 0.05, **P < 0.01, ***P < 0.001. WT vs. ob/ob: *P < 0.05, **P < 0.01, ***P < 0.001. Data are presented as mean ± SEM.
We also examined Srebp1c and Insig1 in WAT from genetically obese and IR ob/ob mice. At 4 months of age, when ob/ob mice are severely obese, hyperglycemic, and IR (26), expression of both Srebp1c and Insig1 mRNA in WAT is decreased compared with lean, insulin-sensitive control mice (Fig. 1E). Of note, this is not a leptin deficiency–dependent effect because levels of Srebp1c and Insig1 mRNA are increased in young 1-month-old ob/ob mice (Fig. 1F), when they are still insulin-sensitive and glucose-tolerant, as indicated by increased markers of insulin sensitivity (e.g., Glut4 [Fig. 1F] and Adiponectin (26)). At 1 month of age, ob/ob mice show active expansion of their WAT, and as observed previously in the diet-induced obesity (DIO) model, the WAT of ob/ob mice at 1 and 4 months of age showed similar levels of SREBP1 active protein with respect to wild-type (WT) littermates (Fig. 1G and H and Supplementary Fig. 1E and F) irrespectively of their gene expression pattern. Further support for discordant regulation of SREBP1 in lean and obese mice came from analyses of adipocyte sizes in 10-week-old ob/ob mice. In line with the concept that larger adipocytes are associated with IR, we found a negative correlation between adipocyte size and SREBP1 mature protein levels (Supplementary Fig. 10E) in both the ob/ob and WT mice. However, the levels of SREBP1 for a given size of adipocyte were five times higher in the ob/ob mice.
Insig1 and Srebp1 mRNA are specifically downregulated in adipocytes of morbidly obese humans.
We next determined whether the same Insig1/SREBP1 adaptive changes were observed in human WAT. We analyzed Srebp1 and Insig1 in WAT from lean (BMI 22.2 ± 0.8 kg/m2), obese (BMI 30.2 ± 1.1 kg/m2), and morbidly obese (MO) (BMI 48.9 ± 1.8 kg/m2) human subjects (Supplementary Table 1, cohort 1). Expression of Insig1 mRNA was severely decreased in omental (Om) and subcutaneous (SC) isolated adipocytes from obese and MO compared with lean individuals (Fig. 2A). In the same isolated adipocyte samples, Srebp1c and Srebp1a mRNAs were also specifically reduced in MO compared with lean and obese individuals (Fig. 2B). In an independent set of human samples (Supplementary Table 1, cohort 2), we confirmed that INSIG1 protein levels were reduced in SC WAT from MO (BMI 50.5 ± 5.0 kg/m2) compared with lean subjects (BMI 25.1 ± 0.9 kg/m2) (Fig. 2C).
FIG. 2.

Insig1 and Srebp1 mRNA and protein levels in white adipocytes of non-IR and IR MO subjects. RT-PCR analysis was performed on RNA obtained from mature adipocytes isolated from SC or Om depots from lean (BMI 22.2 ± 0.8 kg/m2), obese (BMI 30.2 ± 1.1 22.2 ± 0.8 kg/m2), and MO (BMI 48.9 ± 1.8 22.2 ± 0.8 kg/m2) human subjects (Supplementary Table 1, cohort 1), examining Insig1/2 (A) and Srebp1c/a (B) mRNA levels, standardized to 18S mRNA within each group (n = 10 lean, n = 10 obese, n = 12 morbid obese). MO vs. lean: *P < 0.05, **P < 0.01, ***P < 0.005, ****P < 0.0005. MO vs. obese: &P < 0.05. ANOVA: $P < 0.05, $$P < 0.005. C: Western blot analysis of INSIG1 protein in SC white adipocytes from lean (BMI 25.1 ± 0.9 kg/m2) and MO (BMI 50.5 ± 5.0 kg/m2) (Supplementary Table 1, cohort 2). The blots from SC samples were analyzed by densitometry, and INSIG1 protein levels were normalized to β-actin as a loading control (n = 6 lean, n = 6 MO). MO vs. lean: *P < 0.05 **P < 0.01. D: RT-PCR analysis was performed on RNA obtained from SC WAT from overweight (BMI 26.1 ± 1.19 kg/m2, HOMA 1.57 ± 1.19), MO non-IR (BMI 39.0 ± 4.27 kg/m2, HOMA 2.31 ± 0.44), and MO IR (BMI 41.9 ± 1.84 kg/m2, HOMA 4.40 ± 0.51) human subjects (Supplementary Table 1, cohort 1) examining Srebp1/Insig1 mRNA expression (n = 6 overweight, n = 4 MO non-IR, n = 4 MO IR). MO non-IR and IR vs. overweight: *P < 0.05, **P < 0.005. MO non-IR vs. IR: &P < 0.05. ANOVA: $P < 0.005. E: Mature SREBP1 protein levels were determined in these groups by Western blot analysis and normalized to β-actin. Blots were analyzed by densitometry (n = 4 overweight, n = 3 MO non-IR, n = 3 MO-IR). Data are presented as mean ± SEM.
IR leads to Insig1 downregulation in SC adipose tissue, resulting in maintenance of mature SREBP1 protein.
