

Glycosylated and nonglycosylated proteins misfolded in the ER are degraded by discrete but interchangeable pathways in the ERAD system. Disulfide reductase ERdj5 plays a central role in both pathways through the complex formation with EDEM and/or BiP. The nonglycoprotein ERAD pathway serves as a backup system under ER stress conditions.
Abstract
During endoplasmic reticulum (ER)–associated degradation (ERAD), terminally misfolded proteins are retrotranslocated from the ER to the cytosol and degraded by the ubiquitin-proteasome system. Misfolded glycoproteins are recognized by calnexin and transferred to EDEM1, followed by the ER disulfide reductase ERdj5 and the BiP complex. The mechanisms involved in ERAD of nonglycoproteins, however, are poorly understood. Here we show that nonglycoprotein substrates are captured by BiP and then transferred to ERdj5 without going through the calnexin/EDEM1 pathway; after cleavage of disulfide bonds by ERdj5, the nonglycoproteins are transferred to the ERAD scaffold protein SEL1L by the aid of BiP for dislocation into the cytosol. When glucose trimming of the N-glycan groups of the substrates is inhibited, glycoproteins are also targeted to the nonglycoprotein ERAD pathway. These results indicate that two distinct pathways for ERAD of glycoproteins and nonglycoproteins exist in mammalian cells, and these pathways are interchangeable under ER stress conditions.
INTRODUCTION
Proteins entering the secretory pathway are translocated across the endoplasmic reticulum (ER) membrane in an unfolded form. Cotranslational modifications, including N-linked glycosylation and formation of disulfide bonds, facilitate proper folding of nascent polypeptides in the ER. Proteins that are correctly folded and assembled exit the ER and are transported to their final destinations, whereas misfolded and unassembled proteins are retained in the ER through quality control mechanisms. Terminally misfolded proteins are retrotranslocated into the cytosol and subsequently degraded by the ubiquitin-proteasome system in a process called ER-associated degradation (ERAD; Ellgaard and Helenius, 2003; Hoseki et al., 2010; Ushioda and Nagata, 2011).
Productive folding of N-glycosylated proteins in the ER depends on the lectin-like molecular chaperones calnexin (CNX) and calreticulin, both of which recognize the monoglucose form of N-glycans and facilitate polypeptide folding. Mannose trimming plays a key role in targeting misfolded glycoproteins to the ERAD pathway. ER degradation-enhancing mannosidase-like protein 1 (EDEM1) receives terminally misfolded glycoproteins from CNX and promotes their degradation in a mannose trimming–dependent manner (Hosokawa et al., 2001); the precise molecular function of EDEM1 is controversial (Molinari et al., 2003; Oda et al., 2003; Olivari et al., 2006; Cormier et al., 2009). The lectin-like proteins OS9 and XTP3-B also recognize misfolded glycoproteins after mannose trimming and transfer them to the ERAD complex (Hosokawa et al., 2009; Bernasconi et al., 2010).
During productive folding of nascent polypeptides in the ER, many secretory and membrane-associated proteins acquire disulfide bonds that stabilize the tertiary structure of the molecules. The disulfide bond–linked three-dimensional structure of misfolded proteins, however, interferes with efficient transport of misfolded proteins through retrotranslocation channels. The EDEM1-binding protein ERdj5, which contains an N-terminal J domain and four thioredoxin (Trx)-like domains containing CXXC motifs, acts as a disulfide reductase in the oxidative environment of the ER (Ushioda et al., 2008; Hagiwara et al., 2011). Misfolded glycoproteins are transferred from CNX to EDEM1 and then to ERdj5, which cleaves disulfide bonds through the reductase activity of its C-terminal cluster (Hagiwara et al., 2011). The J domain of ERdj5 interacts with BiP in an ATP-dependent manner, and BiP may recruit substrates to the retrotranslocation channel after conversion of ATP to ADP (Ushioda et al., 2008; Hagiwara et al., 2011).
Components involved in ERAD of misfolded nonglycoproteins include BiP and the Herp complex, which includes p97, Hrd1, and Derlin-1 (Plemper et al., 1997; Nishikawa et al., 2001; Okuda-Shimizu and Hendershot, 2007; Otero et al., 2010). ERdj4 and ERdj5 also accelerate ERAD of nonglycosylated proteins (Dong et al., 2008). The mechanisms by which misfolded nonglycosylated proteins are recognized and recruited to the degradation machinery are unknown. Here we show that ERAD of glycoproteins and nonglycoproteins occurs via two distinct pathways and these pathways can be switched under conditions of ER stress.
