

Abstract
Objectives:
To determine the genetic distribution and the phenotypic correlation of an extensive series of patients with Charcot-Marie-Tooth disease in a geographically well-defined Mediterranean area.
Methods:
A thorough genetic screening, including most of the known genes involved in this disease, was performed and analyzed in this longitudinal descriptive study. Clinical data were analyzed and compared among the genetic subgroups.
Results:
Molecular diagnosis was accomplished in 365 of 438 patients (83.3%), with a higher success rate in demyelinating forms of the disease. The CMT1A duplication (PMP22 gene) was the most frequent genetic diagnosis (50.4%), followed by mutations in the GJB1 gene (15.3%), and in the GDAP1 gene (11.5%). Mutations in 13 other genes were identified, but were much less frequent. Sixteen novel mutations were detected and characterized phenotypically.
Conclusions:
The relatively high frequency of GDAP1 mutations, coupled with the scarceness of MFN2 mutations (1.1%) and the high proportion of recessive inheritance (11.6%) in this series exemplify the particularity of the genetic distribution of Charcot-Marie-Tooth disease in this region.
Charcot-Marie-Tooth (CMT) disease refers to the genetically heterogeneous group of hereditary motor and sensory neuropathies. It is one of the most common inherited neurologic disorders, with a prevalence of 15.2 to 40 cases per 100,000.1–3 Molecular studies have provided an ever-growing list of more than 40 involved genes and loci (http://www.molgen.ua.ac.be/CMTMutations/, http://neuromuscular.wustl.edu/, both accessed June 24, 2013). Most of the patients with CMT disease have autosomal dominant (AD) inheritance, but many have X-linked or autosomal recessive (AR) inheritance. CMT disease can be classified according to clinical, electrophysiologic, and nerve pathology findings into demyelinating forms (CMT1, CMT4), with a median motor nerve conduction velocity (MMNCV) of <38 m/s and pathologic evidence of nerve fiber demyelination; and axonal forms (CMT2), with preserved conduction velocities (MMNCV >38 m/s) and pathologic signs of axonal degeneration and regeneration.4 An intermediate type (CMT-I) is accepted in which MMNCV lies between 25 and 45 m/s and nerve pathology shows axonal and/or demyelinating features.5
Clinically, the most frequent CMT phenotype is characterized by progressive distal weakness and sensory loss appearing toward the second decade, with foot deformities and absent reflexes. However, other patients develop a much more severe form with onset in infancy or early childhood and great disability within a few years, or a milder course with few symptoms until adulthood. This clinical heterogeneity, coupled with the expanding genetic diversity, is the complex scenario of the inherited neuropathies. Comprehensive clinical series, in which at least the most frequent genes have been studied, are needed to shed light on the populational genetic distribution and genotype-phenotype correlation in CMT disease.6,7 Herein, we present the genetic distribution and phenotypic characterization of an extensive series of CMT disease after an exhaustive genetic screening in the Region of Valencia, a geographically well-defined Mediterranean area.
METHODS
Subjects.
This is a longitudinal descriptive study, which includes all of the patients with the diagnosis of CMT disease and evaluated at the inherited neuropathy clinic of Hospital Universitari i Politècnic La Fe in Valencia from 2000 to 2012. Patients with sensory-motor neuropathy were considered to have CMT disease if a) a causative genetic defect was determined, b) family members with similar characteristics were detected, or c) sporadic cases were included if their medical history, examination, and neurophysiology were compatible with CMT disease, and other known causes of acquired neuropathy were reasonably discarded. Patients with inherited neuropathies with exclusive motor (distal hereditary motor neuropathies) or sensory and autonomic (hereditary sensory and autonomic neuropathies) signs were excluded from this study, as well as those with hereditary neuropathy with liability to pressure palsies, and those with complex disorders in which neuropathy was not the most predominant phenotypic feature. Patients were subclassified with demyelinating or axonal CMT disease according to MMNCVs of the proband, except when the amplitudes of median compound motor action potentials were reduced >90%. In those cases, the conduction velocities to nerves innervating proximal muscles were measured (palmaris longus for the median nerve, flexor carpi ulnaris for the ulnar nerve, etc.), and occasionally latencies of other proximal nerves such as the axillary nerve, or pathologic evidence were considered.
Standard protocol approvals, registrations, and patient consents.
This study protocol was approved by the Institutional Review Board of the Hospital Univesitari i Politècnic La Fe. Written informed consents were obtained from all of the members included in this study.
Clinical assessments.
The clinical assessment included strength, muscular atrophy, sensory loss, reflexes, foot deformities, as well as a general and neurologic examination. Muscle strength was graded using the standard Medical Research Council scale. CMT neuropathy score was recorded in all patients followed since 2006,8 and the Functional Disability Scale score in those after 20009; previous clinical data were extrapolated to CMT neuropathy score and Functional Disability Scale score when possible. Comprehensive electrophysiologic studies were performed in 401 of 438 patients (91.6%), and were not performed only when the genetic diagnosis of another family member was already available. Lower limb muscle MRI and sural nerve biopsy were performed only when there were reasonable doubts regarding the clinical diagnosis or for investigational purposes, and followed the protocols described previously.10
Mutational analysis.