We then compared SC WAT from MO subjects showing different degrees of IR (MO non-IR, BMI 39.0 ± 4.27 kg/m2, homeostasis model assessment [HOMA] 2.31 ± 0.44; and MO IR, BMI 41.9 ± 1.84 kg/m2, HOMA 4.40 ± 0.51, respectively) versus slightly overweight control subjects (BMI 26.1 ± 1.19 kg/m2, HOMA 1.57 ± 0.19) (Supplementary Table 1, cohort 3) to assess whether different degrees in obesity (BMI) and IR status affected Insig1/SREBP1. Srebp1 mRNA level was preserved in adipocytes from MO non-IR versus control subjects, yet was significantly decreased in MO adipocytes from IR subjects (Fig. 2D). In addition, Insig1 expression was significantly decreased in MO non-IR and further decreased in MO IR (Fig. 2D) versus overweight individuals, and this was associated with maintenance of similar levels of mature SREBP1 protein among the three experimental groups (Fig. 2E). Assessment of INSIG1 and SREBP1 protein expression in SC WAT of an independent cohort (Supplementary Table 1, cohort 4) of MO subjects with more extreme BMI and a different degree of IR, MO non-IR (BMI 56.72 ± 1.71 kg/m2, HOMA 3.60 ± 0.32), and IR (BMI 56.30 ± 1.65 kg/m2, HOMA 12.76 ± 1.25) showed significantly lower INSIG1 protein levels in the more IR patients (Supplementary Fig. 2A), whereas the mature form of SREBP1 was present in similar amount between the two groups (Supplementary Fig. 2B). A similar analysis performed in Om adipose tissue of the same cohort (Supplementary Fig. 3A and B) revealed a comparable level of expression of mature SREBP1 and INSIG1 proteins among lean, MO non-IR, and MO IR. Globally considered, these results indicate that the changes in Insig1 may be predominantly driven by IR and that changes in Insig1 protein levels are preferentially affected in the human SC adipose tissue, suggesting that the SC depot may be more affected by metabolic stress than the Om WAT depot (35,36).
Using an in vitro model of induced IR in 3T3-L1 adipocytes that showed decreased levels of p-AKT (Supplementary Fig. 4A), Glut4, and Adiponectin mRNA (Supplementary Fig. 4B), we recapitulated the changes in Srebp1 and Insig1 observed in vivo (Supplementary Fig. 4C and D), further supporting a primary mechanistic role for IR in Insig1 and SREBP1 changes.
Maintained mature SREBP1 levels in MO-derived adipocytes maintain expression of genes regulating unsaturation and TG re-esterification.
Expression analysis of SREBP1-target genes in isolated Om- and SC-derived human adipocytes from lean, obese, and MO individuals (Fig. 3A and B, respectively, and Supplementary Table 1, cohort 1) showed that the de novo FA synthesis genes—Acc1 and Fas—were significantly reduced in Om and SC adipocytes from MO subjects (BMI 48.9 ± 1.8 kg/m2) compared with lean (BMI 22.2 ± 0.8 kg/m2) or obese (BMI 30.2 ± 1.1 kg/m2) adipocytes. Of relevance, the expression of Scd1 was relatively conserved in obese adipocytes, only showing a moderate decrease when lean and MO SC adipocytes were compared. Similarly, Cctα expression was conserved in obese adipocytes and only significantly reduced in the MO-derived Om adipocytes compared with lean. Expression of genes involved in FA re-esterification (e.g., Gpat1 and Dgat1 [diacylglycerol O-acyltransferase 1]) was maintained in the obese population in both adipose depots independently of their IR status. As previously shown (37,38), Glut4 mRNA was markedly decreased in adipocytes from IR obese and MO subjects when compared with lean.
FIG. 3.

SREBP1 target genes expression in omental and subcutaneous white adipocytes of lean, obese, and MO. RT-PCR analysis was performed on RNA obtained from mature adipocytes isolated from lean (BMI 22.2 ± 0.8 kg/m2), obese (BMI 30.2 ± 1.1 kg/m2), and MO (BMI 48.9 ± 1.8 kg/m2) (Supplementary Table 1, cohort 1). Adipocytes derived from Om (A) and SC depots (B) were examined and standardized to the average 18S mRNA value within each group (n = 10 lean, n = 10 obese, n = 12 MO). Obese and MO vs. lean: *P < 0.05, **P < 0.01, ***P < 0.005, ****P < 0.001. MO vs. obese: #P < 0.05, ##P < 0.01, ###P < 0.005. ANOVA: $P < 0.05, $$P < 0.005, $$$P < 0.001. Data are presented as mean ± SEM.
The same pattern of SREBP1 target genes expression was observed in our IR 3T3-L1 in vitro model (Supplementary Fig. 4E) and our DIO and ob/ob murine models (Supplementary Fig. 5A and C) when they developed IR. When still insulin-sensitive, WAT from mice fed the HFD for 15 days and 1-month-old ob/ob mice exhibited a similar lipogenic gene expression pattern compared with their control littermates (Supplementary Fig. 5B and D).
Altogether, these data suggest that the combination of reduced Insig1 expression and optimized maturation of SREBP1 in WAT of the MO population was associated with maintained FA desaturation and re-esterification SREBP1-dependent pathways.
Decreased expression of Insig1 facilitates TG accumulation and accelerates differentiation of 3T3-L1 cells.