RESULTS
ERdj5 promotes ERAD of nonglycosylated substrates
To elucidate the molecular mechanisms involved in the degradation of misfolded nonglycoproteins, we mutated the N-glycosylation sites of the null Hong Kong (NHK) variant of α1-antitrypsin (A1AT), which is a model ERAD substrate (Figure 1A). In the QQQ mutant, which is degraded through an EDEM-independent pathway (Ye et al., 2005; Cormier et al., 2009), the asparagine residues in the three N-glycosylation sites of NHK were mutated to glutamine. A QQQ/CS mutant, in which a cysteine residue in the QQQ mutant of NHK was mutated to serine, was also generated; this mutant was unable to form homodimers (Figure 1A). As shown in Figure 1B, pulse-chase analyses of HEK293T cells transfected with the QQQ and QQQ/CS mutants revealed that the degradation rate of the QQQ mutant (t1/2 ≈ 60 min) was faster than the previously reported degradation rate of wild-type NHK (t1/2 ≈ 100 min), which was consistent with previous reports (Hosokawa et al., 2001; Hirao et al., 2006; Ushioda et al., 2008). In addition, the degradation rate of the QQQ/CS mutant was much faster than that of the QQQ mutant (Figure 1B). Degradation of the QQQ mutant was markedly accelerated by overexpression of ERdj5 (Figure 1C), whereas degradation of the QQQ/CS mutant was not (Figure 1D). The level of overexpressed ERdj5 was around 10-fold higher than for the endogenous one (Supplemental Figure S1A), and those of overexpressed ERdj5 mutants were confirmed to be almost same as for wild-type ERdj5 (Supplemental Figure S1B). SDS–PAGE analyses detected the formation of mixed disulfide intermediates of ERdj5 bound to the QQQ mutant under nonreducing conditions (Figure 1E); however, these intermediates were not detected for the QQQ/CS mutant (data not shown). These results suggest that cleavage of intermolecular disulfide bonds is necessary for nonglycoprotein ERAD.
FIGURE 1:

ERdj5 accelerates ERAD of misfolded nonglycosylated proteins by cleaving their disulfide bonds. (A) Top, molecular features of wild-type, QQQ mutant, and QQQ/CS mutant NHK. The QQQ mutant lacked the three N-glycosylation sites. In the QQQ/CS mutant, a cysteine residue was replaced by serine. Bottom, nonreducing (NR) immunoblot analyses of HEK293T cells expressing wild-type and mutant NHK. (B) Twenty-four hours after transient transfection with expression plasmids containing the QQQ or QQQ/CS mutant, HEK293T cells were metabolically labeled with [35S]methionine/cysteine for 15 min and then chased for the indicated times. The QQQ and QQQ/CS mutants were immunoprecipitated with an anti-A1AT antibody. (C, D) Pulse-chase analyses of the effect of overexpression of ERdj5 on degradation of the QQQ (C) and QQQ/CS (D) mutants. (E) Twenty-four hours after the transfection of HEK293T cells with expression plasmids containing ERdj5 and the QQQ mutant, cell lysate supernatants were immunoprecipitated (IP) with an anti-FLAG antibody and then immunoblotted (IB) with anti-FLAG and anti-A1AT antibodies. NR, nonreducing condition; R, reducing condition. (F) Pulse-chase analysis of the effect of the overexpression of ERdj5/WT and ERdj5/AA on degradation of the QQQ mutant in transiently transfected HEK293T cells. (G) Effect of siRNA-induced knockdown of endogenous ERdj5 on QQQ ERAD. Pulse-chase experiments were performed 24 h after transfection of HEK293T cells with the QQQ mutant expression plasmid (72 h after transfection with either nonspecific [NS] or ERdj-5-specific siRNA). (B–D, F, G) Data represent the mean ± SD of n = 3 independent experiments.
Replacement of the cysteine residues in the four CXXC motifs of ERdj5 with alanines (ERdj5/AA) results in loss of reductase activity (Ushioda et al., 2008). When overexpressed in HEK293T cells, ERdj5/AA did not accelerate ERAD of the QQQ mutant, but instead showed a dominant-negative effect compared with control cells (Figure 1F). As expected, degradation of the QQQ mutant was delayed in cells transfected with ERdj5-specific siRNA (Figure 1G). Taken together, these results indicate that ERdj5 accelerates the degradation of nonglycosylated substrates by cleaving their disulfide bonds.
EDEM1 is not involved in recruitment of nonglycosylated proteins to ERdj5
We previously demonstrated that EDEM1 is necessary for glycoprotein ERAD (Hosokawa et al., 2001; Oda et al., 2003; Ushioda et al., 2008; Hagiwara et al., 2011). To examine whether EDEM1 is also required for nonglycoprotein ERAD, we identified the EDEM-binding region of ERdj5 by preparing domain-deletion mutants of ERdj5. We previously demonstrated that the Trx3 and Trx4 domains in the C-terminal clusters of ERdj5 are involved in both reductase activity and ERAD activation (Hagiwara et al., 2011); therefore we generated deletion mutants of the C-terminal region. When the Trx4 domain was deleted, hemagglutinin (HA)-tagged EDEM1 did not coimmunoprecipitate with FLAG-tagged ERdj5 (Figure 2A), whereas EDEM1 did coimmunoprecipitate with the isolated Trx4 domain (Figure 2B), indicating that the Trx4 domain of ERdj5 interacts with EDEM1.
FIGURE 2:

ERdj5 promotes ERAD of misfolded nonglycosylated proteins in an EDEM1-independent manner. (A, B) Twenty-four hours after transfection of HeLa cells with EDEM1-HA and the wild type (WT), ΔTrx4, ΔTrx34, or isolated Trx4 domain of ERdj5-FLAG, cell lysate supernatants were immunoprecipitated (IP) with an anti-FLAG antibody and then immunoblotted (IB) with anti-FLAG or anti-HA antibodies. (C, D) Twenty-four hours after transfection with NHK or the QQQ mutant, and the WT or Trx4 ERdj5, HEK293T cells were labeled for 15 min with [35S]methionine/cysteine and then chased for the indicated times. The labeled substrates were immunoprecipitated with an anti-A1AT antibody. Data represent the mean ± SD of n = 3 independent experiments.
