

Abstract
Photosystem II supports four manganese centers through nine oxidation states from manganese(II) during assembly through to the most oxidized state before O2 formation and release. The protein-based carboxylate and imidazole ligands allow for significant changes of the coordination environment during the incorporation of hydroxido and oxido ligands upon oxidation of the metal centers. We report the synthesis and characterization of a series of tetramanganese complexes in four of the six oxidation states from MnII3MnIII to MnIII2 MnIV2 with the same ligand framework (L) by incorporating four oxido ligands. A 1,3,5-triarylbenzene framework appended with six pyridyl and three alkoxy groups was utilized along with three acetate anions to access tetramanganese complexes, Mn4Ox, with x = 1, 2, 3, and 4. Alongside two previously reported complexes, four new clusters in various states were isolated and characterized by crystallography, and four were observed electrochemically, thus accessing the eight oxidation states from MnII4 to MnIIIMnIV3. This structurally related series of compounds was characterized by EXAFS, XANES, EPR, magnetism, and cyclic voltammetry. Similar to the ligands in the active site of the protein, the ancillary ligand (L) is preserved throughout the series and changes its binding mode between the low and high oxido-content clusters. Implications for the rational assembly and properties of high oxidation state metal-oxido clusters are presented.
1. Introduction
In biological systems, the oxidation of water to dioxygen is performed by the oxygen-evolving center (OEC) of photosystem II (PSII).1 In the catalytic cycle, or Kok cycle,2, 3 four photo-generated oxidizing equivalents sequentially oxidize the OEC, which releases O2 upon the fourth oxidation. Although the oxidation states are still debated,4 a common assignment of the intermediates, denoted Sn states (n = 0–4), range in oxidation state from S0, MnIII3MnIV, to S4, MnIV4-ligand radical or MnIV3MnV.1, 5–7 While highly efficient, the OEC must be reassembled frequently under full solar flux due to photooxidative damage to the inorganic cluster and D1 peptide.8 The process by which the OEC is assembled, called photoactivation,9 uses Mn2+, Ca2+, Cl−, water, and oxidizing equivalents generated by light absorption.10–12 Although bicarbonate is not coordinated to the fully assembled OEC,13, 14 spectroscopic evidence supports a role for bicarbonate in photoactivation of the cluster.15–17 Kinetically distinct species in photoactivation have been defined,17–22 and reduction of the intermediates in the Kok cycle have led to species in oxidation states lower than S0 (Sn with n = −1, −2, −3) with S−3 corresponding to a putative MnIIMnIII3 oxidation state.23–26
A remarkable characteristic of PSII is its ability to support four manganese centers over a considerable span of oxidation states from four MnII at the beginning of photoactivation through to four MnIV before O–O bond formation utilizing the same, protein-defined set of carboxylate and imidazole ligands. To do so, water is concurrently incorporated and deprotonated to form hydroxido and oxido donors that stabilize the higher oxidation states of the Mn4CaOx cluster. Moreover, during catalytic turnover, lower oxidation state and lower oxygen-atom content moieties must be generated upon loss of O2. Thus, the protein-derived ligand set available must stabilize the constant reorganization, reoxidation, and reoxygenation of the Mn4CaOx cluster.
A large effort to synthetically model the OEC has produced a variety of di-, tri-, and tetramanganese-oxido clusters.27–29 These models have provided invaluable spectroscopic benchmarks in the characterization of the OEC and have helped elucidate the chemistry of multinuclear clusters in general. No single system has been shown to stabilize manganese oxidation states as widely as PSII. We report herein a synthetic framework with a well-defined ligand set that supports tetranuclear, manganese-oxido clusters over the eight oxidation states from MnII4 to MnIIIMnIV3. Stepwise oxido incorporation stabilizes the increasing oxidation state, which vary from MnII4 to MnIIIMnIV3 with one, two, three, and four oxido donors. Structural (XRD) and spectroscopic (XAS, EPR) characterization, interconversion of these clusters, the role of the ancillary ligands, and the relation to the OEC are discussed.
2. Results and Discussion
2.1 Synthesis of Tetramanganese Clusters
We have reported a ligand framework – 1,3,5-tris(2-di(2′-pyridyl)hydroxymethylphenyl)benzene (H3L) (Scheme 1) – that supports multimetallic complexes of a variety of first-row transition metals.30–32 Starting from H3L, trimanganese(II) complex 1 was synthesized upon in situ deprotonation and reaction with three equivalents of MnII(OAc)2 (Scheme 1).31 Trinuclear species 1 has been used as precursor for the synthesis of complexes displaying Mn4O4 (6) and Mn3CaO4 cubane moieties as structural models of the OEC.32 In order to access tetramanganese clusters in a range of oxidation states and oxido content, 1 was treated with oxygenating reagents and MnII precursors under a variety of reaction conditions.
Scheme 1.

Synthesis and interconversion of tetramanganese complexes 2–6. Dashed arrows represent conceptual conversions involving water, proton and electron transfers. Curved lines represent 2-pyridyl groups. OAc = acetate. OTf = trifluoromethanesulfonate. Fc+ = ferrocenium.
Addition of a solution of Mn(OTf)2•CH3CN (OTf = trifluoromethanesulfonate) in tetrahydrofuran (THF) to a suspension of 1 in THF followed by one equivalent of iodosobenzene (PhIO) led to a color change from yellow to brown/purple over two hours (Scheme 1). Electrospray ionization mass spectrometry (ESI-MS) analysis of purified purple product suggests oxygen and manganese incorporation, with peaks at 1417.0 and 1268.3 m/z – [LMn4O1(OAc)3(OTf)]+ and [LMn4O1(OAc)3]+. A single crystal X-ray diffraction (XRD) study is consistent with the ESI-MS findings and supports the structure of 3 as a tetramanganese monooxido species (vide infra).
The addition of KO2 to an equimolar mixture of 1 and Mn(OAc)2 or Mn(OTf)2•CH3CN allowed the isolation of different complexes as a function of reaction solvent. A brown solid 2 precipitated from the heterogeneous mixture of 1, Mn(OAc)2, and KO2 in THF upon stirring for four days. ESI-MS of 2 shows a single peak at 1268.4 m/z, corresponding to [LMn4O1(OAc)3]+, and an XRD study confirms the structure of 2 as a tetramanganese monooxido species similar to complex 3 (Figure S9).
The reaction of 1 with KO2 and Mn(OAc)2 in acetonitrile generated tan precipitate 4 after 3.5 days of stirring. As observed in THF, 2 was the major species in solution at early reaction times (<10h, 1H NMR spectroscopy, NMR), yet as the reaction progressed, 4 became dominant. The ESI-MS spectrum displays major peaks at 1225.2, 1268.2, and 1285.0 m/z, assigned to [LMn4O2(OAc)2]+, [LMn4O1(OAc)3]+, and [LMn4O2(OAc)3H]+, respectively. Vapor diffusion of Et2O into a DMF solution of 4 gave crystals amenable to XRD (vide infra).
Complexes 2–6 are 1H NMR active with paramagnetically broadened and shifted peaks. While the resonances could not be assigned to specific protons, NMR was still useful in observing the reaction mixtures and purity of the samples. The synthesis and structure of complexes 5 and 6 have been reported elsewhere.32, 33 The cubane moiety of 6 could be further functionalized by selective removal of an acetate ligand with one equivalent of trimethylsilyl triflate followed by addition of nitrogenous donors such as dimethylpyrazole (Eqn. 1). This reaction sequence allowed isolation of a cationic MnIII2MnIV2O4 cubane complex (7).
