

Abstract
Recent work has provided compelling evidence that increased levels of acetylcholine (ACh) can be protective in heart failure, whereas reduced levels of ACh secretion can cause heart malfunction. Previous data show that cardiomyocytes themselves can actively secrete ACh, raising the question of whether this cardiomyocyte derived ACh may contribute to the protective effects of ACh in the heart. To address the functionality of this non-neuronal ACh machinery, we used cholinesterase inhibitors and a siRNA targeted to AChE (acetylcholinesterase) as a way to increase the availability of ACh secreted by cardiac cells. By using nitric oxide (NO) formation as a biological sensor for released ACh, we showed that cholinesterase inhibition increased NO levels in freshly isolated ventricular myocytes and that this effect was prevented by atropine, a muscarinic receptor antagonist, and by inhibition of ACh synthesis or vesicular storage. Functionally, cholinesterase inhibition prevented the hypertrophic effect as well as molecular changes and calcium transient alterations induced by adrenergic overstimulation in cardiomyocytes. Moreover, inhibition of ACh storage or atropine blunted the anti-hypertrophic action of cholinesterase inhibition. Altogether, our results show that cardiomyocytes possess functional cholinergic machinery that offsets deleterious effects of hyperadrenergic stimulation. In addition, we show that adrenergic stimulation upregulates expression levels of cholinergic components. We propose that this cardiomyocyte cholinergic signaling could amplify the protective effects of the parasympathetic nervous system in the heart and may counter-act or partially neutralize hypertrophic adrenergic effects.
Keywords: acetylcholine, cardiomyocyte, hypertrophy, VAChT, cholinergic machinery
1. Introduction
Extrinsic control of heart function is primarily regulated by the autonomic nervous system. The parasympathetic branch, which releases the neurotransmitter acetylcholine (ACh), is well known to control heart rate [1] by predominantly regulating atrial function [2]. Rich cholinergic innervations are found in the sinoatrial node, atrial myocardium, atrioventricular node and in the ventricular conducting system of many species [3]. Although less abundant, parasympathetic fibers are also found throughout the ventricles, where stimulation of type 2 muscarinic acetylcholine receptor (M2-AChR) by ACh leads to L-type calcium channel inhibition and, consequently, reduced cardiomyocyte contractility [4]. Recently, novel physiological functions of ACh in the heart have emerged and new data suggest that this neurotransmitter plays unanticipated long-term roles in cardiac protection that may not be necessarily linked to its role in regulating the electrical properties of the heart [5].
ACh is synthesized in the cytoplasm of nerve terminals through the action of choline acetyltransferase (ChAT) and stored within acidic synaptic vesicles for release. Activity coupled transport of ACh into synaptic vesicles in nerve endings is mediated by the vesicular acetylcholine transporter (VAChT) [6-8]. Released ACh, which activates muscarinic receptors on cardiac cells, is rapidly degraded by the enzyme acetylcholinesterase (AChE) to form acetate and choline. Choline is then recycled through the action of the high-affinity choline transporter (CHT1), and reused as a substrate for the synthesis of new ACh molecules [9].
Previously, we have shown that VAChT knockdown homozygous mice (VAChT KDHOM mice), which express only 30% of normal VAChT levels, present a number of cholinergic deficits due to reduced ACh release [8], including a remarkable dysfunction in ventricular myocytes [10]. This is characterized by depressed ventricular contractile function, calcium (Ca2+) signaling dysfunction and altered gene expression in ventricular myocytes [10]. Confirming these findings, another model of cholinergic dysfunction, mice with reduced CHT1 expression showed age-dependent ventricular dysfunction [11]. Along these lines, M2-AChR knockout (KO) mice presented increased susceptibility to cardiac stress [12]. Taken together, these results suggest that decreased cholinergic function can affect outcomes for proper heart activity. Complementary to these findings, increased availability of ACh can be protective in heart disease. For example, in vivo stimulation of the vagus nerve [13] or treatment with a cholinesterase inhibitor [14] led to improved outcome in experimental heart failure in rats. Recent data from Kanazawa et al. [15] have provided further evidence for cholinergic mediated cardioprotection. These authors found that cholinergic transdifferentiation of sympathetic neurons takes place during heart failure in rats, mice and humans. Moreover, inhibition of transdifferentiation in mice increased mortality in a model of heart failure.
Multiple mechanisms may contribute to this protective effect of ACh on ventricular myocyte function. For example, increased parasympathetic tone can directly modulate heart function and also increase anti-inflammatory activity [16], which would be highly beneficial in heart failure. Furthermore, previous works have shown that ventricular cardiomyocytes express all the required components involved in ACh synthesis and release, and actively secrete ACh [17,18]. Yet, the relevance of this non-neuronal cardiomyocyte derived cholinergic system in ventricular function is poorly understood. Here, we address the functional significance of this machinery in cardiac cells and show that cardiomyocyte released ACh can act in an autocrine/paracrine manner to mitigate the effects of adrenergic overstimulation. Together, our results point to another level of cholinergic control of cardiac function, which goes beyond the cardiac parasympathetic system, and it is intrinsic to ventricular myocytes.
2. Materials and Methods
For a detailed description of immunofluorescence, Western blot and qPCR protocols, see the expanded materials and methods section in the supplementary material available online.
2.1 Animal models
In this study we used adult and neonatal C57BL/6 mice and Wistar rats. Rat and mouse neonatal cardiomyocytes were isolated from 2-4 and 2-day old animals, respectively. Adult ventricular myocytes were isolated from 10-12 weeks old male mice and from male Wistar rats weighing 220-250g. VAChT knockdown homozygous mouse line (VAChT KDHOM mice) [8] was previously described. Animals were maintained at UFMG, Brazil, and at the University of Western Ontario in accordance with NIH guidelines and the Canadian Council for Animal Care for the care and use of animals. Experiments were performed according to approved animal protocols from the Institutional Animal Care and Use Committee at UFMG (protocol# 016/11) and at the University of Western Ontario (2008-089).
2.2 Neonatal cardiomyocyte culture
Rat and mouse neonatal cardiomyocytes were cultured as previously described [19,20]. Briefly, cardiac cells were plated in dishes containing M199 medium supplemented with 100 units/ml penicillin, 100μg/ml streptomycin, 10% Fetal Bovine Serum and 2 mmol/L L-glutamine. To prevent growth of fibroblasts, medium was supplemented with 20 μg/mL cytosine-D-arabinofuranoside (ARA-c). After 48 hours, neonatal cardiomyocytes were exposed to isoproterenol (10 μmol/L) and/or pyridostigmine or neostigmine (10 μmol/L or 1 mmol/L) for 48 hours at the indicated concentrations. The cells were then used for immunofluorescence, Western blotting or qPCR analyses. When necessary, cells were incubated with atropine (10 μmol/L), vesamicol (5 μmol/L) or Nω-Nitro-L-Arginine Methyl Ester Hydrochloride (L-NAME, 10 μmol/L) for 48 hours. ACh (10 μmol/L) and phenylephrine (50 μmol/L) were also used in these experiments.
2.3 Preparation of siRNA
Potential target sites within the acetylcholinesterase gene were selected and then searched with NCBI Blast to confirm specificity for the enzyme. The siRNA for AChE was prepared as previously described by our group [21]. The sense and antisense oligonucleotides of siRNA were respectively, asfollows: 5′- AAAAGGTGGTAGCATCCAATACCTGTCTC- 3′and 5′-AATATTGGATGCTACCACCTTCCTGTCTC-3′. In some studies we designed and tested a second siRNA targeting AChE (sense) 5′-AACGTATTGGTAGCAGACATTCCTGTCTC- 3′ and (antisense) 5′-AAAATGTCTGCTACCAATACGCCTGTCTC -3′. For siRNA studies, neonatal cardiomyocyte cultures at day 4 were transfected with 100nM of siRNA. The cells were incubated at 37°C in an atmosphere of 5% CO2 for 30 hours and then exposed to isoproterenol (10 μmol/L) for another 24 hours prior to use.
2.4 Ventricular cardiomyocyte isolation and Ca2+ recording
Adult ventricular myocytes were freshly isolated as previously described [22] and stored in M199 (Sigma), supplemented with 100 units/ml penicillin, 100 μg/ml streptomycin, L-carnitine (2 mmol/L), taurine (5 mmol/L), creatine (5 mmol/L), glucose (5.5 mmol/L), selenium (300 nmol/L) and transferrin (70 nmol/L). Cardiomyocytes were plated and intracellular Ca2+ (Ca2+i) measurements were performed 20 hours after isoproterenol (1 μmol/L) or isoproterenol/pyridostigmine (1 μmol/L/ 500 μmol/L) treatment in Fluo-4/AM (10 μmol/L; Invitrogen) loaded cells (30 min). After loading, cells were subsequently washed with normal Tyrode solution (in mmol/L: NaCl, 140; KCl, 4; MgCl2, 1; CaCl2, 1.8; Glucose, 10 and HEPES, 5; pH = 7.4 adjusted with NaOH) to remove the excess dye. Cells were electrically stimulated at 1 Hz to produce steady-state conditions. Experiments were performed at room temperature. The confocal line-scan imaging was done in a Zeiss LSM 510 Meta confocal microscope located at CEMEL (UFMG).