Overexpression of the ΔTrx4 mutant in HEK293 cells did not accelerate degradation of NHK (Figure 2C) or the tyrosinase mutant Tyr/T373K, another glycoprotein substrate, which was previously shown to undergo ERAD (Supplemental Figure S2A; Toyofuku et al., 2001). The ERdj5/mC4 mutant, in which the CXXC motif in the Trx4 domain was replaced by AXXA, retained reductase activity because the Trx3 domain was intact in this mutant and was able to accelerate ERAD of NHK in a manner similar to that of wild-type ERdj5 (Supplemental Figure S3). This result suggests that the inability of the ΔTrx4 mutant to accelerate degradation of glycosylated NHK was due to a defect in EDEM binding rather than a loss of reductase activity. In contrast, ERdj5-ΔTrx4, EDEM-nonbinding mutant, facilitated ERAD of the nonglycosylated QQQ mutant in a manner similar to that of wild-type ERdj5 (Figure 2D). These results indicate that interaction of EDEM1 with the Trx4 domain of ERdj5 is necessary for glycoprotein ERAD but not for nonglycoprotein ERAD.
Binding of BiP to the J domain of ERdj5 is necessary for ERAD of both glycosylated and nonglycosylated substrates
ERdj5 binds to the ATP-bound form of BiP via the histidine-proline-aspartic acid (HPD) motif in the J domain of ERdj5 (Ushioda et al., 2008). The requirement of the association between BiP and ERdj5 for ERAD of glycosylated and nonglycosylated substrates was examined using an HPD mutant of ERdj5 (ERdj5/H63A) in which the histidine in the HPD motif was replaced with alanine. In HEK293T cells overexpressing ERdj5/WT or ERdj5/H63A and NHK or Tyr/T373K, the degradation rates of glycosylated NHK (Figure 3A) and Tyr/T373K (Supplemental Figure S2A) were not affected by mutation of the HPD motif. Degradation of the QQQ mutant in the ERdj5/H63A mutant-transfected cells, however, was slower than that in the ERdj5/WT-transfected cells (Figure 3B). These results apparently suggested that BiP was not involved in glycoprotein ERAD. This was not the case, however, as will be shown.
FIGURE 3:

Interaction between BiP and the J domain of ERdj5 is required for recruitment of nonglycosylated but not glycosylated substrates. (A, B) Twenty-four hours after transfection with NHK (A) or the QQQ mutant (B) and ERdj5/WT or ERdj5/H63A, HEK293T cells were labeled for 15 min with [35S]methionine/cysteine and then chased for the indicated times. The labeled substrates were immunoprecipitated with an anti-A1AT antibody. (C–E) Pulse-chase experiments performed 24 h after transfection of HEK293T cells with NHK and ERdj5/WT or ERdj5/H63A: 72 h after transfection with NS (C), ERdj5-specific (D), or ERdj4/ERdj5-specific siRNA (E). Data represent mean ± SD of n = 3 independent experiments.
It is possible that endogenous ERdj5 or other ERdj proteins were substituted for the lack of BiP-binding ability in ERdj5/H63A. To investigate this possibility, we performed small interfering RMA (siRNA)–mediated knockdown of ERdj5 and ERdj4, which is also involved in ERAD (Dong et al., 2008). Transfection of cells with nonspecific siRNA did not affect the ERdj5/H63A-induced or ERdj5/WT-induced degradation of NHK (Figure 3C); specific knockdown of ERdj5, however, reduced the effect of ERdj5/H63A on the degradation of NHK (Figure 3D). Double knockdown of ERdj5 and ERdj4 caused a complete loss of the ability of ERdj5/H63A to promote degradation of NHK, whereas the degradation by ERdj5/WT was not affected (Figure 3E). These data suggest that endogenous ERdj4 and ERdj5 can be substituted for the inability of ERdj5/H63A to bind to BiP and that BiP is required for glycoprotein ERAD, which is supported by our observation that the interaction of BiP with the J domain of ERdj5 facilitates transfer of substrates from the ERdj5/EDEM complex (Hagiwara et al., 2011).
Although overexpression of ERdj5/AA in HEK293T cells had a dominant-negative effect on degradation of the QQQ mutant (Figure 1F), there was no effect of overexpression of ERdj5/AA mutant when endogenous ERdj5 was knocked down (Supplemental Figure S4, A−C), suggesting that the dominant-negative effect of ERdj5/AA is caused by competition for substrate binding between endogenous ERdj5 and exogenously transfected ERdj5/AA. This hypothesis was confirmed by introducing the H63A mutation into ERdj5/AA (ERdj5/H63A/AA), which canceled the dominant-negative effect because this double mutant did not compete with endogenous ERdj5 in binding the substrates (Supplemental Figure S4D).
