

Abstract
Serine hydrolases are one of the largest and most diverse enzyme classes in Nature. Inhibitors of serine hydrolases are used to treat many diseases, including obesity, diabetes, cognitive dementia, and bacterial and viral infections. Nonetheless, the majority of the 200+ serine hydrolases in mammals still lack selective inhibitors for their functional characterization. We and others have shown that activated carbamates, through covalent reaction with the conserved serine nucleophile of serine hydrolases, can serve as useful inhibitors for members of this enzyme family. The extent to which carbamates, however, cross-react with other protein classes remains mostly unexplored. Here, we address this problem by investigating the proteome-wide reactivity of a diverse set of activated carbamates in vitro and in vivo using a combination of competitive and click chemistry (CC)-activity-based protein profiling (ABPP). We identify multiple classes of carbamates, including O-aryl, O-hexafluoroisopropyl (HFIP), and O-N-hydroxysuccinimidyl (NHS) carbamates that react selectively with serine hydrolases across entire mouse tissue proteomes in vivo. We exploit the proteome-wide specificity of HFIP carbamates to create in situ imaging probes for the endocannabinoid hydrolases monoacylglycerol lipase (MAGL) and alpha-beta hydrolase-6 (ABHD6). These findings, taken together, designate the carbamate as a privileged reactive group for serine hydrolases that can accommodate diverse structural modifications to produce inhibitors that display exceptional potency and selectivity across the mammalian proteome.
Serine hydrolases comprise about ~1% of all proteins in most eukaryotic and prokaryotic organisms, including humans,(1) and perform a diverse array of crucial physiological functions including the regulation of bacterial cell wall biosynthesis,(2) viral replication,(3) inflammation,(4) nutrient digestion(5) and metabolism,(6) blood clotting(7) and neuronal signaling.(8, 9) Because of this, serine hydrolases have been the focus of drug discovery programs, which have yielded new medicines to treat human disorders such as obesity,(10) diabetes,(11, 12) dementia associated with Alzheimer's disease(13) and infectious diseases.(3, 14) Despite these advances, selective and in vivo-active inhibitors are still lacking for most of the 200+ serine hydrolases in the human proteome(15) and, consequently many of these enzyme remain poorly characterized in terms of their endogenous biochemical and cellular functions. Thus, there is a great need for new pharmacological tools that target serine hydrolases with good selectivity in vivo, which would serve not only as chemical probes to investigate the function of these enzymes in cell and animal models, but also as potential leads for drug development.(15, 16)
Both reversible and irreversible classes of serine hydrolase inhibitors have been developed that target members of this enzyme family with excellent selectivity.(15) Irreversible inhibitors, however, offer several potential advantages, especially as first-generation chemical probes, in that they can more readily achieve sustained and complete target inhibition in vivo without requiring extensive optimization of their physicochemical and pharmacokinetic properties.(17, 18) Irreversible inhibitors are also straightforward to evaluate using a versatile suite of activity-based protein profiling (ABPP(19-21)) methods to confirm target engagement and proteome-wide selectivity in cell and animal models(22). A primary strategy for developing irreversible serine hydrolase inhibitors exploits the intrinsic reactivity of active-site serine nucleophiles, a unifying feature of this enzyme class, by designing complementary electrophiles, such as activated lactams,(23, 24) lactones,(25) carbamates(26, 27) and ureas.(28, 29) Mechanistically, irreversible serine hydrolase inhibitors mimic the natural ester, amide, or thioester substrates of serine hydrolases, undergoing initial nucleophilic attack to form a covalent-enzyme adduct (Figure 1); but, unlike natural substrates, which form transient intermediates (i.e. acyl-enzyme complexes) that are rapidly hydrolyzed to restore the active enzyme, irreversible inhibitors generate a stable (covalent) inhibitor-enzyme adduct that traps the enzyme in an inactive state. Thus, the design of irreversible serine hydrolase inhibitors has profited from not only a consideration of the affinity of an inhibitor for the enzymes active site, but also from an understanding of the reactivity of the electrophilic group itself.
Figure 1.
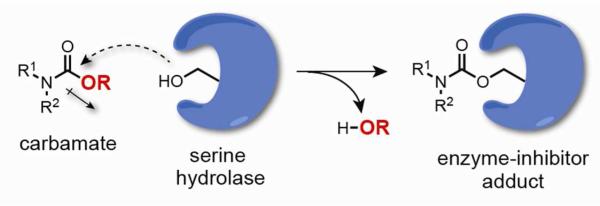
Mechanism of serine hydrolase inhibition by carbamates.
The parameters of affinity and reactivity must be coordinately optimized to create potent and selective irreversible inhibitors. The process of tuning inhibitor electrophilicity, however, often proceeds without a full knowledge of the broader impact on proteome reactivity. It is generally appreciated, at least on a theoretical level, that there is a “ceiling” of acceptable reactivity at which point further enhancing inhibitor potency through increased electrophilicity is counteracted by covalent modification of other proteins. Indeed, non-specific proteome reactivity is considered a major liability for drugs and drug metabolites, as this process can lead to impairments in biochemical pathways and the formation of immunogenic conjugates that promote toxicity.(30) Assuring that covalent inhibitors have maximal proteome-wide selectivity is, therefore, an important goal.(17) Historically, this problem has proven difficult to address, especially in physiologically relevant contexts; but, chemoproteomic technologies, such as competitive(27, 31, 32) and click chemistry (CC)-ABPP,(33, 34) have recently emerged that provide robust and general platforms for evaluating the proteome-wide reactivity of irreversible inhibitors directly in living systems.
Since the extent of non-specific proteome reactivity for the majority of covalent inhibitors remains unknown, we wanted to shed light on this subject by analyzing one of the most versatile chemotypes for serine hydrolases inhibition, the carbamate. We synthesized a focused library of probes bearing carbamates of varying reactivity and a common binding group that directed these agents to the endocannabinoid hydrolases monoacylglycerol lipase (MAGL) and alpha, beta hydrolase-6 (ABHD6). Competitive and CC-ABPP assays revealed that O-aryl, O-hexafluoroisopropyl (HFIP), and O-N-hydroxysuccinimidyl (NHS) carbamates selectively inhibit serine hydrolases in vivo, showing negligible cross-reactivity with other proteins in mouse tissue proteomes. The NHS carbamate was a particularly intriguing case, in that it showed evidence of non-specific proteome reactivity at high concentrations in vitro, but not in mice, where the inhibitor inactivated MAGL with good potency and selectivity. Finally, we took advantage of the remarkably high selectivity that HFIP carbamates displayed for MAGL and ABHD6 to develop an activity-based imaging probe for localizing the activity of these enzymes in mouse and human cells. These studies thus designate the carbamate as a versatile chemotype for creating irreversible inhibitors and functional imaging probes for serine hydrolases that show exquisite proteome-wide selectivity in vivo.
