

Abstract
Obesity is a major risk factor for type 2 diabetes and cardiovascular diseases. And overnutrition is a leading cause of obesity. After most nutrients are ingested, they are absorbed in the small intestine. Signals from β-catenin are essential to maintain development of the small intestine and homeostasis. In this study, we used a hyperphagia db/db obese mouse model and a high-fat diet (HFD)-induced obesity mouse model to investigate the effects of overnutrition on intestinal function and β-catenin signaling. The β-catenin protein was upregulated along with inactivation of glycogen synthase kinase (GSK)-3β in the intestines of both db/db and HFD mice. Proliferation of intestinal epithelial stem cells, villi length, nutrient absorption, and body weight also increased in both models. These changes were reversed by caloric restriction in db/db mice and by β-catenin inhibitor JW55 (a small molecule that increases β-catenin degradation) in HFD mice. Parallel, in vitro experiments showed that β-catenin accumulation and cell proliferation stimulated by glucose were blocked by the β-catenin inhibitor FH535. And the GSK-3 inhibitor CHIR98014 in an intestinal epithelial cell line increased β-catenin accumulation and cyclin D1 expression. These results suggested that, besides contribution to intestinal development and homeostasis, GSK-3β/β-catenin signaling plays a central role in intestinal morphological and functional changes in response to overnutrition. Manipulating the GSK-3β/β-catenin signaling pathway in intestinal epithelium might become a therapeutic intervention for obesity induced by overnutrition.
Obesity, affecting ~30% of the world population, is a major risk factor for metabolic syndrome, inflammation, type 2 diabetes (T2D), and cardiovascular diseases (1). Epidemiological evidence suggests that body weight is regulated by complex physiological mechanisms (2,3). However, environmental factors, especially when the energy intake from food exceeds normal physiological needs, are considered to be culprits for becoming overweight and then obese. Conversely, caloric restriction (CR) significantly reduces obesity and incidences of T2D and cardiovascular disease in rodents, primates, and humans (4–6).
Appetite and food intake are a complex physiologic process. Regulation of appetite involves numerous hormones and signals, and defects of these appetite-related molecules and related signaling pathways cause severe obesity (7–9). These findings strongly suggest a prominent role for excess food intake and an oversupply of nutrients in obesity and related diseases. Studies show that high-fat diet (HFD) could induce intestinal epithelial proliferation, absorption, and adiposity (10–12). However, the underlying mechanisms remain poorly understood.
The internal surface of the mammalian intestine is covered by a single layer of epithelial cells that protrude into the intestinal lumen to form finger-like villi that absorb nutrients from food. This single layer of cells is renewed every 3–5 days. Besides these villi, other specialized structures have evolved in the intestinal epithelium, termed crypts, which contain multipotent stem cells and are responsible for intestinal epithelial cell renewal. This cell-renewal process is strictly controlled through a series of coordinated signaling pathways (13,14).
In mammals, the canonical Wnt signaling pathway is essential for maintaining intestinal crypt cell proliferation during development and for intestinal epithelium homeostasis during adulthood (14–16). As a core effector of the Wnt signaling pathway, β-catenin is regulated mainly at the protein level by a proteolytic degradation complex that consists of adenomatous polyposis coli, casein kinase I, glycogen synthase kinase (GSK)-3β, and axin. When the complex is assembled, the GSK-3β will effectively phosphorylate β-catenin, leading to β-catenin protease hydrolysis (17). However, GSK-3β is inactivated by phosphorylation at Ser9, leading to cytoplasmic β-catenin accumulation and nuclear translocation, resulting in an increase of β-catenin target gene, such as cyclin D1 expression and cell proliferation (18).
To better understand whether and how small intestine homeostasis is involved in its morphological and functional changes induced by excess food intake and HFD, we used a hyperphagic db/db obese mouse model and a model of obesity induced by HFD to investigate the changes in absorptive surface area and related signaling in the small intestine during the occurrence of obesity. We found that intestinal epithelial cell proliferation induced by excess food intake was correlated with activation of the GSK-3β/β-catenin signaling pathway, suggesting that nutrient-induced activation of GSK-3β/β-catenin signaling in the intestinal epithelium may contributes to increased nutrient absorption and obesity development.
RESEARCH DESIGN AND METHODS
Male and female db/+ mice of a hyperphagia db/db mouse model, obtained from The Jackson Laboratory (Bar Harbor, ME), were mated to generate db/db mice. Mice were fed with a standard chow diet containing 4% fat and 50% carbohydrate. Age-matched male ad libitum db/+ and db/db mice were used for further studies (n = 8 per group). For embryonic intestine sample collection, the pregnant female db/+ mice were killed at the time that embryos became 18.5 days old.
In the CR group, 4-week-old male db/db mice (n = 5) were housed in individual cages and fed twice daily with a restricted amount (60% of ad libitum) of the standard chow diet; these mice were killed after 4 weeks of CR, and intestinal samples were harvested for further studies.
For the HFD-induced obesity model, at the age of 4 weeks, male C57BL/6 mice were fed an HFD (cat. no. D12492; Research Diets, New Brunswick, NJ), that contained 35 g (60 kcal) fat, for 8 weeks. Age- and sex-matched controls were fed a standard chow diet (n = 8 per group).
For forming the β-catenin inhibition group, 4-week-old male C57BL/6 mice were randomized into four groups and fed for 3 weeks: standard chow diet (n = 8), HFD (n = 8), HFD + DMSO (n = 8), or HFD + JW55 in DMSO (100 mg/kg, daily oral application) (n = 4). JW55 (Tocris Bioscience) is a small molecule that increases β-catenin degradation (19).
All mice were maintained under specific pathogen-free conditions in the Association for Assessment and Accreditation of Laboratory Animal Care International–accredited animal facility at Peking University. Mice had free access to water, and body weight was measured weekly or twice a week. The current study was approved by the Institutional Animal Care and Use Committee of Peking University and was in accordance with the principles of laboratory animal care of the National Academy of Sciences/National Research Council.