BiP plays a role in transferring substrates to and from ERdj5
Binding of the QQQ mutant to FLAG-tagged ERdj5 was examined by immunoprecipitation using [35S]methionine-labeled QQQ. The ability of the QQQ mutant to bind to ERdj5/H63A/AA was much lower than its ability to bind to ERdj5/AA (Figure 4A). Conversely, NHK bound to ERdj5/AA and ERdj5/H63A/AA with similar affinities (Figure 4A). These data suggest that the interaction between BiP and the J domain of ERdj5 is required for the recruitment of nonglycosylated but not glycosylated substrates to ERdj5. This hypothesis was confirmed by the fact that BiP coprecipitated twice as much of the QQQ mutant compared with that of NHK (Supplemental Figure S5) and is consistent with a previous report that NHK is directly transferred from CNX to the EDEM1/ERdj5 complex (Oda et al., 2003).
FIGURE 4:
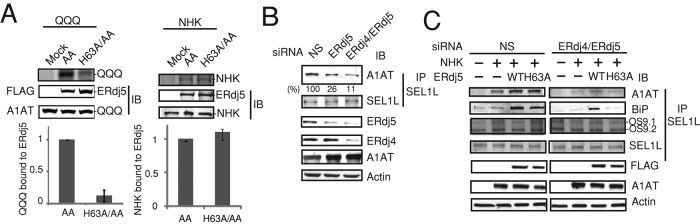
BiP transfer the substrates from ERdj5 to ERAD machinery. (A) Twenty-four hours after transfection, HEK293T cells were radiolabeled for 15 min and then chased for 30 min in the presence of 10 μM MG132. The ERdj5 mutants were immunoprecipitated with an anti-FLAG antibody, and the levels of coimmunoprecipitated NHK or QQQ mutant were quantified. (B, C) Immunoblot analyses of anti-SEL1L antibody coimmunoprecipitates from HEK293T cells transfected with NS, ERdj5-specific, or ERdj4/ERdj5-specific siRNA. Coimmunoprecipitation experiments were performed 24 h after transfection of cells with NHK and ERdj5/WT or ERdj5/H63A (72 h after transfection with siRNA). After pretreatment with 10 μM MG132 for 3 h, HEK293T cells were lysed in 1% NP40 lysis buffer, and then immunoprecipitation and immunoblotting with the indicated antibodies were performed. (B) Percentage binding of A1AT normalized to that in cells transfected with NS-siRNA. (C) OS9 isoforms that stably associate with SEL1L are also shown as control after immunoprecipitation with anti-SEL1L antibody.
Retrotranslocation of misfolded proteins requires a complex set of ERAD components, including factors that form the dislocon channel. In mammalian cells, SEL1L is a scaffold for the HRD1/SEL1L ERAD complex, which contains p97, VIMP, and Derlin-1 (Hirao et al., 2006; Mueller et al., 2008), and this complex also associates with OS9 and BiP for delivering the substrates to the dislocon channel (Christianson et al., 2008). BiP also interacts with the SEL1L ERAD complex (Hosokawa et al., 2008); therefore we examined the role of BiP in the transfer of substrates from ERdj5 to SEL1L. An anti-SEL1L antibody coprecipitated NHK, but this interaction was attenuated by knockdown of ERdj5 (Figure 4B) and almost completely abolished by double knockdown of ERdj5 and ERdj4 (Figure 4B), suggesting that ERdj4 and ERdj5 are required for the recruitment of NHK to the SEL1L complex. Overexpression of ERdj5/WT or ERdj5/H63A in HEK293T cells increased binding of NHK to SEL1L (Figure 4C). Overexpression of ERdj5/WT in cells transfected with ERdj4- and ERdj5-specific siRNAs also promoted recruitment of substrates to SEL1L; overexpression of ERdj5/H63A, however, did not (Figure 4C). The interaction between BiP and SEL1L was also promoted by the overexpression of ERdj5 in ERdj4/ERdj5 double-knockdown cells (Figure 4C). Taken together, these results indicate that the interaction between BiP and the J domain of ERdj5 is a prerequisite for the recruitment of substrates to the SEL1L complex.
Glycoproteins are degraded by the nonglycoprotein ERAD pathway under ER stress conditions
Treatment of cells with the α-glucosidase inhibitor castanospermine (Cst) to prevent entry of nascent glycopeptides into the CNX cycle transfers the folding of glycoproteins from the CNX pathway to the BiP pathway (Zhang et al., 1997; Molinari and Helenius, 2000). Treatment of cells with CST clearly caused decreased binding of NHK with EDEM1 (Figure 5A), suggesting that degradation of NHK in the presence of CST is EDEM1 independent. In the presence of Cst, ERdj5/WT transiently transfected in HEK293T cells accelerated degradation of glycosylated NHK and Tyr/T373K, but ERdj5/H63A did not (Figure 5B and Supplemental Figure S2B). When such experiments were performed under the condition of ERdj4/ERdj5 double knockdown, the ERAD of NHK was completely abolished in nontransfected control cells and ERdj5/H63A mutant–transfected cells (Figure 5C). Inhibition of mannose trimming by treating cells with kifunensine (Kif) prevents degradation of misfolded glycoproteins (Hosokawa et al., 2001). Kif-induced inhibition of glycoprotein ERAD was abolished when cells were also treated with Cst (Figure 5D). Compared with cells incubated in the absence of Cst, overexpression of BiP in HEK293 cells markedly enhanced the degradation of NHK in the presence of Cst (Figure 5E).
