

Abstract
Objective:
To identify the genetic cause of a syndrome causing cerebellar ataxia and eye movement abnormalities.
Methods:
We identified 2 families with cerebellar ataxia, eye movement abnormalities, and global developmental delay. We performed genetic analyses including single nucleotide polymorphism genotyping, linkage analysis, array comparative genomic hybridization, quantitative PCR, and Sanger sequencing. We obtained eye movement recordings of mutant mice deficient for the ortholog of the identified candidate gene, and performed immunohistochemistry using human and mouse brain specimens.
Results:
All affected individuals had ataxia, eye movement abnormalities, most notably tonic upgaze, and delayed speech and cognitive development. Homozygosity mapping identified the disease locus on chromosome 4q. Within this region, a homozygous deletion of GRID2 exon 4 in the index family and compound heterozygous deletions involving GRID2 exon 2 in the second family were identified. Grid2-deficient mice showed larger spontaneous and random eye movements compared to wild-type mice. In developing mouse and human cerebella, GRID2 localized to the Purkinje cell dendritic spines. Brain MRI in 2 affected children showed progressive cerebellar atrophy, which was more severe than that of Grid2-deficient mice.
Conclusions:
Biallelic deletions of GRID2 lead to a syndrome of cerebellar ataxia and tonic upgaze in humans. The phenotypic resemblance and similarity in protein expression pattern between humans and mice suggest a conserved role for GRID2 in the synapse organization between parallel fibers and Purkinje cells. However, the progressive and severe cerebellar atrophy seen in the affected individuals could indicate an evolutionarily unique role for GRID2 in the human cerebellum.
The cerebellum is responsible for the integration of afferent signals from the periphery and efferent output from the cerebral cortex to produce precise, coordinated movements. It is also a critical component of motor learning1–3 and participates in cognitive processes.4,5 Diseases of the cerebellum typically present with ataxia, which is characterized by discoordination of movement, gait instability, impairment of articulation, and abnormalities of eye movement and swallowing.6 Other findings, such as hypotonia and intention tremor, as well as changes in cognition and mood, can be seen. There are numerous genetic disorders having cerebellar ataxia as a prominent feature, and some are inherited as autosomal recessive traits. These autosomal recessive cerebellar ataxias are highly heterogeneous and include conditions with structural abnormalities of the cerebellum (e.g., pontocerebellar hypoplasia, RELN-associated lissencephaly, and cerebellar hypoplasia), cerebellar atrophy (e.g., ataxia telangiectasia), or grossly normal cerebellar structure (e.g., Friedreich ataxia).7,8 Here we describe a cerebellar ataxia syndrome in 2 unrelated families. The affected individuals presented with ataxia, gross motor delay, eye movement abnormalities characterized by tonic upgaze and nystagmus, and cognitive delay. We found that biallelic deletions in the gene GRID2 are responsible for the disease phenotype.
METHODS
Study subjects.
Affected individuals were examined by at least one pediatric neurologist (A.M., G.H.M.), developmental pediatrician (R.N.), or clinical geneticist (E.L.M., J.M.S.).
Standard protocol approval, registrations, and patient consents.
All study participants or their guardians provided written informed consent. The protocols were approved by the participating institutions, and were in accordance with the ethical standards of the responsible national and institutional committees on human subject research.
Genetic analysis.
Genomic DNA was extracted from whole blood in acid-citrate-dextrose buffer or saliva in Oragene collection kits (DNA Genotek, Kanata, Canada). DNA samples from family CH-4900 were genotyped using Illumina OmniExpress single nucleotide polymorphism (SNP) microarray or Illumina 660w microarrays. The data were visualized using dChip software package.9,10 Multipoint lod scores were calculated using MERLIN,11 assuming a recessive mode of disease inheritance, full penetrance, and a disease allele frequency of 0.0001.
For comparative genomic hybridization (CGH), a custom microarray was designed on the 8 × 60K platform using eArray software (Agilent, Santa Clara, CA), and the experiment was conducted according to the manufacturer's protocol. For microsatellite marker analysis, markers that are adjacent to the GRID2 deletions found in the CH-5400 family were selected using the UCSC Genome Browser, and primers were synthesized with the forward primers labeled with 6-FAM. Quantitative real-time PCR experiments were performed using the StepOnePlus real-time PCR system (Applied Biosystems, Foster City, CA). PCR amplification and Sanger sequencing across the deletion boundaries were performed according to standard methods. Additional details on assays, reagents, and primer sequences are provided in e-Methods and table e-1 on the Neurology® Web site at www.neurology.org.
Recording spontaneous eye movements in mice.
Eye movements were monitored in the resting state under room light conditions (300–400 lux) from homozygous engineered Grid2 knockout (C57BL/6 background)12 or ho-15J (C3HJ background)13 mice at postnatal day 52 or older. Wild-type littermates of each mutant line were used as controls. Eye movements were continuously monitored with an infrared camera and the traces were recorded for 15 seconds, 5 times. Additional details on assays are provided in e-Methods.
