

Abstract
Objective:
To identify potential genetic modifiers contributing to the phenotypic variability that is detected in patients with repeat expansions in chromosome 9 open reading frame 72 (C9ORF72), we investigated the frequency of these expansions in a cohort of 334 subjects previously found to carry mutations in genes known to be associated with a spectrum of neurodegenerative diseases.
Methods:
A 2-step protocol, with a fluorescent PCR and a repeat-primed PCR, was used to determine the presence of hexanucleotide expansions in C9ORF72. For one double mutant, we performed Southern blots to assess expansion sizes, and immunohistochemistry to characterize neuropathology.
Results:
We detected C9ORF72 repeat expansions in 4 of 334 subjects (1.2% [or 1.8% of 217 families]). All these subjects had behavioral phenotypes and also harbored well-known pathogenic mutations in either progranulin (GRN: p.C466LfsX46, p.R493X, p.C31LfsX35) or microtubule-associated protein tau (MAPT: p.P301L). Southern blotting of one double mutant with a p.C466LfsX46 GRN mutation demonstrated a long repeat expansion in brain (>3,000 repeats), and immunohistochemistry showed mixed neuropathology with characteristics of both C9ORF72 expansions and GRN mutations.
Conclusions:
Our findings indicate that co-occurrence of 2 evidently pathogenic mutations could contribute to the pleiotropy that is detected in patients with C9ORF72 repeat expansions. These findings suggest that patients with known mutations should not be excluded from further studies, and that genetic counselors should be aware of this phenomenon when advising patients and their family members.
Hexanucleotide repeat expansions in chromosome 9 open reading frame 72 (C9ORF72) have been described in patients with phenotypic heterogeneity.1,2 They are the major genetic cause of disease in patients with motor neuron disease (MND) and frontotemporal dementia (FTD), but they have also been reported in patients clinically diagnosed with memory disorders, and patients presenting with parkinsonism or psychosis.3 It is currently unknown which modifiers determine its variable disease onset, progression, and other manifestations. Recently, however, evidence pointed toward an oligogenic pathogenesis of MNDs.4 In addition, more than 20 patients have been reported with both C9ORF72 expansions and a mutation in another MND- and/or FTD-associated gene.5–7 A large cohort of cases with known mutations would be ideal to investigate whether an oligogenic disease model could explain the pleiotropy detected in patients with C9ORF72 expansions. We therefore assessed the frequency of these expansions in a heterogeneous cohort of 334 subjects previously found to carry pathogenic mutations in genes associated with neurodegenerative diseases.
METHODS
Subject selection.
Two study cohorts were analyzed for C9ORF72 repeat expansions. Our initial study cohort consisted of 149 probands with mutations in genes known to be associated with a spectrum of neurodegenerative diseases (table 1). These subjects were of North American descent and obtained through the Mayo Clinic (n = 84), University of Western Ontario (n = 3), University of British Columbia (n = 12), University of Texas Southwestern Medical Center (n = 2), University of California (n = 16), The David Geffen School of Medicine at University of California (n = 2), Northwestern University Feinberg School of Medicine (n = 7, E.H.B.), Drexel University College of Medicine (n = 3), Robarts Research Institute (n = 5), Banner Sun Health Research Institute (n = 4), Coriell Research Institute (n = 8), and Harvard Brain Bank (n = 3).
Table 1.