2.5 NO measurement
For these experiments we used freshly isolated adult ventricular myocytes from wild-type and VAChT KDHOM mice aged 10-12 weeks. Measurement of NO production in living ventricular myocytes was done using the membrane permeable fluorescent indicator 4-amino-5-methylamino- 2′,7′-difluorofluorescein diacetate (DAF-FM diacetate, Invitrogen). To detect cytosolic NO, cardiomyocytes were loaded at 37°C with 5 μmol/ L DAF-FM diacetate for 30 min and then washed for 30 min with Tyrode solution. Cardiomyocytes were then incubated with ACh (10 μmol/L) or pyridostigmine (1 mmol/L) for 30 min. After this time, the fluorescence generated by NO production was recorded using a confocal microscope (CEMEL, UFMG). Analyses were performed with ImageJ software (NIH). In a separate group of experiments, cardiomyocytes were pre-incubated with atropine (10 μmol/L) or hemicholinium-3 (10 μmol/L) for 30 min before the addition of acetylcholine or pyridostigmine to the cells. In another set of experiments, we incubated mouse ventricular myocytes with different concentrations of ACh (0.1 to 10 μmol/L) or PYR (0.1 to 1000 μmol/L).
2.6 FM 1-43 FX
Freshly isolated adult ventricular myocytes were loaded with FM1-43 FX dye (4 μM, Invitrogen) for 1 hour at 37°C in order to establish the localization of recycling vesicles. Before being fixed in a 4% paraformaldehyde (PFA) solution, the cells were ressuspended in Tyrode solution and washed for approximately 20 min to remove excess dye. Cells were then submitted to our immunofluorescence protocol (see expanded Methods section in supplemental material).
2.7 Statistical analysis
All data are expressed as mean ± SEM, and the number of cells or experiments is shown as n. Significant differences between groups were determined with a Student's t-test or ANOVA followed by the Bonferroni post hoc test. Values of p < 0.05 were considered to be statistically significant.
3. Results
3.1 Cardiomyocytes present functional ACh synthesis and release machinery
Previous work has shown that adult and neonatal rat cardiomyocytes express the three proteins involved in ACh synthesis and storage (ChAT, VAChT and CHT1) [17]. To further assess how ubiquitous this phenomenon is, we extended these findings and compared the expression of these three cholinergic markers in freshly isolated ventricular myocytes from adult and neonatal mice. As shown in Fig. 1A, both neonatal and adult cardiomyocytes express VAChT, observed as a 75 kDa band. For these experiments we used an anti-VAChT antibody that has previously been validated by using striatal specific VAChT knockout mice [23]. Our data show that the major band identified by this antibody is highly expressed in brain, and shows lower levels of expression in cardiomyocytes, as expected (Fig. 1A). ChAT, the enzyme responsible for ACh synthesis, was also detected in both neonatal cardiomyocytes and in adult ventricular cells (Fig. 1B). Interestingly, in cardiomyocytes, ChAT was observed mainly as a 52 kDa protein, in contrast with the more abundant 80 kDa protein found in the brain. The 52 kDa protein was also found in the brain, albeit in lower levels. We also detected CHT1 in both adult and neonatal cardiomyocytes (Fig. 1C). We further analysed these cholinergic markers by using immunofluorescence and confocal microscopy to ascertain their localization in isolated adult mouse ventricular myocytes. Importantly, in cardiomyocytes, VAChT staining was found mainly in a perinuclear compartment, although VAChT punctuate staining was also found in the cytoplasm (Fig. 1D). Because VAChT is a transmembrane protein usually located in endosomal, synaptic and recycling vesicles, we also tested whether this transporter would colocalize with the vital dye FM1-43 FX, a fixable analogue of FM1-43 which can be used to visualize recycling vesicles [24]. FM1-43 FX staining was observed all over the cardiac cell, but was enriched at the perinuclear region, where it extensively colocalized with VAChT (Fig. 1E). This finding is in agreement with previous data showing the presence of vesicle-associated membrane proteins (VAMP) in a punctate perinuclear pattern in cardiomyocytes [25]. In addition, we observed ChAT immunostaining in the nuclear region in close proximity to where VAChT is localized (Fig. 1F). The nuclear localization of ChAT matches previous findings in neurons [26]. In agreement with the Western blot data, CHT1 was also found in isolated cardiomyocytes. However, in contrast with the localization of VAChT and ChAT, CHT1 was mostly localized to the plasma membrane (Fig. 1G), where it presumably plays an active role in the reuptake of choline.
Figure 1. Mouse cardiomyocytes express prototypical cholinergic markers.
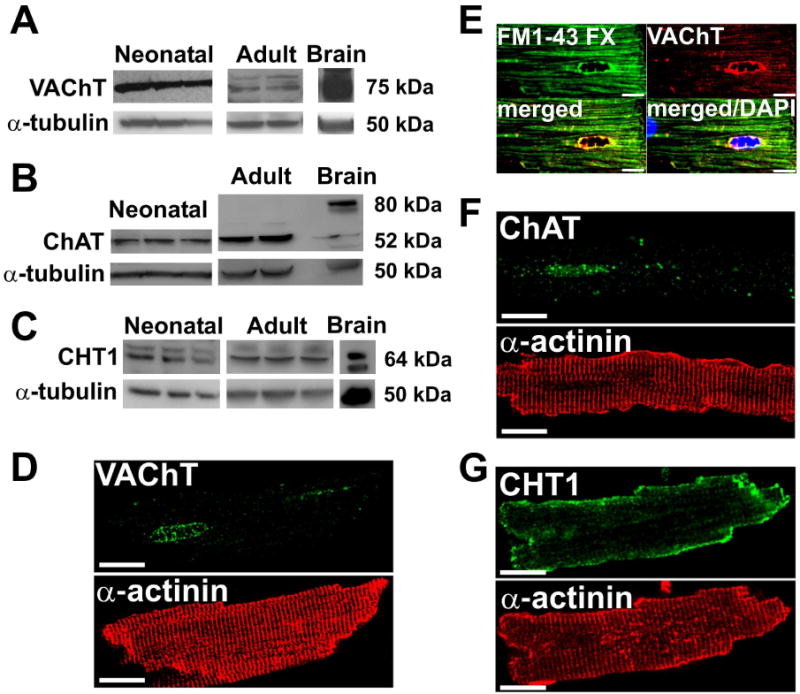
A-C. Representative Western blots of immunoreactivity for VAChT, ChAT, and CHT1 in neonatal and adult mouse ventricular myocytes. Brain samples were used as a positive control. α-tubulin was used as a loading control. D-G. VAChT, FM1-43 FX, ChAT and CHT1 staining in adult mouse ventricular myocytes. Cardiomyocytes were also labeled with antibody against the sarcomeric protein α-actinin to show cellular organization. Scale Bar=10 μm.
A critical issue in identifying ACh secretion in non-neuronal cell culture is its rapid degradation by AChE, an enzyme that has very high catalytic activity and is abundantly expressed in cardiomyocytes [17]. Furthermore, detection of very low levels of ACh is usually a challenge. One of the many responses of cardiomyocytes to ACh is an increase in NO levels [27]. Therefore, we next sought to determine whether we could use the generation of NO as a biological sensor for the effects of autocrinally released ACh. Thus, freshly isolated ventricular myocytes from adult wild-type mice were loaded with the fluorescent indicator DAF-FM to assess NO generation in response to ACh (Fig. 2A-F). Fig. 2A shows representative images of DAF fluorescence in mouse ventricular myocytes. Average values (Fig. 2B) show that exogenously added ACh (10 μmol/L) induced a significant increase in NO generation in ventricular myocytes, which was blocked by atropine, a muscarinic antagonist. As shown in Fig. 2B, atropine alone had no effect on NO generation. In order to test if ACh released by cardiomyocytes could act in these cells in an autocrine/paracrine manner, we used a reversible cholinesterase inhibitor (pyridostigmine) as a way to increase the availability of cardiomyocyte derived ACh, by preventing its degradation by AChE. Wild-type cardiomyocytes were incubated with pyridostigmine (1 mmol/L; 30 minutes), and DAF fluorescence was assessed. As shown in Fig. 2C-D, adult mouse ventricular myocytes treated with pyridostigmine alone presented increased NO generation to levels similar to that found in cells exposed to exogenous ACh. This effect was completely blocked by atropine. To investigate if the effect of pyridostigmine on NO generation was dependent on CHT1 activity, we treated the cells with hemicholinium-3 (HC-3, 10 μmol/L), a CHT1 inhibitor that abolishes ACh synthesis. Fig. 2E-F shows that pre-incubation of cardiomyocytes with HC-3 also prevented the effects of pyridostigmine on DAF fluorescence. Similar to atropine, HC-3 alone had no effect on DAF fluorescence.
Figure 2. NO levels can be used as a biosensor to detect ACh release in cardiomyocytes.
