

Abstract
Objective:
To describe the hearing loss in facioscapulohumeral muscular dystrophy (FSHD) and examine the relationship to genotype.
Methods:
Medical records of all individuals with FSHD seen at the University of Iowa neuromuscular clinic between July 2006 and July 2012 (n = 59) were reviewed. Eleven had significant hearing loss and no non-FSHD cause. All available audiology records for these individuals were analyzed. The relationship between the FSHD mutation (EcoRI/BlnI fragment size) and hearing loss was evaluated using a logistic regression analysis.
Results:
In patients with hearing loss, recalled age at onset of facial weakness ranged from birth to 5 years and shoulder weakness was 3 to 15 years. The age at diagnosis of hearing loss ranged from birth to 7 years. Only 2 were identified by newborn hearing screen. Most audiograms demonstrated a bilateral, sloping, high-frequency sensorineural hearing loss. Of the 4 patients with more than 5 years of data, 3 had progression of hearing loss. Logistic regression showed statistically significant negative association between the presence of hearing loss and EcoRI/BlnI fragment size (p = 0.0207).
Conclusions:
FSHD with a small EcoRI/BlnI fragment is associated with a bilateral, progressive, sloping, high-frequency hearing loss with onset in childhood. Patients with FSHD and small EcoRI/BlnI fragment sizes should have hearing screened, even if the child passed newborn hearing screening.
Facioscapulohumeral muscular dystrophy (FSHD) is an autosomal dominant muscular dystrophy caused by contraction of a region of DNA on chromosome 4 containing repeated DNA sequences called D4Z4 repeats. People without the disease have >10 D4Z4 repeats, while people with the disease have 1–10 D4Z4 repeats (EcoRI fragment <38 kb, EcoRI/BlnI fragments <35 kb).1 The most severe form has onset in childhood and is associated with fewer D4Z4 repeats. High-frequency sensorineural hearing loss and retinal vasculopathy can be part of this severe phenotype.
Published guidelines for hearing screening in FSHD were based on expert opinion and recommend hearing screening until school age.1 Here we describe the hearing phenotype associated with FSHD and its relationship with genetic testing in a series from one clinic. Our results provide data to support the current testing recommendations.
METHODS
Participants.
Census data from the pediatric neuromuscular clinic at the University of Iowa Hospitals and Clinics were reviewed to identify patients with FSHD seen between July 2006 and July 2012. The clinic at University of Iowa Hospitals and Clinics has a special interest in pediatric FSHD and hosted a one-time clinic for early-onset FSHD during this time period. Participants from this clinic are included in the analysis. All analyses were retrospective. FSHD genetic test results were abstracted for the whole cohort. The fragment size was defined as the shortest EcoRI/BlnI allele reported. One patient had only an EcoRI fragment size reported and EcoRI/BlnI fragment size was extrapolated to be 3 kb shorter than the EcoRI fragment size, as would be expected unless there had been a translocation between chromosomes 4 and 10.
The hearing loss cohort consisted of those with symptomatic hearing loss documented in the medical record most likely related to FSHD. Exclusion criteria were hearing loss possibly due to presbycusis (defined as bilateral high-frequency sensorineural hearing loss diagnosed after age 50 years) and hearing loss with onset after significant loud noise exposure. Those who had family members with hearing loss were included to avoid excluding participants with FSHD-associated hearing loss present in the family.
Standard protocol approvals, registrations, and patient consents.
Retrospective review of patient charts to identify patients with FSHD and hearing loss was approved by the University of Iowa institutional review board (IRB). Written informed consent was obtained from those seen after initiation of the IRB protocol in 2010. All subjects with hearing loss were contacted to provide written informed consent to allow collection of outside audiology records.
Audiology reports.
We collected pure tone audiometry, acoustic reflex data, and tympanometry from the University of Iowa medical record and local audiologists for all participants with hearing loss. Audiograms were compiled sequentially to analyze progression over time. Progression was defined as hearing loss described as progressive in the audiology note, loss of 10 dB between 2 consecutive audiograms, or loss of 5 dB between 2 consecutive audiograms that was maintained in the next consecutive audiogram.