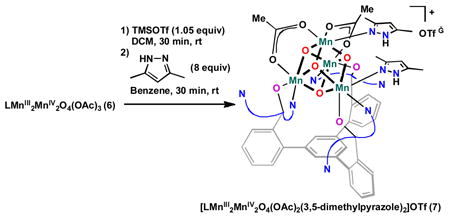 |
(1) |
Although a large variety of manganese cluster models of the OEC have been reported,27, 28 the family of compounds reported here is unique in displaying significant variation in oxidation state and the number of oxido ligands for four manganese centers supported by the same set of ancillary ligands. Previously reported tetramanganese-oxido complexes vary in shape, containing butterfly,34 planar,35 linear,36 cubic,37, 38 adamantane,39 and dimer-of-dimer40, 41 geometries. The isolation of oxido-content homologs is rare.42,43
2.2 Solid-State Structures
The crystal structure of 3 shows three basal metal centers bridged by three alkoxides forming a six-membered ring, and the pyridines of each dipyridyloxymethyl moiety coordinate to adjacent metal centers. This motif is conserved from the trinuclear 1.31 A centrally located μ4-oxido and three κ2-acetates bridge these three basal manganese centers to a fourth manganese that is capped by a trifluoromethanesulfonate (Fig. 1). An outer sphere trifluoromethanesulfonate is present in the solid-state, consistent with a MnII2MnIII2 oxidation state assignment. Comparison to structurally related Mn4-μ4-O motifs that have been previously characterized44–51 supports the presence of MnII and/or MnIII in 3. Elemental analysis results indicate the presence of two triflate anions in the isolated crystalline powder, supporting the oxidation state assignment as MnII2MnIII2. In agreement, the cyclic voltammogram of 3 shows two reduction events (Figure S7), accessing MnII3MnIII and MnII4 oxidation states.
Fig. 1.

Solid-state structures of complexes 2–5 and 7. Top: Depictions of full molecules 2 and 7; Bottom: Truncated Mn4Ox cores. Thermal ellipsoids are drawn at 50% probability. Hydrogen atoms, outersphere anions, and solvent molecules are not shown for clarity. Bolded bonds highlight the Mn4Ox cores. See SI for detailed crystallographic information. The structure of complex 5 has been reported previously.33
In contrast to 3, 2 has the apical metal center capped by an acetate rather than trifluoromethanesulfonate and no outer sphere anion, indicating a one-electron reduced state, assigned as MnII3MnIII (Figure 1). The acetate bridging Mn3-Mn4 adopts two bridging modes, one mode is the usual κ2 mode as in the other acetates and the other with a single oxygen atom forming the bridge, as shown in the figure. Mn1 has a shorter bond to O4 (the central μ4-oxido) than the other three Mn centers: 1.919(4) versus 2.041(4), 2.049(4), and 2.157(3). This short distance is most consistent with the literature MnIII-oxido distances in Mn4-μ4-O motifs,44–51 while the other three distances match MnII-oxido distances. Furthermore, the successful conversion of 3 to 2 with one equivalent of a one-electron reductant indicates that 3 and 2 differ by a single electron in oxidation state, consistent with oxidation state assignments of 2 and 3 as stated (vide infra).
For 4, the trimanganese-tris-μ2-alkoxide core as found in 1–3 is present, and similar to 2, a μ4-oxide and two κ2-acetates connect the fourth manganese center to the basal trinuclear core. Unlike 2 and 3, however, a second oxido ligand is present, bridging the apical manganese to a basal manganese (O11 of 4 in Fig. 1). Analysis of the structural parameters supports an oxidation state assignment as MnII2MnIII2. Within the diamond core, the Mn–O bond distances are Mn4-O11, 1.8576(17) Å; Mn4-O10, 1.8295(17) Å; Mn1-O11, 1.8638(19) Å; Mn1-O10, 1.9639(16)Å; and the Mn1-Mn4 distance is 2.7921(6)Å. These parameters are consistent with MnIII. The other manganese centers in the base have similar Mn-alkoxide and Mn-pyridine distances to 1, and are thus assigned as MnII ions. The Mn4(μ4-O)(μ2-O) structural motif has not been previously described, although Mn4O2 complexes of butterfly34, planar35, fused-cubane52, and ladder-like53 shapes have been reported, for example.
The solid state structure of 7 parallels the structures of complexes 5 and 6 that have been reported previously.32, 33 Different from 1–4, the three alkoxides bind terminally to three Mn centers, three pyridines are unbound, and the Mn4 unit moves away from the triarylbenzene framework. In 7, one of the bridging acetates is replaced with two 3,5-dimethylpyrazoles that π-stack with a pyridine of the ligand framework (Fig 1). The N–H groups of the pyrazoles H-bond to a triflate counterion. As in 6, two manganese centers show axial distortion as expected for psuedo-octahedral MnIII, and two have shorter Mn-O distances consistent with MnIV (Table 1).
Table 1.
Selected Structural Parameters of 2–7.
| Complex | Metal ion | Average Mn-oxo distance (Å) | Average Mn–Mn distance (Å) | Oxidation state |
|---|---|---|---|---|
| Mn1 | 1.919(4) | 3.2 | Mn(III) | |
| 2 | Mn2 | 2.157(3) | 3.3 | Mti(II) |
| MnII3MnIIIO | Mn3 | 2.041(4) | 3.2 | Mn(II) |
| Mn4 | 2.049(3) | 3.5 | Mti(II) | |
|
| ||||
| Overall Average | 2.04 ± 0.10 | 3.31 ± 0.22 | ||
|
| ||||
| Mn1 | 2.068(2) | 3.3 | Mn(III) | |
| 3 | Mn2 | 2.095(3) | 3.2 | Mri(II) |
| MnII2MnIII2O | Mn3 | 1.980(3) | 3.2 | Mn(III) |
| Mn4 | 2.048(3) | 3.5 | Mn(II) | |
|
| ||||
| Overall Average | 2.05 ± 0.05 | 3.32 ± 0.21 | ||
|
| ||||
| Mn1 | 1.91 ± 0.07 | 3.0 | Mn(III) | |
| 4 | Mn2 | 2099(2) | 3.3 | Mn(II) |
| MnII2MnIII2O2 | Mn3 | 2.174(2) | 3.3 | Mti(II) |
| Mn4 | 1.84 ± 0.02 | 3.3 | Mn(III) | |
|
| ||||
| Overall Average | 1.96 ±0.14 | 3.24 ± 0.29 | ||
|
| ||||
| Mn1 | 1.88 ±0.03 | 3.1 | Mn(III) | |
| 5 | Mn2 | 1.89 ±0.05 | 3.1 | Mn(III) |
| MnIII4O3 | Mn3 | 1.884 ± 0.004 | 3.1 | Mn(III) |
| Mn4 | 2.02 ± 0.12 | 2.8 | Mn(III) | |
|
| ||||
| Overall Average | 1.93 ± 0.09 | 3.03 ± 0.21 | ||
|
| ||||
| Mn1 | 2.04 ± 0.19 | 3.0 | Mn(III) | |
| 6 | Mn2 | 1.86 ±0.02 | 2.9 | Mn(IV) |
| MnIII2MnIV2O4 | Mn3 | 1.93 ± 0.07 | 2.9 | Mn(IV) |
| Mn4 | 2.01 ±0.16 | 2.9 | Mn(III) | |
|
| ||||
| Overall Average | 1.96 ± 0.13 | 2.92 ± 0.10 | ||
|
| ||||
| Mn1 | 1.87 ± 0.02 | 2.9 | Mn(IV) | |
| 7 | Mn2 | 1.88 ± 0.01 | 2.9 | Mn(IV) |
| MnIII2MnIV2O4 | Mn3 | 2.00 ± 0.10 | 2.9 | Mn(III) |
| Mn4 | 2.06 ± 0.15 | 2.9 | Mn(III) | |
|
| ||||
| Overall Average | 1.95 ± 0.09 | 2.91 ± 0.09 | ||
The bond distance with the corresponding XRD esd in parentheses are given for Mn centers with one Mn–O bond. The average Mn–O distance with a standard deviation are given for Mn centers with multiple Mn–O bonds. Complexes 5 and 6 have been reported previously.32, 33 These metrics are included here for comparison. The Mn labels for 6 are equivalent to those given in ref. 32, Figure 2c.