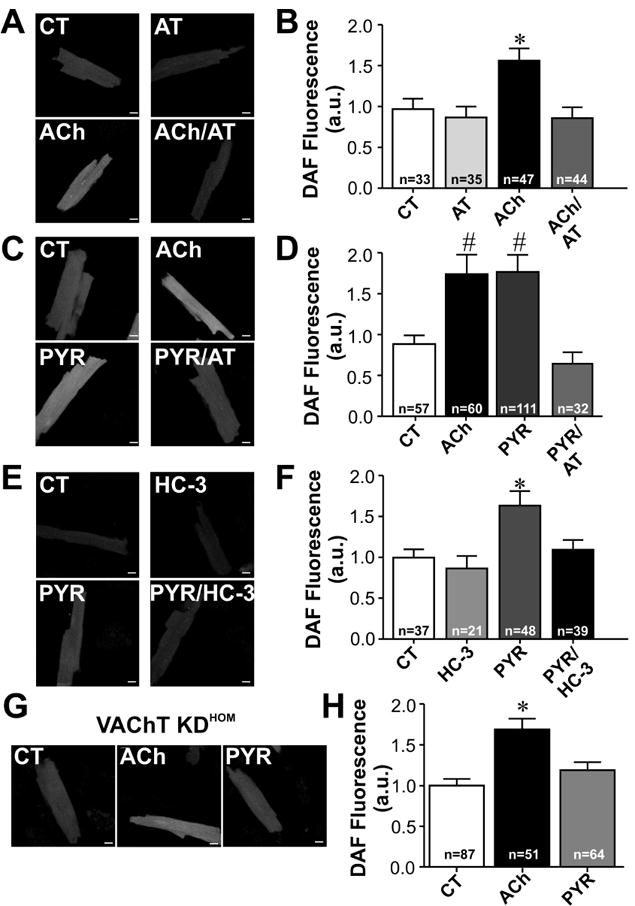
A-F. DAF fluorescence measurements performed in wild-type (WT) cardiomyocytes. A. Sample confocal images show cellular increase in DAF fluorescence in cardiomyocytes from WT mice treated with ACh. B. Averaged DAF fluorescence increase in adult ventricular myocytes following acute treatment with exogenously added ACh for 30 minutes. The pre-incubation of muscarinic receptor antagonist, atropine (AT), inhibits the increase in DAF fluorescence in ACh treated cardiomyocytes. C. Sample images of DAF fluorescence of cardiomyocytes incubated with ACh, pyridostigmine (PYR) or PYR/AT. D. Effect of cholinesterase inhibition on DAF fluorescence in the presence or absence of atropine (AT) pre-treatment. Cardiomyocytes exposed to pyridostigmine presented an increase in DAF fluorescence, which was blunted by atropine. E. Sample images of DAF loaded cardiac myocytes treated with hemicholinium-3 (HC-3) and/or PYR for 30min. F. HC-3 blunted PYR induced increase in DAF fluorescence in ventricular myocytes. G. Sample images of DAF fluorescence in ventricular myocytes of VAChT KDHOM mice incubated with ACh or PYR. H. Cholinesterase inhibition in cardiomyocytes with reduced VAChT expression levels does not lead to a significant increase in NO generation. n= number of cells analysed. * p < 0.05 when compared to the other groups. # p<0.05 when compared to CT and PYR/AT groups. All cells were loaded with fluorescent dye following the same protocol, and imaging was done preserving the same parameters in both control and drug treated cardiomyocytes. Scale Bar= 10 μm.
To assess the specificity of pyridostigmine effect on NO production we compared the concentration dependency of ACh and pyridostigmine on DAF loaded ventricular myocytes. Cardiac cells were exposed to increased concentrations of ACh and pyridostigmine and DAF fluorescence was assessed. As shown in Fig. S1 A-B, both ACh and pyridostigmine produced a concentration dependent increase in NO levels, thus eliminating the possibility that pyridostigmine exerts off target effects.
We then examined the effects of pyridostigmine on DAF fluorescence in cardiomyocytes from VAChT KDHOM mice, an animal model that presents 70% reduction in VAChT levels and concomitant decrease in ACh release [8]. Fig. 2G-H shows that cardiomyocytes obtained from VAChT KDHOM mice fail to respond to pyridostigmine. This finding contrasted with the significant response to pyridostigmine found in wild-type myocytes (Fig. 2C-F). Moreover, exogenously added ACh elicited a significant increase in DAF fluorescence in cardiomyocytes from VAChT KDHOM mice. The response to ACh found in these cells was similar to the one observed in wild-type cardiomyocytes (Fig. 2B and D). Therefore, by using a cholinesterase inhibitor, it is possible to increase the availability of cardiomyocyte derived ACh, which in turn activates muscarinic receptors to increase NO levels. Taken together, these data show that cardiomyocytes are capable of synthesizing and releasing ACh confirming the functionality of cholinergic machinery in these cells. Moreover, it seems that both choline reuptake and ACh storage in VAChT positive organelles are required for ACh secretion from adult cardiomyocytes.
3.2 Cholinesterase inhibition prevents adrenergic hypertrophic signaling
Our data indicate that cholinesterase inhibition increases the availability of ACh secreted by cardiomyocytes. Therefore, we investigated the possibility that this cardiomyocyte cholinergic signaling can exert protective effects. Thus, the next group of experiments was conducted in mouse neonatal cardiomyocytes as these cells are easier to maintain in culture than isolated adult cells and their phenotype is highly stable [28,29]. In order to simulate disease conditions, neonatal cardiomyocytes were treated with isoproterenol (ISO, 10 μmol/L) for 48 hours and cellular hypertrophy was assessed by direct measurement of myocyte surface area. Fig. S2A shows representative immunofluorescence images from α-actinin stained cardiomyocytes. Cardiomyocytes treated with isoproterenol demonstrated increased cell surface area by 40% (Fig. S2B), whereas concomitant treatment with vesamicol (5 μmol/L), a specific VAChT inhibitor, caused an even greater increase in cell surface area (80%), thus implicating released ACh in the endogenous protection against cellular hypertrophy. Importantly, pyridostigmine (10 μmol/L) prevented the hypertrophic effect of isoproterenol. Similar effects were observed in cells treated with 1mmol/L pyridostigmine (data not shown). The action of pyridostigmine was dependent on VAChT activity, and ACh secretion, as isoproterenol was again able to effectively induce hypertrophy when vesamicol was present in the media. We further confirmed these data in a second cohort of cells that were treated with isoproterenol and another cholinesterase inhibitor, neostigmine (10 μmol/L). Fig. S2C-D shows that neostigmine was as effective as pyridostigmine in preventing the hypertrophic actions of isoproterenol. We again found that vesamicol was able to block the protective effect of neostigmine. Together, these data suggest that ACh released by cardiomyocytes can offset hypertrophic effects of isoproterenol on mouse neonatal cardiac cells.
3.3 Mechanisms involved in mitigation of isoproterenol-induced hypertrophy
In order to further understand the mechanisms involved in this protective response to endogenously secreted ACh, we used rat cardiomyocytes, which also have an intrinsic cholinergic system, as previously demonstrated by others [17,18], and are easier to culture than mouse cells [30]. Initially, we assessed expression levels of cholinergic markers in rat cardiomyocytes (Fig. S3). Overall these data match previous findings in the literature [17,18], and also confirm our data obtained from mouse cardiomyocytes, as we show that rat cardiomyocytes express VAChT, ChAT, M2-AChR and AChE. In adult rat ventricular cells, VAChT is detected as a 75 kDa and a 47 kDa band (Fig. S3A). VAChT expression was also observed in neonatal rat cardiomyocytes isolated from 2-4 day old rats, although in these cells VAChT was observed at an intermediate size band, as previously reported [17]. These differences in molecular mass are probably due to different glycosylation patterns of VAChT molecules [31,32]. ChAT was also detected in both cellular systems (Fig. S3B). In addition, we show that adult and neonatal cardiomyocytes express AChE (Fig. S3C), as shown by others [33].
Our finding that VAChT is presented at the perinuclear region is potentially important since it suggests that cardiomyocytes segregate the exocytotic machinery at the nuclear region, as observed for other granules in these cells [25,34]. In order to confirm this finding, we stained isolated rat adult ventricular myocytes and imaged them by confocal microscopy. As previously observed, VAChT is mainly found at the perinuclear region where it colocalized with recycling vesicles stained with FM1-43 FX (Fig. S3D, arrow). Some sparse VAChT positive vesicles were also observed along the T-tubular system (Fig. S3D, arrowhead). In addition, we observed ChAT immunostaining in the nuclear and intercalated disk regions, as well as in T-tubules, with some presence in the perinuclear area (Fig. S3E). M2-AChR receptor immunostaining was also enriched at the perinuclear region, with some staining found at the T-tubules (Fig.S3F). Corroborating the data obtained from adult ventricular myocytes, neonatal rat cardiomyocytes also showed intense staining and similar subcellular localization for both VAChT (Fig. S3G) and ChAT (Fig. S3H). Taken together, our data from mice and rats confirm the presence of prototypical cholinergic markers in adult and neonatal cardiomyocytes and show for the first time that VAChT positive vesicles segregate to the nuclear periphery along with other cholinergic markers.
In order to gain mechanistic insight into the functionality of this system, and to verify whether its anti-hypertrophic action is conserved between mice and rats we exposed neonatal rat cardiomyocytes to isoproterenol and pyridostigmine. As shown in Fig. 3A-B, pyridostigmine was again effective in preventing the hypertrophic effects of isoproterenol. Importantly, neither pyridostigmine nor ACh (10 μmol/L) alone altered the cell surface area of neonatal rat cardiomyocytes (Fig. 3C-D). To further confirm these findings, we depleted endogenous acetylcholinesterase in rat neonatal cardiomyocytes using specific siRNA targeting AChE. Real-time PCR shows the efficiency of siRNA induced reduction of AChE mRNA (by approximately 80%, Fig. 4A). Treatment of cardiomyocytes with isoproterenol is well known to increase expression levels of atrial natriuretic peptide (ANP) [35] and β-myosin heavy chain (β-MHC) transcripts [36]. Therefore, the expression levels of these transcripts was analysed by qPCR. As expected, we observed a significant upregulation of ANP and β-MHC levels upon isoproterenol stimulation of neonatal rat cardiomyocytes. Importantly, AChE silencing partially abolished the effect of isoproterenol on ANP and β-MHC expression (Fig. 4B-C). Similar findings were observed with a distinct siRNA targeted to AChE (data not shown). Thus these data further support the hypothesis that preservation of endogenously secreted ACh prevents the activation of the hypertrophic signaling cascade induced by isoproterenol in cardiomyocytes.