Statistical analysis.
A simple logistic regression model was used to analyze the relationship between hearing loss and size of FSHD-associated mutation using the SAS v9.3 statistical software package. The outcome variable was presence of hearing loss and the independent variable was EcoRI/BlnI fragment size.
RESULTS
Study population.
Our initial cohort included 59 individuals with FSHD. Thirteen of the 59 individuals had hearing loss. Two were excluded from further analyses, due to presbycusis and loud noise exposure in military service, respectively, leaving 11 unrelated individuals with hearing loss.
Evaluation of hearing loss.
Audiometry data were available for 10 subjects, and the subjective report was available for one. The clinical data are summarized in the table. Age at detection of hearing loss ranged from birth to 7 years. Three participants underwent newborn hearing screening, and 2 failed. Audiograms showed sloping sensorineural hearing loss in 21/22 ears (patient 7 had normal hearing at 4,000 Hz in the right ear only, figure 1C). Four participants had hearing loss that was most severe at 4,000 Hz and slightly improved at 8,000 Hz.
Table.
Characteristics of participants with hearing loss
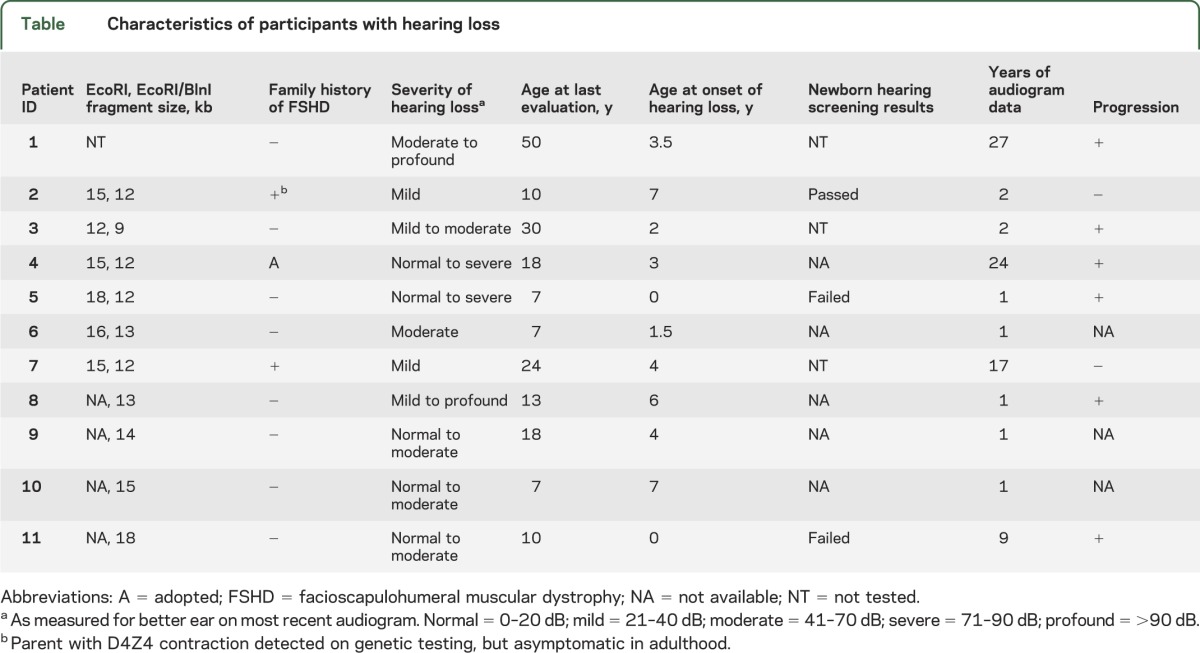
Figure 1. Pure tone audiometry data from participants with more than 5 years of data.