Blood samples were drawn and genomic DNA was obtained by standard methods from peripheral white blood cells. In all of the probands, the CMT1A duplication was analyzed by MLPA (Multiplex Ligation–dependent Probe Amplification, SALSA kit P033 CMT1; MRC-Holland, Amsterdam, the Netherlands) in a genetic analyzer ABI Prism 3130xl (Applied Biosystems, Foster City, CA). Once the CMT1A duplication was discarded, a mutational screening of genes involved in CMT disease was performed taking into account the ethnicity of the proband and the phenotype. In patients with Gypsy ethnicity, the genetic testing strategy was planned as described previously.11 In Caucasian patients, the mutational screening was clinically oriented, and included the genes detailed in table 1 until the causative mutation was identified or all of the genes had been analyzed.
Table 1.
Genes analyzed in the mutational screening
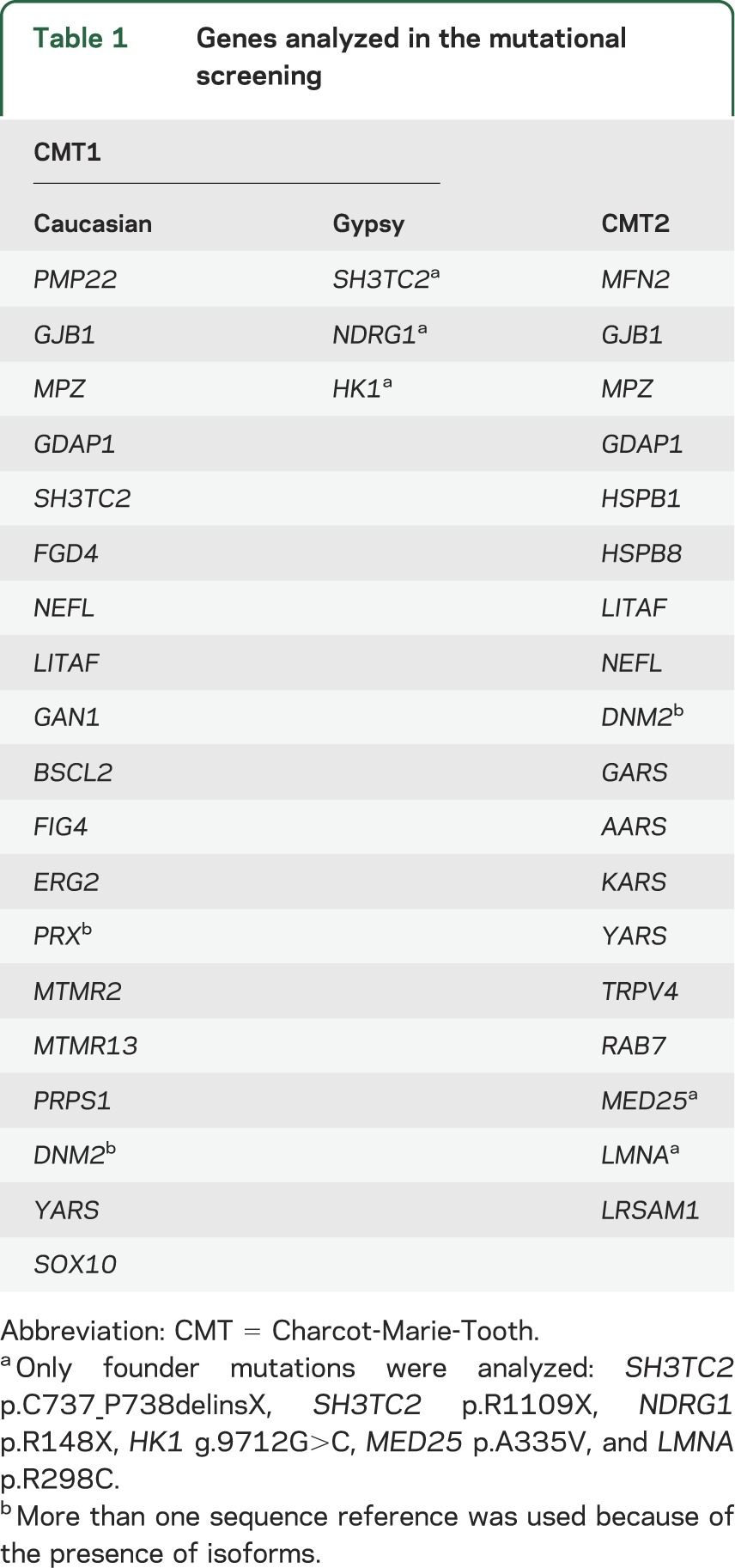
The mutational screening was performed by amplification of all exons and their intronic flanking sequences, except in the GJB1 gene in which the promoter sequence has also been analyzed. The Gene Runner version 3.05 software was used for designing primers (available on request). The PCR products were analyzed by using denaturing high-performance liquid chromatography (WAVE System; Transgenomic Inc., Omaha, NE), and the anomalous patterns were investigated by Sanger sequencing (ABI Prism 3130xl). Finally, in both the MPZ and the GJB1 genes, large deletions and/or duplications were investigated by MLPA using the SALSA kits P143 and P129 (MRC-Holland) in an ABI Prism 3130xl autoanalyzer. We did not screen MT-ATP6, PDK3, DHTKD1, GNB4, or TRIM2 genes because they had not been described when this project was concluded.12–16
When possible, segregation analyses within the families were performed, and novel mutations were analyzed in 200 chromosomes from healthy controls of Spanish ancestry. The biological relevance of the amino acid changes was studied using both SIFT (http://blocks.fhcrc.org/sift/SIFT.html, accessed June 24, 2013) and PolyPhen (http://genetics.bwh.harvard.edu/pph, accessed June 24, 2013) programs. When the detected alteration modified a splicing sequence, we used the NNSPLICE (http://fruitfly.org:9005/seq_tools/splice.html, accessed June 24, 2013) and the Splice View (http://zeus2.itb.cnr.it/∼webgene/wwwspliceview_ex.html, accessed June 24, 2013) software.