Figure 3. Cholinesterase inhibition suppresses hypertrophy induced by isoproterenol in rat neonatal cardiomyocytes.
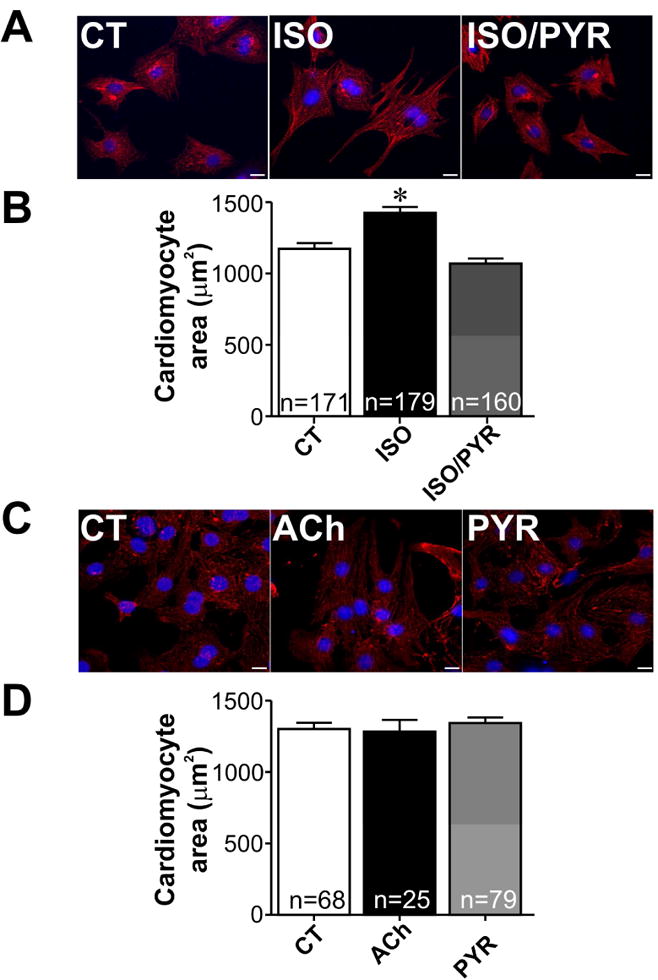
A. Representative immunofluorescence images from α-actinin/DAPI stained neonatal cardiomyocytes in control (CT), isoproterenol (ISO) and isoproterenol/pyridostigmine (ISO/PYR) treated groups. B. Quantification of cardiomyocyte surface area from experiments shown in A. C-D. Representative immunofluorescence images and quantification of cellular surface area from α-actinin/DAPI stained neonatal cardiomyocytes from CT, ACh and PYR treated groups. *p<0.05 when compared to control and ISO/PYR groups. n= number of cells analysed in each group. Scale bar=10 μm.
Figure 4. AChE silencing attenuates isoproterenol induced fetal gene expression in neonatal cardiomyocytes.
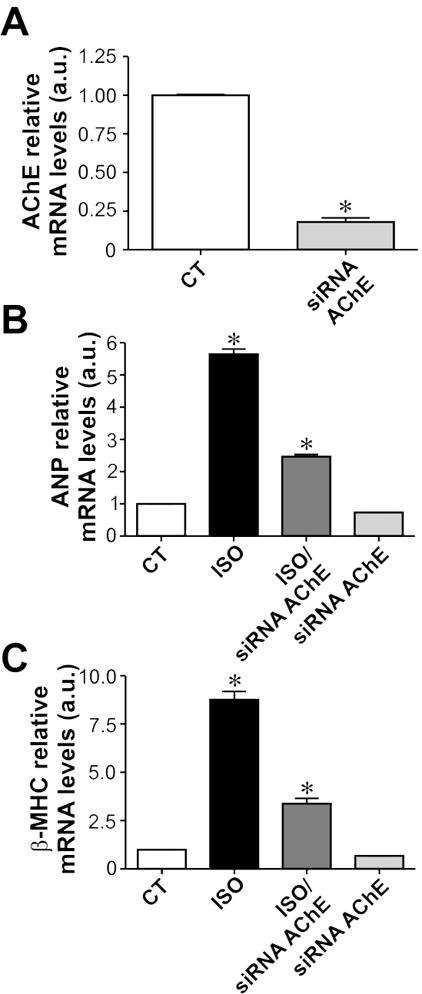
A. siRNA targeting AChE significantly reduces mRNA expression levels of this enzyme. B-C. Isoproterenol treatment significantly increases ANP and β–MHC mRNA levels in neonatal cardiomyocytes, an effect that is partially abolished in cells transfected with siRNA targeting AChE. n= 4 to 8 samples from each group. *p<0.05 when compared to the other groups.
Similarly, the effect of isoproterenol on β-MHC mRNA was also abolished in cardiomyocytes treated with the pharmacological cholinesterase inhibitor, pyridostigmine (Fig 5A). In order to gain mechanistic insight into how cardiomyocyte derived ACh exerts its anti-hypertrophic effects, we investigated whether nitric oxide was involved in this process. Thus, we assessed the effect of L-NAME on β-MHC mRNA levels in neonatal rat cardiomyocytes treated with isoproterenol and pyridostigmine. Inhibition of nitric oxide production with L-NAME significantly blunted pyridostigmine induced β-MHC transcript downregulation in isoproterenol treated cardiomyocytes, indicating a role for NO in the anti-hypertrophic response elicited by intrinsic ACh (Fig. 5A). Moreover, isoproterenol induced ANP upregulation was also abolished in neonatal cardiomyocytes incubated with pyridostigmine (Fig. 5B), supporting our findings obtained with AChE-siRNA. Interestingly, the effect of pyridostigmine in preventing ANP transcript upregulation was inhibited by atropine (Fig. 5B), confirming the finding that available ACh after pyridostigmine treatment uses muscarinic receptors. ANP protein localization was also analysed in the presence of isoproterenol by immunostaining (Fig. 5C). Isoproterenol stimulation resulted in significantly increased perinuclear ANP staining, an effect that was prevented by pyridostigmine. Similar results were observed when isoproterenol treated cells were exposed to neostigmine (Fig. S4). The ability of pyridostigmine to prevent ANP increase under isoproterenol stimulation was lost when cardiomyocytes were exposed to atropine (Fig. 5C).
Figure 5. Cholinesterase inhibition prevents isoproterenol induced remodeling in neonatal rat cardiomyocytes.
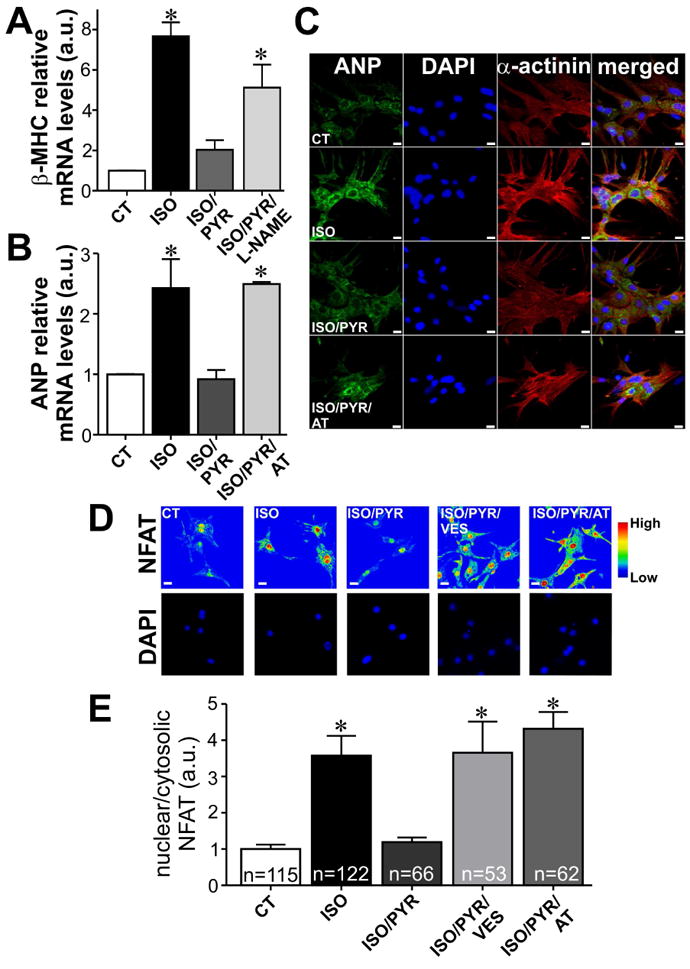
A. Cholinesterase inhibition in neonatal cardiomyocytes prevents isoproterenol (ISO) induced upregulation of β– MHC mRNA, an effect that is lost in the presence of L-NAME. n= 4 to 5 samples from each group. B. qPCR experiments revealed a significant upregulation in ANP transcript levels in ISO treated cells. Pyridostigmine prevented ANP upregulation induced by ISO treatment. The effect of ISO on ANP transcription in pyridostigmine (PYR) treated cells was restored upon exposure to atropine (AT). n= 3 to 5 samples from each group. C. ISO stimulation increased perinuclear ANP staining with levels of staining in ISO/PYR treated cardiomyocytes comparable to that in control cells. The ability of PYR to prevent ANP increase under ISO stimulation was blunted by atropine. D. In control cells, NFAT is found mainly in the cytosol, and upon ISO stimulation this pro-hypertrophic transcription factor translocates to the nucleus. ISO induced NFAT translocation was completely suppressed by PYR. Addition of either vesamicol or atropine significantly inhibited PYR effects on NFAT localization in ISO treated cells. E. Bar graph showing the relative amount of nuclear/cytosolic NFAT. n= number of cells analysed. *p<0.05 when compared to control and ISO/PYR groups. Scale bar=10 μm.