It has previously been shown that Insig1 mRNA levels increase during differentiation of 3T3-L1 and that its overexpression decreases lipid accumulation during adipogenesis (16). We examined whether decreasing Insig1, as observed in SC WAT of obese IR individuals, facilitated lipid accumulation in adipocytes. To address this question, stable Insig1 KD and control cell lines were generated. Gene expression analysis performed in these cells cultured at low glucose (5 mmol/L) and high glucose (25 mmol/L) concentrations showed a reduction of Insig1 mRNA of 60–80% (Fig. 4A and Supplementary Fig. 6A). Notably, Insig2 mRNA levels were unchanged (Fig. 4A and Supplementary Fig. 6A). In the absence of Insig1, Srebp1c was upregulated, regardless of glucose concentration (Fig. 4A and Supplementary Fig. 6A), whereas Srepb1a mRNA levels were significantly increased only at 5 mmol/L glucose (Fig. 4A). Srebp1 upregulation was associated with a significant increase in mature SREBP1 in I1KD cells versus controls (Fig. 4B and Supplementary Fig. 6B).
FIG. 4.

Insig1 knockdown results in increased TG accumulation, accelerated differentiation, and augmented Srebp1 and lipogenic gene expression. A: I1KD and control 3T3-L1 cells were differentiated at 5 mmol/L glucose concentration for 8 days when RNA was isolated and Srebp1c, Srebp1a, Insig1, and Insig2 gene expression analyzed using quantitative RT-PCR (n = 6). I1KD vs controls: *P < 0.01, **P < 0.001. B: Western blot analysis was performed in I1KD and control cells to assess mature SREBP1 levels. Mature SREBP1 was normalized to α-tubulin. The blot was quantified by densitometry (n = 6). I1KD vs. controls: *P < 0.05. C: Photomicrographs of Oil-Red-O–stained I1KD and control 3T3-L1 cells, 8 days after induction of differentiation at 5 mmol/L glucose. D: TG levels were measured 10 days after induction of differentiation at 5 mmol/L glucose of I1KD and control cells (n = 3). I1KD vs. 5 mmol/L controls: **P < 0.001. E: Real time PCR analysis was performed on RNA, and gene expression levels of lipid-related SREBP1 target genes were analyzed in cells differentiated in 5 mmol/L glucose (n = 6). I1KD vs. controls: *P < 0.05, **P < 0.01,***P < 0.001,****P < 0.0001. F: De novo FA synthesis radioactive assay was carried out in Insig1 KD and control cells at day 8 of differentiation after overnight serum starvation and at the condition indicated (n = 8–9). I1KD vs. control: *P < 0.005. I1KD treated with C75 vs. control treated with C75: &P < 0.001. Control vs. control treated with C75 and I1KD vs. I1KD treated with C75: #P < 0.01. Control vs control treated with insulin and I1KD vs. I1KD treated with insulin: ^P < 0.005. Control treated with insulin vs. I1KD treated with insulin: $P < 0.005. G: Lipidomic analysis and subsequent evaluation of SCD1 activity were performed in I1KD and control cells. Cells were harvested at day 8 of differentiation and processed as described in research design and methods (n = 12). I1KD vs. controls: *P < 0.05, **P < 0.01. Data are presented as mean ± SEM.
I1KD cells showed increased lipid accumulation compared with controls, regardless of glucose concentration, as shown by Oil-Red-O staining (Fig. 4C and Supplementary Fig. 6C) and measurement of TG content (Fig. 4D and Supplementary Fig. 6D). Increased TG levels in I1KD cells corresponded with increased expression of lipogenic genes (Fig. 4E and Supplementary Fig. 6E), particularly Scd1, at both low and high glucose concentrations. The primary effects of decreased Insig1 on fat deposition and Scd1 expression were confirmed by de novo lipogenesis assay and lipidomic analysis performed on I1KD cells versus controls. The functional relevance of Insig1 knockdown was confirmed because the de novo lipogenesis functional assay showed that I1KD cells incorporated more 13C-sodium acetate than control cells after 1 h incubation, an effect that was further enhanced by insulin (Fig. 4F). In support of a preferential effect on desaturation, global free FA profile performed on Insig1 KD cells showed that Insig1 depletion led to a significant increase in the desaturation indexes (C18:1n9/18:0, C16:1n7/16:0, and C14:1n5/14:0) associated with SCD1 activity (Fig. 4G).
Insig1 knockout WAT and adipocyte primary culture show increased Srebp1, SREBP1 lipogenic target gene expression, and augmented lipid accumulation.
The results of the Insig1 KD in vitro model were validated in vivo using an Insig1 knockout (KO) mouse (generation strategy described in Supplementary Fig. 11). Although this murine model has a complex phenotype characterized by being lighter (Supplementary Fig. 7A–C) and shorter (Supplementary Fig. 7B), it is useful to study the effects of Insig1 genetic ablation without the confounding effect of IR on lipogenesis. In support of a facilitated fat deposition phenotype, Insig1 KO mice exhibit an increased percentage of fat mass (Fig. 5A) and increased gonadal and subcutaneous depot mass (% of body weight) (Supplementary Fig. 7D). Concerning the main metabolic parameter, such as food consumption (Supplementary Fig. 7E), glucose tolerance (Supplementary Fig. 8A), insulin sensitivity (Supplementary Fig. 8B), respiratory exchange ratio (Supplementary Fig. 8C), and energy expenditure (Supplementary Fig. 8D), the mice in this model do not present any difference with respect to their WT littermates. Concerning blood biochemistry, the Insig1 KO mice show decreased TG compared with controls (Supplementary Table 3). Moreover, Insig1 KO mice show an increased number of smaller adipocytes in the gonadal WAT depot (Supplementary Fig. 9).