Intestinal sample collection.
For evaluation of epithelial cell proliferation and migration in the small intestinal villi, 1 h or 24 h after injection of BrdU (Sigma, St. Louis, MO) at 100 mg/kg body wt i.p., mice were anesthetized with sodium pentobarbital (100 mg/kg body wt) and killed by cervical dislocation. The entire intestine was then removed, weighed, and rinsed rapidly with iced Ringer buffer (in mM: NaCl 115, NaHCO3 25, MgCl2 1.2, CaCl2 1.2, K2HPO4 2.4, and KH2PO4 0.4, pH 7.3) supplemented with phenylmethylsulfonyl fluoride. Small intestinal tissue was dissected, and the jejunum was snap frozen in liquid nitrogen for isolation of total RNA and protein or was fixed in ice-cold 4% paraformaldehyde (PFA) overnight for histological and immunohistochemical analysis. Embryonic intestinal sample collection was performed as previously described (20). In brief, embryos were removed from time-mated females at E18.5 1 h after intraperitoneal injection of BrdU. For histological and immunohistochemical analysis, the intestines were fixed in 4% PFA for paraffin section preparation. For isolation of total RNA and protein, the small intestine was frozen in liquid nitrogen immediately after dissection.
Histology and immunohistochemistry.
The PFA-fixed intestines were embedded in paraffin, and 5-μm sections were prepared. The sections were stained with hematoxylin-eosin (H-E) for histological analysis. For immunohistochemistry, sections were incubated with anti-BrdU (BD) or anti–Cyclin D1 (Santa Cruz Biotechnology, Santa Cruz, CA) antibodies overnight at 4°C, followed by anti-rabbit or mouse horseradish peroxidase–labeled secondary antibodies (Dako Diagnostics) for 1 h at room temperature. Slides were developed using 3,3′-diaminobenzidine tetrahydrochloride and counterstained with hematoxylin.
For measurement of length of villi, the H-E–stained sections were imaged with a light microscope BX51 (Olympus, Japan). One hundred well-orientated villi were randomly chosen from ten ×100 images and measured with Image-Pro MC 6.0 software (Olympus). For cell proliferation assays, the BrdU- or Cyclin D1–positive cells were imaged within 100 well-oriented crypts from ten ×100 images and counted per crypt using Image-Pro.
Western blot.
Total intestinal protein was extracted using a radioimmunoprecipitation assay lysis buffer supplemented with a protease inhibitor cocktail (Sigma), phosphorylation protease inhibitor (Roche), and phenylmethylsulfonyl fluoride. After rapid homogenization, the homogenate was incubated in ice for 30 min and centrifuged at 12,000g for 15 min at 4°C. Protein samples (30–100 µg) were separated by 10% SDS-PAGE. Blots were incubated at 4°C overnight with primary antibodies: rabbit anti-mouse GLUT2 (1:1,000; Millipore), rabbit total GSK-3β and anti–phosphorylated Ser9–GSK-3β (1:1,000; Cell Signaling Transduction), mouse anti–β-catenin (1:1,000; BD), rabbit anti–cyclin D1 (1:1,000, Santa Cruz Biotechnology), and mouse anti-mouse β-actin (1:1,000, Santa Cruz Biotechnology), followed by a 1-h incubation with anti-rabbit or mouse horseradish peroxidase–labeled secondary antibodies (Santa Cruz Biotechnology) at room temperature. An ECL detection system (Bio-Rad) was used to reveal the peroxidase label. Relative abundance was quantified by densitometry using Quantity One 4.6.7 software (Bio-Rad).
Intestinal glucose absorption.
Glucose uptake was performed as previously described (21). Briefly, intestinal rings were incubated in an oxygenated Ringer solution containing 50 mmol/L D-[U-3H]glucose (0.1 µGi/mL phycoerythrin) for 10 min. Then, the intestine was lysed overnight in 10% HNO3 at 4°C. The radioactivity of the lysate was measured by liquid scintillation. Counts were expressed as micromoles per liter per centimeter ± SEM.
Cell culture and cell proliferation assay.
The mouse colon epithelial cell line (CT26) was from the American Type Culture Collection (Manassas, VA). Cells were cultured at 37°C under 5% CO2 and 95% O2 in RPMI 1640 medium containing 10% heat-inactivated FBS, 100 units/mL penicillin, and 100 μg/mL streptomycin (Gibco). For determination of the effect of high-glucose exposure on cell proliferation, after 24-h culturing, cells were serum starved for 24 h and then incubated for another 48 h in fresh medium supplemented with 0 or 20 mmol/L d-glucose. For testing the function of GSK-3β/β-catenin signaling in cell proliferation, 5 μmol/L FH535 (an inhibitor of β-catenin/T cell factor (TCF)-mediated transcription) (Sigma) (22) or CHIR98014 (GSK-3 inhibitor) (Selleckchem, Houston, TX) (23) was added to culture medium containing 20 mmol/L d-glucose. For proliferation assays, MTT assay was performed as previously described (24,25). Absorbance was measured at 490 nm and converted into cell number. Three independent experiments were performed.
For determination of the signaling pathways involved in CT26 cell proliferation in response to high glucose or free fatty acid (FFA) (palmitic acid; Sigma), cells were seeded in six-well plates in RPMI 1640 medium to reach 70–80% confluence. After 24 h of serum starvation, the cells were incubated with serum-free RPMI 1640 medium containing 20 mmol/L d-glucose or serum-free RPMI 1640 medium containing 2% BSA and 0.5 mmol/L FFA for 15, 30, 60, 120, or 240 min. The cells then were lysed in radioimmunoprecipitation assay buffer to extract protein.
Statistical analysis.
All data are expressed as mean ± SEM. Student t test was used to compare the differences between groups. A P value <0.05 was considered statistically significant.
RESULTS
Intestinal growth and glucose absorption increased in db/db mice and was reversed by CR.