Baseline characteristics
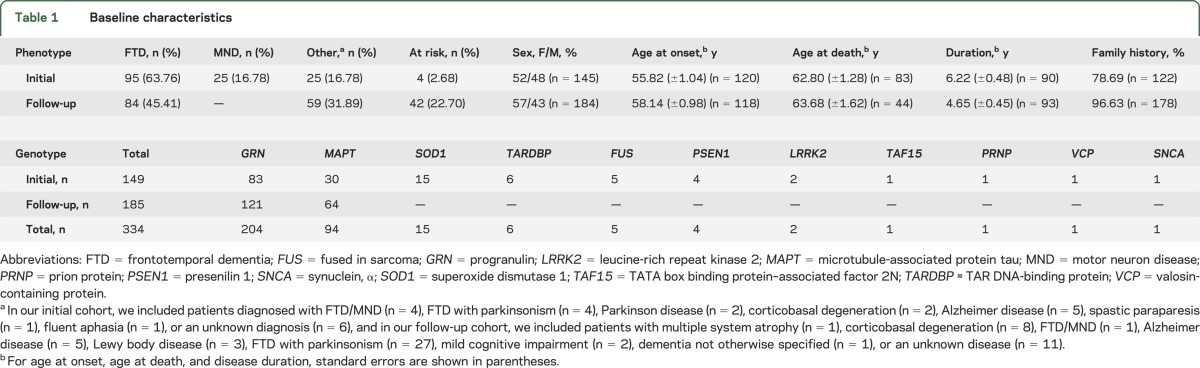
Based on the identification of C9ORF72 repeat expansions in progranulin (GRN) and microtubule-associated protein tau (MAPT) mutation carriers in our initial study cohort, we selected a follow-up cohort with additional GRN and MAPT mutation carriers. This follow-up cohort consisted of 102 family members of probands from our initial study cohort, and 83 subjects from 68 new families of North American and Italian origin. These latter families were provided by other collaborators at IRCCS Istituto Centro San Giovanni di Dio–Fatebenefratelli (n = 68), University of Pennsylvania School of Medicine (n = 3), Northwestern University Feinberg School of Medicine (n = 2, M.M.), National Institute of Neurological Disorders and Stroke and NIH (n = 3), and University of Washington School of Medicine (n = 7).
Standard protocol approvals, registrations, and patient consents.
All subjects agreed to be in the study, and biological samples were obtained after informed consent with ethical committee approval from the respective institutions.
Genetic analysis.
The GGGGCC repeat in C9ORF72 was assessed using a 2-step protocol.1 Briefly, genomic DNA was PCR-amplified with genotyping primers and one fluorescently labeled primer, followed by fragment length analysis on an ABI3730 DNA analyzer (Applied Biosystems, Foster City, CA) and visualized using GeneMapper v4.0 software (Applied Biosystems). For individuals who were shown to be homozygous for C9ORF72 repeats, we performed a repeat-primed PCR, and PCR products were analyzed with an ABI3730 DNA Analyzer and GeneMapper software. A characteristic stutter pattern was considered evidence of a C9ORF72 repeat expansion.
Southern blot.
Southern blotting was performed as described by DeJesus-Hernandez et al.,1 with minor modifications. DNA was isolated from frozen frontal cortex, temporal cortex, and cerebellum. A total of 10 µg of genomic DNA was digested with XbaI, and electrophoresed in an agarose gel. DNA was then transferred to a positively charged nylon membrane (Roche, Penzberg, Germany.) and cross-linked by UV irradiation. After prehybridization in digoxigenin (DIG) EasyHyb solution (Roche), hybridization with a DIG-labeled probe in hybridization solution was performed. Anti-DIG antibody (1:10,000; Roche) was used to detect the probe, which was subsequently visualized with CDP-star substrate (Roche) on X-ray film after an exposure of 30 to 60 minutes.
Immunohistochemistry.
Immunohistochemistry was performed for 3 patients in a blinded fashion, including patient F with double mutations, and for 2 patients from our initial study cohort with only GRN mutations (p.Thr382SerfsX30, c.IVS1+1G>A). Immunohistochemistry for TAR DNA-binding protein 43 (TDP-43) (1:2,500, rabbit polyclonal, Mayo Clinic),8 repeat-associated non-ATG translation peptides (C9RANT, Rb5823, 1:5,000),9 and p62 (1:1,000, lck ligand; BD Bioscience, Franklin Lakes, NJ) was performed on 5-µm-thick sections from the frontal cortex, hippocampus, and cerebellum. These sections were cut from formalin-fixed paraffin-embedded blocks, deparaffinized in xylene, rehydrated in a graded series of ethanol, and washed in distilled water. DAKO Autostainer Plus (DAKO, Carpinteria, CA) and DAKO EnVision+ System–horseradish peroxidase (diaminobenzidine) were used to process stains. To block nonspecific binding, normal goat serum (1:20 in Tris-buffered saline and Tween 20; Sigma, St. Louis, MO) was added to slides before the primary antibody.