RESULTS
A total of 1,009 patients were evaluated at our inherited neuropathy clinic during the timeframe 2000 to 2012; 438 of them were considered to have CMT disease and met our inclusion criteria. All were Spanish, and 401 of them (91.6%) were currently living or had ancestral roots in the Region of Valencia, in the Western Mediterranean area. Initially, 275 (62.8%) were classified as demyelinating CMT, and 163 (37.2%) as axonal CMT. Regarding the inheritance pattern, 242 (55.3%) were considered as AD, 51 (11.6%) were AR, 52 (11.9%) were X-linked, and 93 (21.2%) were considered sporadic. Genetic diagnosis was achieved in 365 of 438 patients (83.3%), with a higher success rate in the demyelinating forms (263/275; 95.6%) over the axonal forms (102/163; 62.6%). The causative mutations were detected in 214 of 242 patients (88.4%) with AD inheritance, 45 of 51 (88.2%) with AR inheritance, 52 of 52 (100%) with X-linked inheritance, and in only 54 of 93 (58.1%) with a sporadic presentation. In table 2, the detailed genetic diagnosis can be analyzed and compared with the latest published data, and in the figure, the distribution according to CMT subtype is shown. All of the genetic and clinical information has also been recorded in a readily accessible mutation database (http://www.treat-cmt.es/db, accessed June 24, 2013).
Table 2.
Genetic distribution and comparison to other series
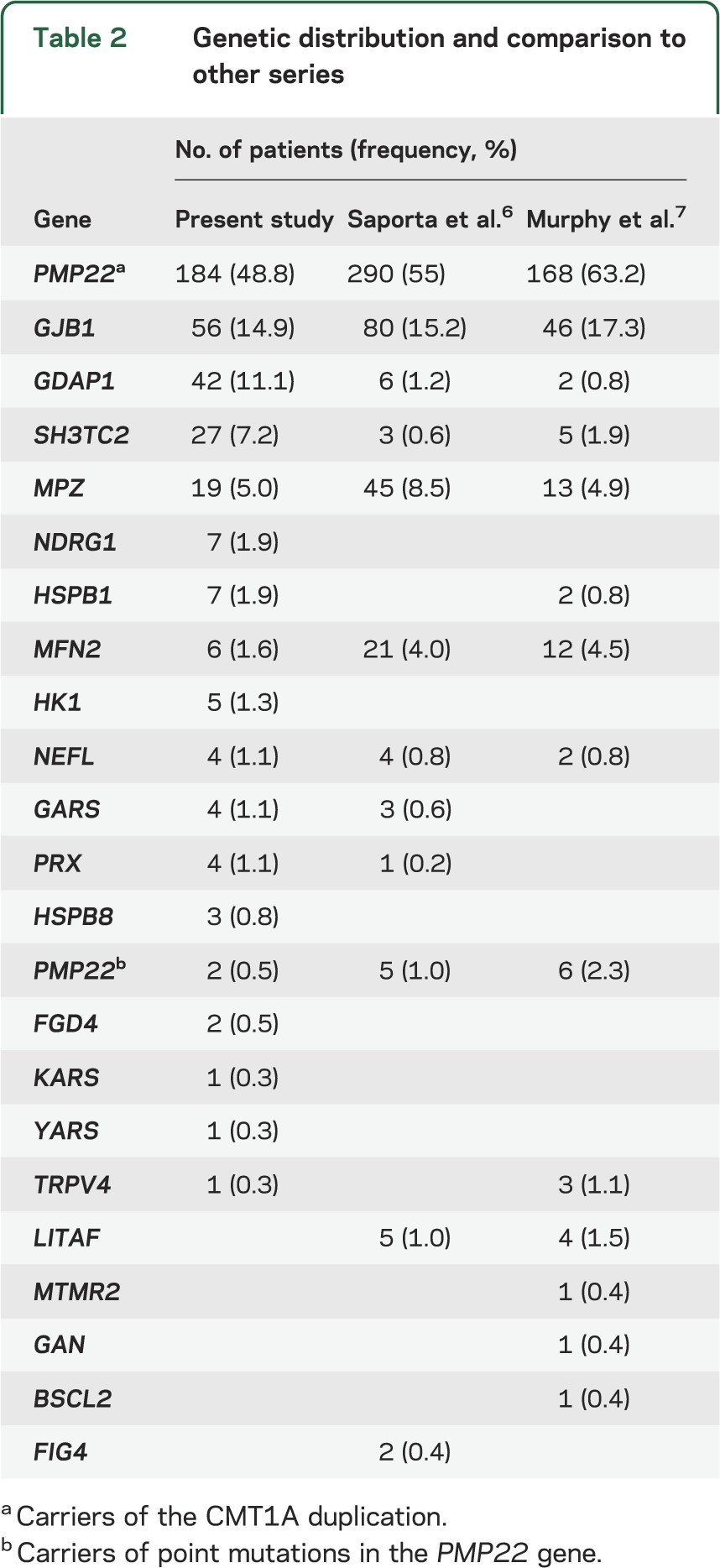
Figure. Genetic characterization of CMT disease subtypes.

Patients evaluated at the inherited neuropathy clinic during the timeframe 2000–2012. a Carriers of the CMT1A duplication. b Carriers of point mutations in the PMP22 gene. AD = autosomal dominant; AR = autosomal recessive; CMT = Charcot-Marie-Tooth.