A prominent signaling cascade stimulated by isoproterenol, and whose overactivation is associated with hypertrophic remodeling in cardiomyocytes, is the nuclear factor of activated T cells (NFAT) pathway. In neonatal cardiomyocytes, isoproterenol treatment leads to NFAT translocation to the nucleus, as previously described by others [37]. Fig. 5D-E shows the localization of NFAT in non-treated neonatal rat cardiomyocytes and demonstrates that isoproterenol stimulation leads to increased immunoreactivity for NFAT in the nucleus. The isoproterenol induced nuclear translocation of NFAT was completely prevented by pyridostigmine. Similar finding was observed in isoproterenol treated neonatal cardiomyocytes that were exposed to ACh (Fig. S5). Addition of vesamicol, a specific inhibitor of the VAChT, or atropine, significantly blunted the effects of pyridostigmine on NFAT translocation in isoproterenol treated cells (Fig. 5D-E).
To further assess the possible mechanism through which ACh released by cardiomyocytes prevents isoproterenol induced hypertrophy, we examined Ca2+ transients in isolated adult rat cardiomyocytes kept in culture for 20 hours. Activation of β-adrenergic receptors by isoproterenol in ventricular myocytes is known to enhance the magnitude of the intracellular Ca2+ transient, [Ca2+]i, which contributes to the alterations in intracellular signaling. It is also known that ACh binds to M2-AChR which couple to Gi/0 family of G proteins [38] and inhibit adenylyl cyclase. In many ways, therefore, the effects of ACh are opposed to those of sympathetic activation. Freshly isolated rat ventricular myocytes were plated in laminin-coated dishes and treated with isoproterenol (1 μmol/L). After a 20 hours treatment, cells were loaded with Fluo-4/AM for 30 min, and Ca2+ transient parameters were examined in a confocal microscope. Fig. 6A displays typical line scan fluorescence images recorded from electrically stimulated ventricular myocytes. Chronic activation of β-adrenergic receptor by isoproterenol increases Ca2+ transient amplitude, and this, in part, is responsible for the adrenergic regulation of myocytes (Fig. 6B). Concomitant addition of pyridostigmine (500 μmol/L) to isoproterenol treated myocytes significantly attenuated the increase in Ca2+ transient amplitude induced by isoproterenol thus suggesting an antagonistic effect of pyridostigmine. Collectively, these data support the functionality of ACh synthesis and release machinery in ventricular myocytes, and indicate that non-neuronal ACh can activate muscarinic receptors to counteract the effect of isoproterenol at the calcium signaling level.
Figure 6. Cardiomyocyte cholinergic signaling prevents isoproterenol effects on Ca2+ transients.

A. (Top) Sample Ca2+ transient images recorded in Fluo-4/AM loaded-ventricular myocytes kept in culture for 20h. (Bottom) Ca2+ transient profile. B. Averaged bar graph showing the increase in Ca2+ transient following isoproterenol stimulation. Cholinesterase inhibition by pyridostigmine prevented isoproterenol effects on Ca2+ transient amplitude. n= number of cells analysed. *=p<0.05 when compared to the other groups.
We next investigated whether pyridostigmine could also antagonize the hypertrophic response induced by another adrenergic stimulus, phenylephrine, which is known to activate a Gq signaling pathway through α-adrenergic receptors stimulation [39]. Neonatal rat cardiomyocytes treated with 50 μmol/L phenylephrine for 48 hours presented a 39% increase in cell surface area when compared to control cardiomyocytes (Fig. 7A-B). This hypertrophic effect was partially prevented by pyridostigmine. Moreover, pyridostigmine significantly attenuated phenylephrine-induced upregulation of ANP transcript levels (Fig. 7C). Once again, atropine and L-NAME were effective on preventing pyridostigmine effects on phenylephrine treated neonatal cardiomyocytes. Collectively, our data show that increased availability of released ACh prevents phenylephrine-induced pathological remodeling in cardiomyocytes.
Figure 7. Phenylephrine-induced hypertrophy is partially abolished by cholinesterase inhibition in neonatal cardiac myocytes.
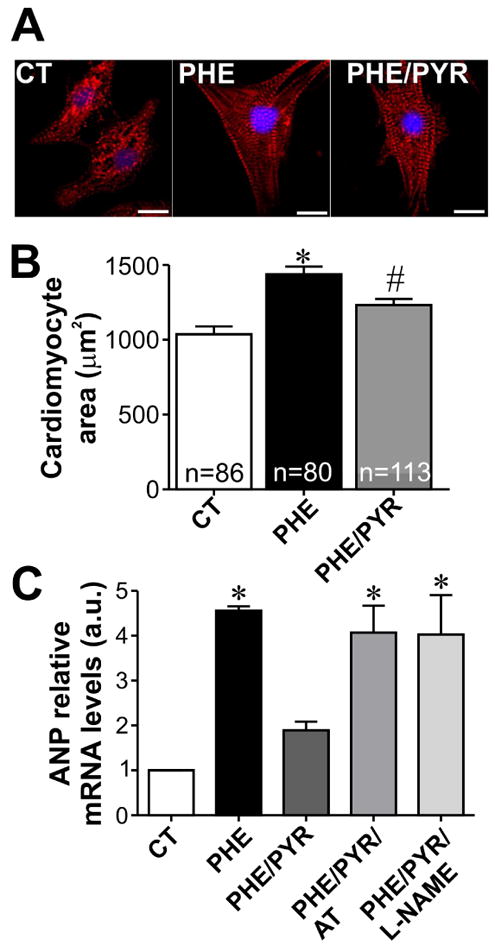
A. Sample images of α-actinin/DAPI stained neonatal rat cardiomyocytes treated for 48 hours with phenylephrine (PHE) or PHE/PYR. Scale bar=10 μm. B. Bar graph showing partial inhibition of PHE induced hypertrophy in PYR-treated neonatal cardiomyocytes. n=number of cells analysed. C. PYR treatment of PHE exposed cardiomyocytes significantly prevents ANP mRNA upregulation. Addition of either atropine or L-NAME blunted PYR effects on ANP mRNA in PHE-exposed cells. n=3-7 samples from each group. *p<0.05 when compared to control and PHE/PYR group. # p<0.05 when compared to control group .
3.4 Cardiomyocyte cholinergic machinery is upregulated by adrenergic stimulation
Previous studies using neuronal cells [40] have implicated protein kinase A as an important modulator of VAChT and ChAT mRNA levels, which indicates that adrenergic stimulus could regulate expression of cholinergic components. In order to better understand how this intrinsic cholinergic system is modulated and to explore at the mechanistic level how cholinergic signaling counteracts the adrenergic effects, we next assessed the expression levels of cholinergic machinery components in response to isoproterenol and phenylephrine. In neonatal rat cardiomyocytes, ChAT, VAChT and M2-AChR mRNA were upregulated by isoproterenol (Fig. 8A) and phenylephrine (Fig. 8B). Similar effect was seen at the protein level for ChAT, VAChT and M2-AChR on isoproterenol treated cardiomyocytes (Fig. 8C-E). We then investigated the effect of isoproterenol on ChAT, VAChT and M2-AChR localization in neonatal cardiomyocytes by performing immunofluorescence experiments. As shown in Fig. 8F, isoproterenol treatment significantly increased ChAT levels at both cytoplasm and nuclei of neonatal cardiomyocytes. Moreover, neonatal cardiomyocytes treated with isoproterenol presented increased VAChT and M2-AChR cytoplasmic staining (Fig. 8G and H). In conclusion, these data show that adrenergic stimulation upregulates the expression levels of cholinergic components in cardiac myocytes.
Figure 8. Adrenergic stimulation upregulates expression of cholinergic machinery in cardiomyocytes.
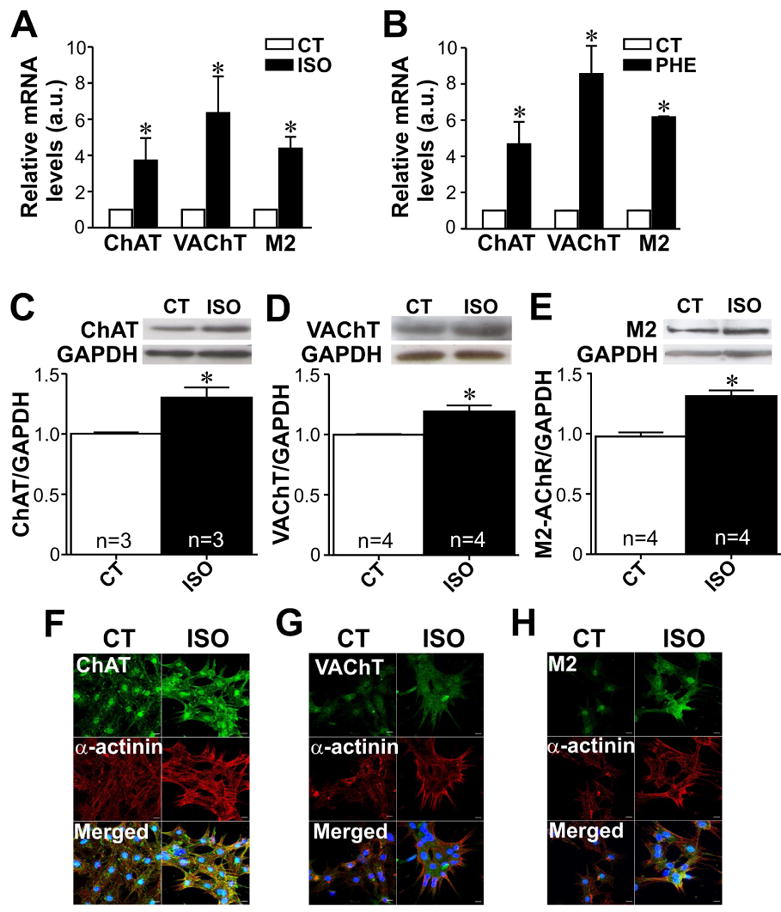
A-B. Either isoproterenol (ISO) or phenyleprine (PHE) upregulates mRNA levels of ChAT, VAChT and M2-AChR in neonatal cardiac cells. n= 2 to 5 samples from each group. C-E. Top, representative western-blot. Bottom, bar graph showing the effect of isoproterenol on ChAT, VAChT and M2-AChR protein levels. n= number of cardiomyocyte samples analysed from each group. F-H. Immunofluorescence images showing ChAT, VAChT and M2-AChR changes upon isoproterenol stimulation. The nucleus was stained with DAPI (blue). *p<0.05 when compared to control. Scale bar=10 μm.