RESULTS
Within our initial study cohort of 149 subjects with known pathogenic mutations in neurodegenerative disease genes, we identified 3 individuals with an additional C9ORF72 repeat expansion (2.0%). These expansions were present in 2 subjects with a GRN mutation (p.C466LfsX46 [c.1395_1396insC], p.C31LfsX35 [c.90_91insCTGC]) and in one subject with a MAPT mutation (p.P301L [c.902C>T]). In our follow-up cohort of 185 subjects with GRN or MAPT mutations, we detected another individual with both a GRN mutation (p.R493X [c.1477C>T]) and a C9ORF72 repeat expansion (0.5%).
In total, we therefore identified double mutations in 4 of 334 subjects (1.2%) or 4 of 217 families (1.8%) studied. The C9ORF72 repeat expansions were present in 3 of 204 subjects from our GRN subgroup (1.5% [or 2.0% of 152 GRN families]), and in 1 of 94 subjects from our MAPT subgroup (1.1% [or 3.3% of 30 MAPT families]). Importantly, these subjects with double mutations all showed a behavioral phenotype, and they did not demonstrate signs of MND. Furthermore, our subjects with double mutations appeared to be relatively young when they presented with behavioral impairment (53 years or younger; table 2), as compared with the mean of our initial and follow-up cohorts (56 and 58 years; table 1). All subjects with double mutations had a positive family history for dementia, and interestingly, 2 of 4 of these subjects had family members with MND (50%). Chromatograms and electropherograms of identified subjects are displayed in figure 1, and their pedigrees are shown in figure e-1 on the Neurology® Web site at www.neurology.org. DNA of relatives was unavailable; therefore, cosegregation of the mutations in families could not be assessed.
Table 2.
Clinical characteristics of subjects with 2 mutations

Figure 1. Chromatograms and electropherograms of identified subjects.

(A) Chromatograms of subjects included in this study. Shown are GRN mutations p.C466LfsX46 (c.1395_1396insC), p.R493X (c.1477C>T), and p.C31LfsX35 (c.90_91insCTGC), and MAPT mutation p.P301L (c.902C>T). (B) C9ORF72 repeat expansions detected in the subjects with GRN and MAPT mutations. PCR products of repeat-primed PCR reactions separated on an ABI3730 DNA Analyzer and visualized by GeneMapper software, showing 4 double mutation carriers detected in this study with their characteristic stutter amplification. C9ORF72 = chromosome 9 open reading frame 72; GRN = progranulin; MAPT = microtubule-associated protein tau.
Brain autopsy material, however, was available for one of the identified subjects (F). We obtained DNA from the frontal cortex, temporal cortex, and cerebellum, and performed a Southern blot (in duplicate). Patient F clearly demonstrated an expanded repeat (figure 2), as shown by a smear of high-molecular-weight bands, suggestive for somatic repeat instability.1 The longest repeat expansion of this double mutant was present in the frontal cortex and was estimated to be approximately 23 kb in size, bearing more than 3,000 hexanucleotide repeats.
Figure 2. Southern blot of patient F.

Southern blot demonstrating 3 brain regions of patient F. DIG-labeled DNA Molecular Weight Marker II (Roche) was used with fragments of 2,027; 2,322; 4,361; 6,557; 9,416; and 23,130 base pairs. A positive control that harbors a C9ORF72 expansion, but no additional mutation in GRN or MAPT, is shown in lanes 7, 8, and 9. In lane 10, a negative control without a C9ORF72 expansion is shown; this patient only displays the 2.3-kb wild-type allele. C9ORF72 = chromosome 9 open reading frame 72; DIG = digoxigenin; GRN = progranulin; MAPT = microtubule-associated protein tau.