Patients with demyelinating CMT disease.
Of the 275 patients with demyelinating CMT disease, 241 were of Caucasian ethnicity and 34 were of Gypsy origin. Of the Caucasian patients with the demyelinating form, 184 (76.3%) carried the CMT1A duplication, which is the most frequent cause of CMT disease. In the remaining 57 Caucasian patients, the disease causing mutation was identified in 45 with the following distribution: 25 mutations in GJB1, 9 in MPZ, 4 in PRX, 2 point mutations in PMP22, 2 in FGD4, 2 in SH3TC2, and 1 in NEFL. Six novel mutations were detected in demyelinating CMT (table 3). Once the genetic screening was performed, the causative change remained unknown in 12 patients (4.9%). No mutations were identified in any of the following genes: LITAF, EGR2, GDAP1, MTMR2, MTMR13, FIG4, PRPS1, DNM2, YARS, and SOX10. In the Gypsy population, the disease-causing mutation was identified in all cases, and consisted exclusively of founder mutations related to CMT disease in the Gypsy population.11
Table 3.
Novel mutations with detailed assessments and conduction velocities of the probands, and phenotypic peculiarities
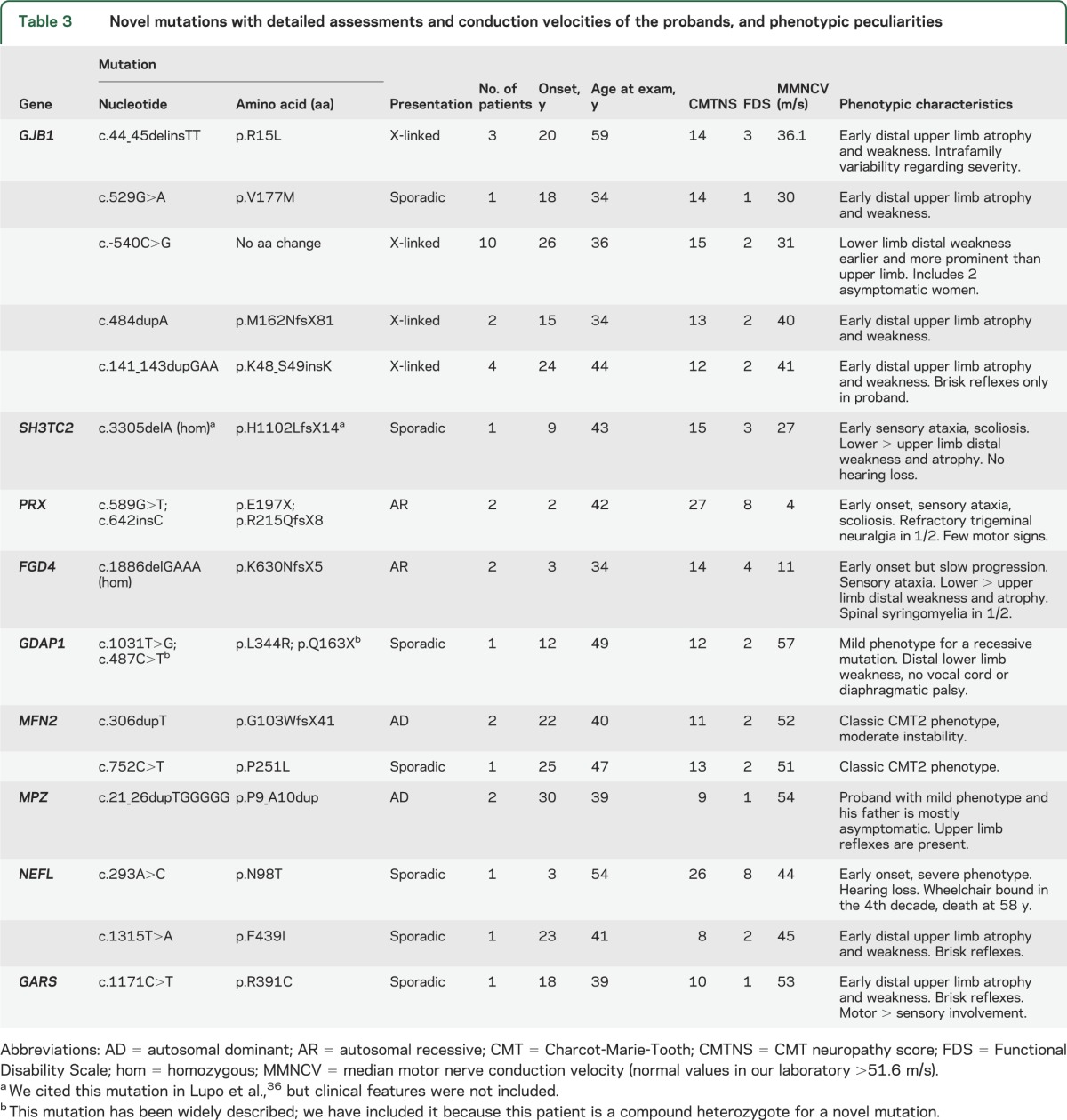
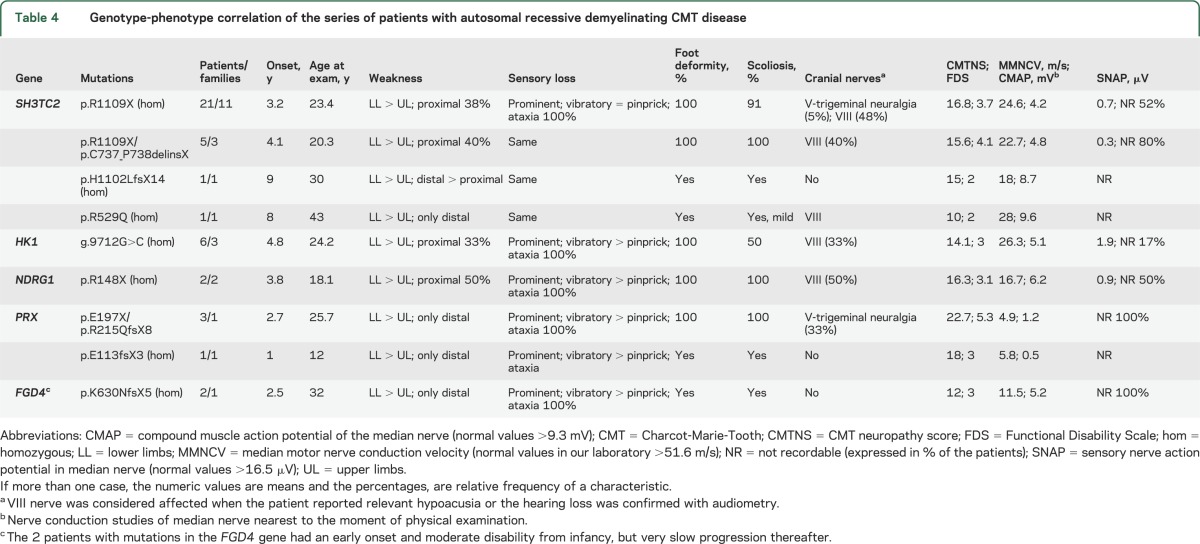
Table 4 shows the relevant clinical features associated with AR forms of demyelinating CMT disease (CMT4). These forms have certain common characteristics such as early onset, delayed motor development, and severe disability, but other features differ between the CMT4 subtypes.
Table 4.
Genotype-phenotype correlation of the series of patients with autosomal recessive demyelinating CMT disease
Patients with axonal CMT disease.
The mutational screening detailed in table 1 led to identification of the disease-causing mutation in 102 of 163 patients with axonal CMT disease (62.6%). In this set of patients, there is a marked genetic heterogeneity, with mutations in the GDAP1 and GJB1 genes being the 2 most frequent causes of axonal CMT disease. Mutations in the GDAP1 gene correspond to 24 patients (14.7% of CMT2) with AD inheritance (caused by the p.R120W mutation in all cases except one) and 18 patients (11.0%) with AR inheritance and diverse genotype. All of our patients with GDAP1 mutations were defined as CMT2 because the neurophysiologic findings were clearly axonal, although the pathology included both axonal features (fiber loss, axonal degeneration, few regenerative clusters, etc.) and myelin abnormalities (thin myelin sheaths, abnormal myelin folding, occasional onion bulb–like formations). Patients with AR inheritance developed a severe phenotype with important disability, vocal cord, and diaphragmatic palsies whereas patients with dominant GDAP1 mutations presented with a mild to moderate phenotype with certain clinical and MRI particularities reported previously.10