Results and Discussion
Design of a Carbamate Scaffold for Chemoproteomic Analysis
A limited set of O-aryl carbamates was examined previously by competitive and CC-ABPP and found to display promising selectivity for serine hydrolases in proteomes and in vivo.(35) However, whether other types of activated carbamates also exhibited proteome-wide specificity for serine hydrolases remained an important and unanswered question. We first set out to develop a series of chemical probes that could be used to assess the proteome-wide reactivity of various carbamates against a background of serine hydrolase inhibition. We chose to target monoacylglycerol lipase (MAGL) as a representative serine hydrolase because we could capitalize on established structure-activity relationships for carbamate inhibitor design, which included irreversible inactivation by various carbamate subtypes.(36-38) Considering MAGL's prominent role as a regulator of both endocannabinoid and eicosanoid signaling pathways in a number of disease models,(39-43) we also anticipated that our studies could impact the design of next-generation inhibitors and functional probes for studying the biological activities and biomedical relevance of this enzyme.
Our previously optimized carbamate inhibitors of MAGL, namely JZL184(37) and KML29,(36) featured a benzhydrylpiperidine “carbamylating” motif coupled to O-p-nitrophenyl (PNP) and O-HFIP leaving groups, respectively (Figure 2a). Our first goal was to modify these inhibitors with a simplified, common carbamylating group that would retain inhibitory activity against MAGL and also be amenable to incorporation of an alkyne group for future CC-ABPP experiments. On the basis of previous structure-activity relationship studies,(37, 44, 45) we reasoned that the 1-(4,4'-dichlorobenzhydryl)piperazine scaffold might serve this dual purpose, not only because of its predicted potency and selectivity for MAGL over most other serine hydrolases,(37) but also because the chlorine substituents provided a prime location to install a sterically equivalent ethynyl group. We first prepared the HFIP carbamate JW651 (Figure 2a), which could be synthesized in a single step from commercially available materials, and assessed its inhibitory activity against serine hydrolases in the mouse brain proteome in vitro using competitive ABPP with the broad-spectrum, serine hydrolase-directed probe fluorophosphonate-rhodamine (FP-Rh(46)). JW651 was found to potently inhibit MAGL with an IC50 of 38 nM and did not exhibit cross-reactivity with other brain serine hydrolases off-targets up to 10 μM, where a partial blockade of ABHD6 was observed (Figure 2b and Supplementary Table S1). MAGL inhibition by JW651 was confirmed using a 2-arachidonylglycerol (2-AG) substrate assay (IC50 of 4.5 nM; Supplementary Figure S1). JW651 also potently and selectively inhibited MAGL in vivo. C57Bl/6J mice were administered JW651 by oral gavage (1.0 to 40 mg•kg-1), and, after 4 h, mice were sacrificed and their brain tissue harvested to measure serine hydrolase activities and lipid levels. Near complete and selective inhibition of MAGL was observed at doses as low as 5 mg•kg-1, and this profile was maintained to a dose of 40 mg•kg-1, where partial inhibition of ABHD6 was observed (Figure 2c). Consistent with these competitive ABPP data, brain levels of 2-AG in JW651-treated mice were elevated 10-fold with concomitant reductions in the MAGL product arachidonic acid (AA) (Supplementary Figure S1). On the other hand, levels of other brain lipids, such as the fatty acid amide hydrolase (FAAH) substrate N-arachidonoylethanolamide (AEA), were unaffected by JW651 treatment, indicating that FAAH, a common off-target for MAGL inhibitors,(36, 47) was not affected by JW651. These data, taken together, suggested that the simplified 1-(4,4'-dichlorobenzhydryl)piperazine scaffold was well-suited for carbamate reactivity profiling in vitro and in vivo.
Figure 2.

Design and in vitro and in vivo characterization of JW651. (a) Structures of previously developed MAGL inhibitors JZL184 and KML29 leading to the simplified benzhydrylpiperazine scaffold of JW651. (b) In vitro competitive ABPP of JW651 using the serine hydrolase-directed probe FP-Rh in the membrane fraction of the mouse brain proteome. JW651 potently and selectively inhibits FP-Rh labeling of MAGL, with ABHD6 being the only detectable off-target (up to 100 μM JW651). (c) In vivo competitive ABPP of brain proteomes isolated from JW651-treated mice (1.0 – 40 mg•kg–1, p.o.) 4 h after administration. JW651 completely inhibits MAGL in the brain at doses as low as 5 mg•kg–1 (See also Supplementary Figure S1 for endocannabinoid levels in the brain for JW651 treated mice).
In Vitro Evaluation of Carbamate Chemotypes
Having established that JW651 maintains activity and selectivity towards MAGL, we next prepared a series of 1-(4,4'-dichlorobenzhydryl)piperazine probes bearing alternative carbamate leaving groups (Figure 3a). O-aryl carbamates are known to inhibit numerous serine hydrolases,(27) and we therefore synthesized unsubstituted O-phenyl carbamate JW843 and its more reactive PNP counterpart (JW842). We also prepared O-trifluoroethyl (TFE) carbamate JW814 as a less activated variant of JW651. Finally, we prepared NHS carbamate MJN110, the NHS leaving group of which we postulated might mimic the head group of 2-AG and possibly engage in favorable H-bonding interactions with His131 and Tyr204 within MAGL's active site.(45)
Figure 3.

Development of carbamate probes and profiling their reactivity against brain serine hydrolases by competitive ABPP. (a) Structures of parent (R = Cl) and clickable (R = alkyne) carbamate probes with varying leaving groups. (b) In vitro competitive ABPP of mouse brain showing that leaving groups have a significant effect on serine hydrolase reactivity. See also Supplementary Table S1 for calculated IC50 values for MAGL, ABHD6 and FAAH.
Each carbamate was initially evaluated by competitive ABPP with FP-Rh in mouse brain proteomes in vitro (Figure 3b, Supplementary Table S1). As expected, the PNP (JW842) and HFIP (JW651) carbamates inhibited MAGL (and ABHD6) with much greater potency than their less reactive phenyl (JW843) and trifluoroethyl (JW814) carbamate counterparts, respectively. These data underscore the importance of carbamate electrophilicity for MAGL inhibition. Notably, the NHS carbamate MJN110 displayed the highest MAGL and ABHD6 inhibitory activity and was surprisingly selective, with LYPLA1/2 being the only other off-targets observed below 100 μM.