Immunohistochemistry.
Paraffin sections (4 μm in thickness) of the human and C57BL/6 mouse cerebella were used for immunohistochemistry. Additional details on methods and reagents are provided in e-Methods.
RESULTS
Clinical presentation and phenotype.
We identified a consanguineous pedigree (CH-4900) of Jordanian heritage that had 3 children from 2 branches of the family with a unique neurologic condition (figure 1A). The parents of each branch were first cousins, and the mothers were sisters. The proband (CH-4901) initially presented at 9 months of age with global developmental delay, hypotonia, and intermittent tonic upgaze (figure 1B). On subsequent examination at 5 years, frequent tonic upgaze persisted and occasional horizontal nystagmus on primary gaze was noted (video 1). She had truncal and appendicular ataxia, and walked with support. She spoke in 2-word sentences. Her cousins (CH-4904 and CH-4911) were found to have similar presentations (figure 1B). CH-4911 (14 years) had occasional tonic upgaze and horizontal nystagmus on primary gaze (video 2). She had truncal and appendicular ataxia, developed contractures of the shoulder and ankle joints, and was wheelchair-dependent. She spoke in full sentences and could name a few body parts and colors. Her comprehension was estimated to be the equivalent of 6–7 years. CH-4904 (12 years) had rather persistent tonic upgaze, frequent bursts of upward or diagonal nystagmus, and gaze-evoked nystagmus (video 3). He had truncal and appendicular ataxia, and walked with support. He spoke in single words and some short sentences. His comprehension was estimated to be the equivalent of 3 years. A fourth affected individual (CH-5401) from an unrelated, nonconsanguineous family (CH-5400; figure 1A) of Mexican heritage was also identified and presented with a similar phenotype. On examination at 4 years, she had occasional tonic upgaze and bursts of horizontal nystagmus. She had truncal and appendicular ataxia and could walk only with support. She spoke in 2-word sentences and could follow one-step commands.
Figure 1. Family pedigrees and phenotypic characterization.

(A) Pedigrees of families CH-4900 and CH-5400. Two horizontal lines connecting individuals indicate consanguinity. (B) Facial pictures of the affected individuals in family CH-4900. There are no obvious dysmorphic features. They often show involuntary tonic upgaze, which is captured in individuals CH-4901 and CH-4904. (C) Midsagittal T1-weighted MRI of 2 affected individuals and an age-matched neurologically normal control. At approximately 1 year of age, volume reduction of the cerebellar vermis is already apparent (arrows) in individuals CH-4901 and CH-5401. At 4 years of age, progression of cerebellar atrophy and lack of normal pontine enlargement are evident (arrows). Scale bar = 2 cm.
Brain MRI studies were obtained for 2 of the affected individuals: CH-4901 at 10 months and again at 4 years of age, and CH-5401 at 14 months and again at 4 years of age. Both of these individuals showed progressive cerebellar atrophy (figure 1C, video 4), but no cerebral abnormalities were noted. The cerebellar flocculus appeared to be particularly severely affected (figure e-1). At 4 years of age, the cerebellar vermal area (as measured on a sagittal midline MRI and adjusted to the total intracranial area on the same plane) was decreased by about 80% in CH-4901 and about 96% in CH-5401 compared to a normal control (figure e-2).
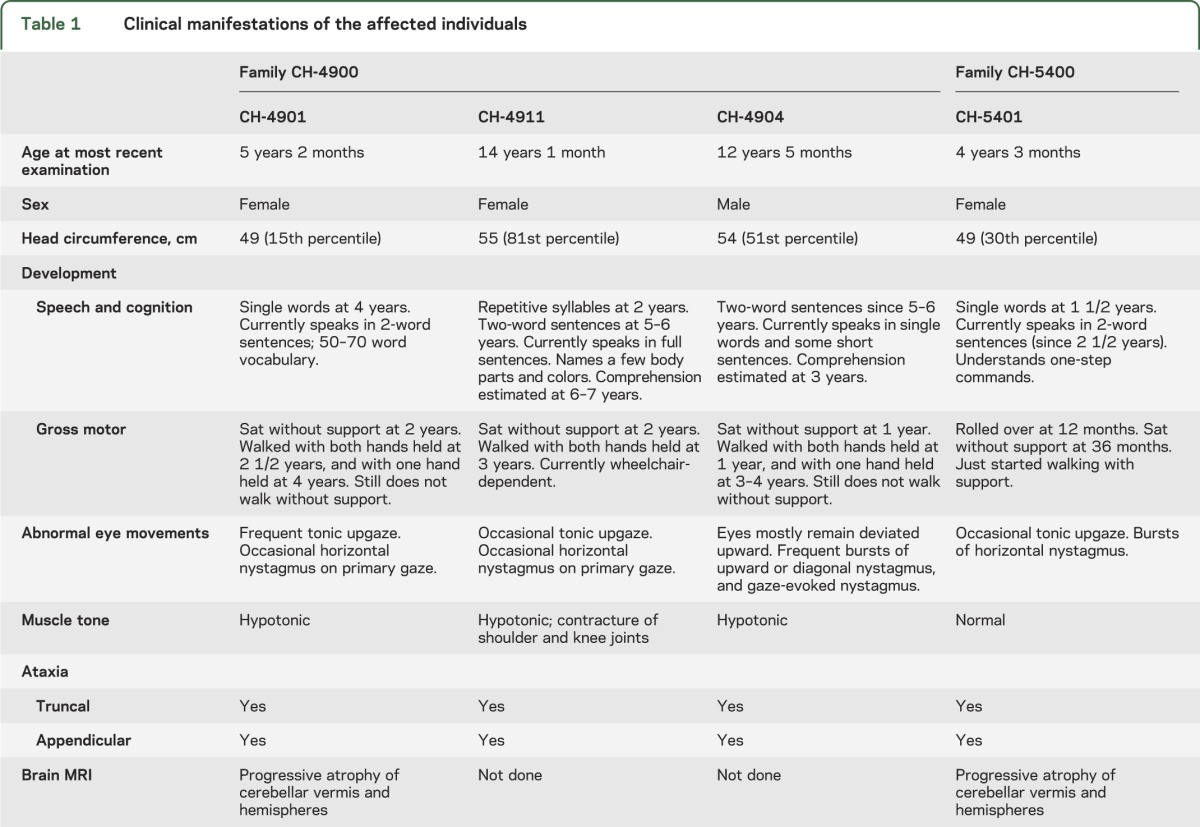
No developmental regression was noted in any of the individuals, except for CH-4911, who had a decline in motor function due to progression of joint contractures. None of the affected individuals had seizures, and none had dysmorphic facial features. A summary of the clinical findings is presented in table 1.