Immunohistochemistry was also performed for patient F and 2 control patients with only GRN mutations (figure 3). Patient F showed type A TDP-43 pathology, including neuronal cytoplasmic inclusions and neuritic pathology. In addition, p62 and C9RANT immunohistochemistry showed many neuronal cytoplasmic inclusions, consistent with a mutation in C9ORF72. Blinded investigations of all 3 patients successfully identified the double mutant; whereas all patients demonstrated type A neuronal TDP-43 inclusions,10,11 only patient F showed these inclusions in combination with C9RANT immunoreactivity, and ubiquitin-positive, TDP-43–negative, neuronal cytoplasmic inclusions in the cerebellum, which have been shown to be unique for C9ORF72 repeat expansion carriers.9,12,13 Patient F, therefore, demonstrated mixed pathology with characteristics of both GRN mutations and C9ORF72 expansions.
Figure 3. Neuropathology of patient F.

Neuropathology of the GRN/C9ORF72 double mutation in a pathologically confirmed frontotemporal lobar degeneration case. (A) Brain MRI revealed dilated lateral ventricles and flattening of the caudate nucleus (arrow) in a T1-weighted coronal MRI scan acquired 3 years before death. MRI findings were supported (B) at autopsy with marked flattening of the caudate (arrow) and cerebral atrophy most significant in the frontal cortex (arrowhead). Superficial spongiosis in layer II of the cortical ribbon (C) is associated with type A TDP-43 pathology, including neuronal cytoplasmic inclusions (arrow) and neuritic pathology (arrowhead). The inset demonstrates a “lentiform” intranuclear inclusion (arrow) in the dentate gyrus of the hippocampus. (D) p62 and (E) C9RANT immunohistochemistry of the cerebellar granule cell layer shows many neuronal cytoplasmic inclusions, consistent with a mutation in C9ORF72. Bar = 25 µm for C, D, and E, and 10 µm for inset. C9ORF72 = chromosome 9 open reading frame 72; GRN = progranulin; TDP-43 = TAR DNA-binding protein 43.
DISCUSSION
In our present study, we discovered C9ORF72 repeat expansions in 4 of 217 families with previously identified mutations in neurodegenerative disease genes. Since the identification of C9ORF72 expansions,1,2 researchers all over the world have reported case series with expansion frequencies.5 On average, these expansions appear to account for 34% of familial MND cases and 26% of familial FTD cases.5 They have, however, also been detected in control subjects; for instance, they were present in 4 of 4,368 control subjects of American and Italian descent.5 Hence, the expansion frequency is significantly higher in our study population than in control subjects of the same origin (1.8% vs 0.1%: p value = 0.0003, Fisher exact test). To date, the sizes of C9ORF72 repeat expansions in brain are largely unknown. Southern blots have proven to be challenging, and good-sized well-characterized cohorts have not been assessed to determine expansion sizes in brain. A recent study did investigate 57 patients with a range of neurodegenerative diseases, and demonstrated great variability in repeat sizes and smear morphologies, but this study focused on blood.14 Thus far, brain tissues of only a few patients have been investigated, demonstrating sizes between approximately 600 and 4,000 repeats.1,14–20 We were able to perform a Southern blot for one of our double mutation carriers, patient F, and detected more than 3,000 hexanucleotide repeats. Our findings therefore indicate that this double mutation carrier has a long repeat expansion, in the same range as other patients with C9ORF72 expansions.
The function of C9ORF72 is presently unknown, although recent reports have suggested that C9ORF72 belongs to a group of DENN (differentially expressed in normal and neoplasia) proteins, which are GEFs (guanosine diphosphate/guanosine triphosphate exchange factors) that activate Rab-GTPase switches and regulate vesicular trafficking processes.21,22 Consequently, repeat expansions in C9ORF72 could result in a loss of function and impair these processes. However, an RNA-mediated gain-of-function mechanism could also contribute to disease due to generation of toxic RNA foci.1 Furthermore, recently it was revealed that transcripts of patients with C9ORF72 repeat expansions are also prone to non-ATG translation.9,13 This unconventional method of translation can cause an accumulation of poly(glycine-proline), poly(glycine-alanine), or poly(glycine-arginine) peptides in neurons throughout the CNS, and result in neuropathology specific to C9ORF72-associated MND and/or FTD.9 In our present study, we have shown that one of our subjects with double mutations, patient F, exhibited C9RANT immunoreactivity, consistent with these novel reports. Patient F also harbored ubiquitin-positive, TDP-43–negative, neuronal cytoplasmic inclusions in the cerebellum, which have been shown to be typical for C9ORF72 repeat expansions.12 In addition, patient F demonstrated TDP-43 type A pathology, characteristic for GRN mutations3,10,11,23; thus, this double mutation carrier displayed pathology distinctive for both C9ORF72 expansions and GRN mutations.