FIG. 5.

Insig1 depletion in vivo and in vitro shows increased fat accumulation, SREBP1 levels, and SREBP1 target gene expression. A: Percentage of fat mass with respect to body weight was established for Insig1 KO males (n = 6) vs. WT littermates (n = 8). B and D: Real-time PCR analysis was performed on gonadal WAT from 11-week-old male Insig1 KO (n = 5) and WT littermates (n = 4). C: Mature SREBP1 levels in gonadal WAT of Insig1 KO (n = 5) and WT littermates (n = 4). E: Oil-Red-O staining of WT and Insig1 KO primary adipocytes at day 4 and 8 of differentiation. Cells were induced to differentiate with a standard MDI induction cocktail using Dulbecco’s modified Eagle’s medium with 25 mmol/L glucose. F: Gene expression analysis of Insig1 KO and WT WAT primary culture after 4 days of differentiation (n = 3–4). Insig1 KO vs. WT: *P < 0.05, **P < 0.01, ***P < 0.0001. Data are presented as mean ± SEM.
We also performed transcriptional analysis of gonadal WAT from the Insig1 KO model that revealed increased Srebp1c and a trend for increased Srebp2 mRNA levels (Fig. 5B). Protein analysis confirmed that the SREBP1 mature protein form was also increased (Fig. 5C). As seen in I1KD 3T3-L1 adipocytes, no compensatory alteration in Insig2 was observed, and in the context of preserved insulin sensitivity, expression levels of Fas, Acc1, and Scd1 were increased in parallel with Srebp1c (Fig. 5D).
In vitro differentiation of primary cultures of Insig1 KO WAT preadipocytes showed increased lipid accumulation (Fig. 5E) and augmented expression of lipid accumulation markers (e.g., adipose differentiation-related protein [Adrp] and Perilipin) at day 4 of differentiation (Fig. 5F) compared with WT counterparts. As shown for gonadal WAT gene profiling performed on Insig1 KO, WAT primary culture confirmed an increase in the expression of SREBP1 target genes, including Scd1, Fas, and Acc1 (Fig. 5F).
Increased SCD1 activity in the WAT of Insig1 KO mice associated with increased Scd1 expression.
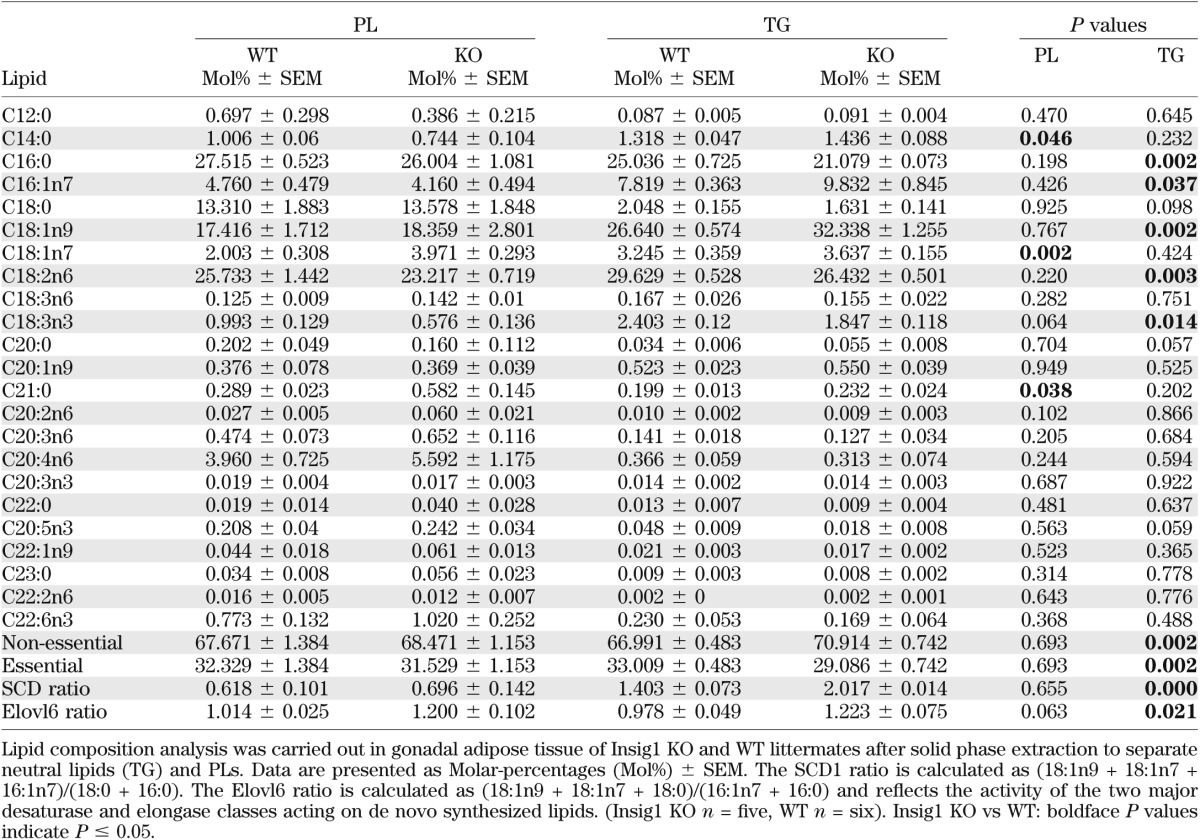
Next, we assessed whether the depletion in Insig1 expression and associated upregulation of Scd1 affected the FA composition of WAT in vivo. FA profiling of TGs revealed a significant increase in the levels of oleate and palmitoleate as well as an increase in the SCD1 ratio of the essential free FAs of the Insig1 KO mouse compared with WT mice (Table 1). Of note, we did not observe changes in products of SCD1 or a change in the SCD1 ratio of the PLs of the Insig1 WAT, consistent with defense of the PL compartment. These analyses also identified an increased amount of nonessential FAs in the TGs, consistent with higher rates of de novo FA synthesis in this model. A similar phenotype of preserved PL-to-SCD1 ratio with an elevated TG-to-SCD1 ratio was also observed in the WAT of ob/ob mice via two independent analyses (Supplementary Fig. 10A–D). Similarly, we checked if the observed change in the Insig1/SREBP1 set point in WAT of MO patients versus lean controls was associated with maintenance of the pool of unsaturated FAs. Lipid composition of SC WAT from lean (BMI 23.2 ± 1.2 kg/m2) and MO (BMI 54.0 ± 4.9 kg/m2, HOMA 4.0 ± 1.4) (Supplementary Table 1, cohort 5) revealed that the absolute amount of unsaturated FAs was conserved between the two groups. Moreover, we observed significant reduction of specific saturated FAs (20:0, 22:0) in WAT of MO versus lean control subjects (Supplementary Table 2). This result agrees with our recent analysis of WAT of weight-discordant monozygotic cotwins, showing that obese twins have a reduction in specific saturated FAs (e.g., 12:0, 18:0, and 20:0) and increased proportion of specific unsaturated FAs such as palmitoleic (16:1n-7) and arachidonic acid (20:4n-6) (2).
TABLE 1.
Lipid composition of PLs and TGs as well as major elongases and desaturase activity ratios
DISCUSSION