The four manganese centers in 2, 3, 5, 6, and 7 form a tetrahedron with the pseudo-C3 axis of the ligand architecture coincident with one of the C3 axes of the tetrahedron. Complex 4 is more asymmetric, with the top manganese leaning off-axis to accommodate the MnIII2O2 diamond core. It is remarkable that the four manganese centers hold the same relative geometry in four different oxidation states (eight including electrochemically observed species; see below and SI) and the incorporation of four oxides. This tetrahedron contracts as the oxido content increases: this translates into shorter average metal-oxido and metal-metal distances (Table 1). For example, the average Mn–Mn distances decreases from 3.32±0.21 Å in 3 to 2.92±0.10 Å in 6.
2.3 XAS
Mn XANES
Mn K-edge X-ray absorption near-edge spectroscopy (XANES) was used to further characterize the metal oxidation states for complexes 3–6. In addition to the oxidation state, XANES is also sensitive to the coordination environment surrounding the metal site.54 Figure 2 compares the Mn XANES spectra of these four complexes with the spectrum of the OEC in the S1-state. Although the spectral features may be influenced by the variation in geometry and the number of oxido ligands, the absorption energy shifts and edge shapes are consistent with the Mn oxidation state assignments summarized in Table 1. There is a clear trend of the rising edge position shifting to higher energy with increasing Mn oxidation.
Fig. 2.

Normalized Mn K-edge XANES spectra from complexes 3–6 compared with the spectrum from spinach PS II in the S1-state.
To determine the absorption edge positions for complexes 3–6, inflection-point energies (IPE) from second-derivative zero crossings were obtained. The following values are the calculated IPEs for the four complexes: 6547.7 ± 0.1 eV (3), 6548.0 ± 0.1 eV (4), 6549.7 ± 0.1 eV (5), and 6551.2 ± 0.1 eV (6). Previous studies on sets of model Mn complexes with similar ligands have shown that the IPE increases by 1–2 eV to higher energy upon Mn oxidation.55, 56 Changing from complexes 4 to 5 and 5 to 6 gives an observed IPE shift of ~1.5–1.7 eV to higher energy, providing support for Mn oxidation state increase. In contrast, the IPE shift is only ~0.3 eV between complexes 3 and 4, suggesting that no Mn-based oxidation occurs in this conversion. The small IPE shift observed here is likely due to the additional oxido ligand. The absorption edge energy position of complex 6 is closest to that for the S1-state of PS II, where the formal Mn oxidation state is assigned as MnIII2MnIV2 57–59 Taken together with the structural analysis and chemical reactivity of these four complexes, the Mn XANES results support the formal oxidation state assignments of 3–6 as MnII2MnIII2(3), MnII2 MnIII2(4), MnIII4 (5), and MnIII2MnIV2 (6).
Mn EXAFS
Complexes 3–6 have also been characterized by Mn K-edge extended X-ray absorption fine structure (EXAFS), and the detailed curve-fitting parameters are summarized in Table S1. In general, the EXAFS results for these four complexes agree well with the solid-state structure distances reported in Table 1 (See also Table S1; See SI for discussion of the Mn EXAFS results).
2.4 Magnetism
Magnetic susceptibility measurements were performed on powdered crystalline samples of 3–6 in the temperature range 4–300 K. At room temperature, the χMT values approach 10.3, 10.5, 5.0, and 6.0 cm3 K mol−1, respectively (Fig. 3). All are below the spin-only values at 300 K and decrease as temperature decreases, indicative of dominant antiferromagnetic coupling. Antiferromagnetically coupled manganese clusters in the literature have also shown significant deviation at 300 K from the expected spin-only value.60–64
Fig. 3.
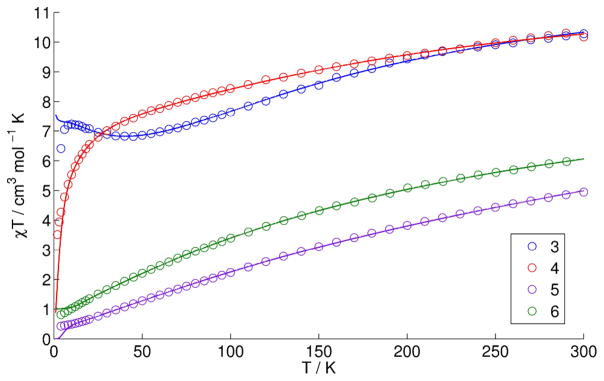
χMT vs. T data (circles) and fits (lines) for compounds 3–6. See Table 2 for fit parameters.
For 3, the χMT value decreases to 6.8 cm3 mol−1 K at 45 K and then increases to a maximum value of 7.2 at 10 K before dropping again, likely due to low temperature effects such as intermolecular exchange interactions. Upon cooling, the χMT value of 4 decreases gradually and then drops sharply below 30 K and does not approach an obvious limiting value at 4 K. The χMT value of 5 and 6 decrease gradually to 0.4 and 0.8 cm3 mol−1 K at 4 K, respectively.
To determine the magnitude of exchange between neighboring metal centers in 3, 4, and 6, the magnetic behavior of the compounds was analyzed using the isotropic spin Hamiltonian considering the three exchange pathways of an asymmetric tetrahedron (See SI for coupling scheme). Ideally, a four J coupling scheme would be used to model all of the different Mn-Mn interactions; however, to determine the eigenvalues using the Kambe65 method, we modeled the four MnIIMnIII interactions in each of 3 and 4 with one J (Jnm in Table 2). Similarly, the four MnIIIMnIV interactions in 6 were modeled with one coupling constant. The J values for all coupling pathways are relatively small (<25 cm−1, ca. 0.07 kcal/mol) with antiferromagnetic values greater than ferromagnetic values, consistent with the decreasing χMT values.
Table 2.
Magnetic susceptibility fitting parameters
| Compound | 3 | 4 | 6 | 5 |
|---|---|---|---|---|
| Diamagnetic Correction (×10−6 cm3/mol) | −777 | −681 | −722 | −691 |
| Jnma (cm−1) | −1.97 | +0.91 | −13.9 | JTOPb= −30.7 |
| Jnn (cm−1) | −9.90 | −0.28 | −6.3 | JBOT = −39.3 |
| Jmm (cm−1) | +3.61 | −22.9 | −18.2 | -- |
| g | 1.87 | 1.81 | 2.02 | 2.01 |
| R (× 10−5) | 1.9 | 1.4 | 1.6 | 4.1 |
n and m refer to the oxidation states of the Mn centers whose interaction the coupling constant is representing. n=2 and m=3 for complexes 3 and 4, whereas n=3 and m=4 for 6.
JTOP describes to the three interactions of the six-coordinate MnIII center with the five-coordinate MnIII centers in 5. JBOT describes the interactions between the three five-coordinate MnIII centers.