While competitive ABPP with FP-Rh is ideal for monitoring inhibition across the serine hydrolase class, this strategy does not provide a complete picture of reactivity across the entire proteome. Click chemistry (CC)-ABPP (33, 34) is better suited for this purpose, as it gives a direct readout of covalent probe-protein interactions through the use of an alkyne-bearing inhibitor, which can be detected by conjugation with a rhodamine-azide (Rh-N3) reporter tag using copper-catalyzed azide-alkyne cycloaddition chemistry.(48) We therefore next prepared clickable analogues of each carbamate agent by replacing a single chloro group with an alkyne group. Mouse brain proteomes were then treated with varying concentrations of each click probe for 30 min at 37 °C, split into two fractions and reacted with either FP-Rh to assess serine hydrolase activity or Rh-N3 to provide a more comprehensive portrait of proteome reactivity. Proteomes treated with FP-Rh revealed that the exchange of the chloro substituent for the alkyne had only modest effects on probe activity against MAGL, ABHD6, and other serine hydrolases, although a slight increase in inhibitory activity against FAAH was observed for JW842yne (compare Figure 3b and 4a). CC-ABPP of proteomes confirmed that MAGL and ABHD6 were indeed the primary targets of each carbamate (Figure 4b), with JW842yne also displaying some cross-reactivity with FAAH, as expected from the competitive ABPP analysis (Figure 4a). Click probes based on the most potent carbamate inhibitors (JW651yne and MJN110yne) could detect MAGL reactivity at probe concentrations as low as 10 nM (Figure 4b). Most revealing were differences in the background labeling profiles observed at high concentrations of each probe. MJN110yne showed the highest degree of background proteome reactivity that became apparent at 10 μM probe and more dramatic at 100 μM probe. Notably, this degree of background proteome reactivity could not have been predicted from its competitive ABPP profiles, which were limited to analyzing serine hydrolases (Figure 4a). Coomassie staining of the brain proteome gels revealed that MJN110yne's off-target reactivity aligned, at least in some cases, with abundant proteins (Supplementary Figure S2), which may indicate that the broader proteome reactivity reflects low-level modification of highly expressed proteins. Other carbamates showed much more limited background proteome reactivity (even at concentrations up to 100 μM) that also corresponded to weak signals comigrating with highly abundant brain proteins.
Figure 4.

Competitive and CC-ABPP of clickable carbamate probes in vitro. (a, b) In vitro competitive ABPP (a) and CC-ABPP (b) of mouse brain membrane proteomes treated with the indicated concentration of clickable carbamate probes. Each inhibitor labels MAGL and ABHD6—and in the case of JW842yne, FAAH—to varying degrees and also shows a limited number of off-targets (denoted with red asterisks and brackets in (b)), which were not detected by competitive ABPP with the FP-Rh probe (a). See also, Supplementary Figure S2 for Coomassie stain of brain proteomes showing that these off-targets comigrate with highly abundant proteins. Note further that MJN110yne exhibits a more extensive off-target labeling profile at high (10 μM or greater) concentrations of probe.
In Vivo Evaluation of PNP, HFIP and NHS Carbamates
To obtain a more physiologically relevant portrait of inhibitor-proteome interactions, we orally administered the three most MAGL-active clickable probes, JW842yne, JW651yne and MJN110yne (1-40 mg•kg–1, 4 h), to mice and analyzed their brain and liver proteomes by competitive and CC-ABPP. Competitive ABPP experiments showed that JW651yne and MJN110yne substantially inhibited MAGL in the brain at doses as low as 5.0 mg•kg–1, while JW842yne proved to be less active (Figure 5a). The only detectable off-target activity for JW651yne and MJN110yne was ABHD6. JW842yne, on the other hand, produced strong inhibition of FAAH at doses of 10 mg•kg–1 or greater. CC-ABPP provided the expected complementary portrait of direct labeling of MAGL, ABHD6, and FAAH by each probe, although low-level cross-reactivity with 60 kDa and 20-25 kDa proteins was observed at higher doses (≥ 10 mg•kg–1) of MJN110yne (Figure 5b). Based on the in vitro competitive ABPP profile for MJN110yne (Figure 5a), we believe that these cross-reactivities correspond to FAAH and LYPLA1/2, respectively. That labeling of FAAH and LYPLA1/2 was not detected in MJN110yne-treated mice by competitive ABPP further indicates that these cross-reactivities likely constitute low target occupancy events in vivo. Importantly, our CC-ABPP experiments did not reveal broader background proteome labeling in vivo for MJN110yne or the other carbamate probes across the entire tested dose range (Figure 4a).
Figure 5.

Competitive and CC-ABPP of clickable carbamate probes in vivo. (a–d) Competitive ABPP of brain (a) and liver (c) membrane proteomes isolated from mice treated with indicated doses of each carbamate (1.0 – 40 mg•kg–1, p.o.). CC-ABPP for the same brain (b) and liver (d) proteomes derived from vehicle- or inhibitor-treated mice. Each inhibitor shows clear labeling of MAGL and ABHD6 in the brain, while FAAH is labeled by JW842yne and, to lesser extent, by MJN110yne. Liver profiles reveal differing degrees of off-target cross-reactivities for each carbamate with JW651yne exhibiting the highest selectivity across the proteome.
We also examined liver proteomes from probe-treated mice to assess cross-reactivity with carboxylesterases (CESs), a sub-family of 50-65 kDa serine hydrolases that are common off-targets for carbamate inhibitors.(27) Competitive ABPP confirmed potent inactivation of MAGL in liver by JW651yne and MJN110yne, but not JW842yne, with little or no evidence of inhibition of CESs by any of the probes (Figure 5c). CC-ABPP, however, revealed that each carbamate probe showed some degree of cross-reactivity with CESs, with JW842yne exhibiting the strongest CES labeling profile, followed by MJN110yne, and JW651yne displaying very limited CES interactions (Figure 5d). Once again, because these CES interactions were not detected by competitive ABPP, we suspect that they reflect low or partial target engagement events or, alternatively, the inhibition of lower abundance targets that are obscured by co-migrating serine hydrolases in competitive ABPP gels.
Taken together, our in vivo assessment revealed that clickable carbamate probes show excellent overall selectivity for MAGL in mice, exhibiting only modest and incomplete cross-reactivity with a handful of additional serine hydrolases in brain and liver and very little, if any evidence of broader reactivity across the proteome. The HFIP carbamate, in particular, was distinguished by high potency and exquisite selectivity for MAGL and ABHD6. We therefore next explored whether the HFIP carbamate could be modified to create imaging probes for visualizing the activity of these endocannabinoid hydrolases in living cells.
Development of an In situ Imaging Probe for MAGL and ABHD6
MAGL and ABHD6, despite both exhibiting 2-AG hydrolytic activity, are predicted to diverge in their subcellular distributions, as ABHD6 is an integral membrane enzyme with a single N-terminal transmembrane domain and MAGL is a soluble enzyme that appears to peripherally associate with membranes. Indeed, initial immunofluorescence studies have indicated that MAGL and ABHD6 are localized to the pre-(49) and post-synaptic(50) regions of neurons in the mouse brain, respectively. Tools that could more precisely define the distribution of MAGL and ABHD6 activity in cells could further help to differentiate the distinct functions of these enzymes.