Table 1.
Clinical manifestations of the affected individuals
Linkage and copy number analysis.
SNP genotyping was performed on all individuals of pedigree CH-4900 shown in figure 1A and identified 6 genomic regions over 1 cM that were shared by all affected children, with the largest being a 7.0-cM region on chromosome 4q22 (table e-2). Genome-wide linkage analysis yielded a maximum multipoint lod score of 3.608 for the 4q22 region, which was the only locus with a significant lod score, thus identifying it as the candidate disease locus (figure e-3). Within this candidate interval, there was a block where 6 consecutive SNPs failed to be called in the affected children by dChip software (from rs13105522 to rs17264265). This raised the possibility of a homozygous deletion in these individuals. The interval included exon 4 of the gene GRID2. Furthermore, clinical chromosome microarray testing of the affected child in pedigree CH-5400 revealed compound heterozygous deletions of exon 2 in GRID2.
To evaluate genomic copy number in this region in more detail, we designed a custom CGH microarray, primarily targeting the region of these deletions. All affected children in the CH-4900 family were found to have a homozygous deletion of approximately 37 kb including GRID2 exon 4 (figure 2, A and B), which is expected to result in a frameshift and early truncation of the protein (p.Asp177Glyfs*5). The parents of the proband (CH-4902 and CH-4903) were also examined, and the expected heterozygous deletion was found in both. In the CH-5400 family, the affected individual (CH-5401) inherited a 50-kb deletion involving GRID2 exon 2 from her mother (figure 2, A and B). She also had a larger 335-kb deletion including GRID2 exon 2, resulting in biallelic deletion of this exon (figure 2, A and B). However, this larger deletion was not present in her father. Microsatellite marker analysis eliminated the possibility of nonpaternity and uniparental disomy (figure e-4), and therefore the 335-kb deletion appears to have arisen de novo on the paternally derived chromosome. Deletion of GRID2 exon 2 is expected to result in an in-frame deletion of 52 amino acids (p.Gly30_Glu81del). The genomic GRID2 deletions in both families were confirmed by qPCR (figure e-5). PCR amplification and Sanger sequencing were performed across each of the identified exonic deletions and thus eliminated the possibility of complex rearrangements, such as an inversion (figure e-6).
Figure 2. Exonic deletions in GRID2.

(A) Schematic of the gene GRID2 on chromosome 4q22.1-q22.2. Exons are represented by vertical lines. Deletions are indicated with red bars. (B) Array comparative genomic hybridization (aCGH) results highlight the deletions shown in panel A. Individual CH-5401 inherited a 50-kb deletion that abolishes exon 2 from her mother (CH-5402). CH-5401 also has a larger 335-kb deletion that abolishes exon 2 on the paternally inherited chromosome, but it was not present in her father (CH-5403), suggesting a de novo deletion. Individual CH-4901 inherited identical 37-kb deletions abolishing exon 4 from her mother (CH-4902) and father (CH-4903), who are first cousins. Probes with a log2 ratio of approximately 0 indicate a normal copy number of 2, those with a log2 ratio of approximately −1 indicate a copy number of 1, and those with a log2 ratio of approximately −4 indicate a copy number of zero. Probes hybridized to the reference DNA are indicated in red while those hybridized to the DNA of subjects are indicated in green.