Interestingly, 2 patients have already been described with both C9ORF72 repeat expansions and a GRN variant (p.Y294C),24 or a MAPT variant (p.A239T).6 The p.Y294C GRN variant is novel, and has not been detected in other patients or in controls; it was present in a patient with behavioral variant FTD and it is predicted to be damaging.24 The p.A239T MAPT variant was also identified in a patient with behavioral variant FTD.6 She had 2 brothers with C9ORF72 expansions without the MAPT variant, who demonstrated signs of MND. The index case showed mixed pathology with both tauopathy and ubiquitin-positive, TDP-43–negative, neuronal cytoplasmic inclusions in the cerebellum. Nonetheless, accumulation of tau has also been reported in patients with only C9ORF72 expansions, and this accumulation could have resulted from disrupted protein degradation that favors accumulation of multiple different proteins, including tau.25 Although the pathogenicity of the p.Y294C GRN variant and the p.A239T MAPT variant are not entirely clear, the 4 subjects that we identified with C9ORF72 expansions do harbor well-known pathogenic FTD-associated mutations. In tables e-1 and e-2, we have provided a detailed overview of more than 80 families carrying these relatively common mutations. Worldwide, the p.C466LfsX46 mutation accounts for 0.4% of all families with GRN mutations, the p.R493X mutation accounts for 18.6%, and the p.C31LfsX35 mutation accounts for 2.6%; the p.P301L mutation is present in 23.9% of families with MAPT mutations (http://www.molgen.ua.ac.be/FTDmutations/). Therefore, these 4 mutations are frequently detected, and are responsible for approximately 22% of patients with GRN mutations and approximately 24% of patients with MAPT mutations.
Phenotypes of C9ORF72 repeat expansion carriers, in general, appear to differ from patients with mutations in GRN or MAPT.26–30 It was shown that age at onset is earlier in patients with C9ORF72 expansions compared to patients with GRN mutations,26–29 but later compared to patients with MAPT mutations.27–29 Moreover, MND is frequently detected in patients with C9ORF72 expansions, whereas signs of MND are scarce in patients with GRN or MAPT mutations.26,28–30 Behavioral variant FTD is the predominant phenotype of all 3 groups; primary progressive aphasia and corticobasal syndrome phenotypes are rare in patients with C9ORF72 expansions, and are more frequently detected in patients with GRN or MAPT mutations.26,29
The co-occurrence of C9ORF72 expansions and mutations in GRN or MAPT in patients with FTD could have several explanations. First, it could be argued that only C9ORF72 expansions are pathogenic, and that the GRN or MAPT mutations are rare benign variants or mere risk factors. This explanation, however, is not supported by ample studies that have revealed a strong association with FTD and indicated that they are definitely disease-causing mutations (tables e-1 and e-2). Second, it is possible that C9ORF72 expansions are not sufficient to develop disease, and that an additional mutation or environmental exposure is needed. This would provide an explanation for the increased frequency of C9ORF72 repeat expansions in cases with other genetic mutations. It is also in accordance with studies that have detected C9ORF72 expansions in control subjects,5 and with studies that reported incomplete or age-dependent penetrance; approximately 50% of expansion carriers were clinically symptomatic by an age of 48 to 58 years, and almost full penetrance was seen at an age of 75 to 80 years.27,31,32 This variable penetrance could be caused by differences in repeat sizes: long repeat sizes may represent clear pathogenic mutations, whereas intermediate sizes may act as risk factors that require a second factor, either genetic or environmental, to cause disease. Our current findings, however, oppose this explanation, while Southern blotting of one of our subjects with double mutations, patient F, revealed a relatively long repeat expansion, comparable to a patient carrying only a C9ORF72 repeat expansion. An age-dependent penetrance has also been reported for GRN, with only 50% of mutation carriers affected by the age of 60 years, and 90% of