The remarkable specificity displayed by HFIP carbamates for MAGL and ABHD6 suggested that simple tethering of this reactive group to a hydrophobic fluorophore could engender construction a dual-enzyme imaging probe for these endocananbinoid hydrolases. Based on previous success using the BODIPY fluorophore to create activity-based imaging probes,(51, 52) we synthesized the fluorescent HFIP carbamate probe JW912 (Figure 6a). To confirm JW912's activity and selectivity for MAGL and ABHD6, we first treated mouse brain homogenates with increasing concentrations of the fluorescent probe and divided each sample into two parts, one that was directly quenched with SDS loading buffer, and the other that was treated with FP-Rh. Both samples were then analyzed by gel-based ABPP, which revealed selective labeling and inhibition of MAGL and ABHD6 at JW912 concentrations as low as 100 nM (Figure 6b, c). Importantly, no significant cross-reactivity was observed by either competitive (imaging of the Rh fluorophore) or direct (imaging of the BODIPY fluorophore) ABPP up to 10 μM JW912.
Figure 6.

Development of an activity-based imaging probe for MAGL and ABHD6. (a) Structure of JW912 imaging probe highlighting the HFIP carbamate group, which directs this probe to MAGL and ABHD6, and the BOPIDY fluorophore, which allows visualization. (b) Competitive ABPP for JW912 showing selective inhibition of MAGL and ABHD6 over other serine hydrolases in the brain. (c) BODIPY channel gel image revealing that JW912 selectively labels MAGL and ABHD6 at concentrations below 10 μM across the proteome.
While JW912 did not discriminate between MAGL and ABHD6, we reasoned that the respective subcellular distributions of these enzymes could be imaged with this single probe by competition with selective inhibitors. We first set out to test whether JW912 could image MAGL and ABHD6 by studying cell lines that endogenously express one, but not both of these enzymes. Analysis of published gene expression(53) and activity-based proteomic(54) data sets indicated that H29 and Neuro2A cells selectively express MAGL and ABHD6, respectively. Consistent with these predicted expression profiles, we observed selective labeling of proteins in H29 and Neuro2A cells by JW912 (100 nM, 2 h) with molecular masses that matched MAGL and ABHD6, respectively, and, more importantly, these labeling events were selectively blocked by the MAGL and ABHD6 inhibitors JW651 and KT195 (54), respectively (Figure 7a, b).
Figure 7.

In situ activity-based imaging of MAGL and ABHD6 in cancer cells with JW912. (a-g) Competitive ABPP (FP-Rh) and direct labeling (BODIPY) profiles for H29 (a), Neuro2A (c), and PC3 (f) cells labeled with JW912 (100 nM) following treatment with either DMSO, KT195 (10 nM), or JW651 (10 nM). Confocal imaging of H29 (b), Neuro2A (d), and PC3 (g) cells treated with JW912 ± JW651 (10 nM), KT195 (10 nM) or both to inhibit labeling of MAGL, ABHD6 or both, respectively. (e) Strategy used to selectively image either MAGL or ABHD6 with JW912 in PC3 cells which express both enzymes. Scale bars = 5 μm.
We next imaged MAGL and ABHD6 activities in H29 (Figure 7c) and Neuro2A (Figure 7d) cells, respectively, using confocal fluorescence microscopy. Using the BODIPY and DAPI channels to detect JW912 staining and nuclear DNA, respectively, we observed prominent JW912-dependent labeling localized to intracellular membrane compartments in both H29 and Neuro2A cells. These signals were completely blocked by pretreatment with JW651 and KT195, respectively. Conversely, pretreatment of H29 cells with KT195 or Neuro2A cells with JW651 had no effect on JW912-labeling. These results demonstrate that JW912 can be used to image both MAGL and ABHD6 activity in cells.
Of course, many cell types express both MAGL and ABHD6, and, while JW912 reacts with both of these enzymes, we reasoned that their respective subcellular distributions could potentially be imaged with this single probe by competition with selective inhibitors (Figure 7e). Using the human prostate cancer cell line PC3 —which expresses both enzymes(55)—as a model system, we first treated cells with either DMSO, JW651 (10 nM), KT195 (10 nM), or both inhibitors for 2 h and then with JW912 (100 nM) for an additional 2 h. Competitive and direct ABPP profiles confirmed that ABHD6 or MAGL could be selectively labeled with JW912 by pre-blocking with the complementary inhibitor (Figure 7f). Note here that the relative gel migration patterns for human MAGL and ABHD6 differ slightly from those observed in mouse tissues, with human ABHD6 migrating between (rather than below) the two MAGL isoforms. We next imaged PC3 cells treated under these same conditions using confocal fluorescence microscopy (Figure 7g). We observed BODIPY fluorescence on intracellular membranes distributed throughout PC3 cells treated with JW912, and these signals were blocked by pre-treatment with both JW651 and KT195, but not by either inhibitor alone. Cells pretreated with KT195 or JW651 both exhibited punctate JW912-staining patterns, indicating that MAGL and ABHD6 might partially overlap in their respective subcellular distributions in prostate cancer cells. While further studies will be needed to define the types of membranous structures that harbor ABHD6 and MAGL, the predominant perinuclear staining pattern of ABHD6 appears to be consistent with distribution to the endoplasmic reticulum, while the more diffuse, punctate staining for MAGL may indicate additional localization to endosomal or other intracellular organelles.(51)
Conclusions
Our aim in this study was to determine the proteome-wide reactivity profiles for a series of irreversible serine hydrolase inhibitors based on the carbamate electrophile. Using a combination of competitive and CC-ABPP, we found that each of the carbamates tested maintained good selectivity for inhibiting serine hydrolases and, in general, displayed very limited reactivity across the greater mammalian proteome. This selectivity profile was especially evident in vivo. While we do not yet fully understand why the background proteome reactivity observed for the NHS carbamates was avoided in vivo, we believe that this finding underscores the importance of evaluating the selectivity of enzyme inhibitors directly in living systems, where protein complexes and other subcellular structures are preserved. That the NHS carbamate MJN110yne also showed excellent potency for inhibiting MAGL in mice designates this compound, and its parent agent MJN110, as worthy of future investigation as a pharmacological probe for studying endocannabinoid pathways in vivo. The HFIP carbamate stood out in terms of showing exceptional selectivity for the 2-AG hydrolases MAGL and ABHD6 and minimal cross-reactivity with other serine hydrolases or the greater mammalian proteome in vitro and in vivo. We leveraged the specificity of HFIP carbamates to create a fluorescent activity-based imaging probe for MAGL and ABHD6 and used this reagent JW912, in combination with selective inhibitors, to visualize the subcellular distributions of endocannabinoid hydrolases in prostate cancer cells. We should also note that tailored activity-probes like JW912 also provide a means to detect enzyme activities by gel-based ABPP that may be difficult to visualize using broad-spectrum probes to the presence of more abundant, co-migrating enzymes (e.g., see the ABHD6 signals in the FP-Rh and JW912 profiles for Neuro2A cells; Figure 7b). JW912 thus joins a growing collection of small-molecule probes that can be used to image enzyme activities with good selectivity in living systems.(51, 56, 57)
Projecting forward, our study establishes benchmark reactivity profiles for various carbamates, which should guide future efforts to develop selective, irreversible inhibitors and activity probes for serine hydrolases. Recent work from our lab and others has also identified other chemotypes, such as heterocyclic ureas(28, 58-61) and activated lactam,(24) for the irreversible inhibition of serine hydrolases, and it would be interesting, in the future, to more thoroughly evaluate these alternative scaffolds by competitive and CC-ABPP. From a methodological perspective, our results emphasize the complementary value of competitive and CC-ABPP methods for evaluating irreversible inhibitors. Competitive ABPP can be used to quantify the extent of inhibition of enzymes in proteomes and living systems, while CC-ABPP enables a global assessment of the reactivity of inhibitors across the proteome. Together, competitive and CC-ABPP provide a general platform to determine target engagement and proteome-wide specificity for any irreversible probe in a wide range of biological systems. In this manner, irreversible inhibitors can be optimized to ensure that they display the requisite potency and selectivity for use as chemical probes and possibly even therapeutic agents.