Mutations in the GJB1 gene were detected in 31 patients with axonal CMT disease (19.0%). It is interesting to note that although the patients were classified as having demyelinating or axonal CMT disease according to the MMNCVs of the proband, more than 80% of these families would be classified as having intermediate forms of CMT disease.
The remaining mutations were actually quite rare, accounting for only 29 cases (17.8%), and are distributed among several genes: 10 patients with mutations in MPZ, 7 in HSPB1, 4 in MFN2, 3 in HSPB8, 3 in NEFL, 1 in GARS, and 1 in KARS. In the aggregate, 25 different mutations were identified in the CMT2 series and 10 of them were novel (table 3). Once the mutational screening was performed, the disease-causing mutation remained unknown in 61 patients (37.4%). No change was identified in the following genes: RAB7, DNM2, YARS, AARS, LRSAM1, and TRPV4, nor the founder mutations MED25 p.A335V or LMNA p.R298C.
DISCUSSION
A thorough genetic screening has been performed in an extensive clinical series of patients with CMT disease in a Western Mediterranean area. Overall, a molecular diagnosis was achieved in 83.3%, with a higher success rate in demyelinating than in axonal CMT disease. In demyelinating patients, these rates are comparable to the other series in which a comprehensive genetic screening was performed (table 2),6,7,17 suggesting that few genes involved in this form of CMT disease remain undiscovered. However, in CMT2, although the success rate is higher than in other series, 37.4% of patients remain without genetic diagnosis. The mutational distribution described confirms the extensive heterogeneity intrinsic to this disease; 56 different mutations have been detected in this series, and 16 had not been described previously. This comprehensive study depicts the genetic distribution of a large CMT series in the Mediterranean basin, and there are certain distinctive features compared with other geographic areas.
The CMT1A duplication is by far the most common mutation detected, and all patients were classified as demyelinating CMT; in fact, none had MMNCV >30 m/s. CMT1A accounts for 66.9% of the demyelinating forms, which is somewhat lower than other series that report slightly more than 70%.18 Actually, these results are biased by the presence of 34 Gypsy patients affected by demyelinating CMT disease who harbored the previously described founder mutations associated with the Gypsy population as we have previously reported.11,19 These 34 Gypsy patients and 8 others of Caucasian ethnicity (4 with mutations in PRX, 2 in SH3TC2, and 2 in FGD4; table 4) comprise the 11.6% of demyelinating CMT with an AR inheritance (CMT4). The percentage of patients with AR or sporadic presentation is in fact greater than in other series6 and may reflect certain populational peculiarities, as the Region of Valencia hosts a numerous Gypsy population (more than 50,000), and certain isolated areas have a high consanguinity rate.