Here, we present strong supporting evidence for an adaptive mechanism designed to maintain cellular FA unsaturation when WAT is under adverse metabolic conditions imposed by obesity and IR. This adaptive response involves facilitated lipid biosynthesis mediated by downregulation of Insig1 and optimized activation of SREBP1 and SCD1. This change in the set point of the Insig1/SREBP1 regulatory feedback loop helps to maintain lipid desaturation of adipocytes in a situation of metabolic stress.
Using genetically modified in vitro and in vivo models, we have shown that downregulation of Insig1 per se facilitates TG accumulation in parallel with favored biosynthesis of unsaturated lipids through preferential induction of SCD1. We observed this downregulation of Insig1 in WAT of dietary and genetic mouse models of obesity-induced IR, in obese and MO IR humans, and in cellular models of IR.
Our data indicate that it is the IR associated with obesity, more than the amount of fat stored, that is the main factor responsible for the decreased Srebp1c WAT mRNA expression observed in MO individuals. Insig1 mRNA expression is decreased in the SC depot of MO individuals compared with obese patients and is further reduced in the presence of IR. On the basis of the role of Insig1 controlling SREBP1 activation, the decrease in Insig1 is expected to help to maintain the levels of the active form of SREBP1. From the clinical perspective, this change in set point may be important to reconcile the perceived paradox between the excessive amounts of lipids that MO individuals are able to store in their WAT despite their IR and decreased mRNA expression of fundamental prolipogenic genes such as Srebp1 (39–41).
We found that reducing Insig1 mRNA to a similar degree observed in MO human WAT accelerates adipocyte differentiation and enhances lipogenesis, and consequently, lipid accumulation. Notably, the effect of Insig1 KD enhancing lipid accumulation in 3T3-L1 adipocytes could be reproduced in conditions that mimicked both physiological and pathophysiological glucose levels. In our opinion, these data indicate that the regulation of Insig1 in WAT may be more important under conditions of IR and metabolic stress than for the adipose tissue expansion observed in anabolic states.
Maintenance of SREBP1 mature protein levels in morbid obesity or under IR conditions is not associated with conserved global expression or restoration of all known SREBP1 target genes. Indeed, genes involved in the early stages of de novo lipogenesis from glucose, such as Acc1 and Fas, were downregulated in WAT of MO IR patients, in our DIO and ob/ob IR mouse models, and in our IR in vitro model, despite maintained levels of mature SREBP1. This suggests that other transcription factors regulated by glucose, such as ChREBP, may be required for their full expression. Moreover, it has been shown that in livers of HFD-fed C57BL6 mice, there is upregulation of SREBP1 and preferentially SCD1 as its target gene compared with unaltered FAS expression (42). Conversely, pathways involved in modification of the FA chain or FA storage, such as Scd1, Cctα, Gpat1, and Dgat1, were maintained to a greater extent in obese subjects, being closer to those observed in lean individuals.
One of the most revealing findings, both in vivo and in vitro, is the identification of SCD1 as a preferentially regulated target for Insig1/SREBP1. From an adaptive perspective, maintenance of a proper degree of unsaturation could be the main homeostatic function of this regulatory loop. This view is supported by the fact that in our in vitro studies, inactivation of Insig1 under both physiological and high glucose conditions preferentially induced Scd1 gene expression and consequently an increase in SCD1-dependent lipid unsaturation indexes such as C18:1n9/18:0, C16:1n7/16:0 (43,44), and C14:1n5/14:0 (45).