Nutrient absorption in the intestine after food intake is the gateway for the body’s energy supply. Energy overload in excess of expenditure is the basis of obesity, while CR significantly reduces the incidence of obesity and its related diseases (4,5,26). To investigate changes in intestinal absorption related to excess food intake and developing obesity, we first looked at hyperphagic db/db mice. We found that, from the age of 4 weeks, food intake in db/db mice was increased significantly compared with age-matched lean db/+ mice (Fig. 1A) in concordance with body weight (Fig. 1B). Restricting food intake caused a distinct reduction in body weight in db/db mice compared with their ad libitum db/db littermates (Fig. 1B). Intestinal length and weight also dramatically increased in db/db mice. CR did not alter db/db mouse intestinal length in db/db mice but robustly decreased intestine weight (Fig. 1C and D).
FIG. 1.

Intestinal growth and glucose absorption are increased in adult db/db mice and can be reversed by CR. A: Food intake in db/db and db/+ mice. B: Body weight in db/db, db/+, and db/CR mice. C: Whole length of small intestine in db/db, db/+, and db/CR mice. D: Small intestine weight per cm length in db/db, db/+, and db/CR mice. E: Glucose uptake in jejunal rings of 8-week-old db/db, db/+, and db/CR mice. Counts are expressed as μmol/L per cm. F and G: Intestinal GLUT2 protein expression in 8-week-old db/db, db/+, and db/CR mice. Western blot data from at least three independent experiments and are expressed as mean ± SEM; mice n = 5–8 per group. *P < 0.05, **P < 0.01.
To test whether the absorption capacity per unit length of intestine also increased in db/db mice, we carried out glucose uptake assays in jejunal rings of 8-week-old db/db, db/+, and db/CR mice using a 3H-labeled glucose tracer (21). The data showed that the intestinal glucose absorption of the jejunal rings increased in db/db mice compared with the same length of db/+ mouse intestine (Fig. 1E). Consistently, the GLUT2 protein, which mediates glucose transport from the intestinal lumen to the blood stream (21,27), was increased dramatically in db/db intestines (Fig. 1F and G). CR significantly reduced glucose uptake and GLUT2 expression in db/db mice (Fig. 1E, F, and G). These results suggested that excess food intake, along with the increased intestinal absorption capacity, may contribute to obesity progression and that this can be reversed by CR.
Intestinal absorption area increased in db/db mice, and CR reduced absorption area in db/db mice.
The intestinal mucosa is the main site of nutrient absorption. Villi in the mucosa increase the surface area over which absorption takes place. We studied morphological changes to determine whether the overall absorption surface area of the small intestine in db/db mice was increased and the influence of CR on the absorption surface area. H-E staining of the jejunum showed that the villi lengthened by approximately one-third (36.65%, P < 0.001) in db/db mice, and CR significantly reduced the length of villi (Fig. 2A and B), indicating that the increased absorption area may have contributed to the increased absorption capacity in db/db mice, and CR reduced absorption by reducing growth of villi.
FIG. 2.

Enhanced epithelial proliferation in the small intestine of adult db/db mice. A: H-E staining of paraffin-embedded jejunum section shows longer villi length in db/db mice and decreased length in db/CR mice. B: Quantification of the villi length. C: Cell proliferation identified by BrdU immunohistochemical staining of paraffin section. Brown-color nucleus indicates BrdU-positive cells. D: Quantification of the BrdU-positive cell. E: Epithelial cell proliferation and migration detected by BrdU immunohistochemical staining. A brown nucleus indicates BrdU-positive cells. F: Quantification of BrdU-positive cells. G: Quantification of BrdU(+) cell migration. Data are expressed as mean ± SEM; mice n = 5–8 per group. **P < 0.01. Scale bar, 50 µm.
In adult mice, epithelial cells in the villi are renewed every 3–5 days through cell proliferation, differentiation, migration, and apoptosis from multipotent stem cells located in the intestinal crypts. During the cell-renewal process, these stem cells proliferate, differentiate, and convert into several types of functional mature epithelial cells that migrate along the villi (15). To evaluate the epithelial cell proliferation and migration in the small intestinal villi, we injected BrdU (intraperitoneally) into 8-week-old mice to label S-phase cells. One hour after BrdU was injected, the BrdU-positive rate in the jejunal crypts was significantly higher in db/db mice. The increase in BrdU-positive cells was reduced by CR in db/db mice (Fig. 2C and D). In addition, we performed immunohistochemistry 24 h after BrdU was injected to trace the proliferation and migration of BrdU-labeled cells. After 24 h, the number of BrdU-positive cells increased significantly in db/db mice (Fig. 2E and F) and the distance of BrdU-positive cells from the crypt/villi axis was significantly longer in the db/db mice. Nearly 100% of BrdU-positive cells in the control mice were distributed within 25 cells from the base of the crypt, while more than half of the BrdU-positive cells were located between cell positions 25 and 40 in db/db mice (Fig. 2G). These data suggest that increased proliferation and migration of stem cells from the crypts were likely responsible for the observed villi elongation and increased absorption in db/db mice.
Activation of GSK-3β/β-catenin axis in the mucosa of small intestine of db/db mice.
Cellular proliferation regulated by β-catenin plays a pivotal role in crypt stem cells and epithelial progenitor cells during gut development and in colon cancer (14,15,28). To assess whether β-catenin signaling also is involved in villi elongation in db/db mice, we examined β-catenin expression and signaling in the jejunum of 8-week-old db/db and db/+ mice. β-catenin mRNA expression within the intestine did not differ between these mice when assayed by quantitative RT-PCR (Fig. 3A). However, the protein level was significantly increased in db/db mice compared with db/+ mice (Fig. 3B and C). This suggested that β-catenin proteins accumulation is increased in the db/db mouse intestine and that this accumulation was reduced with CR (Fig. 3B and C). Analysis of GSK-3β phosphorylation showed that the ratio of Ser9 phosphorylation (p-GSK-3β) to total GSK-3β levels was upregulated in intestines of db/db. While the total GSK-3β (t-GSK-3β) protein expression did not change, the ratio of p-GSK-3β to t-GSK-3β was reduced by CR (Fig. 3B and D). As expected, when using immunohistochemistry to labeling the β-catenin target gene cyclin D1, positive cells also increased in the intestine of db/db mice and decreased after CR (Fig. 3E and F). These results mean that excess food intake–stimulated GSK-3β/β–catenin signaling activation might be involved in proliferation of intestinal epithelium.