The exchange parameters of 5 were analyzed using a two exchange pathway model based on the psuedo-C3 symmetry of 5 (See SI).66 Both types of MnIIIMnIII interactions were antiferromagnetic, with couplings of −39.3 cm−1 between the three, basal five-coordinate Mn and −30.7 cm−1 between the top, six-coordinate Mn and the basal, five-coordinate Mn. These values are similar to MnIII2(μ3-O)2 systems with similar Mn-O-Mn angles.34
2.5 EPR
X-band continuous-wave (CW) EPR studies were performed on frozen solution samples of complexes 3–7 (Figure 4). All species explored exhibit significant temperature-dependent lineshape changes. These spectral changes are diagnostic of exchange-coupled spin systems with (at least some) rather small exchange coupling constants (i.e. on the order of kBT), consistent with magnetic susceptibility results (Table 2). That the overall integrated intensity of the EPR spectrum increases with increasing temperature also indicates that anti-ferromagnetic couplings, in particular, are dominant. At higher temperatures, states with larger S-values are populated and the transition between spin levels of these states have larger transitions probabilities giving rise to increased signal intensity.
Fig. 4.

Temperature-dependent (see legends for temperatures employed) CW X-band EPR spectra of the frozen solutions of (from top to bottom) 3(dissolved in 1:1 dichloromethane/acetonitrile), 4 (dissolved in pure dichloromethane), 5 – 6 (dissolved in 1:1 dichloromethane/toluene), and 7 (dissolved in pure toluene). Experimental parameters: microwave frequency = 9.33 – 9.37 GHz; power = 2.0 mW for all complexes except power = 1.0 mW for complex 3, 0.02 mW for complex 4, and 0.5 mW for complex 7; modulation amplitude = 10 G; modulation frequency = 100 kHz. Data for complex 6 are staggered for ease of comparison. The signal from a small amount of contaminating mononuclear MnII was subtracted from the data for complex 7.
Each of the complexes 3–7 is expected to have an even number of unpaired electrons and, in the case of maximal antiferromagnetic coupling, we would expect an ST = 0 ground state. Nonetheless, all of the complexes give rise to EPR signals owing to thermal population of paramagnetic excited states that are quite low in energy. In addition, exchange-coupled systems with three or more spin centers can exhibit “spin-frustration” in which all pair-wise couplings that are antiferromagnetic (i.e. negative J-value) are not achievable. This leads to ground state total spin quantum numbers that are greater than zero.
In the example of complex 3, neglecting any contribution to the eigenvalue from zero-field splitting, using the J-values in Table 2 predicts that the ground state is S = 3 with an S = 4 excited state approximately 0.2 cm−1 higher in energy at zero field. These manifolds are roughly equally populated at 5 K. The next excited state—predicted to be an S = 4 manifold—is 13.3 cm−1 higher in energy and should be appreciably populated at 15–20 K. This state could give rise to the temperature-dependent signals discussed next. The 5 K CW EPR spectrum of 3 contains weak signals at low field. As the temperature is increased up to 25 K, a negative feature becomes more prominent at g = 15.2 that then starts to diminish at 40 K. A corresponding feature appears in the parallel-mode spectrum (data not shown). These two properties confirm the formulation of 3 as being an integer spin system with a large value for the ground state S.
The 5 K EPR spectrum of 4 is weak and broad with two clear resonances at g = 7.4 and 2.9 that become slightly broader as the temperature is raised until 50 K when they begin to disappear. The g = 7.4 feature also has a corresponding negative peak in the parallel-mode spectrum (data not shown). The relatively large value for J3 pairs the spins of the two MnIII ions thus the lowest energy states of 4 consist of the 36 microstates of the exchange-coupled MnII ions.
A multiline feature containing 11 peaks centered at g = 1.98 and split by 9.5–12.9 mT is faintly visible at all temperatures explored. This signal is reminiscent of spectra for weakly coupled MnIII,IV dimers in which the exchange coupling is small compared to the zero-field splitting of the MnIII ion. The amount to which this signal contributed to the spectrum varied with different preparations of 4 leading to the conclusion that the corresponding species is likely a degradation product.
The spectrum of 5 collected at 5 K reveals two peaks at g = 7.57 and 4.95. The feature at g = 4.95 disappears quickly when the temperature is raised from 5 K whereas the feature at g = 7.57 exhibits Curie-type behavior (i.e. is proportional to 1/T). An additional feature appears at g = 2.5 at temperatures above 25 K. Overall, the spectrum of 5 is very reminiscent of that for a mononuclear MnIV center (cf. for example, the spectrum of MnIV 3,5-di-tert-butylquinone67). However, we interpret the marked increase in integrated spectral intensity with increasing temperature as being diagnostic of these resonances arising from an exchange-coupled system, not a mononuclear one.
Complex 6 yields a spectrum at 5 K that is somewhat reminiscent of the multiline signal corresponding to the S = 1/2 form of the S2 state of the [Mn4CaO5] cluster in PSII. Namely, there is a broad feature (spanning 250 mT) centered at g = 1.98; however the 55Mn hyperfine contributions are unresolved in the case of complex 6. As the temperature is increased from 5 K to 20 K the g = 1.98 feature grows slightly in intensity until 20 K and it begins to decrease in intensity and split into two peaks. That this feature is similar to the multiline signal for photosystem II could suggest that some of 6 became oxidized by one electron to give a MnIII,IV,IV,IV complex, the oxidation state scheme employed to rationalize the S2 EPR and X-ray absorption results. Alternatively, the spectrum can be rationalized as coming from a S = 1 or S = 2 state (these are the two lowest spin states for 6 based on the exchange couplings in Table 2, see SI) with a relatively small zero-field splitting (ZFS) parameter (D ~ 1000 and 600 MHz, respectively) and convolved by broad, unresolved 55Mn hyperfine features. Such small ZFS is generally unexpected for a MnIII-containing compound; however structural considerations provide a rationale. XRD data for 6 show a highly symmetric cube especially compared to complex 3, 4 or 5, and the Jahn-Teller (JT) axes of the two MnIII ions are perpendicular to each other. This orthogonality effectively cancels the contributions of the two MnIII site-specific ZFS tensors to the molecular ZFS tensor.68
The lowest-temperature EPR spectrum of complex 7 is fairly similar to that for 6, though a bit broader indicating a slightly larger ZFS for this lowest energy state. This behavior is not surprising given the modest difference in ligand set between the two species. However, there is a much more dramatic temperature dependence in which features at g = 2.16 and g =1.81 grow in as the temperature is raised.
2.6 Cluster Reactivity and Interconversion
The present series of clsuters of varying oxidation state and oxido content provides a unique opportunity to investigate cluster reactivity conceptually related to the PSII photoactivation. PSII binds four Mn centers, photooxidizing the Mn and incorporating oxido/hydroxido ligands to stabilize the increasing Mn oxidation states up through MnIV4. As shown in Scheme 1 by dashed arrows, oxidative water incorporation formally interconverts many of the complexes 2–6, and thus cluster interconversion was explored here to conceptually model these key steps of photoactivation and turnover. Complex 2 formally differs from 3 by one electron and a ligand exchange from trifluoromethanesulfonate to acetate. Complex 4 corresponds formally to water incorporation and double deprotonation from 3. Complex 5 is the product of a formal oxygen-atom addition to 4 – a process comprised of water incorporation, a two-electron oxidation, and double deprotonation. Complex 6 represents a similar oxygen-atom addition to 5.
The conversion of 3 to 2 involves a one-electron reduction, without change in the oxygen content of the cluster. Cyclic voltammetry studies of 3 in CH2Cl2 with 0.1 M [nBu4N][PF6] showed two irreversible reductions at −0.2 V and −1.0 V versus the Fc/Fc+ couple, suggesting the formation of MnII3MnIII and MnII4 species (Fc = ferrocene) (Figure S7). Indeed, if one equivalent of bis(pentamethylcyclopentadienyl)iron(II) was added to 3, a new species was formed that upon isolation, dissolution in CH3CN, and addition of excess [nBu4N][OAc] cleanly precipitated complex 2 (1H NMR; Figure S2).