To consolidate the concept that downregulation of Insig1 promotes a prolipogenic effect in WAT, we generated an Insig1 KO mouse. This murine model allowed us to study the effect of Insig1 depletion without the confounding effects of obesity and/or IR. Mice lacking intact Insig1 are characterized by an increased percentage of fat mass with respect to WT littermates. Also primary cultures of Insig1 KO preadipocytes showed increased and accelerated lipid accumulation after induction of differentiation, as previously observed in the Insig1 KD 3T3-L1. These previous results led us to hypothesize that the adaptive changes in Insig1/SREBP1 set point will maintain the pool of unsaturated FAs in WAT. Lipid composition analysis performed on WAT of MO versus lean control subjects supports this concept, because the absolute amount of unsaturated FA was maintained at similar levels in both groups. Most interestingly, profiling of specific lipid species in WAT of our models of genetic-induced obesity and IR, as well as our Insig1 KO mouse, indicated that the level of unsaturation and SCD1 activity is specifically maintained in the membrane PL fraction.
This result strengthens our previous observation that in weight-discordant monozygotic twin pairs (2), the obese cotwins displayed reduced specific saturated FA and augmented proportions of specific unsaturated Fas, such as palmitoleate, in membrane PL, alongside increases in indexes reflecting augmented activity of desaturation enzymes.
In summary, our results indicate the existence of an adaptive response in human WAT involving changes in the set point of the Insig1/SREBP1/SCD1 axis in order to optimize lipogenic gene expression programs that ensure availability of structural and biologically essential unsaturated lipids under disadvantageous metabolic conditions such as obesity or IR. Thus, defects in insulin-regulated lipid metabolism associated with IR may at least be partially ameliorated by a reduction of Insig1 and optimized maturation of the active form of SREBP1 as a consequence of a resetting of the Insig1/SREBP1 negative regulatory feedback loop (Supplementary Fig. 12).
ACKNOWLEDGMENTS
This work was funded by MRC Programme grant, FP7 Etherpath, FP7 MITIN (Integration of the System Models of Mitochondrial Function and Insulin Signalling and its Application in the Study of Complex Diseases), Biotechnology and Biological Sciences Research Council, European Foundation for the Study of Diabetes grant, Integrative Pharmacology Fund, and Fritz Thyssen Foundation fellowship (Germany). The authors thank the National Institute for Health Research for its support.
No potential conflicts of interest relevant to this article were reported.
S.C., R.M.H., and C.J.L. performed the experiments, analyzed the data, designed the experiments, discussed the manuscript, coordinated and directed the project, developed the hypothesis, and wrote the manuscript. M.S. performed the experiments, analyzed the data, designed the experiments, and discussed the manuscript. G.M.-G. discussed the manuscript. C.-Y.T. developed analytical platforms and performed and analyzed lipidomic experiments. A.S., N.B., A.T., E.L., M.La., R.V.C., H.V., D.L., F.J.T., and J.-M.F.R. characterized human cohorts and provided human Om and SC adipose tissue samples. M.Ló. performed the experiments and analyzed the data. H.J.A., S.V., M.O., and J.L.G. developed analytical platforms and performed and analyzed lipidomic experiments. M.B., M.B.-Y., and T.W. contributed to the generation of the Insig1 KO mouse. J.K.S. discussed the manuscript and coordinated and directed the project. M.Ló. designed the experiments and discussed the manuscript. A.V.-P. designed the experiments, discussed the manuscript, coordinated and directed the project, developed the hypothesis, and wrote the manuscript. A.V.-P. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
The authors thank Dr. Wendy Kimber (University of Cambridge, Metabolic Research Laboratories, Institute of Metabolic Science) for helping in I1KD generation and Dr. Andrew Whittle and Dr. Sergio Rodriguez-Cuenca (University of Cambridge, Metabolic Research Laboratories, Institute of Metabolic Science) for their useful comments and suggestions. The authors would like to acknowledge all staff at the transgenic center, AstraZeneca, Mölndal, Sweden, who contributed to the development of the Insig1 KO mouse model. In particular, Marie Jönsson and Anna-Karin Gerdin are acknowledged for in vivo phenotypic analyses. Mark Campbell and Martin Dale (University of Cambridge, Metabolic Research Laboratories, Institute of Metabolic Science) are acknowledged for their technical help. The authors also thank Janice Carter, Dan Hart, Sarah Grocott, Haidee Pitt, and Helen Westby (University of Cambridge, Metabolic Research Laboratories, Institute of Metabolic Science) for the excellent husbandry of the Insig1 KO mice.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db12-1748/-/DC1.