FIG. 3.
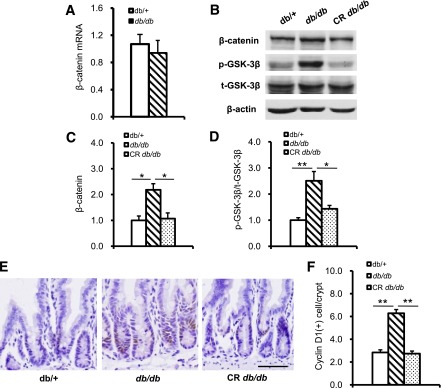
GSK-3β/β-catenin signaling is activated in db/db mouse intestinal epithelium. A: β-Catenin transcription level was detected by real-time RT-PCR. mRNA expression was calculated relative to β-actin. B and C: β-Catenin accumulation was increased in db/db mice compared with db/+ mice, and the β-Catenin accumulation in db/db mice was reduced by CR. B and D: GSK-3β Ser9 phosphorylation was increased in db/db mice compared with db/+ mice, and the increased GSK-3β Ser9 phosphorylation in db/db mice was reversed by CR. E: β-Catenin target gene cyclin D1 was detected by immunohistochemical staining. F: β-Catenin target gene cyclin D1 was increased in db/db mice intestine and was decreased by CR. PCR or Western blot data are from at least three independent experiments and are expressed as mean ± SEM. *P < 0.05, **P < 0.01. Scale bar, 50 µm.
Leptin signaling defects did not affect proliferation of intestinal epithelium in the embryonic stage.
Because db/db mice are leptin receptor mutant mice, we investigated whether increased intestinal epithelial proliferation was derived from abnormal gut development caused by the loss of leptin signaling. E18.5 db/db and db/+ littermates showed no difference in body weight or intestinal length (Fig. 4A and B). H-E staining also did not show gross differences in intestinal histology and villi length (Fig. 4C and D). In addition, there was no difference in cryptal BrdU labeling between the two groups (Fig. 4E and F). β-Catenin protein level, GSK-3β phosphorylation (Fig. 4G, H, and I), and cyclin D1–positive cells (data not shown) were similar in embryonic db/db and db/+ mouse intestines. These findings indicated that the genetic defect in the leptin receptor did not affect β-catenin signaling and cell proliferation in the embryonic intestinal epithelium. Combined with the earlier results, these results support a model in which increased intestinal epithelial cell proliferation was due to the excess food intake stimulation in adult db/db mice and suggested that compensatory intestinal epithelial cell proliferation/migration was important in increasing intestinal absorption and might contribute to obesity.
FIG. 4.

Embryonic intestinal epithelium proliferation did not differ between db/db and db/+ mice. A: Body weight of E18.5 db/db and db/+ embryos. B: Intestinal length of E18.5 db/db and db/+ embryos. C: H-E staining of E18.5 embryonic intestines. D: Length of embryonic intestine villi. E: E18.5 embryonic intestinal epithelial cell proliferation identified by BrdU staining. F: Quantification of BrdU-positive cells in E18.5 embryonic intestine. G, H, and I: β-Catenin, GSK-3β Ser9 phosphorylation, and total GSK-3β expression detected by Western blot in E18.5 db/db and db/+ embryonic jejunum; β-actin was used as a loading control. Western blot data are expressed as mean ± SEM from at least three independent experiments; embryos n = 10 per group. Scale bar, 20 µm.
HFD increased intestinal epithelial cell proliferation through activation of GSK-3β/β-catenin signaling pathway.
As described above, we observed that proliferation of epithelial cells in the intestine of db/db mice was increased significantly and could be reversed by CR. Because the db/db mice were leptin receptor mutant and hyperphagia, the intestinal changes in adult mice also could have been caused either by genetic background or physical stimulation due to the increased volume of food consumed. To confirm this, we subjected 4-week-old, wild-type C57BL/6 mice to HFD. After 8 weeks of HFD, the mice had significantly greater body weight than mice fed a standard chow diet (Fig. 5A and B). Concurrently, increased villi length and epithelial cell proliferation were found in the small intestines of HFD mice (Fig. 5C–F). Moreover, GSK-3β phosphorylation, β-catenin protein, and cyclin D1 level all were increased (Fig. 5G–J). These results confirmed that it was the energy density in the food behind the pronounced signaling of GSK-3β/β-catenin and stimulated intestinal epithelial proliferation and absorption rather than genetic background and volume of food intake.
FIG. 5.

Enhanced jejunal epithelial proliferation and GSK-3β/β-catenin signaling activation in HFD mice. A: Body weight of CD and HFD mice. B: Food intake in CD and HFD mice. C: H-E staining CD and HFD mice intestine. D: Quantification of the villi length. E: BrdU immunohistochemical staining of CD and HFD mice intestine. F: Quantification of BrdU-positive cells. G: β-Catenin, GSK-3β Ser9 phosphorylation, total GSK-3β, and cyclin D1 expression were detected by Western blot in CD and HFD mice jejunum; β-actin was used as a loading control. H, I, and J: Quantification of β-catenin, GSK-3β Ser9 phosphorylation, and cyclin D1 expression. Western blot data from at least three independent experiments and expressed as mean ± SEM; mice n = 6–8 per group. *P < 0.05, **P < 0.01. Scale bar, 50 µm.