Eye movement recordings of Grid2-deficient mice.
Spontaneously occurring loss-of-function mutations in the GRID2 ortholog (Grid2, also known as GluD2) are common in mice and generally referred to as hotfoot.14 Interestingly, one of the hotfoot mutants, ho-15J, is caused by an in-frame deletion of exon 2, as observed in individual CH-5401.13 Indeed, kinematic analyses of limb movements indicate that ho-15J mice show ataxia analogous to the ataxic gait of individuals with cerebellar disorders.15 Thus, to examine whether the eye movement abnormalities seen in humans with GRID2 mutations are also observed in ho-15J mice, we monitored eye movements using an infrared camera under room light conditions. Significantly larger spontaneous and random eye movements were observed in ho-15J mice (19.9 ± 2.8 mm in 15 seconds, n = 5) than in littermates (10.3 ± 1.3 mm in 15 seconds, n = 5, p = 0.015 by Student t test; figure 3, A and B). Since the genetic background of ho-15J (C3HJ) is associated with retinal degeneration, we examined eye movements in engineered Grid2 knockout mice on the C57BL/6 background.12 As reported previously,16 these mice also showed larger spontaneous eye movements (16.2 ± 2.2 mm in 15 seconds, n = 3) than littermates (7.7 ± 0.8 mm in 15 seconds, n = 3, p = 0.023 by Student t test; figure 3, C and D). These results indicate that some of the ocular motor dysfunctions in the affected individuals are recapitulated in ho-15J and Grid2 knockout mice.
Figure 3. Eye movements in Grid2-KO and ho-15J mice.
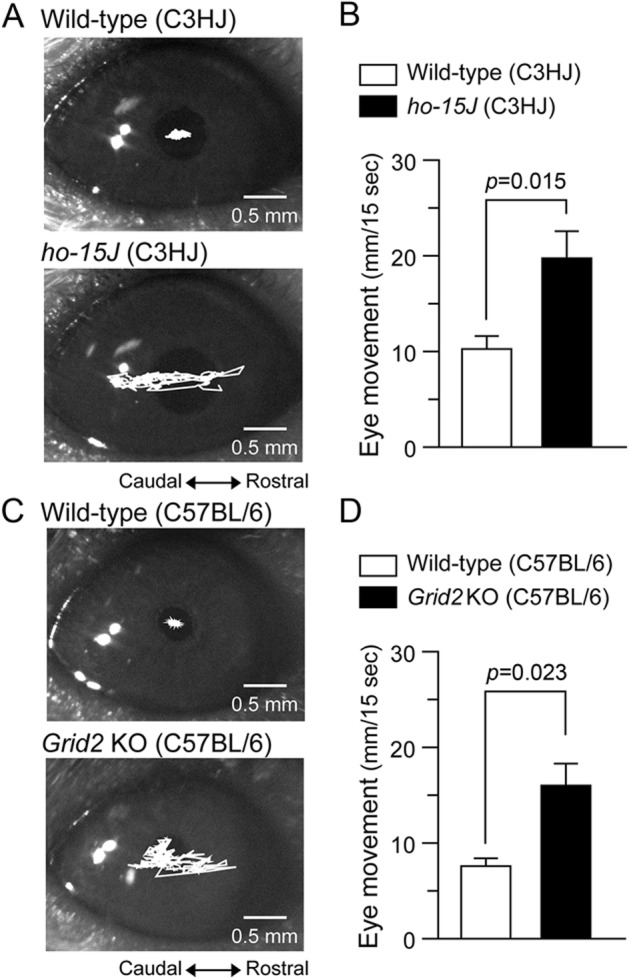
(A) Representative traces of eye movements in an adult ho-15J mouse (bottom) and its littermate (top) monitored for 15 seconds. (B) Averaged data show the total distance of eye movements in 15 seconds in ho-15J mice and littermates; n = 5 for each genotype. (C) Representative traces of eye movements in an adult Grid2-KO mouse (bottom) and its littermate (top) monitored for 15 seconds. (D) Averaged data show the total distance of eye movements in 15 seconds in Grid2-KO mice and littermates; n = 3 for each genotype. Data are presented as the mean ± SEM and statistical significance was assessed by 2-tailed Student t test. KO = knockout.
Immunohistochemistry for GRID2 in developing and adult human cerebella.