Water incorporation/deprotonation was investigated for the conversion of 3 to 4. However, reaction of 3 with water in the presence of amines or hydroxide as bases generated complicated mixtures that did not contain 4 by 1H NMR spectroscopy. Cationic 3 is soluble in water, generating a green solution rather than purple-brown as observed in organic solvents. Removal of water in vacuo after thirty minutes and dissolution in CH3CN regenerated 3 (1H NMR) as a purple-brown solution. Water coordination likely occurs at the five-coordinate apical metal center rather than the six-coordinate, basal metal centers. The weakly coordinating OTf− anion might be displaced by the incoming Lewis base; if water binds at this axial position, isomerization to the equatorial position and displacement of a κ2-acetate must occur before bridging to the basal metal centers as in 4. The sterically open apical manganese could be prone to oligomerization by hydroxide or acetate bridges, precluding conversion to 4.
Oxygen-atom transfer in the context of cluster reorganization was studied for the conversion of 4 to 5. Complex 5 was successfully generated from the reaction of 4 with PhIO, albeit as a minor species in a mixture of products. Reaction of 4 with excess PhIO leads to the generation of complex 6 in low yield. The conversion of 4 to 5 is remarkable given the extent of reorganization that the cluster has to undergo due to the change in the binding mode of the ligand L. The generation of a mixture of products is consistent with an incomplete reorganization leading to unidentified species. The observed conversion of 4 to 6 corresponds to the addition of two oxygen atoms, analogous to incorporation and full reduction of one equivalent of dioxygen. Correspondingly, reaction of a dilute solution of 4 in DMF with one atmosphere of O2 leads to the generation of complex 6 over thirteen days (Figure S5). This one-step conversion is notable because it involves the reduction of O2 to two O2− moieties by a tetramanganese site, which represents the microscopic reverse of the O–O bond forming reaction performed by the OEC. The low oxidation states of manganese in precursor 4 allow for the reaction to proceed in the O2-reduction direction.
Oxido-ligand incorporation into partial cubane 5 to form 6 was reported previously,33 with detailed mechanistic studies performed. For both oxidative water incorporation and reductive oxygen-atom transfer, μ3-oxido migration within the Mn4 cluster was found to be a key mechanistic step, with implications for the structure and turnover of the OEC.
2.7 Electrochemistry and Potential Leveling
The redox properties of 5, 6, and 7 were studied by cyclic voltammetry (Figure 5). Although voltammograms of 5 and 6 have been reported,33 no comparative discussion was presented in the context of potential leveling and are thus included here alongside complex 7. Complex 5 irreversibly reduces to a proposed MnIIMnIII3 oxidation state ca. −1.0 V vs ferrocene/ferrocenium (Fc/Fc+) in THF with a coupled return oxidation and oxidizes quasireversibly at +100 mV vs Fc/Fc+ to the MnIII3MnIV oxidation state. In contrast, 6 reduces to the MnIII3MnIV state at −870 mV in THF. This oxidation state is ca. 1 V more negative in 6 because of the presence of the fourth oxide versus the three oxides of 5. An oxidation of 6 was also observed at +250 mV in THF, proposed to correspond to the MnIII2MnIV2/MnIII3MnIV couple. Complex 7, a cationic rather than neutral MnIII2MnIV2 complex, quasireversibly oxidizes at +340 mV in THF and irreversibly reduces at −510 mV. The oxidation event varies little (ca. 90 mV) in potential from 6 despite the buildup of charge at the now dicationic complex. Although the product of chemical oxidation has not yet been fully characterized, the electrochemical oxidation of 6 and 7 indicates that the MnIIIMnIV3 species is accessible. Only one MnIIIMnIV3O4 cubane has been previously reported.69, 70
Fig. 5.

Redox properties of 5–7. Cyclic voltammograms of 5 (bottom), 6 (middle) and 7 (top) referenced to Fc/Fc+. The scan rate was 50 mV/s in the positive direction. The analyte concentration was 1.0 mM. The electrolyte was 0.1M [nBu4N][PF6] in THF. Open-circuit potential for 5 = − 460 mV, for 6 = − 200 mV, and for 7 = −50 mV. E1/2 values: 5+/5 = +0.10 V; 5/5− = −1.0 V; 6+/6 = +0.25 V; 6/6− = −0.87 V; 7/7+ = +0.34 V; EC of 7/7− = −0.51 V.
The reduction and oxidation events of 5 parallel those of 6, with the reductions only differing by ca. 130 mV and the oxidations by 150 mV (Fig. 5, Scheme 2). With only a slight increase in potential, a MnIIIMnIV3 oxidation state can be accessed in 6 where only a MnIII3MnIV oxidation state is accessible in 5. This can be explained by the neutralization of charge buildup on the cluster by incorporation of an O2− donor (Scheme 2). The water incorporated into 5 to give 6 is deprotonated twice, allowing access to oxidation states two units more oxidized at close to the same potential. Neutralization of charge buildup has been demonstrated for a dimanganese catalase model system with a terminal water/hydroxide.71 Notably, that system was able to span four oxidation states – MnII2 through MnIIIMnIV – with little change in the chelating ligand framework as observed herein.72, 73 This redox leveling of the cluster upon formal water incorporation and deprotonation is relevant to the OEC, as the oxidizing equivalents come at the same potential for all four oxidations during catalysis to generate O2.
Scheme 2.
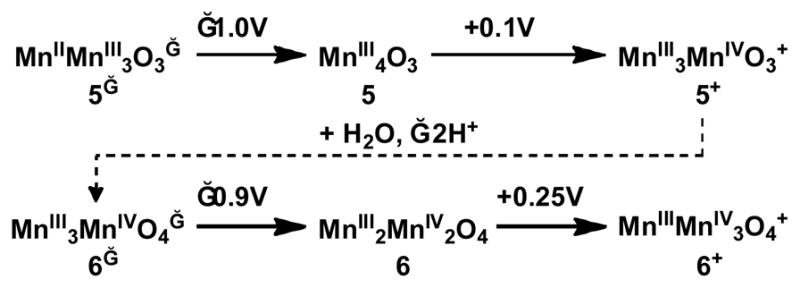
Effect of oxidation state and oxido ligand content on leveling of reduction potentials for 5 and 6.
2.8 Ligand Flexibility as Design Element
The propensity of the present supporting multinucleating ligand to allow for different binding modes is instrumental for supporting the wide span of metal oxidation states and oxido content. The lability of the dipyridylalkoxymethyl moiety is well documented in the coordination chemistry of dipyridylketone and the gem-diol or hemiacetal form thereof, which chelate and bridge metal ions in a wide variety of binding modes.74 Bridging three dipyridylalkoxymethyl units through a triarylbenzene scaffold therefore provides rich possibilities for coordination that benefit the formation of complexes 2 through 7. Clusters rich in labile MnII are coordinated by nine donors from L, binding to twelve coordination sites (counting three μ-alkoxides) while the higher oxidation state species, displaying MnIII and MnIV, require only six donors (Fig. 6). The switch in coordination mode is likely due to the strong Mn-oxido bonds that lead to the displacement of the pyridine and μ2-alkoxide donors. The three acetates from precursor 1 complete the metal coordination spheres by bridging the Mn3(μ-OR)3 core of 2–4 to the fourth manganese and the three diamond core motifs in 5–7. The versatility of carboxylate ligation in manganese cluster chemistry has also been documented.75 The present compounds show conservation of the ancillary ligands (L and acetates) over a large set of oxidation states and oxide contents, indicating that donor flexibility is an important factor in the design of ligands for clusters in multielectron chemistry involving transfers of oxygenous moieties.