To investigate further whether β-catenin was responsible for proliferation and morphological changes of intestinal cell from over nutrition, we treated mice fed the HFD with JW55. JW55 is a small-molecule inhibitor of the β-catenin signaling pathway. It stabilizes AXIN2, a member of the β-catenin destruction complex, and increases degradation of β-catenin (19). After 3 weeks’ treatment with JW55, β-catenin and cyclin D1 were significantly lower compared with the HFD DMSO control treatment group, but no differences were found with the CD group (Fig. 6A–D). Also, villi length and BrdU-positive cells were lower in the JW55-treated group (Fig. 6E–H). The absolute increased body weight in mice treated with JW55 was lower, but not significantly, compared with mice treated with DMSO (8.25 ± 0.37 g and 9.23 ± 0.46 g for JW55 and DMSO treatments, respectively). In our prior studies, significant differences in body weight between chow diet– and HFD-fed C57BL/6 mice usually appeared after 4 weeks. The present result might be due, in part, to the shorter duration of HFD feeding and treatment. These data indicated that an activated β-catenin signaling pathway was essential for proliferation intestinal epithelium in response to HFD feeding.
FIG. 6.

Increased degradation of β-catenin attenuated proliferation of intestinal epithelium in HFD-fed mice. A: β-Catenin, GSK-3β Ser9 phosphorylation, total GSK-3β, and cyclin D1 expression detected by Western blot; β-actin were used as a loading control. B, C, and D: Quantification of β-catenin, GSK-3β Ser9 phosphorylation, and cyclin D1 expression. E: H-E staining show longer villi length in HFD and HFD + DMSO mice and shorter villi length in HFD + JW55 mice. F: Quantification of villi length. G: Cell proliferation identified by immunohistochemical staining with BrdU. Brown nucleus indicates BrdU-positive cells. H: Quantification of the BrdU-positive cells. Western blot data from at least three independent experiments and are expressed as mean ± SEM; mice n = 4–8 per group. *P < 0.05, **P < 0.01. Scale bar, 20 µm.
High glucose and FFA induce GSK-3β phosphorylation and β-catenin accumulation in epithelial cells.
Carbohydrate is the main component of regular mouse chow. Most ingested carbohydrate is digested and broken down into glucose in the small intestine and transported into the blood stream. Previous studies have reported that exposure to high glucose levels induces proliferation in some cell types (29,30). In addition, high-sucrose diets promote intestinal epithelial cell proliferation and tumorigenesis in APCmin mice (31). Therefore, we performed an in vitro study to determine whether glucose directly stimulates intestine epithelial cell proliferation. When CT26 epithelial cells were exposed to high glucose (20 mmol/L), phosphorylation of GSK-3β increased, and the expression of β-catenin and its target gene, cyclin D1, was upregulated after 1 h of high-glucose stimulation (Fig. 7A and B). After mice were fed an HFD, the primary nutrient absorbed in the intestine was FFA. To confirm whether FFA directly activates GSK-3β/β-catenin signaling, we added FFA to CT26 cell culture medium. GSK-3β phosphorylation and β-catenin accumulation were increased after 1 h of FFA stimulation. The expression of the β-catenin target gene, cyclin D1, also was upregulated after FFA stimulation (Fig. 7C and D). However, treating CT26 cells with FH535, an inhibitor of β-catenin/TCF transcription, reduced β-catenin and significantly blocked expression of cyclin D1 (Fig. 8A–C) and cell growth induced by high glucose (Fig. 8D). Similarly, when the GSK-3 inhibitor CHIR98014 was add to CT26 cells, the phosphorylation of GSK-3β, accumulation of β-catenin, and expression of Cyclin D1 all were upregulated significantly (Fig. 8E–H). These data support the supposition that high nutrient levels directly activate GSK-3β/β-catenin signaling and stimulate intestinal epithelial cell proliferation.
FIG. 7.
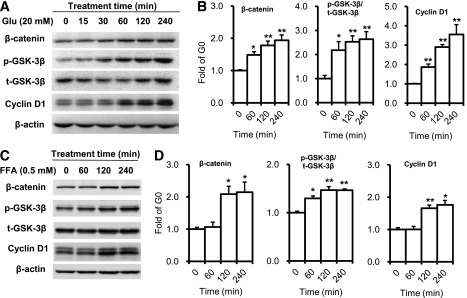
High-concentration glucose (Glu) and FFA activate GSK-3β/β-catenin signaling pathway in epithelial cell. A: Expression of β-catenin, GSK-3β Ser9 phosphorylation, and cyclin D1 were detected by Western blot in CT26 cells after stimulation with glucose. B: Expression of β-catenin, GSK-3β Ser9 phosphorylation, and cyclin D1. C: Expression of β-catenin, GSK-3β Ser9 phosphorylation, total GSK-3β, and cyclin D1 were detected by Western blot in CT26 cell after FFA stimulation. D: Expression of β-catenin, GSK-3β Ser9 phosphorylation, and cyclin D1. Data are expressed as mean ± SEM; n = 3 independent experiments. *P < 0.05, **P < 0.01 vs. time 0.
FIG. 8.
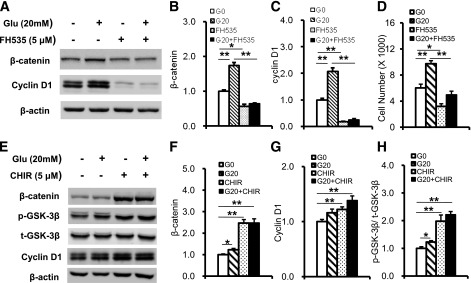
β-Catenin inhibitor FH535 reduces expression of cyclin D1, and GSK-3 inhibitor increases cyclin D1 expression. A, B, and C: FH535 blocked β-catenin accumulation and expression of cyclin D1 induced by high glucose. D: FH535 blocked intestinal epithelial cell proliferation induced by high glucose. E, F, G, and H: CHIR98014 alone or combined with high glucose increases GSK-3β Ser9 phosphorylation, β-catenin accumulation, and cyclin D1 expression in CT26 cells. Data are expressed as mean ± SEM; n = 3 independent experiments. *P < 0.05, **P < 0.01.