Mutations in GJB1 were the second most common genetic diagnosis after CMT1A, accounting for 12.8% of the CMT series. These patients were classified according to the MMNCV of the proband, but clinically all patients had a consistent phenotype that was not so much influenced by conduction velocities, as by sex.20,21 Only 5 patients (9%) had signs of CNS involvement (brisk reflexes and Babinski sign in 2 of them) with normal encephalic and spinal MRI. It is worth noting that in 2 of these patients after a long follow-up (>20 years), the pyramidal signs became less prominent as the neuropathy progressed, becoming overshadowed by the neuropathic signs. More than 300 mutations have been described in the GJB1 gene, throughout the coding region and exceptionally, in the 5'-UTR (untranslated region). A very extensive family of our series was found to be carriers of a novel c.-540C>G mutation in this region. Its pathogenicity was demonstrated by a luciferase assay (data not shown).
Mutations in MPZ were detected in only 4.3% of the series; 9 were classified as demyelinating CMT and 10 as axonal CMT. In this case, there was important phenotypical variability, as has been reported in this gene.22,23 Except for one family, demyelinating patients were more severely affected, with earlier disease onset (first decade), prominent sensory loss, and moderate to severe disability with progression. One of these patients, carrier of the MPZ p.S121F mutation, developed a severe congenital hypomyelinating neuropathy.24 Other genes were actually quite scarcely affected in our CMT1 series (NEFL, point mutations in PMP22, PRX, SH3TC2, and FGD4).
There is a great genetic diversity in axonal forms of CMT disease, as 25 different mutations were detected in 9 genes. The success rate of our series in these patients (62.6%) is one of the highest that has been published, probably because of the ample genetic screening that has been performed, and the high relative frequency of GDAP1. The genetic distribution in CMT2 shows that the 2 most frequent causes of axonal CMT disease were mutations in the GDAP1 and GJB1 genes, which combined accounted for 44.8% of patients who had axonal CMT disease. However, 37.4% of the patients with CMT2 remained undiagnosed, and this constitutes a great challenge for the near future.
Our series of 42 patients with mutations in the GDAP1 gene is to date the most extensive one published and all of them presented neurophysiologic features of axonal CMT disease. Patients with apparently demyelinating or intermediate nerve conduction studies have been reported,25,26 but in our patients, the only ones with slow conduction velocities were those in which compound motor action potential was <0.5 mV, and nerve conduction velocity was clearly normal if measured to nerves innervating proximal muscles. Although the neurophysiologic findings in these patients were unequivocally axonal, the pathology included both axonal degeneration and myelin abnormalities.10,27 Eighteen patients with recessive GDAP1 mutations were detected, with an early disease onset and rapid progression, and were wheelchair-bound in the second or third decade in all cases except 2 (associated with p.L344R/p.Q163X compound heterozygote, and p.R282C/p.R282C homozygote genotypes) who had a relatively milder phenotype.27 Twenty-four of 25 patients with dominant GDAP1 mutations carried the p.R120W substitution, which is to date the most frequent dominant mutation detected in the GDAP1 gene. Although this mutation has been described in families with different geographic origins,28–30 the GDAP1 p.R120W probably has a founder effect in our population, and presents with a mild to moderate phenotype.10
Apart from the high prevalence of GDAP1 mutations, the other notable factor in the axonal CMT series is the low number of cases with mutations in the MFN2 gene (2.5%). MFN2 has been identified as the most common gene in axonal CMT disease in many series,7,8 accounting consistently for 10% to 33%31–33 of this CMT form, even in other Spanish Mediterranean areas.34 Certain other European series have described even lower frequencies35 than our own, suggesting that the distribution of MFN2 mutations may be quite heterogeneous within Europe. The remaining mutations identified in axonal patients were even less frequent, including MPZ, HSPB1, NEFL, GARS, HSPB8, and YARS genes (15.3% of the CMT2 series).
The knowledge derived from thoroughly screened CMT series is essential to comprehend the global picture of this disease, as there may be relevant changes in the genetic distribution of different areas. A clear example of this is the relatively high prevalence of recessive forms and the predominance of GDAP1 over MFN2 in this clinical series. More information about the genetic distribution in other Spanish or Mediterranean areas is needed to discern whether this is only a local characteristic, or can be extrapolated to other areas.
ACKNOWLEDGMENT
The authors are grateful to Paula Sancho for helping to generate the database of CMT mutations in the Spanish population, to Susana Rovira for performing the study of the GJB1 gene promoter region, and to Itziar Llopis for the sample management.
GLOSSARY
- AD
autosomal dominant
- AR
autosomal recessive
- CMT
Charcot-Marie-Tooth
- MMNCV
median motor nerve conduction velocity
AUTHOR CONTRIBUTIONS
Dr. Sivera: acquisition of data, analysis and interpretation, initial manuscript elaboration. Dr. Sevilla: study concept and design, initial manuscript elaboration. Dr. Vílchez: critical revision of the manuscript for important intellectual content. Ms. Martínez-Rubio: genetic studies, acquisition of data. Dr. Chumillas: nerve conduction studies, acquisition of data. Dr. Vázquez: acquisition of data, analysis and interpretation. Dr. Muelas and Dr. Bataller: critical revision of the manuscript for important intellectual content. Dr. Millán: genetic studies (CMT1A duplication), acquisition of data. Dr. Palau: study concept and design, critical revision of the manuscript for important intellectual content. Dr. Espinós: study concept and design, study supervision, genetic screening.