FIGURE 5:

Glycosylated ERAD substrates are degraded via BiP-mediated pathway under ER stress. (A) The binding of NHK with EDEM1 was analyzed by immunoprecipitation. Twenty-four hours after transfection with EDEM-HA and NHK, HEK293T cells were radiolabeled with [35S]methionine/cysteine for 15 min and then chased for 1 h in the presence of 10 μM MG132. The amount of NHK bound to EDEM1 with or without CST was estimated by immunoprecipitation with anti-HA antibody. (B) HEK293T cells overexpressing ERdj5/WT or ERdj5/H63A were pretreated with 1 mg/ml Cst for 1 h and then pulse chased with [35S]methionine/cysteine in the presence of Cst. (C) the same experiment as in B was performed for cells under double knockdown of ERdj4/ERdj5. (D) Pulse-chase analysis of NHK after treatment of HEK293T cells with 1 mg/ml Cst or Kif for 1 h before radioisotope labeling. Twenty-four hours after transfection, HEK293T cells were metabolically labeled with [35S]methionine/cysteine for 15 min and chased for the indicated times. (E) Effect of overexpression of BiP on the degradation of NHK in the presence or absence of Cst. Pulse-chase analyses were performed as described for B. (B–E) Labeled NHK was immunoprecipitated with an anti-A1AT antibody. Data represent mean ± SD of n = 3 independent experiments. (F, G) Coimmunoprecipitations of A1AT with CNX and BiP under ER stress conditions induced by 1 mM DTT and 10 μg/ml BFA for indicated periods were performed after 24 h of transfection. Transfected cells were lysed in 1% digitonin lysis buffer containing 2.27 U/ml apyrase for removal of ATP.
The foregoing results suggest that the prevention of glucose trimming switches degradation of glycosylated substrates from the N-glycan trimming–dependent (CNX-EDEM1-ERdj5) pathway to the N-glycan trimming–independent (BiP-ERdj5) pathway. Pathway switching after Cst treatment was previously reported to be a backup system in which glycoproteins are retained by BiP (Ye et al., 2005). The results also provide an explanation for the complete inhibition of glycoprotein ERAD by Kif and the comparably modest effect of Cst; when mannose trimming is inhibited after glucose residues on the A chain of N-glycans are trimmed, the substrates are trapped in association with CNX and cannot be reloaded to the BiP-ERdj5 pathway.
Next we examined substrate recruitment to BiP or CNX under ER stress by immunoprecipitation combined with pulse-chase experiments in A1AT-transfected cells. Although a large amount of A1AT was immunoprecipitated with CNX in the absence of ER stress (time zero), binding of A1AT to CNX was decreased during the chase period for 2–6 h in the presence of dithiothreitol (DTT) as ER stressor (Figure 5, F and G). Conversely, binding of A1AT to BiP increased during this chase period. This observation was similarly observed in the presence of brefeldin A (BFA), another ER stressor, with a different stress-inducing mechanism (Figure 5, F and G). Under the ER stress condition, increases in the amount of misfolded glycoproteins overflowed the capacity of CNX. At the same time, the induction of BiP by ER stress facilitated the transfer of glycoproteins overflowed from CNX to ERdj5 in an N-glycan trimming–independent manner. Thus the N-glycan trimming–independent pathway might serve as a backup system under severe ER stress to maintain proteostasis in the ER.
DISCUSSION
Here we report the existence of two distinct ERAD pathways for glycoproteins and nonglycoproteins in mammalian cells. Whereas glycoproteins misfolded in the ER are degraded by the CNX/EDEM1/ERdj5 pathway, misfolded nonglycoproteins in the ER are processed by the BiP/ERdj5 pathway for degradation (Figure 6).
FIGURE 6:

Schematic of glycoprotein and nonglycoprotein ERAD pathways and the switching of substrate transfer from glycoprotein ERAD pathway to nonglycoprotein ERAD pathway under ER stress.
During ERAD of glycoproteins, terminally misfolded glycoproteins are recognized by CNX after glucose trimming and then transferred to EDEM1 after mannose trimming (Molinari et al., 2003; Oda et al., 2003). EDEM1 interacts with the Trx4 domain of ERdj5, and the disulfide bonds of substrates are then reduced by the C-terminal Trx3 and/or Trx4 domains of ERdj5 (Hagiwara et al., 2011). BiP binds to the J domain of ERdj5; its release requires J domain–dependent hydrolysis of BiP-bound ATP. During this releasing process of BiP from the J domain, substrates that form strong interactions with BiP are pulled out of the ERdj5/EDEM1 complex and transferred to the SEL1L complex, which contains HRD1, OS9, p97, and Derlin-1, for dislocation through the retrograde transport channel (Figure 6).
Here we show that, in contrast to the glycoprotein ERAD pathway, nonglycoproteins are recognized and recruited to ERdj5 by BiP. After ATP hydrolysis of BiP and cleavage of disulfide bonds in the substrates by ERdj5, the nonglycoproteins are transferred to the dislocation channel by BiP (Figure 6). The Herp complex on the ER membrane, which contains p97, Hrd1, and Derlin-1, facilitates expulsion of BiP-bound nonglycosylated substrates from the ER (Ellgaard and Helenius, 2003). Terminally misfolded nonglycoproteins in the BiP/ERdj5 pathway may be transferred to the Herp complex for dislocation after reduction of disulfide bonds by ERdj5. The glycoprotein NHK was degraded via the BiP/ERdj5 pathway when cells were treated with Cst to prevent glucose trimming. Degradation of NHK was inhibited, however, when cells were treated with the ER mannosidase inhibitor Kif (Hosokawa et al., 2001; Ushioda et al., 2008), suggesting that substrates are not degraded by the alternative BiP/ERdj5 pathway once they enter the CNX/EDEM1/ERdj5 pathway (Figure 6).