DISCUSSION
The increasing incidence of obesity and its association with disease have become a great challenge to global health. Obesity increases the risk of cardiovascular disease, premature death, insulin resistance, T2D, and many types of cancer (32,33). However, few drugs for antiobesity are approved by the U.S. Food and Drug Administration, such as phentermine and orlistat (34). Unfortunately, because of insufficient weight loss and significant gastrointestinal side effects, these antiobesity drugs are less than ideal. So, there is an urgent need to find new therapeutic targets for obesity.
The etiology of obesity is very complex, and pathological mechanisms have been studied widely in adipose tissue, liver, and muscle (35,36). Several genes have been identified that regulate adipose mass and are associated with the development of obesity. Recent findings regarding the role of intestine-secreted hormones in metabolic diseases and the effects of gastric bypass bariatric surgery make the intestine a primary site in the pathophysiology of obesity and T2D (37–39). However, aside from the impact of gut-derived endocrine hormones on T2D, there has been little study of the contribution of the intestine to excess food intake–related obesity.
Some studies describing gene regulation in the intestine during the development of obesity induced by an HFD found that many dietary fat–induced molecular changes are associated with lipid metabolism, the cell cycle, inflammation, and the immune response (40–42). But details of mechanisms that affect the functionality of intestine during the progression of obesity have not been studied. In a regular diet, carbohydrates are the most prevalent nutrient. Monosaccharides in the intestinal lumen interact with the intestinal epithelium after carbohydrate digestion. Whether this interaction affects intestinal function is unknown. β-Catenin–dependent intestinal stem cell proliferation is a prerequisite for the maintenance of epithelial homeostasis and absorption (13).
Free β-catenin proteins accumulating in cells are an important feature of activating β-catenin downstream signaling (16). Chen et al. found that retinal tissue sections from patients with diabetes displayed increased β-catenin expression and nuclear translocation (43). Anagnostou and Shepherd also verified that high glucose induces upregulation of β-catenin in two macrophage cell lines (44). In this study, nutrient overload in animals and high glucose or FFA in cultured intestinal epithelial cells induced β-catenin accumulation and intestinal epithelial cell proliferation. In confirmation of this, high glucose did not stimulate intestinal cell proliferation or cyclin D1 expression after treatment with the inhibitor of β-catenin transcription, FH535. Giving a β-catenin inhibitor, JW55, to mice fed an HFD also prevented intestinal epithelial cell proliferation. Together, these results indicate that nutrients may prevent β-catenin degradation and promote downstream transcriptional activation that increases intestinal epithelial cell proliferation. Functionally, GLUT2, a main transporter of dietary sugar in the intestine (27), increased in hyperphagic obese db/db mice. On the contrary, food restriction in db/db mice reduced intestinal cell proliferation and absorption, the indication being that intestinal absorptive capacity was increasing along with the increased food intake and intestinal epithelial proliferation.
GSK-3β–based protein degradation is a key event in regulating intracellular β-catenin protein accumulation in canonical Wnt signaling (14,16). The ninth serine in the NH2-terminus is very important for GSK-3β activity. Once this serine is phosphorylated, GSK-3β fails to bind and phosphorylate β-catenin for degradation. We found that excess food intake promoted phosphorylation of the ninth serine of GSK-3β in intestine. Adding CHIR98014 (a GSK-3 inhibitor) alone, or with high glucose, to epithelial cells could have increased GSK-3β phosphorylation and then induced β-catenin accumulation and cyclin D1 expression. This suggests that the increased β-catenin protein in intestinal epithelial cells may depend on phosphorylation of the ninth serine of GSK-3β.
Recently, in addition to Wnts, a variety of signaling molecules involved in the regulation of intracellular transcriptional activity of β-catenin/TCF in different contexts have been found, including insulin, IGF-1, platelet-derived growth factor, glucagon-like peptide (GLP)-2, and forkhead box class O (45–47). Studies have shown that, in breast cancer cells, IGF-1 stabilizes intracellular β-catenin and promotes the transcription activity of β-catenin/TCF. IGF-1 and insulin are high in the plasma of obese animals and are closely related to nutrient status. GLP-2 is one of the best studied peptide secreted from enteroendocrine L cells of the intestine that are closely related to intestinal function (48,49). Studies show that GLP-2 could stimulate cell proliferation of intestine epithelium through IGF-1 signaling and β-catenin alterations (46,47,50). And GLP-2 is also involved in intestinal epithelial proliferation after HFD stimulation (49). After 3 weeks of HFD feeding, blood glucose and insulin were increased significantly, though GLP-2 had no clear increase in the HFD group. Phosphorylation of Akt increased significantly in adipose tissue but not in the intestine (data not shown). Nutrients seemed to have a direct and important effect on the proliferation of intestinal epithelial cells, at least in the early period of nutrient overload. However, we cannot exclude that GLP-2, inflammation, hyperglycemia, or hyperinsulinemia might also have contributed to intestinal epithelial cell proliferation in long-term nutrient overload or obesity status.
Taken together, our findings confirm the importance of nutrition on initiating proliferation of intestinal epithelium and provide a possible link between intestinal metabolism and obesity development. Targeting epithelial GSK-3β/β-catenin signaling in the small intestine may provide a novel strategy to prevent obesity related to diet. Further investigation into the mechanisms responsible for interactions between diet and the intestine will advance our knowledge of the pathogenesis of obesity.
ACKNOWLEDGMENTS
This study was supported by the National Natural Science Foundation of China (30870996, 81270883, and 31221002).
No potential conflicts of interest relevant to this article were reported.