In the rat and mouse, Grid2 is exclusively expressed in cerebellar Purkinje cells, and selectively localized at parallel fiber synapses.17–20 We tested whether this expression profile is preserved in human cerebella. At 98 and 308 days of age, intense immunoreactivity for GRID2 was observed in the molecular layer, whereas it was very low in the external and internal granular layers (figure 4, A–D). At 25 years of age, tiny GRID2(+) puncta densely occupied the molecular layer and aligned along Car8(+) Purkinje cell dendrites (figure 4, E and F), similar to the pattern in adult mouse cerebellum (figure 4, G and H). Furthermore, GRID2(+) puncta in adult human cerebellum were often contacted to VGluT1(+) parallel fiber terminals (figure 4, I), never with VGluT2(+) climbing fiber terminals (figure 4, J). These results suggest that exclusive expression of GRID2 at parallel fiber-Purkinje cell synapses is preserved in the human cerebellum. We then inquired whether the territories in Purkinje cell dendrites occupied by parallel fiber and climbing fiber are different between mouse and human. The climbing fiber territory in Purkinje cell dendrites can be defined as Car8(+) dendrites associated with VGluT2, and it appeared to occupy relatively small portions in the molecular layer of adult human cerebellum (figure 4, K) as compared to adult mouse cerebellum (figure 4, L). This was supported by a significantly large decrease in the mean dendritic length of climbing fiber territory per 1 mm2 of the molecular layer in adult human (10.06 ± 0.68 mm) than in adult mouse (55.88 ± 12.05 mm; p < 0.001, Student t test). This suggests that the ratio of parallel fiber territory relative to climbing fiber territory has expanded during the course of evolution.
Figure 4. GRID2 expression in the human and mouse cerebellum.

(A–H) GRID2 immunofluorescence (green) in the human cerebellar cortex at 98 days (A, B), 308 days (C, D), and 25 years (E, F) of age, and Grid2 immunofluorescence in the adult mouse cerebellar cortex (G, H). Sections are counterstained with TOTO3 (blue) in A and C, and costained for Purkinje cell marker Car8 (red) in E and G. (I, J) Double immunofluorescence for GRID2 (green) and VGluT1 (red in panel I) or VGluT2 (red in panel J) in the adult human cerebellar cortex. (K, L) Double immunofluorescence for Car8 and VGluT2 in the cerebellar cortex of adult human (K) and mouse (L). Scale bars = 50 μm (A–F), 20 μm (G, H, K, L), 5 μm (I, J). Dn = shaft dendrite; EGL = external granular layer; IGL = internal granular layer; ML = molecular layer; PCL = Purkinje cell layer.
DISCUSSION
In this study we describe an autosomal recessive syndrome of cerebellar atrophy, eye movement abnormalities that include intermittent tonic upgaze and nystagmus, variable motor abnormalities and cognitive delay, and identify biallelic deletions of the gene GRID2 as its cause.
The function of the GRID2 protein in the human brain has not yet been elucidated, but its mouse ortholog, Grid2, has been extensively studied. It is a member of the ionotropic glutamate receptor family, though it does not bind glutamate.17,19 Grid2 is expressed primarily by cerebellar Purkinje cells and localizes to dendritic spines that synapse with parallel fibers.18,20,21 Recently Cbln1 was identified as its ligand, and the interaction between Grid2 and Cbln1 was shown to be essential for organization of synapses between parallel fibers and Purkinje cells.22,23 Grid2 has also been shown to mediate long-term depression in the developing mouse cerebellum through its interaction with d-serine.24,25 With its dual roles in synapse organization and modulation of synaptic communication, Grid2 is essential for the development and function of the mouse cerebellum.
Grid2 loss-of-function mouse mutants, such as hotfoot14 and Grid2 knockout mice,25 show ataxia and mild volume loss of the cerebellum. Thus, they recapitulate the clinical manifestations observed in individuals with biallelic GRID2 deletions. At least 10 distinct naturally occurring hotfoot alleles have been characterized to date, and most of them are due to deletion of one or more exons.13,26 One of the hotfoot alleles, ho-15J, represents a homozygous deletion of exon 2, the same exon deleted in the CH-5400 family presented herein.13 Removing exon 2 is predicted to lead to an in-frame deletion of 52 amino acids in both mouse and human. However, it has been shown that the resulting protein product in mice is retained in the endoplasmic reticulum and degraded, suggesting it is a null mutation.13 Deletion of exon 4, found in the CH-4900 family, is expected to cause a frameshift and early truncation, and thus it is also a likely null mutation.
One of the unique clinical features shared by all affected individuals described herein is involuntary tonic upgaze. The pathophysiology of this symptom is not well-understood, but it is interesting to note that the flocculus appears to be one of the cerebellar areas most severely affected by atrophy in the affected children. The flocculus has been implicated in the pathogenesis of downbeat nystagmus, which is characterized by slow, upward drift of the eyes.27 Therefore, there could be a shared underlying mechanism between tonic upgaze and downbeat nystagmus that involves floccular dysfunction. The tonic upgaze described also resembles what is seen in a rare clinical syndrome known as paroxysmal tonic upgaze (PTU).28,29 Most reported cases of PTU appear clinically distinct from the individuals we report; for example, PTU is often seen as a transient condition in otherwise neurologically normal children. However, some cases of PTU are associated with ataxia or developmental delay,28 and thus mutations in GRID2 could account for some of the reported cases of PTU.