REFERENCES
- 1.Virtue S, Vidal-Puig A. Adipose tissue expandability, lipotoxicity and the Metabolic Syndrome—an allostatic perspective. Biochim Biophys Acta 2010;1801:338–349 [DOI] [PubMed] [Google Scholar]
- 2.Pietilainen K, Róg T, Seppänen-Laakso T, et al. Association of lipidome remodeling in the adipocyte membrane with aquired obesity in humans. PLoS Biol 2011;9:e1000623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim JY, van de Wall E, Laplante M, et al. Obesity-associated improvements in metabolic profile through expansion of adipose tissue. J Clin Invest 2007;117:2621–2637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Juster RP, McEwen BS, Lupien SJ. Allostatic load biomarkers of chronic stress and impact on health and cognition. Neurosci Biobehav Rev 2010;35:2–16 [Review] [DOI] [PubMed] [Google Scholar]
- 5.Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest 2002;109:1125–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Le Lay S, Lefrère I, Trautwein C, Dugail I, Krief S. Insulin and sterol-regulatory element-binding protein-1c (SREBP-1C) regulation of gene expression in 3T3-L1 adipocytes. Identification of CCAAT/enhancer-binding protein beta as an SREBP-1C target. J Biol Chem 2002;277:35625–35634 [DOI] [PubMed] [Google Scholar]
- 7.Shimano H, Horton JD, Shimomura I, Hammer RE, Brown MS, Goldstein JL. Isoform 1c of sterol regulatory element binding protein is less active than isoform 1a in livers of transgenic mice and in cultured cells. J Clin Invest 1997;99:846–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shimomura I, Shimano H, Korn BS, Bashmakov Y, Horton JD. Nuclear sterol regulatory element-binding proteins activate genes responsible for the entire program of unsaturated fatty acid biosynthesis in transgenic mouse liver. J Biol Chem 1998;273:35299–35306 [DOI] [PubMed] [Google Scholar]
- 9.Moon YA, Shah NA, Mohapatra S, Warrington JA, Horton JD. Identification of a mammalian long chain fatty acyl elongase regulated by sterol regulatory element-binding proteins. J Biol Chem 2001;276:45358–45366 [DOI] [PubMed] [Google Scholar]
- 10.Matsuzaka T, Shimano H, Yahagi N, et al. Cloning and characterization of a mammalian fatty acyl-CoA elongase as a lipogenic enzyme regulated by SREBPs. J Lipid Res 2002;43:911–920 [PubMed] [Google Scholar]
- 11.Ericsson J, Jackson SM, Kim JB, Spiegelman BM, Edwards PA. Identification of glycerol-3-phosphate acyltransferase as an adipocyte determination and differentiation factor 1- and sterol regulatory element-binding protein-responsive gene. J Biol Chem 1997;272:7298–7305 [DOI] [PubMed] [Google Scholar]
- 12.Ridgway ND, Lagace TA. Regulation of the CDP-choline pathway by sterol regulatory element binding proteins involves transcriptional and post-transcriptional mechanisms. Biochem J 2003;372:811–819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kast HR, Nguyen CM, Anisfeld AM, Ericsson J, Edwards PA. CTP:phosphocholine cytidylyltransferase, a new sterol- and SREBP-responsive gene. J Lipid Res 2001;42:1266–1272 [PubMed] [Google Scholar]
- 14.Goldstein JL, Brown MS. The clinical investigator: bewitched, bothered, and bewildered—but still beloved. J Clin Invest 1997;99:2803–2812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eberlé D, Hegarty B, Bossard P, Ferré P, Foufelle F. SREBP transcription factors: master regulators of lipid homeostasis. Biochimie 2004;86:839–848 [DOI] [PubMed] [Google Scholar]
- 16.Li J, Takaishi K, Cook W, McCorkle SK, Unger RH. Insig-1 “brakes” lipogenesis in adipocytes and inhibits differentiation of preadipocytes. Proc Natl Acad Sci U S A 2003;100:9476–9481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kast-Woelbern HR, Dana SL, Cesario RM, et al. Rosiglitazone induction of Insig-1 in white adipose tissue reveals a novel interplay of peroxisome proliferator-activated receptor gamma and sterol regulatory element-binding protein in the regulation of adipogenesis. J Biol Chem 2004;279:23908–23915 [DOI] [PubMed] [Google Scholar]
- 18.Way JM, Harrington WW, Brown KK, et al. Comprehensive messenger ribonucleic acid profiling reveals that peroxisome proliferator-activated receptor gamma activation has coordinate effects on gene expression in multiple insulin-sensitive tissues. Endocrinology 2001;142:1269–1277 [DOI] [PubMed] [Google Scholar]
- 19.Yabe D, Brown MS, Goldstein JL. Insig-2, a second endoplasmic reticulum protein that binds SCAP and blocks export of sterol regulatory element-binding proteins. Proc Natl Acad Sci U S A 2002;99:12753–12758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yabe D, Komuro R, Liang G, Goldstein JL, Brown MS. Liver-specific mRNA for Insig-2 down-regulated by insulin: implications for fatty acid synthesis. Proc Natl Acad Sci U S A 2003;100:3155–3160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goldstein JL, DeBose-Boyd RA, Brown MS. Protein sensors for membrane sterols. Cell 2006;124:35–46 [DOI] [PubMed] [Google Scholar]
- 22.Gong Y, Lee JN, Lee PC, Goldstein JL, Brown MS, Ye J. Sterol-regulated ubiquitination and degradation of Insig-1 creates a convergent mechanism for feedback control of cholesterol synthesis and uptake. Cell Metab 2006;3:15–24 [DOI] [PubMed] [Google Scholar]