Methods
Materials
All commercially available chemicals were obtained from Sigma-Aldrich, Acros, Fisher, Fluka, or Maybridge and were used without further purification, except where noted. FP-Rh and JZL184(37) were prepared according the previously reported methods. LC/MS lipid standards were purchased from Cayman Chemical. Detailed synthetic procedures and experimental data for JW842, JW842yne, JW843, JW843yne, JW814, JW814yne, JW651, JW651yne, MJN110 and MJN110yne are provided in the Supplemental Information.
In vitro competitive activity-based protein profiling
Proteomes (50 μL, 1.0 mg/ml total protein concentration) were preincubated with either DMSO or 1 – 100,000 nM concentrations of inhibitors at 37 °C. After 30 min, FP-Rh (1.0 μL, 50 μM in DMSO) was added and the mixture was incubated for another 30 min at room temperature. Reactions were quenched with SDS loading buffer (17 μL - 4X) and run on SDS-PAGE. Following gel imaging, serine hydrolase activity was determined by measuring fluorescent intensity of gel bands corresponding to MAGL, ABHD6 and FAAH using ImageJ 1.43u software.
Click chemistry-ABPP
Brain membrane and liver membrane proteomes from either naïve (in vitro) or inhibitor-treated (in vivo) mice were diluted to 1.0 mg/mL prior to use. Tissues were harvested and prepared for analysis according to previously reported methods.(36) Note: It is beneficial to remove the soluble fraction from liver proteomes due to its adverse effects on the click reaction. Using previously developed methods,(35) Rh-N3 was conjugated to each alkyne probes for in-gel analysis. Briefly, CuSO4 (1.0 μL/reaction, 50 mM in H2O), TBTA (3.0 μL/reaction, 1.7 mM in DMSO:t-BuOH [1:4]), TCEP (1.0 μL/reaction, 50 mM in H2O [freshly prepared]) and Rh-N3 (1.0 μL/reaction, 1.25 mM in DMSO) were premixed. This click reagent mixture (6.0 μL total volume) was immediately added to each proteome (50 μL, 1.0 mg/mL protein concentration) and the reaction was stirred by briefly vortexing. After 1 h at room temperature, reactions were diluted with 4X SDS loading buffer (17 μL) and resolved by SDS-PAGE.
In vivo administration of carbamate probes
Carbamate inhibitors were administered to C57Bl/6J mice in a vehicle of either saline:emulphor:ethanol (18:1:1) for intraperitoneal injections or PEG300 (Fluka) for administration by oral gavage. After the indicated dosing regimens, the mice were anesthetized using isoflurane and sacrificed by cervical dislocation and tissues were harvested and flash frozen in liquid N2. Tissue proteomes were prepared for competitive and CC-ABPP using the same protocol which has been previously described.(36) The studies were performed with the approval of the Institutional Animal Care and Use Committee at The Scripps Research Institute in accordance with the Guide for the Care and Use of Laboratory Animals.
In situ JW912 treatment of cancer cells
JW912 was dissolved in DMSO and diluted into media or buffer prior to cell or proteome treatment, respectively. For in vitro treatment, final DMSO concentration was 4%. For in situ treatments of cells for ABPP, 2 × 106 cells were seeded in 6 cm dishes (100% confluency) 24 h prior to JW912 pretreatment with or without JW651 and KT195 (in DMSO at 0.1% final concentration) in serum-free media (3 ml) for the designated time before harvesting cells for ABPP.
Competitive ABPP Experiments
For ABPP experiments, cell lysate and tissue proteomes were treated with 1 XM FP-Rh for 30 min at room temperature (50 Xl total reaction volume). Reactions were quenched with one volume of standard 4x SDS/PAGE loading buffer (reducing), separated by SDS/PAGE (10% acrylamide), and visualized in-gel with a Hitachi FMBio IIe flatbed fluorescence scanner (MiraiBio). For experiments involving a preincubation with JW912 in the presence or absence of the non-fluorescent competitor JW651 and KT195, the reactions were prepared without FP-Rh. JW912 were added at the indicated concentration and incubated for the indicated time at 37°C. FP-Rh was then added and the reaction was carried out exactly as described above.
Fluorescence Microscopy Assays
For fluorescence microscopy experiments, 5 x 105 cells were plated on glass coverslips in media containing 10% FBS at 37°C under 5% CO2 and allowed to settle overnight. Cells were washed with PBS twice prior to in situ treatment with JW651 or KT195 at the indicated concentration for 4 h in serum-free media. Following pre-treatment with JW651, KT195 or DMSO control, cells were washed twice with PBS and subsequently treated with the indicated concentration of JW912 in fresh media. After incubation for the indicated time, cells were washed twice with PBS and fixed at 25°C for 15 min in 3.7% (w/v) paraformaldehyde in PBS. Fixed cells were then stained with DAPI (Sigma Aldrich), Far-red Wheat-germ agglutinin (Invitrogen) and/or ER-Tracker Red (Invitrogen) according to manufacturers recommendations for 1 h at 25°C. For image acquisition, processed coverslips were mounted on microscope slides and confocal images were acquired using a fully tunable, filter-based emission collection system (Bio-Rad(Zeiss) Radiance 2100 Rainbow laser scanning confocal microscope) using identical acquisition parameters within experiments. Post-acquisition processing (multi-channel overlay, scale bar addition) was performed using ImageJ software (NIH).