We demonstrated that Grid2-deficient mice show larger spontaneous and random eye movements. These eye movements might be considered equivalent to nystagmus in humans, which, in addition to tonic upgaze, was seen in all affected individuals. This further adds to the phenotypic resemblance between Grid2-deficient mice and humans with GRID2 mutations.
Immunohistochemical studies of GRID2 in the developing human cerebellum showed an expression pattern that is markedly similar to that seen in mice. This suggests that GRID2 in humans plays a similar role in cerebellar synapse formation to that in mice. However, the degree of cerebellar atrophy is more pronounced in the affected individuals (80%–96% decrease in the midline vermal area) compared to Grid2-mutant mice (12% decrease in the midline vermal area).30 Cerebellar atrophy due to mutations in GRID2/Grid2 is likely the result of granule cell death. We showed evidence of significant evolutionary expansion of the parallel fiber territory on Purkinje cell dendrites in humans. Therefore, we speculate that cerebellar granule cells in humans depend more heavily on synapses with Purkinje cells (and thus on GRID2) for their survival than their rodent counterparts.
It is interesting to note that all 4 affected individuals showed delay in speech and cognitive development. Since it is recognized that children with various cerebellar disorders can present with cognitive delay, the delay in speech and cognitive development seen in these individuals could be attributable to loss of GRID2 in the cerebellum.31,32 However, previously unrecognized roles of GRID2 in the cerebral cortex cannot be ruled out, and further investigation is warranted.
During the preparation of this manuscript, an article describing a homozygous deletion of GRID2 exons 3 and 4 in 3 members of a Turkish family was published.33 The affected individuals were described to have cerebellar ataxia, oculomotor apraxia, nystagmus, and pyramidal signs. Interestingly, tonic upgaze was not noted in the phenotype of any of the affected individuals. The proband had cognitive delay, though the level of cognitive functioning of the other 2 affected individuals is less clear. The deletion identified in this family appears different from the 3 independent deletions we describe herein.
GRID2 and Grid2 are genomically located in the common fragile sites FRA4F and Fra6C1, respectively. Common fragile sites are regions of the genome that have a relatively high rate of genomic rearrangement and so could explain why there are many spontaneous hotfoot alleles in mice.34,35 Thus, this locus likely represents a deletion hotspot in humans as well.
Supplementary Material
ACKNOWLEDGMENT
SNP genotyping was performed at The W.M. Keck Foundation Biotechnology Resource Laboratory at Yale University through the NIH Neuroscience Microarray Consortium. Human brain tissue was obtained from the NICHD Brain and Tissue Bank for Developmental Disorders at the University of Maryland, Baltimore (NICHD contract no. HHSN275200900011C, Ref. No. N01-HD-9-0011). The authors thank the families for their participation in this research, Dr. Christopher A. Walsh for discussions and comments on the manuscript, Dr. Paul Caruso for providing control MRI, Dr. David Willmot and Dr. Scott Leppanen (Agilent) for help with custom microarray design and experiments, Dr. Peter R. Papenhausen and Romela Pasion (Laboratory Corporation of America) for help with clinical chromosome microarray interpretation, Dr. Harry Chugani and Dr. Mahabubul Huq for sharing human DNA samples, and Dr. Kenji Sakimura for providing Grid2-deficient mice.
GLOSSARY
- CGH
comparative genomic hybridization
- PTU
paroxysmal tonic upgaze
- SNP
single nucleotide polymorphism
Footnotes
Supplemental data at www.neurology.org
AUTHOR CONTRIBUTIONS
L. Benjamin Hills: genetic studies, writing of the manuscript. Dr. Amira Masri: ascertainment of family CH-4900, acquisition of clinical data. Dr. Kotaro Konno: mouse immunohistochemical studies. Dr. Wataru Kakegawa: mouse eye movement studies. Anh-Thu N. Lam: quantitative analysis of brain MRI, genetic studies. Dr. Elizabeth Lim-Melia: ascertainment of family CH-5400, acquisition of clinical data. Nandini Chandy: ascertainment of family CH-5400, acquisition of clinical data. Dr. R. Sean Hill: linkage analysis, interpretation of genetic data. Jennifer N. Partlow: coordination of human subject research, clinical data and human samples. Dr. Muna Al-Saffar: coordination of human subject research, clinical data and human samples, acquisition of clinical data. Dr. Ramzi Nasir: acquisition of clinical data. Dr. Joan M. Stoler: acquisition of clinical data. Dr. A. James Barkovich: interpretation of brain MRI. Dr. Masahiko Watanabe: mouse immunohistochemical studies, critical revision of the manuscript. Dr. Michisuke Yuzaki: mouse eye movement studies, critical revision of the manuscript. Dr. Ganeshwaran H. Mochida: study design, data analysis and interpretation, writing of the manuscript.
STUDY FUNDING
Supported by grants from NINDS (R01NS035129), the Fogarty International Center (R21TW008223), NHGRI (U54 HG003067), the Simons Foundation, and the Dubai Harvard Foundation for Medical Research.