STUDY FUNDING
This collaborative joint project is awarded by IRDiRC and funded by the Instituto de Salud Carlos III (ISCIII)–Subdirección General de Evaluación y Fomento de la Investigación within the framework of the National R+D+I Plan (grants IR11/TREAT-CMT, PI08/90857, PI08/0889, CP08/00053, PI12/00453, and PI12/0946), cofunded with FEDER funds, and the Generalitat Valenciana (grant Prometeo/2009/051). The Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED) and the Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER) are initiatives from the ISCIII.
DISCLOSURE
R. Sivera reports no disclosures. T. Sevilla is funded by grants from the ISCIII (PI12/0946, PI08/0889) and IRDiRC (IR11/TREAT-CMT). J. Vílchez received research support from the CIBERNED. D. Martínez-Rubio, M. Chumillas, J. Vázquez, N. Muelas, L. Bataller, and J. Millán report no disclosures. F. Palau is funded by grants from Fondo de Investigación Sanitaria (PI08/90857), the Generalitat Valenciana (Prometeo/2009/051), the IRDiRC (IR11/TREAT-CMT), and the CIBERER. C. Espinós is funded by a grant from the ISCIII (PI12/00453) and IRDiRC (IR11/TREAT-CMT). Dr. Espinós has a “Miguel Servet” contract funded by the ISCIII and the CIBERER (CP08/00053). Go to Neurology.org for full disclosures.
REFERENCES
- 1.Combarros O, Calleja J, Polo JM, Berciano J. Prevalence of hereditary motor and sensory neuropathy in Cantabria. Acta Neurol Scand 1987;75:9–12 [DOI] [PubMed] [Google Scholar]
- 2.Foley C, Schofield I, Eglon G, Bailey G, Chinnery PF, Horvath R. Charcot-Marie-Tooth disease in Northern England. J Neurol Neurosurg Psychiatry 2012;83:572–573 [DOI] [PubMed] [Google Scholar]
- 3.Skre H. Genetic and clinical aspects of Charcot Marie Tooth's disease. Clin Genet 1974;6:98–118 [DOI] [PubMed] [Google Scholar]
- 4.Dyck PJ, Lambert EH. Lower motor and primary sensory neuron diseases with peroneal muscular atrophy. I. Neurologic, genetic, and electrophysiologic findings in hereditary polyneuropathies. Arch Neurol 1968;18:603–618 [DOI] [PubMed] [Google Scholar]
- 5.Payreson D, Marchesi C. Diagnosis, natural history, and management of Charcot-Marie-Tooth disease. Lancet Neurol 2009;8:654–667 [DOI] [PubMed] [Google Scholar]
- 6.Saporta AS, Sottile SL, Miller LJ, Feely SM, Siskind CE, Shy ME. Charcot-Marie-Tooth disease subtypes and genetic testing strategies. Ann Neurol 2011;69:22–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Murphy SM, Laura M, Fawcett K, et al. Charcot-Marie-Tooth disease: frequency of genetic subtypes and guidelines for genetic testing. J Neurol Neurosurg Psychiatry 2012;83:706–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shy ME, Blake J, Krajewski K, et al. Reliability and validity of the CMT neuropathy score as a measure of disability. Neurology 2005;64:1209–1214 [DOI] [PubMed] [Google Scholar]
- 9.Birouk N, Gouider R, Le Guern E, et al. Charcot–Marie–Tooth disease type 1A with 17p11.2 duplication: clinical and electrophysiological phenotype study and factors influencing disease severity in 119 cases. Brain 1997;120:813–823 [DOI] [PubMed] [Google Scholar]
- 10.Sivera R, Espinós C, Vílchez JJ, et al. Phenotypical features of the p.R120W mutation in the GDAP1 gene causing autosomal dominant Charcot-Marie-Tooth disease. J Peripher Nerv Syst 2010;15:334–344 [DOI] [PubMed] [Google Scholar]
- 11.Sevilla T, Martínez-Rubio D, Márquez C, et al. Genetics of the Charcot-Marie-Tooth disease in the Spanish Gypsy population: the hereditary motor and sensory neuropathy–Russe in depth. Clin Genet 2013;83:565–570 [DOI] [PubMed] [Google Scholar]
- 12.Pitceathly R, Murphy SM, Cottenie E, et al. Genetic dysfunction of MT-ATP6 causes axonal Charcot-Marie-Tooth disease. Neurology 2012;79:1145–1154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kennerson ML, Yiu EM, Chuang DT, et al. A new locus for X-linked dominant Charcot-Marie-Tooth disease (CMTX6) is caused by mutations in the pyruvate dehydrogenase kinase isoenzyme 3 (PDK3) gene. Hum Mol Genet 2013;22:1404–1416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu W, Gu MM, Sun LH, et al. A nonsense mutation in DHTKD1 causes Charcot-Marie-Tooth disease type 2 in a large Chinese pedigree. Am J Hum Genet 2012;91:1088–1094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Soong BW, Huang YH, Tsai PC, et al. Exome sequencing identifies GNB4 mutations as a cause of dominant intermediate Charcot-Marie-Tooth disease. Am J Hum Genet 2013;92:422–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ylikallio E, Pöyhönen R, Zimon M, et al. Deficiency of the E3 ubiquitin ligase TRIM2 in early-onset axonal neuropathy. Hum Mol Genet 2013;22:2975–2983 [DOI] [PubMed] [Google Scholar]