carriers affected at 70 years,33 comparable to C9ORF72. The penetrance is more than 95% for MAPT.34 It is therefore possible that one of the mutations observed in our double mutation carriers has not yet reached penetrance, and that the current symptoms are solely caused by the other mutation. Finally, it could be hypothesized that both C9ORF72 expansions and GRN or MAPT mutations are pathogenic, that each of these mutations independently causes disease, but that they act as disease modifiers when they co-occur. If we compare the mean age at which symptoms of behavioral impairment occurred between our double mutation carriers (mean: 48.8 ± 3.0, n = 4) and the remainder of our study population (mean: 57.0 ± 0.73, n = 235), then a one-tailed nonparametric Mann-Whitney test results in a p value of 0.0357. Because of the relatively small number of double mutation carriers, this difference is borderline significant, but there is clearly a tendency toward an earlier age at onset. Apart from this relatively young age at onset, all of our patients with double mutations developed signs of behavioral impairment without MND, whereas combinations of C9ORF72 repeat expansions with MND-associated mutations in TARDBP (TAR DNA-binding protein), FUS/TLS (fused in sarcoma/translated in liposarcoma), and SOD1 (superoxide dismutase 1) have previously been detected in patients with MND without FTD.4 However, numerous C9ORF72 expansion carriers without a second mutation in a known dementia gene also developed a pure behavioral variant FTD phenotype. In the future, detailed investigations of multiple patients in families carrying double mutations will be critical to determine the contribution of each mutation to disease.
While previous studies have provided evidence for an oligogenic basis of MND,4 our present findings demonstrate that oligogenicity is not confined to MNDs, and that double mutations can be present in patients with FTD as well. Thus, it is important to realize that patients already diagnosed with mutations in FTD/MND-associated genes could also harbor more recently discovered C9ORF72 repeat expansions, and that they should not be excluded from further tests, which is also highly relevant for genetic counseling, both of patients and of their (unaffected) family members.
Supplementary Material
GLOSSARY
- C9ORF72
chromosome 9 open reading frame 72
- DIG
digoxigenin
- FTD
frontotemporal dementia
- GRN
progranulin
- MAPT
microtubule-associated protein tau
- MND
motor neuron disease
- TDP-43
TAR DNA-binding protein 43
Footnotes
Supplemental data at www.neurology.org
AUTHOR CONTRIBUTIONS
Marka van Blitterswijk: study concept or design, acquisition of data, analysis or interpretation of data, statistical analysis, drafting the manuscript for content, including writing for content, revising the manuscript for content, including writing for content. Matthew C. Baker, Mariely DeJesus-Hernandez, Melissa E. Murray, Nicola J. Rutherford, Patricia E. Brown, and Thomas Ravenscroft: acquisition of data, analysis or interpretation of data. Roberta Ghidoni, Luisa Benussi, Elizabeth Finger, Ging-Yuek R. Hsiung, Brendan J. Kelley, Bianca Mullen, Peter E.A. Ash, Kevin F. Bieniek, Kimmo J. Hatanpaa, Anna Karydas, Elisabeth McCarty Wood, Giovanni Coppola, Eileen H. Bigio, Carol Lippa, Michael J. Strong, Thomas G. Beach, David S. Knopman, Edward D. Huey, Marsel Mesulam, Thomas Bird, Charles L. White III, Andrew Kertesz, Dan H. Geschwind, and Vivianna M. Van Deerlin: contribution of vital reagents/tools/patients, revising the manuscript for content, including writing for content. Ronald C. Petersen, Giuliano Binetti, Bruce L. Miller, Leonard Petrucelli, Zbigniew K. Wszolek, Kevin B. Boylan, Neill R. Graff-Radford, Ian R. Mackenzie, Bradley F. Boeve, and Dennis W. Dickson: revising the manuscript for content, including writing for content, contribution of vital reagents/tools/patients, obtaining funding. Rosa Rademakers: study concept or design, acquisition of data, analysis or interpretation of data, drafting the manuscript for content, including writing for content, revising the manuscript for content, including writing for content, study supervision or coordination, obtaining funding.