DISCLOSURE
L. Hills and A. Masri report no disclosures. K. Konno receives Grant-in-Aid for Scientific Research for Young Scientists (B) provided by the Ministry of Education, Culture, Sports, Science and Technology of Japan. W. Kakegawa is funded by the Japan Society for the Promotion of Science (KAKENHI #23689012) and received research support from the Nakajima Foundation, the Naito Foundation, the Takeda Foundation, and the Inamori Foundation. A. Lam, E. Lim-Melia, N. Chandy, and R. Hill report no disclosures. J. Partlow is an employee of the Howard Hughes Medical Institute, and has received an honorarium from the Canadian Association of Genetic Counsellors for presenting at their annual educational conference. M. Al-Saffar reports no disclosures. R. Nasir received research support for the conduct of clinical trials from Seaside Therapeutics and Novartis (20–50% effort). J. Stoler reports no disclosures. A. Barkovich receives salary support from NIH grants #NS35129, NS062820, EB009756, NS046432, and HD072074, and receives royalties from the publication of the book Pediatric Neuroimaging. M. Watanabe receives Grants-in-Aid for Scientific Research (S) provided by the Ministry of Education, Culture, Sports, Science and Technology of Japan, and also for CREST provided by the Japan Science and Technology Agency of Japan. M. Yuzaki is funded by the Ministry of Education, Culture, Sports, Science and Technology of Japan (KAKENHI #23110009), the Japan Society for the Promotion of Science (KAKENHI #23240053), and the Japan Science and Technology Agency (CREST 2009FY). G. Mochida has been supported by grants from the National Institute of Neurological Disorders and Stroke (R01NS035129), the Fogarty International Center (R21TW008223), the Simons Foundation, and the Dubai Harvard Foundation for Medical Research, and received an honorarium from UCB Japan and Otsuka Pharmaceutical for a non-promotional lecture. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Albus JS. A theory of cerebellar function. Math Biosci 1971;10:25–61 [Google Scholar]
- 2.Carey MR. Synaptic mechanisms of sensorimotor learning in the cerebellum. Curr Opin Neurobiol 2011;21:609–615 [DOI] [PubMed] [Google Scholar]
- 3.Marr D. A theory of cerebellar cortex. J Physiol 1969;202:437–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schmahmann JD. The role of the cerebellum in cognition and emotion: personal reflections since 1982 on the dysmetria of thought hypothesis, and its historical evolution from theory to therapy. Neuropsychol Rev 2010;20:236–260 [DOI] [PubMed] [Google Scholar]
- 5.Schmahmann JD, Sherman JC. The cerebellar cognitive affective syndrome. Brain 1998;121:561–579 [DOI] [PubMed] [Google Scholar]
- 6.Schmahmann JD. Disorders of the cerebellum: ataxia, dysmetria of thought, and the cerebellar cognitive affective syndrome. J Neuropsychiatry Clin Neurosci 2004;16:367–378 [DOI] [PubMed] [Google Scholar]
- 7.Anheim M, Tranchant C, Koenig M. The autosomal recessive cerebellar ataxias. N Engl J Med 2012;366:636–646 [DOI] [PubMed] [Google Scholar]
- 8.Barkovich AJ, Millen KJ, Dobyns WB. A developmental and genetic classification for midbrain-hindbrain malformations. Brain 2009;132:3199–3230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li C, Wong WH. Model-based analysis of oligonucleotide arrays: expression index computation and outlier detection. Proc Natl Acad Sci USA 2001;98:31–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin M, Wei LJ, Sellers WR, Lieberfarb M, Wong WH, Li C. dChipSNP: significance curve and clustering of SNP-array-based loss-of-heterozygosity data. Bioinformatics 2004;20:1233–1240 [DOI] [PubMed] [Google Scholar]
- 11.Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin: rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet 2002;30:97–101 [DOI] [PubMed] [Google Scholar]
- 12.Yamasaki M, Miyazaki T, Azechi H, et al. Glutamate receptor delta2 is essential for input pathway-dependent regulation of synaptic AMPAR contents in cerebellar Purkinje cells. J Neurosci 2011;31:3362–3374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Motohashi J, Kakegawa W, Yuzaki M. Ho15J: a new hotfoot allele in a hot spot in the gene encoding the delta2 glutamate receptor. Brain Res 2007;1140:153–160 [DOI] [PubMed] [Google Scholar]
- 14.Lalouette A, Guenet JL, Vriz S. Hotfoot mouse mutations affect the delta 2 glutamate receptor gene and are allelic to lurcher. Genomics 1998;50:9–13 [DOI] [PubMed] [Google Scholar]