- 17.Lin KP, Soong BW, Yang CC, et al. The mutational spectrum in a cohort of Charcot-Marie-Tooth disease type 2 among the Han Chinese in Taiwan. PLoS One 2011;6:e29393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nelis E, Van Broeckhoven C, De Jonghe P, et al. Estimation of the mutation frequencies in Charcot-Marie-Tooth disease type 1 and hereditary neuropathy with liability to pressure palsies: a European collaborative study. Eur J Hum Genet 1996;4:25–33 [DOI] [PubMed] [Google Scholar]
- 19.Claramunt R, Sevilla T, Lupo V, et al. The p.R1109X mutation in SH3TC2 gene is predominant in Spanish Gypsies with Charcot-Marie-Tooth disease type 4. Clin Genet 2007;71:343–349 [DOI] [PubMed] [Google Scholar]
- 20.Shy ME, Siskind C, Swan ER, et al. CMT1X phenotypes represent loss of GJB1 gene function. Neurology 2007;68:849–855 [DOI] [PubMed] [Google Scholar]
- 21.Siskind CE, Murphy SM, Ovens R, Polke J, Reilly MM, Shy ME. Phenotype expression in women with CMT1X. J Peripher Nerv Syst 2011;16:102–107 [DOI] [PubMed] [Google Scholar]
- 22.Warner L, Hilz M, Appel S, et al. Clinical phenotypes of different MPZ (P0) mutations may include Charcot–Marie–Tooth type 1B, Dejerine–Sottas, and congenital hypomyelination. Neuron 1996;17:451–460 [DOI] [PubMed] [Google Scholar]
- 23.Shy ME, Jani A, Krajewski K, et al. Phenotypic clustering in MPZ mutations. Brain 2004;127:371–384 [DOI] [PubMed] [Google Scholar]
- 24.Sevilla T, Lupo V, Sivera R, et al. Congenital hypomyelinating neuropathy due to a novel MPZ mutation. J Peripher Nerv Syst 2011;16:347–352 [DOI] [PubMed] [Google Scholar]
- 25.Baxter RV, Ben Othmane K, Rochelle JM, et al. Ganglioside-induced differentiation-associated protein-1 is mutant in Charcot-Marie-Tooth disease type 4A/8q21. Nat Genet 2002;30:21–22 [DOI] [PubMed] [Google Scholar]
- 26.Senderek J, Bergmann C, Ramaekers VT, et al. Mutations in the ganglioside-induced-differentiation-associated protein-1 (GDAP1) gene in intermediate type autosomal recessive Charcot-Marie-Tooth neuropathy. Brain 2003;126:642–649 [DOI] [PubMed] [Google Scholar]
- 27.Sevilla T, Jaijo T, Nauffal D, et al. Vocal cord paresis and diaphragmatic dysfunction are severe and frequent symptoms of GDAP1-associated neuropathy. Brain 2008;131:3051–3061 [DOI] [PubMed] [Google Scholar]
- 28.Claramunt R, Pedrola L, Sevilla T, et al. Genetics of Charcot-Marie-Tooth disease type 4A: mutations, inheritance, phenotypic variability, and founder effect. J Med Genet 2005;42:358–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zimoń M, Baets J, Fabrizi GM, Jaakkola E, et al. Dominant GDAP1 mutations cause predominantly mild CMT phenotypes. Neurology 2011;77:540–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ammar N, Nelis E, Merlini L, Barisic N, et al. Identification of novel GDAP mutations causing autosomal recessive Charcot-Marie-Tooth disease. Neuromuscul Disord 2003;13:720–728 [DOI] [PubMed] [Google Scholar]
- 31.Verhoeven K, Claeys KG, Züchner S, et al. MFN2 mutation distribution and genotype/phenotype correlation in Charcot-Marie-Tooth type 2. Brain 2006;129:2093–2102 [DOI] [PubMed] [Google Scholar]
- 32.Calvo J, Funalot B, Ouvrier RA, et al. Genotype-phenotype correlations in Charcot-Marie-Tooth disease type 2 caused by mitofusin 2 mutations. Arch Neurol 2009;66:1511–1516 [DOI] [PubMed] [Google Scholar]
- 33.Auranen M, Ylikallio E, Toppila J, Somer M, Kiuru-Enari S, Tyynismaa H. Dominant GDAP1 founder mutations is a common cause of axonal Charcot-Marie-Tooth disease in Finland. Neurogenetics 2013;14:123–132 [DOI] [PubMed] [Google Scholar]
- 34.Casasnovas C, Banchs I, Cassereau J, et al. Phenotypic spectrum of MFN2 mutations in the Spanish population. J Med Genet 2010;47:249–256 [DOI] [PubMed] [Google Scholar]
- 35.Braathen GJ. Genetic epidemiology of Charcot-Marie-Tooth disease. Acta Neurol Scand Suppl 2012;126:1–22 [DOI] [PubMed] [Google Scholar]
- 36.Lupo V, Galindo MI, Martínez-Rubio D, et al. Missense mutations in the SH3TC2 protein causing Charcot-Marie-Tooth disease type 4C affect its localization in the plasma membrane and endocytic pathway. Hum Mol Genet 2009;18:4603–4614 [DOI] [PubMed] [Google Scholar]