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health Grants DA017259 (to B.F.C.), DA009789 (to B.F.C.), and DA032541 (to M.J.N.), the Toni Rosenberg Memorial Fellowship (to J.W.C.) and the Skaggs Institute for Chemical Biology. B.F.C. is a founder of Abide Therapeutics and a member of its scientific advisory board. This work was supported in part by a grant from Abide Therapeutics.
Footnotes
Supporting Information
Synthetic procedures, 1H NMR, 13C NMR and HRMS data are provided for the compounds reported herein. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Long JZ, Cravatt BF. The metabolic serine hydrolases and their functions in mammalian physiology and disease. Chem. Rev. 2011;111:6022–6063. doi: 10.1021/cr200075y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Macheboeuf P, Contreras-Martel C, Job V, Dideberg O, Dessen A. Penicillin Binding Proteins: key players in bacterial cell cycle and drug resistance processes. FEMS Microbiol. Rev. 2006;30:673–691. doi: 10.1111/j.1574-6976.2006.00024.x. [DOI] [PubMed] [Google Scholar]
- 3.Vermehren J, Sarrazin C. New hepatitis C therapies in clinical development. European journal of medical research. 2011;16:303–314. doi: 10.1186/2047-783X-16-7-303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nomura DK, Morrison BE, Blankman JL, Long JZ, Kinsey SG, Marcondes MCG, Ward AM, Hahn YK, Lichtman AH, Conti B, Cravatt BF. Endocannabinoid Hydrolysis Generates Brain Prostaglandins That Promote Neuroinflammation. Science. 2011;334:809–813. doi: 10.1126/science.1209200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Whitcomb DC, Lowe ME. Human pancreatic digestive enzymes. Digestive diseases and sciences. 2007;52:1–17. doi: 10.1007/s10620-006-9589-z. [DOI] [PubMed] [Google Scholar]
- 6.Bongers J, Lambros T, Ahmad M, Heimer EP. Kinetics of dipeptidyl peptidase IV proteolysis of growth hormone-releasing factor and analogs. Biochimica et biophysica acta. 1992;1122:147–153. doi: 10.1016/0167-4838(92)90317-7. [DOI] [PubMed] [Google Scholar]
- 7.Davie EW, Ratnoff OD. Waterfall Sequence for Intrinsic Blood Clotting. Science. 1964;145:1310–1312. doi: 10.1126/science.145.3638.1310. [DOI] [PubMed] [Google Scholar]
- 8.Lane RM, Potkin SG, Enz A. Targeting acetylcholinesterase and butyrylcholinesterase in dementia. Int. J. Neuropsychopharmacol. 2006;9:101–124. doi: 10.1017/S1461145705005833. [DOI] [PubMed] [Google Scholar]
- 9.Ahn K, McKinney MK, Cravatt BF. Enzymatic pathways that regulate endocannabinoid signaling in the nervous system. Chem. Rev. 2008;108:1687–1707. doi: 10.1021/cr0782067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nelson RH, Miles JM. The use of orlistat in the treatment of obesity, dyslipidaemia and Type 2 diabetes. Expert opinion on pharmacotherapy. 2005;6:2483–2491. doi: 10.1517/14656566.6.14.2483. [DOI] [PubMed] [Google Scholar]
- 11.Thornberry NA, Weber AE. Discovery of JANUVIA (Sitagliptin), a selective dipeptidyl peptidase IV inhibitor for the treatment of type 2 diabetes. Current topics in medicinal chemistry. 2007;7:557–568. doi: 10.2174/156802607780091028. [DOI] [PubMed] [Google Scholar]
- 12.Karagiannis T, Paschos P, Paletas K, Matthews DR, Tsapas A. Dipeptidyl peptidase-4 inhibitors for treatment of type 2 diabetes mellitus in the clinical setting: systematic review and meta-analysis. BMJ. 2012;344 doi: 10.1136/bmj.e1369. [DOI] [PubMed] [Google Scholar]
- 13.Herrmann N, Chau SA, Kircanski I, Lanctôt KL. Current and Emerging Drug Treatment Options for Alzheimer's Disease: A Systematic Review. Drugs. 2011;71:2031–2065. doi: 10.2165/11595870-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 14.Kluge AF, Petter RC. Acylating drugs: Redesigning natural covalent inhibitors. Current opinion in chemical biology. 2010;14:421–427. doi: 10.1016/j.cbpa.2010.03.035. [DOI] [PubMed] [Google Scholar]
- 15.Bachovchin DA, Cravatt BF. The pharmacological landscape and therapeutic potential of serine hydrolases. Nat. Rev. Drug Discov. 2012;11:52–68. doi: 10.1038/nrd3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moellering Raymond E., Cravatt Benjamin F. How Chemoproteomics Can Enable Drug Discovery and Development. Chem. Biol. 2012;19:11–22. doi: 10.1016/j.chembiol.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johnson DS, Weerapana E, Cravatt BF. Strategies for discovering and derisking covalent, irreversible enzyme inhibitors. Future Med. Chem. 2010;2:949–964. doi: 10.4155/fmc.10.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Singh J, Petter RC, Baillie TA, Whitty A. The resurgence of covalent drugs. Nat Rev Drug Discov. 2011;10:307–317. doi: 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
- 19.Liu Y, Patricelli MP, Cravatt BF. Activity-based protein profiling: The serine hydrolases. Proc. Natl. Acad. Sci. U.S.A. 1999;96:14694–14699. doi: 10.1073/pnas.96.26.14694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Berger AB, Vitorino PM, Bogyo M. Activity-based protein profiling: applications to biomarker discovery, in vivo imaging and drug discovery. Am J Pharmacogenomics. 2004;4:371–381. doi: 10.2165/00129785-200404060-00004. [DOI] [PubMed] [Google Scholar]
- 21.Cravatt BF, Wright AT, Kozarich JW. Activity-based protein profiling: from enzyme chemistry to proteomic chemistry. Annu. Rev. Biochem. 2008;77:383–414. doi: 10.1146/annurev.biochem.75.101304.124125. [DOI] [PubMed] [Google Scholar]
- 22.Simon GM, Niphakis MJ, Cravatt BF. Determining target engagement in living systems. Nature chemical biology. 2013;9:200–205. doi: 10.1038/nchembio.1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tew DG, Boyd HF, Ashman S, Theobald C, Leach CA. Mechanism of inhibition of LDL phospholipase A(2) by monocyclic-beta-lactams. Burst kinetics and the effect of stereochemistry. Biochemistry. 1998;37:10087–10093. doi: 10.1021/bi9801412. [DOI] [PubMed] [Google Scholar]