J.M. contributed to the experiments and data analysis and wrote the manuscript. X.H. contributed to the experiments and contributed to all revised experiments. Y.X., C.Y., Y.D., and N.H. contributed to the experiments. J.W. and H.C. contributed to the discussion and reviewed and edited the manuscript. X.Z. contributed to the study design, data collection, and interpretation and wrote the manuscript. X.Z. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
The authors thank Dr. Iain C. Bruce (Institute of Molecular Medicine, Peking University), Dr. David M. Ornitz (Department of Developmental Biology, Washington University School of Medicine), and Jon K. Moon, PhD, for help with editing English. The authors also thank Drs. Ruiping Xiao, Xiaojun Zhu, Chunmei Cao, Ruisheng Song, and Yan Zhang (Institute of Molecular Medicine, Peking University) for scientific discussion and critical comments, and Hui Wang, Jun Zhang, and Yuli Liu (Institute of Molecular Medicine, Peking University) for technical support.
REFERENCES
- 1.Grundy SM. Metabolic syndrome pandemic. Arterioscler Thromb Vasc Biol 2008;28:629–636 [DOI] [PubMed] [Google Scholar]
- 2.Barsh GS, Schwartz MW. Genetic approaches to studying energy balance: perception and integration. Nat Rev Genet 2002;3:589–600 [DOI] [PubMed] [Google Scholar]
- 3.Spiegelman BM, Flier JS. Obesity and the regulation of energy balance. Cell 2001;104:531–543 [DOI] [PubMed] [Google Scholar]
- 4.Redman LM, Ravussin E. Caloric restriction in humans: impact on physiological, psychological, and behavioral outcomes. Antioxid Redox Signal 2011;14:275–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Capel F, Klimcáková E, Viguerie N, et al. Macrophages and adipocytes in human obesity: adipose tissue gene expression and insulin sensitivity during calorie restriction and weight stabilization. Diabetes 2009;58:1558–1567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anderson RM, Shanmuganayagam D, Weindruch R. Caloric restriction and aging: studies in mice and monkeys. Toxicol Pathol 2009;37:47–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Murphy KG, Bloom SR. Gut hormones and the regulation of energy homeostasis. Nature 2006;444:854–859 [DOI] [PubMed] [Google Scholar]
- 8.Karra E, Batterham RL. The role of gut hormones in the regulation of body weight and energy homeostasis. Mol Cell Endocrinol 2010;316:120–128 [DOI] [PubMed] [Google Scholar]
- 9.Trayhurn P, Bing C. Appetite and energy balance signals from adipocytes. Philos Trans R Soc Lond B Biol Sci 2006;361:1237–1249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Keelan M, Cheeseman CI, Clandinin MT, Thomson AB. Intestinal morphology and transport after ileal resection in rat is modified by dietary fatty acids. Clin Invest Med 1996;19:63–70 [PubMed] [Google Scholar]
- 11.Petit V, Arnould L, Martin P, et al. Chronic high-fat diet affects intestinal fat absorption and postprandial triglyceride levels in the mouse. J Lipid Res 2007;48:278–287 [DOI] [PubMed] [Google Scholar]
- 12.Scoaris CR, Rizo GV, Roldi LP, et al. Effects of cafeteria diet on the jejunum in sedentary and physically trained rats. Nutrition 2010;26:312–320 [DOI] [PubMed] [Google Scholar]
- 13.Clevers H. Wnt/beta-catenin signaling in development and disease. Cell 2006;127:469–480 [DOI] [PubMed] [Google Scholar]
- 14.MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell 2009;17:9–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Crosnier C, Stamataki D, Lewis J. Organizing cell renewal in the intestine: stem cells, signals and combinatorial control. Nat Rev Genet 2006;7:349–359 [DOI] [PubMed] [Google Scholar]
- 16.van der Flier LG, Clevers H. Stem cells, self-renewal, and differentiation in the intestinal epithelium. Annu Rev Physiol 2009;71:241–260 [DOI] [PubMed] [Google Scholar]
- 17.Ikeda S, Kishida S, Yamamoto H, Murai H, Koyama S, Kikuchi A. Axin, a negative regulator of the Wnt signaling pathway, forms a complex with GSK-3beta and beta-catenin and promotes GSK-3beta-dependent phosphorylation of beta-catenin. EMBO J 1998;17:1371–1384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Karrasch T, Spaeth T, Allard B, Jobin C. PI3K-dependent GSK3ß(Ser9)-phosphorylation is implicated in the intestinal epithelial cell wound-healing response. PLoS ONE 2011;6:e26340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Waaler J, Machon O, Tumova L, et al. A novel tankyrase inhibitor decreases canonical Wnt signaling in colon carcinoma cells and reduces tumor growth in conditional APC mutant mice. Cancer Res 2012;72:2822–2832 [DOI] [PubMed] [Google Scholar]
- 20.Geske MJ, Zhang X, Patel KK, Ornitz DM, Stappenbeck TS. Fgf9 signaling regulates small intestinal elongation and mesenchymal development. Development 2008;135:2959–2968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gouyon F, Caillaud L, Carriere V, et al. Simple-sugar meals target GLUT2 at enterocyte apical membranes to improve sugar absorption: a study in GLUT2-null mice. J Physiol 2003;552:823–832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tanaka S, Terada K, Nohno T. Canonical Wnt signaling is involved in switching from cell proliferation to myogenic differentiation of mouse myoblast cells. J Mol Signal 2011;6:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ring DB, Johnson KW, Henriksen EJ, et al. Selective glycogen synthase kinase 3 inhibitors potentiate insulin activation of glucose transport and utilization in vitro and in vivo. Diabetes 2003;52:588–595 [DOI] [PubMed] [Google Scholar]
- 24.Hardy S, St-Onge GG, Joly E, Langelier Y, Prentki M. Oleate promotes the proliferation of breast cancer cells via the G protein-coupled receptor GPR40. J Biol Chem 2005;280:13285–13291 [DOI] [PubMed] [Google Scholar]