The degradation of glycoprotein substrates NHK and Tyr/T373K occurs via the BiP/ERdj5 pathway when cells are treated with DTT or BFA to induce ER stress. The substrate-binding ability of CNX may be saturated under these stress conditions, which could cause a shift of misfolded glycoproteins to the BiP/ERdj5 pathway (Figure 5, D and E). Under these conditions, BiP is also induced, which would be of advantage for trapping the misfolded substrates for recruitment to the BiP/ERdj5 pathway. In the presence of DTT, conformational change in the arm or globular domain of calnexin by the cleavage of disulfide bond was reported to result in reduced substrate-binding activity of calnexin (Hebert et al., 1995), which raises the possibility that glycosylated substrates are further recruited to BiP/ERdj5 pathway. Therefore the BiP/ERdj5 pathway may serve as a backup ERAD system under ER stress conditions where the number of substrates exceeds the capacity of CNX and/or EDEM1. The role of EDEM1 in nonglycoprotein ERAD is controversial. Cormier et al. (2009) reported that EDEM1 binds to nonnative proteins in a glycan-independent manner but does not enhance degradation of nonglycoproteins, whereas Shenkman et al. (2013) reported that EDEM1 is involved in the degradation of nonglycoprotein substrates. It is possible that some nonglycoprotein substrates are codegraded with EDEM1, because EDEM1 is rapidly degraded by an autophagy-like mechanism (Cali et al., 2008). The existence of two distinct ERAD pathways and a backup system may contribute to the maintenance of protein homeostasis in the ER of mammalian cells under various stress conditions.
MATERIALS AND METHODS
Cell culture and transfections
HEK293T cells were used in all experiments, with the exception of the use of HeLa cells for coimmunoprecipitation of EDEM with ERdj5. Transfections of cells with plasmids and siRNAs were performed using Lipofectamine 2000 and RNAiMAX (Invitrogen, Carlsbad, CA) reagents, respectively. Stealth RNA Negative Control Low GC and Stealth siRNAs specific to human ERdj5 or ERdj4 were obtained from Invitrogen.
Plasmid construction
Mouse ERdj5/WT-FLAG and ERdj5/AA-FLAG were constructed as described previously (Ushioda et al., 2008). ERdj5/H63A and ERdj5/H63A/AA were generated using the QuikChange site-directed mutagenesis kit (Stratagene, Santa Clara, CA). The ΔTrx4 and ΔTrx34 C-terminal deletion mutants of ERdj5 were amplified from mouse ERdj5-FLAG cDNA by PCR and subcloned into pcDNA3.1 (Invitrogen) at the BamHI and EcoRI sites. The ERdj5 Trx4 mutant was PCR amplified and ligated into AgeI-EcoRI–digested pCDNA3.1-mERdj5-FLAG (just after the cDNA portion encoding the signal sequence). Expression plasmids containing mouse EDEM-HA (pCMV-SPORT2-EDEM-HA) and the NHK, QQQ mutant, and QQQ/CS mutant of human A1AT (pREP9-NHK, QQQ, QQQ/CS; Hosokawa et al., 2001; Hirao et al., 2006) were kindly provided by N. Hosokawa (Kyoto University, Kyoto, Japan). The expression plasmid containing Tyr-YFP (human tyrosinase ligated to pEYFP-N1; Kamada et al., 2004), which was kindly provided by I. Wada (Fukushima Medical University, Fukushima, Japan), was mutagenized to Tyr/T373K using the QuikChange site-directed mutagenesis kit.
Antibodies
Mouse monoclonal anti–FLAG M2, anti-actin and anti-BiP antibodies were purchased from Sigma-Aldrich (St. Louis, MO), Chemicon (Temecula, CA), and BD Biosciences (Franklin Lakes, NJ), respectively. Rabbit polyclonal antibodies against A1AT, HA, and OS9 were obtained from Dako (Carpinteria, CA), Santa Cruz Biotechnology (Santa Cruz, CA), and Sigma-Aldrich, respectively. Mouse polyclonal antibodies against ERdj5 and SEL1L were purchased from Abnova (Taipei City, Taiwan) and Abcam (Cambridge, MA), respectively. Goat polyclonal antibodies against ERdj4 and tyrosinase were purchased from Novus (Littleton, CO) and Santa Cruz Biotechnology, respectively.