- 24.Bachovchin DA, Mohr JT, Speers AE, Wang C, Berlin JM, Spicer TP, Fernandez-Vega V, Chase P, Hodder PS, Schurer SC, Nomura DK, Rosen H, Fu GC, Cravatt BF. Academic cross-fertilization by public screening yields a remarkable class of protein phosphatase methylesterase-1 inhibitors. Proc Natl Acad Sci U S A. 2011;108:6811–6816. doi: 10.1073/pnas.1015248108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bottcher T, Sieber SA. Beta-lactones as privileged structures for the active-site labeling of versatile bacterial enzyme classes. Angew Chem Int Ed Engl. 2008;47:4600–4603. doi: 10.1002/anie.200705768. [DOI] [PubMed] [Google Scholar]
- 26.Kathuria S, Gaetani S, Fegley D, Valino F, Duranti A, Tontini A, Mor M, Tarzia G, La Rana G, Calignano A, Giustino A, Tattoli M, Palmery M, Cuomo V, Piomelli D. Modulation of anxiety through blockade of anandamide hydrolysis. Nat Med. 2003;9:76–81. doi: 10.1038/nm803. [DOI] [PubMed] [Google Scholar]
- 27.Bachovchin DA, Ji T, Li W, Simon GM, Blankman JL, Adibekian A, Hoover H, Niessen S, Cravatt BF. Superfamily-wide portrait of serine hydrolase inhibition achieved by library-versus-library screening. Proc. Natl. Acad. Sci. U.S.A. 2010;107:20941–20946. doi: 10.1073/pnas.1011663107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Adibekian A, Martin BR, Wang C, Hsu K-L, Bachovchin DA, Niessen S, Hoover H, Cravatt BF. Click-generated triazole ureas as ultrapotent in vivo6active serine hydrolase inhibitors. Nat. Chem. Biol. 2011;7:469–478. doi: 10.1038/nchembio.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ahn K, Johnson DS, Mileni M, Beidler D, Long JZ, McKinney MK, Weerapana E, Sadagopan N, Liimatta M, Smith SE, Lazerwith S, Stiff C, Kamtekar S, Bhattacharya K, Zhang Y, Swaney S, Van Becelaere K, Stevens RC, Cravatt BF. Discovery and characterization of a highly selective FAAH inhibitor that reduces inflammatory pain. Chem. Biol. 2009;16:411–420. doi: 10.1016/j.chembiol.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stepan A, Walker D, Bauman J, Price D, Baillie T, Kalgutkar A, Aleo M. Structural alert/reactive metabolite concept as applied in medicinal chemistry to mitigate the risk of idiosyncratic drug toxicity: a perspective based on the critical examination of trends in the top 200 drugs marketed in the United States. Chem. Res. Toxicol. 2011;24:1345–1755. doi: 10.1021/tx200168d. [DOI] [PubMed] [Google Scholar]
- 31.Greenbaum DC, Arnold WD, Lu F, Hayrapetian L, Baruch A, Krumrine J, Toba S, Chehade K, Bromme D, Kuntz ID, Bogyo M. Small molecule affinity fingerprinting. A tool for enzyme family subclassification, target identification, and inhibitor design. Chem Biol. 2002;9:1085–1094. doi: 10.1016/s1074-5521(02)00238-7. [DOI] [PubMed] [Google Scholar]
- 32.Kidd D, Liu Y, Cravatt BF. Profiling serine hydrolase activities in complex proteomes. Biochemistry. 2001;40:4005–4015. doi: 10.1021/bi002579j. [DOI] [PubMed] [Google Scholar]
- 33.Speers AE, Adam GC, Cravatt BF. Activity-based protein profiling in vivo using a copper(I)-catalyzed azide-alkyne [3 + 2] cycloaddition. J. Am. Chem. Soc. 2003;125:4686–4687. doi: 10.1021/ja034490h. [DOI] [PubMed] [Google Scholar]
- 34.Speers AE, Cravatt BF. Profiling enzyme activities in vivo using click chemistry methods. Chem. Biol. 2004;11:535–546. doi: 10.1016/j.chembiol.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 35.Alexander JP, Cravatt BF. Mechanism of carbamate inactivation of FAAH: Implications for the design of covalent inhibitors and in vivo functional probes for enzymes. Chem. Biol. 2005;12:1179–1187. doi: 10.1016/j.chembiol.2005.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chang JW, Niphakis MJ, Lum KM, Cognetta AB, Wang C, Matthews ML, Niessen S, Buczynski MW, Parsons LH, Cravatt BF. Highly Selective Inhibitors of Monoacylglycerol Lipase Bearing a Reactive Group that Is Bioisosteric with Endocannabinoid Substrates. Chem. Biol. 2012;19:579–588. doi: 10.1016/j.chembiol.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Long JZ, Li W, Booker L, Burston JJ, Kinsey SG, Schlosburg JE, Pavon FJ, Serrano AM, Selley DE, Parsons LH, Lichtman AH, Cravatt BF. Selective blockade of 26arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nat. Chem. Biol. 2009;5:37–44. doi: 10.1038/nchembio.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Niphakis MJ, Johnson DS, Ballard TE, Stiff C, Cravatt BF. O-hydroxyacetamide carbamates as a highly potent and selective class of endocannabinoid hydrolase inhibitors. ACS chemical neuroscience. 2012;3:418–426. doi: 10.1021/cn200089j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mulvihill MM, Nomura DK. Therapeutic potential of monoacylglycerol lipase inhibitors. Life sciences. 2012 doi: 10.1016/j.lfs.2012.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Piro JR, Benjamin DI, Duerr JM, Pi Y, Gonzales C, Wood KM, Schwartz JW, Nomura DK, Samad TA. A Dysregulated Endocannabinoid-Eicosanoid Network Supports Pathogenesis in a Mouse Model of Alzheimer's Disease. Cell Rep. 2012;1:617–623. doi: 10.1016/j.celrep.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen R, Zhang J, Wu Y, Wang D, Feng G, Tang Y-P, Teng Z, Chen C. Monoacylglycerol Lipase Is a Therapeutic Target for Alzheimer's Disease. Cell Rep. 2012;2:1329–1339. doi: 10.1016/j.celrep.2012.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cao Z, Mulvihill MM, Mukhopadhyay P, Xu H, Erdelyi K, Hao E, Holovac E, Hasko G, Cravatt BF, Nomura DK, Pacher P. Monoacylglycerol Lipase Controls Endocannabinoid and Eicosanoid Signaling and Hepatic Injury in Mice. Gastroenterology. 2013 doi: 10.1053/j.gastro.2012.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fowler CJ. Monoacylglycerol lipase - a target for drug development? British journal of pharmacology. 2012;166:1568–1585. doi: 10.1111/j.1476-5381.2012.01950.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Long JZ, Jin X, Adibekian A, Li W, Cravatt BF. Characterization of Tunable Piperidine and Piperazine Carbamates as Inhibitors of Endocannabinoid Hydrolases. J. Med. Chem. 2010;53:1830–1842. doi: 10.1021/jm9016976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bertrand T, Augé F, Houtmann J, Rak A, Vallée F, Mikol V, Berne PF, Michot N, Cheuret D, Hoornaert C, Mathieu M. Structural Basis for Human Monoglyceride Lipase Inhibition. J. Mol. Biol. 2010;396:663–673. doi: 10.1016/j.jmb.2009.11.060. [DOI] [PubMed] [Google Scholar]