- 23.Reed BD, Charos AE, Szekely AM, Weissman SM, Snyder M. Genome-wide occupancy of SREBP1 and its partners NFY and SP1 reveals novel functional roles and combinatorial regulation of distinct classes of genes. PLoS Genet 2008;4:e1000133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Walker AK, Jacobs RL, Watts JL, et al. A Conserved SREBP-1/phosphatidylcholine feedback circuit regulates lipogenesis in metazoans. Cell 2011;147:840–852. [DOI] [PMC free article] [PubMed]
- 25.Lim HY, Wang W, Wessells RJ, Ocorr K, Bodmer R. Phospholipid homeostasis regulates lipid metabolism and cardiac function through SREBP signaling in Drosophila. Genes Dev 2011;25:189–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Medina-Gomez G, Gray SL, Yetukuri L, et al. PPAR gamma 2 prevents lipotoxicity by controlling adipose tissue expandability and peripheral lipid metabolism. PLoS Genet 2007;3:e64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jitrapakdee S, Slawik M, Medina-Gomez G, et al. The peroxisome proliferator-activated receptor-gamma regulates murine pyruvate carboxylase gene expression in vivo and in vitro. J Biol Chem 2005;280:27466–27476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lagathu C, Christodoulides C, Virtue S, et al. Dact1, a nutritionally regulated preadipocyte gene, controls adipogenesis by coordinating the Wnt/beta-catenin signaling network. Diabetes 2009;58:609–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roberts LD, Virtue S, Vidal-Puig A, Nicholls AW, Griffin JL. Metabolic phenotyping of a model of adipocyte differentiation. Physiol Genomics 2009;39:109–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shimomura I, Matsuda M, Hammer RE, Bashmakov Y, Brown MS, Goldstein JL. Decreased IRS-2 and increased SREBP-1c lead to mixed insulin resistance and sensitivity in livers of lipodystrophic and ob/ob mice. Mol Cell 2000;6:77–86 [PubMed] [Google Scholar]
- 31.Simmgen M, Knauf C, Lopez M, et al. Liver-specific deletion of insulin receptor substrate 2 does not impair hepatic glucose and lipid metabolism in mice. Diabetologia 2006;49:552–561 [DOI] [PubMed] [Google Scholar]
- 32.Prieur X, Tung YC, Griffin JL, Farooqi IS, O’Rahilly S, Coll AP. Leptin regulates peripheral lipid metabolism primarily through central effects on food intake. Endocrinology 2008;149:5432–5439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang LY, Summerhill K, Rodriguez-Canas C, et al. Development and validation of a robust automated analysis of plasma phospholipid fatty acids for metabolic phenotyping of large epidemiological studies. Genome Med 2013;5:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Prieur X, Mok CY, Velagapudi VR, et al. Differential lipid partitioning between adipocytes and tissue macrophages modulates macrophage lipotoxicity and M2/M1 polarization in obese mice. Diabetes 2011;60:797–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ali AT, Ferris WF, Naran NH, Crowther NJ. Insulin resistance in the control of body fat distribution: a new hypothesis. Horm Metab Res 2011;43:77–80 [DOI] [PubMed] [Google Scholar]
- 36.Alligier M, Gabert L, Meugnier E, et al. Visceral fat accumulation during lipid overfeeding is related to subcutaneous adipose tissue characteristics in healthy men. J Clin Endocrinol Metab 2013;98:802–810 [DOI] [PubMed] [Google Scholar]
- 37.Garvey WT, Maianu L, Huecksteadt TP, Birnbaum MJ, Molina JM, Ciaraldi TP. Pretranslational suppression of a glucose transporter protein causes insulin resistance in adipocytes from patients with non-insulin-dependent diabetes mellitus and obesity. J Clin Invest 1991;87:1072–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lefebvre AM, Laville M, Vega N, et al. Depot-specific differences in adipose tissue gene expression in lean and obese subjects. Diabetes 1998;47:98–103 [DOI] [PubMed] [Google Scholar]
- 39.Sewter C, Berger D, Considine RV, et al. Human obesity and type 2 diabetes are associated with alterations in SREBP1 isoform expression that are reproduced ex vivo by tumor necrosis factor-alpha. Diabetes 2002;51:1035–1041 [DOI] [PubMed] [Google Scholar]
- 40.Kolehmainen M, Vidal H, Alhava E, Uusitupa MI. Sterol regulatory element binding protein 1c (SREBP-1c) expression in human obesity. Obes Res 2001;9:706–712 [DOI] [PubMed] [Google Scholar]
- 41.Oberkofler H, Fukushima N, Esterbauer H, Krempler F, Patsch W. Sterol regulatory element binding proteins: relationship of adipose tissue gene expression with obesity in humans. Biochim Biophys Acta 2002;1575:75–81 [DOI] [PubMed] [Google Scholar]
- 42.Biddinger SB, Almind K, Miyazaki M, Kokkotou E, Ntambi JM, Kahn CR. Effects of diet and genetic background on sterol regulatory element-binding protein-1c, stearoyl-CoA desaturase 1, and the development of the metabolic syndrome. Diabetes 2005;54:1314–1323 [DOI] [PubMed] [Google Scholar]
- 43.Chong MF, Hodson L, Bickerton AS, et al. Parallel activation of de novo lipogenesis and stearoyl-CoA desaturase activity after 3 d of high-carbohydrate feeding. Am J Clin Nutr 2008;87:817–823 [DOI] [PubMed] [Google Scholar]
- 44.Stryjecki C, Roke K, Clarke S, et al. Enzymatic activity and genetic variation in SCD1 modulate the relationship between fatty acids and inflammation. Mol Genet Metab 2012;105:421–427 [DOI] [PubMed] [Google Scholar]
- 45.Miyazaki M, Bruggink SM, Ntambi JM. Identification of mouse palmitoyl-coenzyme A Delta9-desaturase. J Lipid Res 2006;47:700–704 [DOI] [PubMed] [Google Scholar]