Fig. 6.
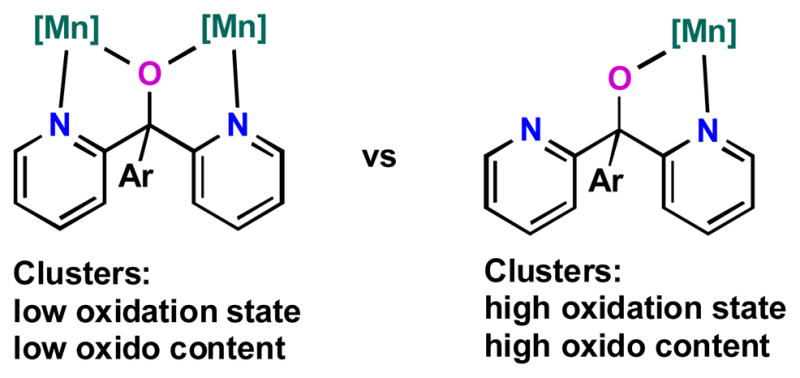
Ligand flexibility as function of cluster oxido content and oxidation state: Binding modes of dipyridylalkoxide arms in clusters 1–7
2.9 Relation to the Assembly and Turnover of the OEC; Design Implications for Oxidized Metal-Oxide Clusters
PSII stabilizes four manganese centers through nine oxidation states. The present compounds comprise eight oxidation states from MnII4 to MnIIIMnIV3, mimicking states from the beginning of photoactivation, a hypothetical S−5, through to S2. As in PSII, the higher oxidation states are stabilized by incorporation of strong oxido ligands. Alongside three acetates, the semilabile ligand framework L is conserved throughout the series with a decrease in the number of coordinating donors from L. The type of change in coordination observed here might be relevant to the assembly of the OEC in PSII. The early biochemical intermediates in photoactivation are reported to proceed to fully assembled OEC in low yields relative to light absorption.20, 76, 77 Additional nitrogen and oxygen donors are present in the protein close to the active site, such as His337 and Asp61. These do not coordinate to the fully assembled cluster, but have been proposed to be important in binding the metal centers in the low oxidation state intermediates of photoactivation (< S0),16 although Asp170 has been shown to coordinate to the first Mn2+ center during assembly and remains coordinated to the full OEC.18, 78 A slow kinetic step after the initial binding and photooxidation of Mn2+ has been proposed to be a protein conformational change, which may involve ligand rearrangements.20, 77
The charge neutralization demonstrated by 5 and 6 conceptually mimics how the OEC can access high oxidation states. In PSII, the oxidizing equivalents, all with the same potential, are provided by the photooxidized chlorophyll P680 via a tyrosine Yz•.79–81,71,82 Therefore, as the OEC is oxidized during turnover, water is incorporated and deprotonated, neutralizing the positive charge built up from oxidation and facilitating access to high oxidation state Mn. Complexes 5 and 6 show that charge neutralization manifests in high oxidation state, tetramanganese-oxido synthetic models of the OEC.
Beyond the assembly of the OEC, the present series of compounds suggests a rational strategy for the synthesis of high oxidation state clusters from reduced precursors. Low oxidation state clusters support fewer oxido ligands due to negative charge buildup. Consequently, additional donors from the multinucleating ligands are necessary to satisfy the coordination sphere of the metal and avoid oligomerization. Upon oxidation, water incorporation and deprotonation, the clusters include additional oxido moieties. These moieties facilitate further oxidation. Increased number of oxido moieties require some supporting ligand dissociation to maintain similar coordination numbers. Labile pendant donors, such as the pyridines in the present case, facilitate not only the isolation of the reduced clusters, but also provide coordination flexibility to support the higher oxidation state, higher oxido content clusters.
3. Conclusions
In summary, a series of tetramanganese complexes of variable oxido-content (one through four) and oxidation state (MnII4 through MnIIIMnIV3) has been obtained. Six clusters in four oxidation states were crystallographically characterized, and four further redox events were accessed electrochemically. XANES data support the assigned oxidation states, and EXAFS were consistent with XRD. Magnetism and EPR studies were performed to elucidate the electronic structures. These complexes conceptually mimic the wide range of tetramanganese-oxido species in photoactivation and the Kok cycle of the OEC in PSII. Cluster interconversion was achieved in several cases. These transformations show that the coordination environment around the metal centers changes as a function of the number of oxido moieties due to their propensity to bridge and form strong bonds. Similar to the OEC active site, several linked donors in a multinucleating ancillary ligand support the four-manganese cluster over a broad range of oxidation states and oxido content. Ligand lability was found instrumental for accommodating the increased number of oxido moieties. The presence of labile pendant donor groups is a design feature that is expected to apply generaly to the rational synthesis of metal-oxido clusters from reduced precursors.
Supplementary Material
Acknowledgments
We are grateful to California Institute of Technology, the Searle Scholars Program, an NSF CAREER (CHE-1151918) award (TA), the Rose Hill Foundation, and the NSF GRFP (J.S.K.) for funding. We thank L. M. Henling for assistance with crystallography. The Bruker KAPPA APEXII X-ray diffractometer was purchased via an NSF CRIF:MU award to Caltech (CHE-0639094). We acknowledge the Gordon and Betty Moore Foundation, the Beckman Institute, and the Sanofi-Aventis BRP for their support of the Molecular Observatory at Caltech. The work carried out by RDB was funded by the Division of Chemical Sciences, Geosciences, and Biosciences, Office of Basic Energy Sciences of the U.S. Department of Energy (DE-FG02-11ER16282). SSRL is operated by Stanford University for the DOE and supported by its Office of Biological and Environmental Research, and by the NIH, NIGMS (including P41GM103393) and the NCRR (P41RR001209). X-ray spectroscopy studies were supported by the NIH (F32GM100595 to R.T.) and by the Director of the Office of Basic Energy Science, Division of Chemical Sciences, Geosciences, and Biosciences, DOE (DE-AC02-05CH11231 to J.Y.).