Hearing loss was progressive in 3 of 4 patients for whom we had more than 5 years of audiometry data. Data are from the following participants: (A) patient 1, (B) patient 4, (C) patient 7, and (D) patient 11. Only right ear data are shown, but results are representative of left ear as well except for patient 7, whose left ear showed mild to moderate sloping hearing loss. Age at audiometry data in years is shown in the key on the right of each chart.
More than 5 years of audiometry data were available for 4 participants. Progression of hearing loss in the high frequencies was documented in 3 of the 4 (figure 1). In one patient, otoacoustic emissions were measured and were absent while ipsilateral acoustic reflexes were present bilaterally. Tympanometry was available for 6 patients and showed normal middle ear function.
Genotype–phenotype association.
EcoRI/BlnI fragment size information was available for 53 participants. We found a statistically significant negative association between hearing loss and fragment size (p value = 0.0207) (figure 2). There was no hearing loss in our cohort with EcoRI/BlnI fragment size >20 kb. For those with EcoRI/BlnI fragment size ≤20 kb, 10/31 (32%) subjects had hearing loss.
Figure 2. Probability of hearing loss as predicted by EcoRI/BlnI fragment size from simple logistic regression analysis.
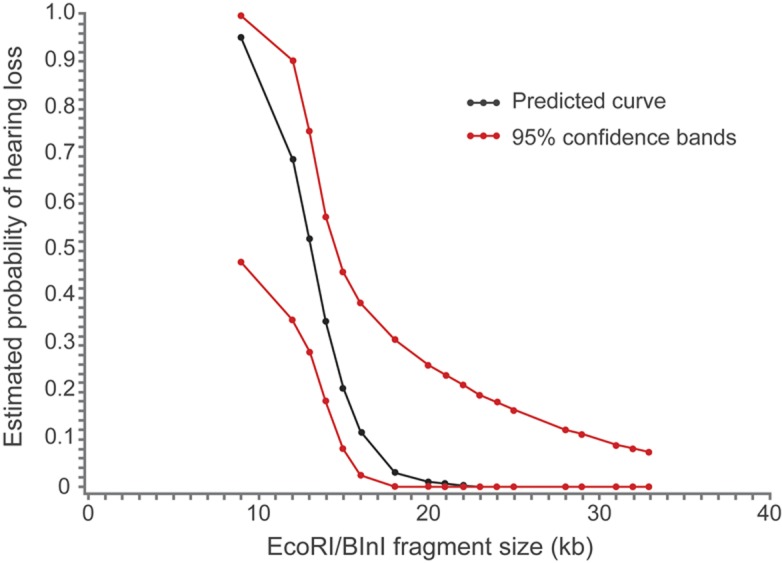
Association between small EcoRI/BlnI fragment size and higher probability of hearing loss was statistically significant (p = 0.0207). The black line represents the logistic regression curve predicted by our cohort. The red lines represent 95% confidence bands.
DISCUSSION
The series presented here, with follow-up ranging from 1 to 27 years, demonstrates that in many cases FSHD-associated hearing loss worsens over time. This is consistent with a previous report, with follow-up of up to 6 years.2 Progression can occur dramatically, as was seen in patient 11, who had a loss of 25 dB at one frequency in 1 year, but is more often slow and may not be noticed when comparing only to the previous year's audiogram. Progression of hearing loss at 4,000 Hz is also seen in noise-induced hearing loss; however, in all of our participants no known noise exposure was documented. We note that one individual with long follow-up had stable hearing loss (figure 1C). This subject is unique in that her left ear did not have sloping high-frequency hearing loss, but instead had normal hearing at 4,000 Hz. It is not clear from this retrospective analysis why in some people hearing loss is progressive and in others it is not, but this mirrors the variability in other aspects of FSHD. These data suggest that when hearing loss is identified, regular monitoring of hearing thresholds should be an important part of management.