- 46.Patricelli MP, Giang DK, Stamp LM, Burbaum JJ. Direct visualization of serine hydrolase activities in complex proteome using fluorescent active site-directed probes. Proteomics. 2001;1:1067–1071. doi: 10.1002/1615-9861(200109)1:9<1067::AID-PROT1067>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 47.Long JZ, Nomura DK, Vann RE, Walentiny DM, Booker L, Jin X, Burston JJ, Sim-Selley LJ, Lichtman AH, Wiley JL, Cravatt BF. Dual blockade of FAAH and MAGL identifies behavioral processes regulated by endocannabinoid crosstalk in vivo. Proc. Natl. Acad. Sci. U.S.A. 2009;106:20270–20275. doi: 10.1073/pnas.0909411106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rostovtsev VV, Green JG, Fokin VV, Sharpless KB. A stepwise Huisgen cycloaddition process: copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem. Int. Ed. Engl. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 49.Straiker A, Hu SS-J, Long JZ, Arnold A, Wager-Miller J, Cravatt BF, Mackie K. Monoacylglycerol Lipase Limits the Duration of Endocannabinoid-Mediated Depolarization-Induced Suppression of Excitation in Autaptic Hippocampal Neurons. Molecular Pharmacology. 2009;76:1220–1227. doi: 10.1124/mol.109.059030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marrs WR, Blankman JL, Horne EA, Thomazeau A, Lin YH, Coy J, Bodor AL, Muccioli GG, Hu SS, Woodruff G, Fung S, Lafourcade M, Alexander JP, Long JZ, Li W, Xu C, Moller T, Mackie K, Manzoni OJ, Cravatt BF, Stella N. The serine hydrolase ABHD6 controls the accumulation and efficacy of 2-AG at cannabinoid receptors. Nat. Neurosci. 2010;13:951–957. doi: 10.1038/nn.2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chang JW, Moellering RE, Cravatt BF. An activity-based imaging probe for the integral membrane hydrolase KIAA1363. Angew. Chem. Int. Ed. 2012;51:966–970. doi: 10.1002/anie.201107236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Greenbaum D, Baruch A, Hayrapetian L, Darula Z, Burlingame A, Medzihradszky KF, Bogyo M. Chemical Approaches for Functionally Probing the Proteome. Molecular & Cellular Proteomics. 2002;1:60–68. doi: 10.1074/mcp.t100003-mcp200. [DOI] [PubMed] [Google Scholar]
- 53.Shankavaram UT, Reinhold WC, Nishizuka S, Major S, Morita D, Chary KK, Reimers MA, Scherf U, Kahn A, Dolginow D, Cossman J, Kaldjian EP, Scudiero DA, Petricoin E, Liotta L, Lee JK, Weinstein JN. Transcript and protein expression profiles of the NCI-60 cancer cell panel: an integromic microarray study. Molecular cancer therapeutics. 2007;6:820–832. doi: 10.1158/1535-7163.MCT-06-0650. [DOI] [PubMed] [Google Scholar]
- 54.Hsu K-L, Tsuboi K, Adibekian A, Pugh H, Masuda K, Cravatt BF. DAGLβ inhibition perturbs a lipid network involved in macrophage inflammatory responses. Nat. Chem. Biol. 2012;8:999–1007. doi: 10.1038/nchembio.1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nomura DK, Lombardi DP, Chang JW, Niessen S, Ward AM, Long JZ, Hoover HH, Cravatt BF. Monoacylglycerol Lipase Exerts Dual Control over Endocannabinoid and Fatty Acid Pathways to Support Prostate Cancer. Chem. Biol. 2011;18:846–856. doi: 10.1016/j.chembiol.2011.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Blum G, Mullins SR, Kernen K, Fonovic M, Jedeszko C, Rice MJ, Sloane BF, Bogyo M. Dynamic imaging of protease activity with fluorescently quenched activity-based probes. Nat. Chem. Biol. 2005;1:203–209. doi: 10.1038/nchembio728. [DOI] [PubMed] [Google Scholar]
- 57.Blum G, von Degenfeld G, Merchant MJ, Blau HM, Bogyo M. Noninvasive optical imaging of cysteine protease activity using fluorescently quenched activity-based probes. Nature chemical biology. 2007;3:668–677. doi: 10.1038/nchembio.2007.26. [DOI] [PubMed] [Google Scholar]
- 58.Aaltonen N, Savinainen JR, Ribas CR, Ronkko J, Kuusisto A, Korhonen J, Navia-Paldanius D, Hayrinen J, Takabe P, Kasnanen H, Pantsar T, Laitinen T, Lehtonen M, Pasonen-Seppanen S, Poso A, Nevalainen T, Laitinen JT. Piperazine and piperidine triazole ureas as ultrapotent and highly selective inhibitors of monoacylglycerol lipase. Chem Biol. 2013;20:379–390. doi: 10.1016/j.chembiol.2013.01.012. [DOI] [PubMed] [Google Scholar]
- 59.Bertrand T, Auge F, Houtmann J, Rak A, Vallee F, Mikol V, Berne PF, Michot N, Cheuret D, Hoornaert C, Mathieu M. Structural basis for human monoglyceride lipase inhibition. Journal of molecular biology. 2010;396:663–673. doi: 10.1016/j.jmb.2009.11.060. [DOI] [PubMed] [Google Scholar]
- 60.Karageorgos I, Zvonok N, Janero DR, Vemuri VK, Shukla V, Wales TE, Engen JR, Makriyannis A. Endocannabinoid enzyme engineering: soluble human thio-monoacylglycerol lipase (sol-S-hMGL) ACS Chem Neurosci. 2012;3:393–399. doi: 10.1021/cn3000263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Morera L, Labar G, Ortar G, Lambert DM. Development and characterization of endocannabinoid hydrolases FAAH and MAGL inhibitors bearing a benzotriazol-1-yl carboxamide scaffold. Bioorganic & medicinal chemistry. 2012;20:6260–6275. doi: 10.1016/j.bmc.2012.09.011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.