- 25.Sun J, Jin T. Both Wnt and mTOR signaling pathways are involved in insulin-stimulated proto-oncogene expression in intestinal cells. Cell Signal 2008;20:219–229 [DOI] [PubMed] [Google Scholar]
- 26.Vieira VJ, Valentine RJ, Wilund KR, Antao N, Baynard T, Woods JA. Effects of exercise and low-fat diet on adipose tissue inflammation and metabolic complications in obese mice. Am J Physiol Endocrinol Metab 2009;296:E1164–E1171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kellett GL, Brot-Laroche E. Apical GLUT2: a major pathway of intestinal sugar absorption. Diabetes 2005;54:3056–3062 [DOI] [PubMed] [Google Scholar]
- 28.van de Wetering M, Sancho E, Verweij C, et al. The beta-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell 2002;111:241–250 [DOI] [PubMed] [Google Scholar]
- 29.Yamamoto M, Acevedo-Duncan M, Chalfant CE, Patel NA, Watson JE, Cooper DR. Acute glucose-induced downregulation of PKC-betaII accelerates cultured VSMC proliferation. Am J Physiol Cell Physiol 2000;279:C587–C595 [DOI] [PubMed] [Google Scholar]
- 30.Li YM, Schilling T, Benisch P, et al. Effects of high glucose on mesenchymal stem cell proliferation and differentiation. Biochem Biophys Res Commun 2007;363:209–215 [DOI] [PubMed] [Google Scholar]
- 31.Wang B, Bobe G, LaPres JJ, Bourquin LD. High sucrose diets promote intestinal epithelial cell proliferation and tumorigenesis in APC(Min) mice by increasing insulin and IGF-I levels. Nutr Cancer 2009;61:81–93 [DOI] [PubMed] [Google Scholar]
- 32.Khandekar MJ, Cohen P, Spiegelman BM. Molecular mechanisms of cancer development in obesity. Nat Rev Cancer 2011;11:886–895 [DOI] [PubMed] [Google Scholar]
- 33.Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 2006;444:840–846 [DOI] [PubMed] [Google Scholar]
- 34.Powell AG, Apovian CM, Aronne LJ. New drug targets for the treatment of obesity. Clin Pharmacol Ther 2011;90:40–51 [DOI] [PubMed] [Google Scholar]
- 35.Samuel VT, Petersen KF, Shulman GI. Lipid-induced insulin resistance: unravelling the mechanism. Lancet 2010;375:2267–2277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Henriksen EJ. Dysregulation of glycogen synthase kinase-3 in skeletal muscle and the etiology of insulin resistance and type 2 diabetes. Curr Diabetes Rev 2010;6:285–293 [DOI] [PubMed] [Google Scholar]
- 37.Takaishi S, Shibata W, Tomita H, et al. In vivo analysis of mouse gastrin gene regulation in enhanced GFP-BAC transgenic mice. Am J Physiol Gastrointest Liver Physiol 2011;300:G334–G344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hansen EN, Tamboli RA, Isbell JM, et al. Role of the foregut in the early improvement in glucose tolerance and insulin sensitivity following Roux-en-Y gastric bypass surgery. Am J Physiol Gastrointest Liver Physiol 2011;300:G795–G802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gautier JF, Choukem SP, Girard J. Physiology of incretins (GIP and GLP-1) and abnormalities in type 2 diabetes. Diabetes Metab 2008;34(Suppl. 2):S65–S72 [DOI] [PubMed] [Google Scholar]
- 40.de Wit NJ, Boekschoten MV, Bachmair EM, et al. Dose-dependent effects of dietary fat on development of obesity in relation to intestinal differential gene expression in C57BL/6J mice. PLoS ONE 2011;6:e19145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Al-Dwairi A, Pabona JM, Simmen RC, Simmen FA. Cytosolic malic enzyme 1 (me1) mediates high fat diet-induced adiposity, endocrine profile, and gastrointestinal tract proliferation-associated biomarkers in male mice. PLoS One 2012;7:e46716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.de Wit NJ, Bosch-Vermeulen H, de Groot PJ, et al. The role of the small intestine in the development of dietary fat-induced obesity and insulin resistance in C57BL/6J mice. BMC Med Genomics 2008;1:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen Y, Hu Y, Zhou T, et al. Activation of the Wnt pathway plays a pathogenic role in diabetic retinopathy in humans and animal models. Am J Pathol 2009;175:2676–2685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Anagnostou SH, Shepherd PR. Glucose induces an autocrine activation of the Wnt/beta-catenin pathway in macrophage cell lines. Biochem J 2008;416:211–218 [DOI] [PubMed] [Google Scholar]
- 45.Rosner G, Rozen P, Bercovich D, et al. A protocol for genetic evaluation of patients with multiple colorectal adenomas and without evidence of APC gene mutation. Isr Med Assoc J 2010;12:549–553 [PubMed] [Google Scholar]
- 46.Rowland KJ, Trivedi S, Lee D, Wan K, Kulkarni RN, Holzenberger M, Brubaker PL. Loss of glucagon-like peptide-2-induced proliferation following intestinal epithelial insulin-like growth factor-1-receptor deletion. Gastroenterology 2011;141:2166–2175 [DOI] [PubMed]
- 47.Dubé PE, Rowland KJ, Brubaker PL. Glucagon-like peptide-2 activates beta-catenin signaling in the mouse intestinal crypt: role of insulin-like growth factor-I. Endocrinology 2008;149:291–301 [DOI] [PubMed] [Google Scholar]
- 48.Sinclair EM, Drucker DJ. Proglucagon-derived peptides: mechanisms of action and therapeutic potential. Physiology (Bethesda) 2005;20:357–365 [DOI] [PubMed] [Google Scholar]
- 49.Baldassano S, Amato A, Cappello F, Rappa F, Mulè F. Glucagon-like peptide-2 and mouse intestinal adaptation to a high-fat diet. J Endocrinol 2013;217:11–20 [DOI] [PubMed] [Google Scholar]
- 50.Dubé PE, Forse CL, Bahrami J, Brubaker PL. The essential role of insulin-like growth factor-1 in the intestinal tropic effects of glucagon-like peptide-2 in mice. Gastroenterology 2006;131:589–605 [DOI] [PubMed] [Google Scholar]