We also demonstrate that newborn screening detects some, but not all, cases of FSHD-associated hearing loss. There is one previous case of FSHD-associated hearing loss detected with newborn screening, but most reports were before the era of routine screening.3 Our finding that 2 of 3 people evaluated failed standard newborn hearing screening has 2 implications. First, FSHD should be included in the differential diagnosis of congenital hearing loss. Second, repeated hearing screening should be routine for children diagnosed with FSHD even if they passed newborn hearing screening. This is further supported by the fact that 6 of our cohort had hearing loss identified after age 3 (when acquisition of language would have already been initiated), suggesting that FSHD-associated hearing loss can have onset in preschool and school-age children. Therefore, continued screening until school age is important for those at risk.
Finally, we described an association between hearing loss and EcoRI/BlnI fragment size. The analysis was limited by the small number of people with hearing loss and FSHD in our cohort; however, this relationship reached statistical significance. The validity of our data is supported by the fact that the range of fragment sizes associated with hearing loss in the literature (8.6–20 kb) is similar to what we report (table e-1 on the Neurology® Web site at www.neurology.org). There was a single case report of a 78-year-old woman with late-onset hearing loss and fragment size of 30 kb; however, it does not appear that age-related hearing loss could be excluded in this case.4 A similar association between small EcoRI/BlnI fragment size and a different non-muscle manifestation of FSHD, Coats disease, was recently reported.5 Thus, our data indicate that identification of a small EcoRI/BlnI fragment size (<20 kb) is an indication for serial monitoring for hearing loss in young children.
Supplementary Material
GLOSSARY
- FSHD
facioscapulohumeral muscular dystrophy
- IRB
institutional review board
Footnotes
Editorial, page 1370
Supplemental data at www.neurology.org
AUTHOR CONTRIBUTIONS
K.L. Lutz contributed to study design, data analysis, and manuscript drafting and revision. Dr. Holte contributed expertise on audiology, data analysis, and manuscript editing. Dr. Kliethermes performed statistical analysis, interpretation, and writing. C. Stephan's contributions included study design and manuscript editing. Dr. Mathews contributed study design and conceptualization, data analysis, and manuscript editing.
STUDY FUNDING
Supported by the Senator Paul D. Wellstone Muscular Dystrophy Cooperative Research Center Fellowship (NIH U54 NS053672).
DISCLOSURE
K. Lutz receives funding by NIH grant #U54 NS053672. L. Holte and S. Kliethermes report no disclosures. C. Stephan receives funding by NIH grant #U54 NS053672. K. Mathews is on the advisory board for the Muscular Dystrophy Association, FSH society, and Parent Project Muscular Dystrophy, receives funding by NIH grant #U54 NS053672 and additional funding from the CDC, receives clinical trial funding from PTC Therapeutics and GSK, and serves on the Data Safety and Review Board for Seaside Therapeutics. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Tawil R, van der Maarel S, Padberg GW, van Engelen BG. 171st ENMC international workshop: standards of care and management of facioscapulohumeral muscular dystrophy. Neuromuscul Disord 2010;20:471–475 [DOI] [PubMed] [Google Scholar]
- 2.Voit T, Lamprecht A, Lenard HG, Goebel HH. Hearing loss in facioscapulohumeral dystrophy. Eur J Pediatr 1986;145:280–285 [DOI] [PubMed] [Google Scholar]
- 3.Ganesh A, Kaliki S, Shields CL. Coats-like retinopathy in an infant with preclinical facioscapulohumeral dystrophy. J AAPOS 2012;16:204–206 [DOI] [PubMed] [Google Scholar]
- 4.Felice KJ, Moore SA. Unusual clinical presentations in patients harboring the facioscapulohumeral dystrophy 4q35 deletion. Muscle Nerve 2001;24:352–356 [DOI] [PubMed] [Google Scholar]
- 5.Statland JM, Sacconi S, Farmakidis C, Donlin-Smith CM, Chung M, Tawil R. Coats syndrome in facioscapulohumeral dystrophy type 1: frequency and D4Z4 contraction size. Neurology 2013;80:1247–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.