4. Discussion
An intrinsic cardiomyocyte cholinergic system has recently been described[17,18]. However, the physiological role of this cardiomyocyte cholinergic machinery and its significance for cardiac function during normal and disease conditions is unknown. Here, we present for the first time evidence that activation of this novel and unexpected machinery has an important role in cardiomyocyte protection in vitro. These data bring into perspective another level of cholinergic control of cardiac function, which goes beyond the cardiac parasympathetic system, and it is intrinsic to ventricular myocytes.
4.1 Localization of cholinergic proteins in cardiomyocytes
We have extended the observations of two previous reports [17,18], and provide evidence that neonatal and adult cardiomyocytes express prototypical cholinergic markers responsible for ACh synthesis and release. Particularly intriguing is the localization of VAChT to the perinuclear region. Our experiments with vital dye FM1-43 show that this region accumulates recycling vesicles that would be required for the release of ACh. Importantly, cardiomyocytes present all the necessary components required for the exocytosis endocytosis cycle, including SNARE molecules such as SNAP-23, Syntaxin-4, VAMP-1, VAMP-2 and VAMP-3, as well as NSF and its co-factor α-SNAP, the calcium sensor synaptogamin, and the GTPases Rab8 and Rab4 [25]. Interestingly, SNARE proteins, VAMP-1, VAMP-2, VAMP-3 and syntaxin-4 are found at the perinuclear region of cardiomyocytes [25], supporting our findings. ANP granules are also present in the perinuclear region of cardiomyocytes, suggesting that secretory granules and vesicles are sequestered away from the cellular periphery in these cells. This may represent a way to segregate the exocytosis machinery required to secrete proteins and small molecules from the constant changes in Ca2+ levels that occur in cardiomyocytes. It is worth mentioning that Inositol 1,4,5-trisphosphate (InsP3) receptors are found in the perinuclear region [41] where they could provide specific Ca2+ signals to activate exocytosis in these cells. However, we cannot discard at the moment the possibility that at least part of the ACh in vesicles is secreted constitutively. The mechanisms involved in triggering and regulating ACh secretion in cardiomyocytes remain to be identified.
Interestingly, in nerve terminals CHT1 is found mainly in synaptic vesicles and endosomal organelles [42] and only a fraction of the protein is present at the plasma membrane [9]. This does not seem to be the case in cardiomyocytes where the protein is found mainly at the sarcolemma. We note that others have also described similar localization of CHT1 in cardiomyocytes [18].
We have also identified M2-AChR at the perinuclear region of cardiac cells in addition to its localization at the T-tubular system. This finding is in agreement with previous data showing the presence of the Gi subunit at the nuclear periphery of ventricular myocytes [43]. In addition, β-adrenergic receptors have also been found at these nuclear structures [43]. Whether this intracellular pool of receptors represents newly-synthesized and recycling receptors or represents functional receptors is still unknown. The possibility that ACh may play unanticipated roles in regulating gene transcription in the cardiac cells by activating nuclear M2 receptors is intriguing. The functional consequences of this nuclear receptor activation, however, have yet to be elucidated.
4.2 ACh release by cardiomyocytes depends on VAChT activity
Although others have shown that ACh is released by cardiomyocytes [17,18], our data extend these findings by demonstrating the functionality of this phenomenon. By using the production of NO as a biosensor for released ACh, and by using the cardiomyocytes themselves as a detection sensor, we showed that inhibition of cholinesterase significantly increases DAF fluorescence i.e. increases NO levels. This effect is similar to that observed when exogenous ACh is added to freshly isolated ventricular myocytes and it is completely abolished by a muscarinic antagonist and by a specific inhibitor of the CHT1. CHT1 is required in nerve terminals for the synthesis of ACh [44], and our data suggest it has similar roles in cardiomyocytes. It is particularly interesting that HC-3 can block the effects of pyridostigmine as it suggests that no other sources of choline can be utilized in order to generate ACh in cardiac cells.
Our data indicate that ACh secretion by cardiomyocytes requires vesicular mechanisms since ventricular myocytes obtained from mice that express substantially less VAChT (VAChT knockdown mice) showed decreased availability of ACh for activation of DAF fluorescence. This result is consistent with our finding that some of the effects of pyridostigmine or neostigmine are inhibited by vesamicol, a selective VAChT inhibitor. VAChT is part of an ancient transport machinery [45] and as long as it is present in acidic vesicles it will be able to transport ACh [46]. Hence, taken together, these experiments provide strong evidence that ACh release by cardiomyocytes depends on VAChT activity. Moreover, our data show that observed effects of pyridostigmine occur via preservation of ACh. It is unlikely that pyridostigmine would have off target effects as similar results were also observed with a second cholinesterase inhibitor, neostigmine, and in cells transfected with a siRNA targeted to AChE. Moreover anti-hypertrophic effects of pyridostigmine were blocked in the absence of ACh transport to vesicles.
4.3 ACh secreted by cardiomyocytes prevents adrenergic hypertrophic effects
Since proteins involved in the synthesis and release of ACh are found in cardiomyocytes, this raises the question as to whether this intrinsic cholinergic system has any direct functional role in regulating cardiomyocyte activity. Considering that we used a cholinesterase inhibitor to observe the effect of cardiomyocyte-derived ACh, it could be argued that this local cholinergic machinery is not essential to cardiac function. However, it should be noted that although parasympathetic innervations in the ventricle are scarce [47], muscarinic receptors are expressed in ventricular myocytes [48]. Therefore, we speculate that this intrinsic cardiomyocyte cholinergic signaling is part of an adaptive defense mechanism against cardiomyocyte insults. This is further supported by our finding that ventricular myocytes from VAChT knockdown mice present a Ca2+ signaling dysfunction that is restored upon pyridostigmine administration in vivo [10], indicating that lack of ACh release has profound effects on ventricular myocytes.
While our data indicates that basal ACh release by cardiomyocytes is low, it is plausible that under certain circumstances ACh release can increase, and exerts protective effects. Modulation of ChAT, VAChT and CHT1 expression levels has already been demonstrated in neuronal cells [49,50]. Accordingly, cAMP leads to a significant increase in ChAT and VAChT mRNA levels in SN56 cells. Here, we show for the first time that chronic adrenergic stimulation upregulates expression levels of cholinergic components VAChT, ChAT and M2-AChR in rat cardiac cells, suggesting that under this condition ACh release is increased. Supporting this assumption, we show that isoproterenol induced increase in cell surface area is exacerbated when VAChT activity and consequently ACh release are inhibited. Together these findings unravel a role for intrinsic ACh as an in situ negative feedback signal that counter-acts adrenergic effects.
At the mechanistic level, the ability of pyridostigmine to prevent the increase in Ca2+ transient induced by prolonged treatment with isoproterenol is of major importance since alterations in Ca2+ may act as a trigger for cardiac remodeling. Accordingly, ACh secreted locally by cardiomyocytes can act in a paracrine/autocrine manner to antagonize the effects of hyperadrenergic stimulation. Taking this into consideration we speculate that under certain circumstances local cardiac cholinergic system could contribute to the protective effects of the parasympathetic nervous system in the heart. This possibility must be considered in future studies of cardiovascular pathology in which treatment with cholinesterase inhibitor is being evaluated in vivo.
The role of non-neuronal ACh synthesis and release machinery in other cell types has also begun to be elucidated. For example, in the immune system, ACh synthesized by a population of T lymphocytes has been recently shown to be required for the function of the cholinergic anti-inflammatory system [51]. In humans, non-neuronal ACh in alpha cells in the pancreas is important for the secretion of insulin [52]. Here we also show a putative role for cholinergic signaling present in cardiomyocytes by opposing the hypertrophic effects of adrenergic stimulation. It is worth mentioning that the only source of ACh in our experiments is the cardiac cell, as evidenced by the absence of neuronal cells in our culture conditions. Moreover, the use of a broad range of pharmacological agents that interfere with the synthesis and storage of ACh in vesicles support the notion that cardiomyocytes have adapted mechanisms used by neurons to secrete ACh. Given that ACh is an ancient and widespread chemical used for cell-cell communication, it should not come as a surprise to find so many distinct non-neuronal roles for ACh. Future experiments, using conditional knockout mice [23,53] will be necessary to further define the role of this non-neuronal cholinergic system in cardiomyocytes in physiological and pathological alterations in the heart in vivo.