Metabolic labeling and pulse chasing
HEK293T cells were preincubated in DMEM lacking methionine and cysteine (Invitrogen) for 30 min and then pulse labeled for 15 min with 8.2 MBq/ml Expre35S35S Protein Labeling Mix (PerkinElmer Life Sciences, Waltham, MA). After washing twice with phosphate-buffered saline (PBS) lacking Ca2+ and Mg2+ (PBS[–]), we incubated the metabolically labeled cells in DMEM during the chase period. Cells were then washed with PBS[–], incubated on ice for 20 min in lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 40 mM iodoacetoamide, and 1% Nonidet P-40 or 1% digitonin) supplemented with protease inhibitors, and then immunoprecipitated with specific antibodies. For coimmunoprecipitation experiments using anti-CNX and anti-BiP antibodies, 2.27 U/ml apyrase was added to remove ATP from the lysate. The immunoprecipitants were separated by SDS–PAGE, and the gels were exposed to an imaging plate. Band radioactivity was detected using a Fujifilm Phosphor Imager (Fujifilm, Tokyo, Japan). Cst (1 mM) was added 1 h before pulse labeling. To inhibit proteasome activity, cells were treated with 10 μM MG132 for 4 h before pulse labeling.
Supplementary Material
Acknowledgments
We thank Yasuko Fukuda for technical support. This work was supported by the Ministry of Education, Culture, Sports, Science and Technology via a Grant-in-Aid for Creative Scientific Research (19GS0314) and for Scientific Research on Priority Areas (19058008) to N.K., a Grant-in-Aid for Scientific Research on Innovative Areas (24121725) to R.U., a Grant-in-Aid for Young Scientists (B) (23770158) to J.H., and a grant from the Takeda Science Foundation to J.H.
Abbreviations used:
- A1AT
alpha 1 antitrypsin
- BFA
brefeldin A
- CNX
calnexin
- CRT
calreticulin
- Cst
castanospermine
- DTT
dithiothreitol
- ER
endoplasmic reticulum
- EDEM
ER degradation–enhancing α-mannosidase–like protein
- ERAD
ER-associated degradation
- HEK
human embryonic kidney
- Kif
kifunensine
- NHK
null Hong Kong
- PBS
phosphate-buffered saline
- siRNA
small interfering RNA
- Trx
thioredoxin
- Tyr
tyrosinase
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E13-03-0138) on August 21, 2013.
*Present address: Research Unit for Physiological Chemistry, Center for the Promotion of Interdisciplinary Education and Research, and Division of Applied Life Sciences, Graduate School of Agriculture, Kyoto University, Kyoto 606–8502, Japan.
REFERENCES
- Bernasconi R, Galli C, Calanca V, Nakajima T, Molinari M. Stringent requirement for HRD1, SEL1L, and OS-9/XTP3-B for disposal of ERAD-LS substrates. J Cell Biol. 2010;188:223–235. doi: 10.1083/jcb.200910042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cali T, Galli C, Olivari S, Molinari M. Segregation and rapid turnover of EDEM1 by an autophagy-like mechanism modulates standard ERAD and folding activities. Biochem Biophys Res Commun. 2008;371:405–410. doi: 10.1016/j.bbrc.2008.04.098. [DOI] [PubMed] [Google Scholar]
- Christianson JC, Shaler TA, Tyler RE, Kopito RR. OS-9 and GRP94 deliver mutant alpha1-antitrypsin to the Hrd1-SEL1L ubiquitin ligase complex for ERAD. Nat Cell Biol. 2008;10:272–282. doi: 10.1038/ncb1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cormier JH, Tamura T, Sunryd JC, Hebert DN. EDEM1 recognition and delivery of misfolded proteins to the SEL1L-containing ERAD complex. Mol Cell. 2009;34:627–633. doi: 10.1016/j.molcel.2009.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong M, Bridges JP, Apsley K, Xu Y, Weaver TE. ERdj4 and ERdj5 are required for endoplasmic reticulum-associated protein degradation of misfolded surfactant protein C. Mol Biol Cell. 2008;19:2620–2630. doi: 10.1091/mbc.E07-07-0674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellgaard L, Helenius A. Quality control in the endoplasmic reticulum. Nat Rev Mol Cell Biol. 2003;4:181–191. doi: 10.1038/nrm1052. [DOI] [PubMed] [Google Scholar]
- Hagiwara M, Maegawa K, Suzuki M, Ushioda R, Araki K, Matsumoto Y, Hoseki J, Nagata K, Inaba K. Structural basis of an ERAD pathway mediated by the ER-resident protein disulfide reductase ERdj5. Mol Cell. 2011;41:432–444. doi: 10.1016/j.molcel.2011.01.021. [DOI] [PubMed] [Google Scholar]
- Hebert DN, Simons JF, Peterson JR, Helenius A. Calnexin, calreticulin, and Bip/Kar2p in protein folding. Cold Spring Harbor Symp Quant Biol. 1995;60:405–415. doi: 10.1101/sqb.1995.060.01.045. [DOI] [PubMed] [Google Scholar]
- Hirao K, et al. EDEM3, a soluble EDEM homolog, enhances glycoprotein endoplasmic reticulum-associated degradation and mannose trimming. J Biol Chem. 2006;281:9650–9658. doi: 10.1074/jbc.M512191200. [DOI] [PubMed] [Google Scholar]
- Hoseki J, Ushioda R, Nagata K. Mechanism and components of endoplasmic reticulum-associated degradation. J Biochem. 2010;147:19–25. doi: 10.1093/jb/mvp194. [DOI] [PubMed] [Google Scholar]