Footnotes
Crystallographic data have been deposited at the Cambridge Crystallographic Database Centre, and copies can be obtained on request, free of charge, by quoting the publication citation and the deposition numbers 858642(2) 858643(3) 842512(4) 938750(7). Electronic Supplementary Information (ESI) available: Synthetic procedures, NMR spectroscopy, cyclic voltammetry, spectroscopic methods and crystallographic details. See DOI: 10.1039/b000000x/
Notes and references
- 1.McEvoy JP, Brudvig GW. Chem Rev. 2006;106:4455–4483. doi: 10.1021/cr0204294. [DOI] [PubMed] [Google Scholar]
- 2.Joliot P. Biochim Biophys Acta. 1965;102:116–134. [PubMed] [Google Scholar]
- 3.Kok B, Forbush B, Mcgloin M. Photochem Photobiol. 1970;11:457–475. doi: 10.1111/j.1751-1097.1970.tb06017.x. [DOI] [PubMed] [Google Scholar]
- 4.Kolling DRJ, Cox N, Ananyev GM, Pace RJ, Dismukes GC. Biophys J. 2012;103:313–322. doi: 10.1016/j.bpj.2012.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pecoraro VL, Baldwin MJ, Caudle MT, Hsieh WY, Law NA. Pure Appl Chem. 1998;70:925–929. [Google Scholar]
- 6.Pecoraro VL, Hsieh WY. In: Manganese and its Role in Biological Systems. Sigel A, Sigel H, editors. Marcel Dekker, Inc; New York: 2000. pp. 429–504. [Google Scholar]
- 7.Sproviero EM, Gascon JA, McEvoy JP, Brudvig GW, Batista VS. J Am Chem Soc. 2008;130:3428–3442. doi: 10.1021/ja076130q. [DOI] [PubMed] [Google Scholar]
- 8.Chow WS, Aro EM. In: The Light-Driven Water: Plastoquinone Oxidoreductase. Wydrzynski TJ, Satoh K, editors. Springer; Dordrecht: 2005. pp. 627–648. [Google Scholar]
- 9.Cheniae GM, Martin IF. Biochem Bioph Res Co. 1967;28:89–95. doi: 10.1016/0006-291x(67)90411-1. [DOI] [PubMed] [Google Scholar]
- 10.Miller AF, Brudvig GW. Biochemistry. 1989;28:8181–8190. doi: 10.1021/bi00446a033. [DOI] [PubMed] [Google Scholar]
- 11.Miller AF, Brudvig GW. Biochemistry. 1990;29:1385–1392. doi: 10.1021/bi00458a007. [DOI] [PubMed] [Google Scholar]
- 12.Burnap RL. Phys Chem Chem Phys. 2004;6:4803–4809. [Google Scholar]
- 13.Shevela D, Su JH, Klimov V, Messinger J. BBA-Bioenergetics. 2008;1777:532–539. doi: 10.1016/j.bbabio.2008.03.031. [DOI] [PubMed] [Google Scholar]
- 14.Ulas G, Olack G, Brudvig GW. Biochemistry. 2008;47:3073–3075. doi: 10.1021/bi8000424. [DOI] [PubMed] [Google Scholar]
- 15.Baranov SV, Tyryshkin AM, Katz D, Dismukes GC, Ananyev GM, Klimov VV. Biochemistry. 2004;43:2070–2079. doi: 10.1021/bi034858n. [DOI] [PubMed] [Google Scholar]
- 16.Dasgupta J, Tyryshkin AM, Dismukes GC. Angew Chem Int Ed. 2007;46:8028–8031. doi: 10.1002/anie.200702347. [DOI] [PubMed] [Google Scholar]
- 17.Dasgupta J, Tyryshkin AM, Baranov SV, Dismukes GC. Appl Magn Reson. 2010;37:137–150. [Google Scholar]
- 18.Campbell KA, Force DA, Nixon PJ, Dole F, Diner BA, Britt RD. J Am Chem Soc. 2000;122:3754–3761. [Google Scholar]
- 19.Tyryshkin AM, Watt RK, Baranov SV, Dasgupta J, Hendrich MP, Dismukes GC. Biochemistry. 2006;45:12876–12889. doi: 10.1021/bi061495t. [DOI] [PubMed] [Google Scholar]
- 20.Zaltsman L, Ananyev GM, Bruntrager E, Dismukes GC. Biochemistry. 1997;36:8914–8922. doi: 10.1021/bi970187f. [DOI] [PubMed] [Google Scholar]
- 21.Dasgupta J, Ananyev GM, Dismukes GC. Coord Chem Rev. 2008;252:347–360. doi: 10.1016/j.ccr.2007.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ananyev GM, Dismukes GC. Biochemistry. 1997;36:11342–11350. doi: 10.1021/bi970626a. [DOI] [PubMed] [Google Scholar]
- 23.Brudvig GW, Beck WF. In: Manganese Redox Enzymes. Pecoraro VL, editor. VCH Publishers, Inc; New York: 1992. pp. 119–140. [Google Scholar]
- 24.Beck WF, Brudvig GW. Biochemistry. 1987;26:8285–8295. doi: 10.1021/bi00399a040. [DOI] [PubMed] [Google Scholar]
- 25.Schansker G, Goussias C, Petrouleas V, Rutherford AW. Biochemistry. 2002;41:3057–3064. doi: 10.1021/bi015903z. [DOI] [PubMed] [Google Scholar]
- 26.Messinger J, Seaton G, Wydrzynski T, Wacker U, Renger G. Biochemistry. 1997;36:6862–6873. doi: 10.1021/bi962653r. [DOI] [PubMed] [Google Scholar]
- 27.Mukhopadhyay S, Mandal SK, Bhaduri S, Armstrong WH. Chem Rev. 2004;104:3981–4026. doi: 10.1021/cr0206014. [DOI] [PubMed] [Google Scholar]
- 28.Mullins CS, Pecoraro VL. Coord Chem Rev. 2008;252:416–443. doi: 10.1016/j.ccr.2007.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mukherjee S, Stull JA, Yano J, Stamatatos TC, Pringouri K, Stich TA, Abboud KA, Britt RD, Yachandra VK, Christou G. Proc Natl Acad Sci USA. 2012;109:2257–2262. doi: 10.1073/pnas.1115290109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tsui EY, Day MW, Agapie T. Angew Chem Int Ed. 2011;50:1668–1672. doi: 10.1002/anie.201005232. [DOI] [PubMed] [Google Scholar]
- 31.Tsui EY, Kanady JS, Day MW, Agapie T. Chem Commun. 2011;47:4189–4191. doi: 10.1039/c0cc05608a. [DOI] [PubMed] [Google Scholar]
- 32.Kanady JS, Tsui EY, Day MW, Agapie T. Science. 2011;333:733–736. doi: 10.1126/science.1206036. [DOI] [PubMed] [Google Scholar]
- 33.Kanady JS, Mendoza-Cortes JL, Tsui EY, Nielson RJ, Goddard WA, Agapie T. J Am Chem Soc. 2013;135:1073–1082. doi: 10.1021/ja310022p. [DOI] [PubMed] [Google Scholar]
- 34.Vincent JB, Christmas C, Chang HR, Li QY, Boyd PDW, Huffman JC, Hendrickson DN, Christou G. J Am Chem Soc. 1989;111:2086–2097. [Google Scholar]
- 35.Kulawiec RJ, Crabtree RH, Brudvig GW, Schulte GK. Inorg Chem. 1988;27:1309–1311. [Google Scholar]
- 36.Philouze C, Blondin G, Girerd JJ, Guilhem J, Pascard C, Lexa D. J Am Chem Soc. 1994;116:8557–8565. [Google Scholar]
- 37.Bashkin JS, Chang HR, Streib WE, Huffman JC, Hendrickson DN, Christou G. J Am Chem Soc. 1987;109:6502–6504. [Google Scholar]
- 38.Ruettinger W, Campana C, Dismukes G. J Am Chem Soc. 1997:6670–6671. [Google Scholar]
- 39.Wieghardt K, Bossek U, Gebert W. Angew Chem Int Ed. 1983;22:328–329. [Google Scholar]
- 40.Chan MK, Armstrong WH. J Am Chem Soc. 1991;113:5055–5057. [Google Scholar]
- 41.Chen HY, Faller JW, Crabtree RH, Brudvig GW. J Am Chem Soc. 2004;126:7345–7349. doi: 10.1021/ja037389l. [DOI] [PubMed] [Google Scholar]