Conclusion
Taken together, our results show that cardiomyocytes express neuronal proteins required for ACh synthesis and release and are capable of secreting ACh. In these cells, VAChT is found mainly at the perinuclear region, where it extensively colocalizes with recycling vesicles that are required for ACh release. ACh secretion by cardiomyocytes depends on both VAChT activity and choline reuptake by CHT1, as observed in neurons. In addition, we show that cardiomyocyte secreted ACh impairs adrenergic hypertrophic signaling, indicating that ACh acting autocrinally has a protective role.
Supplementary Material
Highlights.
Neuronal proteins required for ACh synthesis and release are found in cardiomyocytes
In cardiomyocytes VAChT positive vesicles are mainly found in the perinuclear area
ACh secretion in cardiomyocytes is dependent on VAChT activity and choline reuptake
Cardiomyocyte secreted ACh prevents adrenergic induced hypertrophic signaling Adrenergic stimulation increases expression of cholinergic machinery in cardiac cells
Acknowledgments
This work was supported by grants from NIH R03TW008425 from the Fogarty International Center (SG), Conselho Nacional de Desenvolvimento Científico e Tecnológico (SG, CG, RRR), Fundação de Amparo à Pesquisa do Estado de Minas Gerais (SG, CG and RRR), PRONEX APQ-00746-09 (SG), the Heart and Stroke Foundation of Ontario (MAMP and VFP) and CIHR (RG, VFP and MAMP). C. Rocha-Resende is a recipient of the CNPq PhD fellowship at the Post-graduation Program in Biological Science: Physiology and Pharmacology at UFMG. Ashbeel Roy is a recipient of the Queen Elizabeth II Scholarship in Science and Technology (QEIISST). Robert Gros is supported by a New Investigator Award from the Heart and Stroke Foundation of Canada.
Non-standard abbreviations:
- ACh
acetylcholine
- ChAT
choline acetyltransferase
- VAChT
vesicular acetylcholine transporter
- CHT1
high affinity choline transporter
- VAChT
vesicular acetylcholine transporter
- M2-AChR
type 2 muscarinic acetylcholine receptor
- AChE
acetylcholinesterase
- siRNA
small interfering RNA
Footnotes
Disclosure Statement: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Cibele Rocha-Resende, Email: cibeler@ufmg.br.
Ashbeel Roy, Email: aroy44@uwo.ca.
Rodrigo Resende, Email: rrresende@hotmail.com.
Marina S. Ladeira, Email: marinasladeira@gmail.com.
Aline Lara, Email: lara_bio@yahoo.com.br.
Enéas Ricardo de Morais Gomes, Email: eneasricardo@yahoo.com.br.
Vania F. Prado, Email: vprado@robarts.ca.
Robert Gros, Email: rgros@robarts.ca.
Cristina Guatimosim, Email: cguati@icb.ufmg.br.
Reference List
- 1.Dhein S, van Koppen CJ, Brodde OE. Muscarinic receptors in the mammalian heart. Pharmacol Res. 2001;44:161–82. doi: 10.1006/phrs.2001.0835. [DOI] [PubMed] [Google Scholar]
- 2.Loffelholz K, Pappano AJ. The parasympathetic neuroeffector junction of the heart. Pharmacol Rev. 1985;37:1–24. [PubMed] [Google Scholar]
- 3.Kent KM, Epstein SE, Cooper T, Jacobowitz DM. Cholinergic innervation of the canine and human ventricular conducting system. Anatomic and electrophysiologic correlations Circulation. 1974;50:948–55. doi: 10.1161/01.cir.50.5.948. [DOI] [PubMed] [Google Scholar]
- 4.Nagata K, Ye C, Jain M, Milstone DS, Liao R, Mortensen RM. Galpha(i2) but not Galpha(i3) is required for muscarinic inhibition of contractility and calcium currents in adult cardiomyocytes. Circ Res. 2000;87:903–9. doi: 10.1161/01.res.87.10.903. [DOI] [PubMed] [Google Scholar]
- 5.Katare RG, Ando M, Kakinuma Y, Arikawa M, Yamasaki F, Sato T. Differential regulation of TNF receptors by vagal nerve stimulation protects heart against acute ischemic injury. J Mol Cell Cardiol. 2010;49:234–44. doi: 10.1016/j.yjmcc.2010.03.007. [DOI] [PubMed] [Google Scholar]
- 6.de Castro BM, Pereira GS, Magalhaes V, Rossato JI, De J X, Martins-Silva C, et al. Reduced expression of the vesicular acetylcholine transporter causes learning deficits in mice. Genes Brain Behav. 2009;8:23–35. doi: 10.1111/j.1601-183X.2008.00439.x. [DOI] [PubMed] [Google Scholar]
- 7.de Castro BM, De J X, Martins-Silva C, Lima RD, Amaral E, Menezes C, et al. The vesicular acetylcholine transporter is required for neuromuscular development and function. Mol Cell Biol. 2009;29:5238–50. doi: 10.1128/MCB.00245-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Prado VF, Martins-Silva C, de Castro BM, Lima RF, Barros DM, Amaral E, et al. Mice deficient for the vesicular acetylcholine transporter are myasthenic and have deficits in object and social recognition. Neuron. 2006;51:601–12. doi: 10.1016/j.neuron.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 9.Ribeiro FM, Black SA, Prado VF, Rylett RJ, Ferguson SS, Prado MA. The “ins” and “outs” of the high-affinity choline transporter CHT1. J Neurochem. 2006;97:1–12. doi: 10.1111/j.1471-4159.2006.03695.x. [DOI] [PubMed] [Google Scholar]
- 10.Lara A, Damasceno DD, Pires R, Gros R, Gomes ER, Gavioli M, et al. Dysautonomia due to reduced cholinergic neurotransmission causes cardiac remodeling and heart failure. Mol Cell Biol. 2010;30:1746–56. doi: 10.1128/MCB.00996-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.English BA, Appalsamy M, Diedrich A, Ruggiero AM, Lund D, Wright J, et al. Tachycardia, reduced vagal capacity, and age-dependent ventricular dysfunction arising from diminished expression of the presynaptic choline transporter. Am J Physiol Heart Circ Physiol. 2010;299:H799–H810. doi: 10.1152/ajpheart.00170.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.LaCroix C, Freeling J, Giles A, Wess J, Li YF. Deficiency of M2 muscarinic acetylcholine receptors increases susceptibility of ventricular function to chronic adrenergic stress. Am J Physiol Heart Circ Physiol. 2008;294:H810–H820. doi: 10.1152/ajpheart.00724.2007. [DOI] [PubMed] [Google Scholar]
- 13.Li M, Zheng C, Sato T, Kawada T, Sugimachi M, Sunagawa K. Vagal nerve stimulation markedly improves long-term survival after chronic heart failure in rats. Circulation. 2004;109:120–4. doi: 10.1161/01.CIR.0000105721.71640.DA. [DOI] [PubMed] [Google Scholar]
- 14.Freeling J, Wattier K, LaCroix C, Li YF. Neostigmine and pilocarpine attenuated tumour necrosis factor alpha expression and cardiac hypertrophy in the heart with pressure overload. Exp Physiol. 2008;93:75–82. doi: 10.1113/expphysiol.2007.039784. [DOI] [PubMed] [Google Scholar]
- 15.Kanazawa H, Ieda M, Kimura K, Arai T, Kawaguchi-Manabe H, Matsuhashi T, et al. Heart failure causes cholinergic transdifferentiation of cardiac sympathetic nerves via gp130-signaling cytokines in rodents. J Clin Invest. 2010;120:408–21. doi: 10.1172/JCI39778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huston JM, Tracey KJ. The pulse of inflammation: heart rate variability, the cholinergic anti-inflammatory pathway and implications for therapy. J Intern Med. 2011;269:45–53. doi: 10.1111/j.1365-2796.2010.02321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kakinuma Y, Akiyama T, Sato T. Cholinoceptive and cholinergic properties of cardiomyocytes involving an amplification mechanism for vagal efferent effects in sparsely innervated ventricular myocardium. FEBS J. 2009;276:5111–25. doi: 10.1111/j.1742-4658.2009.07208.x. [DOI] [PubMed] [Google Scholar]
- 18.Rana OR, Schauerte P, Kluttig R, Schroder JW, Koenen RR, Weber C, et al. Acetylcholine as an age-dependent non-neuronal source in the heart. Auton Neurosci. 2010;156:82–9. doi: 10.1016/j.autneu.2010.04.011. [DOI] [PubMed] [Google Scholar]
- 19.Guatimosim S, Amaya MJ, Guerra MT, Aguiar CJ, Goes AM, Gomez-Viquez NL, et al. Nuclear Ca2+ regulates cardiomyocyte function. Cell Calcium. 2008;44:230–42. doi: 10.1016/j.ceca.2007.11.016. [DOI] [PubMed] [Google Scholar]
- 20.Mohler PJ, Schott JJ, Gramolini AO, Dilly KW, Guatimosim S, duBell WH, et al. Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature. 2003;421:634–9. doi: 10.1038/nature01335. [DOI] [PubMed] [Google Scholar]
- 21.Aguiar CJ, Andrade VL, Gomes ER, Alves MN, Ladeira MS, Pinheiro AC, et al. Succinate modulates Ca(2+) transient and cardiomyocyte viability through PKA-dependent pathway. Cell Calcium. 2010;47:37–46. doi: 10.1016/j.ceca.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 22.Guatimosim S, Sobie EA, dos Santos CJ, Martin LA, Lederer WJ. Molecular identification of a TTX-sensitive Ca(2+) current. Am J Physiol Cell Physiol. 2001;280:C1327–C1339. doi: 10.1152/ajpcell.2001.280.5.C1327. [DOI] [PubMed] [Google Scholar]