- 15.Takeuchi E, Sato Y, Miura E, Yamaura H, Yuzaki M, Yanagihara D. Characteristics of gait ataxia in delta2 glutamate receptor mutant mice, ho15J. PLoS One 2012;7:e47553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yoshida T, Katoh A, Ohtsuki G, Mishina M, Hirano T. Oscillating Purkinje neuron activity causing involuntary eye movement in a mutant mouse deficient in the glutamate receptor delta2 subunit. J Neurosci 2004;24:2440–2448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Araki K, Meguro H, Kushiya E, Takayama C, Inoue Y, Mishina M. Selective expression of the glutamate receptor channel delta 2 subunit in cerebellar Purkinje cells. Biochem Biophys Res Commun 1993;197:1267–1276 [DOI] [PubMed] [Google Scholar]
- 18.Landsend AS, Amiry-Moghaddam M, Matsubara A, et al. Differential localization of delta glutamate receptors in the rat cerebellum: coexpression with AMPA receptors in parallel fiber-spine synapses and absence from climbing fiber-spine synapses. J Neurosci 1997;17:834–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lomeli H, Sprengel R, Laurie DJ, et al. The rat delta-1 and delta-2 subunits extend the excitatory amino acid receptor family. FEBS Lett 1993;315:318–322 [DOI] [PubMed] [Google Scholar]
- 20.Takayama C, Nakagawa S, Watanabe M, Mishina M, Inoue Y. Light- and electron-microscopic localization of the glutamate receptor channel delta 2 subunit in the mouse Purkinje cell. Neurosci Lett 1995;188:89–92 [DOI] [PubMed] [Google Scholar]
- 21.Mayat E, Petralia RS, Wang YX, Wenthold RJ. Immunoprecipitation, immunoblotting, and immunocytochemistry studies suggest that glutamate receptor delta subunits form novel postsynaptic receptor complexes. J Neurosci 1995;15:2533–2546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matsuda K, Miura E, Miyazaki T, et al. Cbln1 is a ligand for an orphan glutamate receptor delta2, a bidirectional synapse organizer. Science 2010;328:363–368 [DOI] [PubMed] [Google Scholar]
- 23.Uemura T, Lee SJ, Yasumura M, et al. Trans-synaptic interaction of GluRdelta2 and Neurexin through Cbln1 mediates synapse formation in the cerebellum. Cell 2010;141:1068–1079 [DOI] [PubMed] [Google Scholar]
- 24.Kakegawa W, Miyoshi Y, Hamase K, et al. D-serine regulates cerebellar LTD and motor coordination through the delta2 glutamate receptor. Nat Neurosci 2011;14:603–611 [DOI] [PubMed] [Google Scholar]
- 25.Kashiwabuchi N, Ikeda K, Araki K, et al. Impairment of motor coordination, Purkinje cell synapse formation, and cerebellar long-term depression in GluR delta 2 mutant mice. Cell 1995;81:245–252 [DOI] [PubMed] [Google Scholar]
- 26.Wang Y, Matsuda S, Drews V, Torashima T, Meisler MH, Yuzaki M. A hot spot for hotfoot mutations in the gene encoding the delta2 glutamate receptor. Eur J Neurosci 2003;17:1581–1590 [DOI] [PubMed] [Google Scholar]
- 27.Glasauer S, Stephan T, Kalla R, Marti S, Straumann D. Up-down asymmetry of cerebellar activation during vertical pursuit eye movements. Cerebellum 2009;8:385–388 [DOI] [PubMed] [Google Scholar]
- 28.Ouvrier R, Billson F. Paroxysmal tonic upgaze of childhood: a review. Brain Dev 2005;27:185–188 [DOI] [PubMed] [Google Scholar]
- 29.Ouvrier RA, Billson F. Benign paroxysmal tonic upgaze of childhood. J Child Neurol 1988;3:177–180 [DOI] [PubMed] [Google Scholar]
- 30.Kurihara H, Hashimoto K, Kano M, et al. Impaired parallel fiber–Purkinje cell synapse stabilization during cerebellar development of mutant mice lacking the glutamate receptor delta2 subunit. J Neurosci 1997;17:9613–9623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bolduc ME, Du Plessis AJ, Sullivan N, et al. Spectrum of neurodevelopmental disabilities in children with cerebellar malformations. Dev Med Child Neurol 2011;53:409–416 [DOI] [PubMed] [Google Scholar]
- 32.Tavano A, Grasso R, Gagliardi C, et al. Disorders of cognitive and affective development in cerebellar malformations. Brain 2007;130:2646–2660 [DOI] [PubMed] [Google Scholar]
- 33.Utine GE, Haliloglu G, Salanci B, et al. A homozygous deletion in GRID2 causes a human phenotype with cerebellar ataxia and atrophy. J Child Neurol 2013;28:926–932 [DOI] [PubMed] [Google Scholar]
- 34.Glover TW, Arlt MF, Casper AM, Durkin SG. Mechanisms of common fragile site instability. Hum Mol Genet 2005;14:R197–R205 [DOI] [PubMed] [Google Scholar]
- 35.Helmrich A, Stout-Weider K, Hermann K, Schrock E, Heiden T. Common fragile sites are conserved features of human and mouse chromosomes and relate to large active genes. Genome Res 2006;16:1222–1230 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.