- Hosokawa N, Kamiya Y, Kamiya D, Kato K, Nagata K. Human OS-9, a lectin required for glycoprotein endoplasmic reticulum-associated degradation, recognizes mannose-trimmed N-glycans. J Biol Chem. 2009;284:17061–17068. doi: 10.1074/jbc.M809725200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosokawa N, Wada I, Hasegawa K, Yorihuzi T, Tremblay LO, Herscovics A, Nagata K. A novel ER alpha-mannosidase-like protein accelerates ER-associated degradation. EMBO Rep. 2001;2:415–422. doi: 10.1093/embo-reports/kve084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosokawa N, Wada I, Nagasawa K, Moriyama T, Okawa K, Nagata K. Human XTP3-B forms an endoplasmic reticulum quality control scaffold with the HRD1-SEL1L ubiquitin ligase complex and BiP. J Biol Chem. 2008;283:20914–20924. doi: 10.1074/jbc.M709336200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamada A, Nagaya H, Tamura T, Kinjo M, Jin HY, Yamashita T, Jimbow K, Kanoh H, Wada I. Regulation of immature protein dynamics in the endoplasmic reticulum. J Biol Chem. 2004;279:21533–21542. doi: 10.1074/jbc.M401403200. [DOI] [PubMed] [Google Scholar]
- Molinari M, Calanca V, Galli C, Lucca P, Paganetti P. Role of EDEM in the release of misfolded glycoproteins from the calnexin cycle. Science. 2003;299:1397–1400. doi: 10.1126/science.1079474. [DOI] [PubMed] [Google Scholar]
- Molinari M, Helenius A. Chaperone selection during glycoprotein translocation into the endoplasmic reticulum. Science. 2000;288:331–333. doi: 10.1126/science.288.5464.331. [DOI] [PubMed] [Google Scholar]
- Mueller B, Klemm EJ, Spooner E, Claessen JH, Ploegh HL. SEL1L nucleates a protein complex required for dislocation of misfolded glycoproteins. Proc Natl Acad Sci USA. 2008;105:12325–12330. doi: 10.1073/pnas.0805371105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa SI, Fewell SW, Kato Y, Brodsky JL, Endo T. Molecular chaperones in the yeast endoplasmic reticulum maintain the solubility of proteins for retrotranslocation and degradation. J Cell Biol. 2001;153:1061–1070. doi: 10.1083/jcb.153.5.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda Y, Hosokawa N, Wada I, Nagata K. EDEM as an acceptor of terminally misfolded glycoproteins released from calnexin. Science. 2003;299:1394–1397. doi: 10.1126/science.1079181. [DOI] [PubMed] [Google Scholar]
- Okuda-Shimizu Y, Hendershot LM. Characterization of an ERAD pathway for nonglycosylated BiP substrates, which require Herp. Mol Cell. 2007;28:544–554. doi: 10.1016/j.molcel.2007.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivari S, Cali T, Salo KE, Paganetti P, Ruddock LW, Molinari M. EDEM1 regulates ER-associated degradation by accelerating de-mannosylation of folding-defective polypeptides and by inhibiting their covalent aggregation. Biochem Biophys Res Commun. 2006;349:1278–1284. doi: 10.1016/j.bbrc.2006.08.186. [DOI] [PubMed] [Google Scholar]
- Otero JH, Lizak B, Hendershot LM. Life and death of a BiP substrate. Semin Cell Dev Biol. 2010;21:472–478. doi: 10.1016/j.semcdb.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plemper RK, Bohmler S, Bordallo J, Sommer T, Wolf DH. Mutant analysis links the translocon and BiP to retrograde protein transport for ER degradation. Nature. 1997;388:891–895. doi: 10.1038/42276. [DOI] [PubMed] [Google Scholar]
- Shenkman M, Groisman B, Ron E, Avezov E, Hendershot LM, Lederkremer GZ. A shared ER-associated degradation pathway involving EDEM1 for glycosylated and nonglycosylated proteins. J Biol Chem 288, 2167–2178. 2013 doi: 10.1074/jbc.M112.438275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyofuku K, Wada I, Spritz RA, Hearing VJ. The molecular basis of oculocutaneous albinism type 1 (OCA1): sorting failure and degradation of mutant tyrosinases results in a lack of pigmentation. Biochem J. 2001;355:259–269. doi: 10.1042/0264-6021:3550259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ushioda R, Hoseki J, Araki K, Jansen G, Thomas DY, Nagata K. ERdj5 is required as a disulfide reductase for degradation of misfolded proteins in the ER. Science. 2008;321:569–572. doi: 10.1126/science.1159293. [DOI] [PubMed] [Google Scholar]
- Ushioda R, Nagata K. The endoplasmic reticulum-associated degradation and disulfide reductase ERdj5. Methods Enzymol. 2011;490:235–258. doi: 10.1016/B978-0-12-385114-7.00014-3. [DOI] [PubMed] [Google Scholar]
- Ye Y, Shibata Y, Kikkert M, van Voorden S, Wiertz E, Rapoport TA. Recruitment of the p97 ATPase and ubiquitin ligases to the site of retrotranslocation at the endoplasmic reticulum membrane. Proc Natl Acad Sci USA. 2005;102:14132–14138. doi: 10.1073/pnas.0505006102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JX, Braakman I, Matlack KE, Helenius A. Quality control in the secretory pathway: the role of calreticulin, calnexin and BiP in the retention of glycoproteins with C-terminal truncations. Mol Biol Cell. 1997;8:1943–1954. doi: 10.1091/mbc.8.10.1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.