- 23.Guzman MS, De J X, Raulic S, Souza IA, Li AX, Schmid S, et al. Elimination of the vesicular acetylcholine transporter in the striatum reveals regulation of behaviour by cholinergic-glutamatergic co-transmission. PLoS Biol. 2011;9:e1001194. doi: 10.1371/journal.pbio.1001194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guatimosim C, Romano-Silva MA, Gomez MV, Prado MA. Use of fluorescent probes to follow membrane traffic in nerve terminals. Braz J Med Biol Res. 1998;31:1491–500. doi: 10.1590/s0100-879x1998001100018. [DOI] [PubMed] [Google Scholar]
- 25.Ferlito M, Fulton WB, Zauher MA, Marban E, Steenbergen C, Lowenstein CJ. VAMP-1, VAMP-2, and syntaxin-4 regulate ANP release from cardiac myocytes. J Mol Cell Cardiol. 2010;49:791–800. doi: 10.1016/j.yjmcc.2010.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gill SK, Bhattacharya M, Ferguson SS, Rylett RJ. Identification of a novel nuclear localization signal common to 69- and 82-kDa human choline acetyltransferase. J Biol Chem. 2003;278:20217–24. doi: 10.1074/jbc.M213153200. [DOI] [PubMed] [Google Scholar]
- 27.Kitakaze M, Node K, Komamura K, Minamino T, Inoue M, Hori M, et al. Evidence for nitric oxide generation in the cardiomyocytes: its augmentation by hypoxia. J Mol Cell Cardiol. 1995;27:2149–54. doi: 10.1016/s0022-2828(95)91335-1. [DOI] [PubMed] [Google Scholar]
- 28.Louch WE, Sheehan KA, Wolska BM. Methods in cardiomyocyte isolation, culture, and gene transfer. J Mol Cell Cardiol. 2011;51:288–98. doi: 10.1016/j.yjmcc.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Volz A, Piper HM, Siegmund B, Schwartz P. Longevity of adult ventricular rat heart muscle cells in serum-free primary culture. J Mol Cell Cardiol. 1991;23:161–73. doi: 10.1016/0022-2828(91)90103-s. [DOI] [PubMed] [Google Scholar]
- 30.Zaruba MM, Field LJ. The mouse as a model system to study cardiac regeneration. Drug Discov Today Dis Models. 2008;5:165–71. doi: 10.1016/j.ddmod.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu Y, Edwards RH. Differential localization of vesicular acetylcholine and monoamine transporters in PC12 cells but not CHO cells. J Cell Biol. 1997;139:907–16. doi: 10.1083/jcb.139.4.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Varoqui H, Meunier FM, Meunier FA, Molgo J, Berrard S, Cervini R, et al. Expression of the vesicular acetylcholine transporter in mammalian cells. Prog Brain Res. 1996;109:83–95. doi: 10.1016/s0079-6123(08)62090-7. [DOI] [PubMed] [Google Scholar]
- 33.Nyquist-Battie C, Hagler KE, Love S. Reduced levels of globular and asymmetric forms of acetylcholinesterase in rat left ventricle with pressure overload hypertrophy. Life Sci. 1994;55:653–9. doi: 10.1016/0024-3205(94)00671-7. [DOI] [PubMed] [Google Scholar]
- 34.Peters CG, Miller DF, Giovannucci DR. Identification, localization and interaction of SNARE proteins in atrial cardiac myocytes. J Mol Cell Cardiol. 2006;40:361–74. doi: 10.1016/j.yjmcc.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 35.Morisco C, Zebrowski DC, Vatner DE, Vatner SF, Sadoshima J. Beta-adrenergic cardiac hypertrophy is mediated primarily by the beta(1)-subtype in the rat heart. J Mol Cell Cardiol. 2001;33:561–73. doi: 10.1006/jmcc.2000.1332. [DOI] [PubMed] [Google Scholar]
- 36.Sucharov CC, Mariner PD, Nunley KR, Long C, Leinwand L, Bristow MR. A beta1-adrenergic receptor CaM kinase II-dependent pathway mediates cardiac myocyte fetal gene induction. Am J Physiol Heart Circ Physiol. 2006;291:H1299–H1308. doi: 10.1152/ajpheart.00017.2006. [DOI] [PubMed] [Google Scholar]
- 37.Zou Y, Yao A, Zhu W, Kudoh S, Hiroi Y, Shimoyama M, et al. Isoproterenol activates extracellular signal-regulated protein kinases in cardiomyocytes through calcineurin. Circulation. 2001;104:102–8. doi: 10.1161/hc2601.090987. [DOI] [PubMed] [Google Scholar]
- 38.Lanzafame AA, Christopoulos A, Mitchelson F. Cellular signaling mechanisms for muscarinic acetylcholine receptors. Receptors Channels. 2003;9:241–60. [PubMed] [Google Scholar]
- 39.Fleming JW, Wisler PL, Watanabe AM. Signal transduction by G proteins in cardiac tissues. Circulation. 1992;85:420–33. doi: 10.1161/01.cir.85.2.420. [DOI] [PubMed] [Google Scholar]
- 40.Castell X, Cheviron N, Barnier JV, Diebler MF. Exploring the regulation of the expression of ChAT and VAChT genes in NG108-15 cells: implication of PKA and PI3K signaling pathways. Neurochem Res. 2003;28:557–64. doi: 10.1023/a:1022829608540. [DOI] [PubMed] [Google Scholar]
- 41.Luo D, Yang D, Lan X, Li K, Li X, Chen J, et al. Nuclear Ca2+ sparks and waves mediated by inositol 1,4,5-trisphosphate receptors in neonatal rat cardiomyocytes. Cell Calcium. 2008;43:165–74. doi: 10.1016/j.ceca.2007.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ribeiro FM, ves-Silva J, Volknandt W, Martins-Silva C, Mahmud H, Wilhelm A, et al. The hemicholinium-3 sensitive high affinity choline transporter is internalized by clathrin-mediated endocytosis and is present in endosomes and synaptic vesicles. J Neurochem. 2003;87:136–46. doi: 10.1046/j.1471-4159.2003.01974.x. [DOI] [PubMed] [Google Scholar]
- 43.Boivin B, Lavoie C, Vaniotis G, Baragli A, Villeneuve LR, Ethier N, et al. Functional beta-adrenergic receptor signalling on nuclear membranes in adult rat and mouse ventricular cardiomyocytes. Cardiovasc Res. 2006;71:69–78. doi: 10.1016/j.cardiores.2006.03.015. [DOI] [PubMed] [Google Scholar]
- 44.Ferguson SM, Bazalakova M, Savchenko V, Tapia JC, Wright J, Blakely RD. Lethal impairment of cholinergic neurotransmission in hemicholinium-3-sensitive choline transporter knockout mice. Proc Natl Acad Sci U S A. 2004;101:8762–7. doi: 10.1073/pnas.0401667101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bravo DT, Kolmakova NG, Parsons SM. New transport assay demonstrates vesicular acetylcholine transporter has many alternative substrates. Neurochem Int. 2005;47:243–7. doi: 10.1016/j.neuint.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 46.Prado MA, Reis RA, Prado VF, de Mello MC, Gomez MV, de Mello FG. Regulation of acetylcholine synthesis and storage. Neurochem Int. 2002;41:291–9. doi: 10.1016/s0197-0186(02)00044-x. [DOI] [PubMed] [Google Scholar]
- 47.Hancock JC, Hoover DB, Hougland MW. Distribution of muscarinic receptors and acetylcholinesterase in the rat heart. J Auton Nerv Syst. 1987;19:59–66. doi: 10.1016/0165-1838(87)90145-7. [DOI] [PubMed] [Google Scholar]
- 48.Wei JW, Sulakhe PV. Regional and subcellular distribution of myocardial muscarinic cholinergic receptors. Eur J Pharmacol. 1978;52:235–8. doi: 10.1016/0014-2999(78)90212-1. [DOI] [PubMed] [Google Scholar]
- 49.Berse B, Blusztajn JK. Coordinated up-regulation of choline acetyltransferase and vesicular acetylcholine transporter gene expression by the retinoic acid receptor alpha, cAMP, and leukemia inhibitory factor/ciliary neurotrophic factor signaling pathways in a murine septal cell line. J Biol Chem. 1995;270:22101–4. doi: 10.1074/jbc.270.38.22101. [DOI] [PubMed] [Google Scholar]
- 50.Brock M, Nickel AC, Madziar B, Blusztajn JK, Berse B. Differential regulation of the high affinity choline transporter and the cholinergic locus by cAMP signaling pathways. Brain Res. 2007;1145:1–10. doi: 10.1016/j.brainres.2007.01.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rosas-Ballina M, Olofsson PS, Ochani M, Valdes-Ferrer SI, Levine YA, Reardon C, et al. Acetylcholine-synthesizing T cells relay neural signals in a vagus nerve circuit. Science. 2011;334:98–101. doi: 10.1126/science.1209985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rodriguez-Diaz R, Dando R, Jacques-Silva MC, Fachado A, Molina J, Abdulreda MH, et al. Alpha cells secrete acetylcholine as a non-neuronal paracrine signal priming beta cell function in humans. Nat Med. 2011;17:888–92. doi: 10.1038/nm.2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Martins-Silva C, De J X, Guzman MS, Lima RD, Santos MS, Kushmerick C, et al. Novel strains of mice deficient for the vesicular acetylcholine transporter: insights on transcriptional regulation and control of locomotor behavior. PLoS One. 2011;6:e17611. doi: 10.1371/journal.pone.0017611. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.