- 42.Wang SY, Tsai HL, Hagen KS, Hendrickson DN, Christou G. J Am Chem Soc. 1994;116:8376–8377. [Google Scholar]
- 43.Aromi G, Wemple MW, Aubin SJ, Folting K, Hendrickson DN, Christou G. J Am Chem Soc. 1998;120:5850–5851. [Google Scholar]
- 44.Mckee V, Tandon SS. J Chem Soc Chem Comm. 1988:1334–1336. [Google Scholar]
- 45.Beagley B, Mcauliffe CA, Macrory PP, Ndifon PT, Pritchard RG. J Chem Soc Chem Comm. 1990:309–310. [Google Scholar]
- 46.Gallo E, Solari E, Deangelis S, Floriani C, Re N, Chiesivilla A, Rizzoli C. J Am Chem Soc. 1993;115:9850–9851. [Google Scholar]
- 47.Cotton FA, Daniels LM, Falvello LR, Matonic JH, Murillo CA, Wang X, Zhou H. Inorg Chim Acta. 1997;266:91–102. [Google Scholar]
- 48.Cotton FA, Daniels LM, Jordan GT, Murillo CA, Pascual I. Inorg Chim Acta. 2000;297:6–10. [Google Scholar]
- 49.Millos CJ, Piligkos S, Bell AR, Laye RH, Teat SJ, Vicente R, McInnes E, Escuer A, Perlepes SP, Winpenny REP. Inorg Chem Commun. 2006;9:638–641. [Google Scholar]
- 50.Yang CI, Wernsdorfer W, Tsai YJ, Chung G, Kuo TS, Lee GH, Shieh M, Tsai HL. Inorg Chem. 2008;47:1925–1939. doi: 10.1021/ic7013368. [DOI] [PubMed] [Google Scholar]
- 51.Zaleski CM, Weng TC, Dendrinou-Samara C, Alexiou M, Kanakaraki P, Hsieh WY, Kampf J, Penner-Hahn JE, Pecoraro VL, Kessissoglou DP. Inorg Chem. 2008;47:6127–6136. doi: 10.1021/ic702109c. [DOI] [PubMed] [Google Scholar]
- 52.Mikuriya M, Yamato Y, Tokii T. Chem Lett. 1991:1429–1432. [Google Scholar]
- 53.Sanudo EC, Grillo VA, Knapp MJ, Bollinger JC, Huffman JC, Hendrickson DN, Christou G. Inorg Chem. 2002;41:2441–2450. doi: 10.1021/ic011262k. [DOI] [PubMed] [Google Scholar]
- 54.Yano J, Yachandra VK. Photosynth Res. 2009;102:241–254. doi: 10.1007/s11120-009-9473-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Visser H, Anxolabehere-Mallart E, Bergmann U, Glatzel P, Robblee JH, Cramer SP, Girerd JJ, Sauer K, Klein MP, Yachandra VK. J Am Chem Soc. 2001;123:7031–7039. doi: 10.1021/ja004306h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pizarro SA, Glatzel P, Visser H, Robblee JH, Christou G, Bergmann U, Yachandra VK. Phys Chem Chem Phys. 2004;6:4864–4870. doi: 10.1039/B407513G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yachandra VK, Sauer K, Klein MP. Chem Rev. 1996;96:2927–2950. doi: 10.1021/cr950052k. [DOI] [PubMed] [Google Scholar]
- 58.Messinger J, Robblee JH, Bergmann U, Fernandez C, Glatzel P, Visser H, Cinco RM, McFarlane KL, Bellacchio E, Pizarro SA, Cramer SP, Sauer K, Klein MP, Yachandra VK. J Am Chem Soc. 2001;123:7804–7820. doi: 10.1021/ja004307+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Glatzel P, Bergmann U, Yano J, Visser H, Robblee JH, Gu WW, de Groot FMF, Christou G, Pecoraro VL, Cramer SP, Yachandra VK. J Am Chem Soc. 2004;126:9946–9959. doi: 10.1021/ja038579z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wemple MW, Tsai HL, Wang SY, Claude JP, Streib WE, Huffman JC, Hendrickson DN, Christou G. Inorg Chem. 1996;35:6437–6449. doi: 10.1021/ic9603013. [DOI] [PubMed] [Google Scholar]
- 61.Stoumpos CC, Gass IA, Milios CJ, Lalioti N, Terzis A, Aromi G, Teat SJ, Brechin EK, Perlepes SP. Dalton Trans. 2009:307–317. doi: 10.1039/b810835h. [DOI] [PubMed] [Google Scholar]
- 62.Stoumpos CC, Stamatatos TC, Sartzi H, Roubeau O, Tasiopoulos AJ, Nastopoulos V, Teat SJ, Christou G, Perlepes SP. Dalton Trans. 2009:1004–1015. doi: 10.1039/b813828a. [DOI] [PubMed] [Google Scholar]
- 63.Yang PP, Song XY, Liu RN, Li LC, Liao DZ. Dalton Trans. 2010;39:6285–6294. doi: 10.1039/c0dt00291g. [DOI] [PubMed] [Google Scholar]
- 64.Yang PP, Li LC. Inorg Chim Acta. 2011;371:95–99. [Google Scholar]
- 65.Kambe K. J Phys Soc Jap. 1950;5:48–51. [Google Scholar]
- 66.Hendrickson DN, Christou G, Schmitt EA, Libby E, Bashkin JS, Wang SY, Tsai HL, Vincent JB, Boyd PDW, Huffman JC, Folting K, Li Q, Streib WE. J Am Chem Soc. 1992;114:2455–2471. [Google Scholar]
- 67.Lynch MW, Hendrickson DN, Fitzgerald BJ, Pierpont CG. J Am Chem Soc. 1984;106:2041–2049. [Google Scholar]
- 68.Stamatatos TC, Christou G. Philos Tran Royal Soc A. 2008;366:113–125. doi: 10.1098/rsta.2007.2144. [DOI] [PubMed] [Google Scholar]
- 69.Ruettinger W, Ho D, Dismukes G. Inorg Chem. 1999:1036–1037. doi: 10.1021/ic981145y. [DOI] [PubMed] [Google Scholar]
- 70.Brimblecombe R, Bond AM, Dismukes GC, Swiegers GF, Spiccia L. Phys Chem Chem Phys. 2009;11:6441–6449. doi: 10.1039/b901419e. [DOI] [PubMed] [Google Scholar]
- 71.Caudle MT, Pecoraro VL. J Am Chem Soc. 1997;119:3415–3416. [Google Scholar]
- 72.Gelasco A, Kirk ML, Kampf JW, Pecoraro VL. Inorg Chem. 1997;36:1829–1837. doi: 10.1021/ic970140i. [DOI] [PubMed] [Google Scholar]
- 73.Gelasco A, Bensiek S, Pecoraro VL. Inorg Chem. 1998;37:3301–3309. [Google Scholar]
- 74.Stamatatos TC, Efthymiou CG, Stoumpos CC, Perlepes SP. Eur J Inorg Chem. 2009:3361–3391. [Google Scholar]
- 75.Christou G. Acc Chem Res. 1989;22:328–335. [Google Scholar]
- 76.Radmer R, Cheniae GM. Biochim Biophys Acta. 1971;253:182–186. doi: 10.1016/0005-2728(71)90243-x. [DOI] [PubMed] [Google Scholar]
- 77.Chen CG, Kazimir J, Cheniae GM. Biochemistry. 1995;34:13511–13526. doi: 10.1021/bi00041a031. [DOI] [PubMed] [Google Scholar]
- 78.Debus RJ, Aznar C, Campbell KA, Gregor W, Diner BA, Britt RD. Biochemistry. 2003;42:10600–10608. doi: 10.1021/bi034859f. [DOI] [PubMed] [Google Scholar]
- 79.Debus RJ, Barry BA, Sithole I, Babcock GT, Mcintosh L. Biochemistry. 1988;27:9071–9074. doi: 10.1021/bi00426a001. [DOI] [PubMed] [Google Scholar]
- 80.Metz JG, Nixon PJ, Rogner M, Brudvig GW, Diner BA. Biochemistry. 1989;28:6960–6969. doi: 10.1021/bi00443a028. [DOI] [PubMed] [Google Scholar]
- 81.Baldwin MJ, Pecoraro VL. J Am Chem Soc. 1996;118:11325–11326. [Google Scholar]
- 82.Diner BA, Britt RD. In: Photosystem II: The Light-Driven Water: Plastoquinone Oxidoreductase. Wydrzynski TJ, Satoh K, editors. Springer; Dordrecht: 2005. pp. 207–233. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.