STUDY FUNDING
This work was funded by NIH grants R01 NS080882, R01 NS065782, R01 AG026251, P01 AG017586, P50 NS072187, P50 AG 016574, P30 AG13854, P30 AG12300, and U01 AG006786, the ALS Therapy Alliance, the Consortium for Frontotemporal Dementia Research, and Ricerca Corrente Italian Ministry of Health. Dr. Kelley is the Bob and Sandy Heimann Endowed Chair for Alzheimer's Disease Education and Research at the University of Cincinnati.
DISCLOSURE
M. van Blitterswijk and M. Baker report no disclosures. M. DeJesus-Hernandez holds a patent on methods to screen for the hexanucleotide repeat expansion in the C9ORF72 gene. R. Ghidoni serves on the editorial boards of Frontiers in Bioscience (Managing Editor), Journal of Alzheimer's Disease (Associate Editor), American Journal of Alzheimer's Disease (Associate Editor), American Journal of Neurodegenerative Disease (Senior Editor), International Journal of Clinical and Experimental Medicine, Geriatrics, and Dataset Papers in Medicine. L. Benussi serves on the editorial board of Journal of Alzheimer's Disease (Associate Editor). E. Finger receives research support from the Lawson Health Research Institute, the University of Western Ontario Academic Development Fund, the Hazel Soper Foundation, and the Alzheimer's Society of London and Middlesex. G. Hsiung receives research support from a CIHR clinical genetics investigatorship, from the Alzheimer Society of Canada, and is an investigator in clinical trials sponsored by Baxter, Bristol-Myers Squibb, Genentech, Hoffmann-La Roche, and Pfizer. B. Kelley has served as a consultant to Lilly; has received research support from Novartis and from NIH. M. Murray, N. Rutherford, P. Brown, T. Ravenscroft, B. Mullen, P. Ash, and K. Bieniek report no disclosures. K. Hatanpaa serves as a consultant for Alere Pharmaceuticals, and receives research support from the NIH. A. Karydas and E. McCarty Wood report no disclosures. G. Coppola is funded by NIH/National Institute on Aging (NIA) grant RC1 AG035610, the Consortium for Frontotemporal Dementia Research, the Adelson Medical Research Foundation, the Tau Consortium, and the Easton Consortium. E. Bigio serves on the editorial boards of the Journal of Neuropathology and Experimental Neurology, Acta Neuropathologica, and Brain Pathology. C. Lippa serves on scientific advisory boards for the Association for Frontotemporal Dementias, the Lewy Body Dementia Association, and the Alzheimer's Association (Delaware Valley Chapter); serves as Editor-in-Chief of the American Journal of Alzheimer's Disease and Other Dementias and the editorial boards of Neurology® and Journal of Neuropathology and Experimental Neurology; and has received research support from UCB, Novartis, Elan Corporation, Janssen, Danone, Potamkin Foundation, and the Newmann Foundation. M. Strong reports no disclosures. T. Beach has contractual funding and/or research support from Eli Lilly Pharmaceuticals, Avid Radiopharmaceuticals, Bayer Schering Pharma, GE Healthcare, the National Institute of Neurological Disorders and Stroke (NINDS) and NIA (U24 NS072026 and P30 AG19610), the Arizona Department of Health Services contract 211002, and the Michael J. Fox Foundation for Parkinson's Research, the Arizona Department of Health Services, and the Arizona Biomedical Research Commission. D. Knopman serves as Deputy Editor for Neurology®; serves on a Data Safety Monitoring Board for Lilly Pharmaceuticals, is an investigator in clinical trials sponsored by Janssen Pharmaceuticals, and receives research support from the NIH. E. Huey is supported by the Intramural Program of NIH/NINDS and by NIH/NINDS grant R00 NS060766. M. Mesulam serves on the scientific advisory boards for the Cure Alzheimer Fund and the Association on Frontotemporal Dementia; serves on the editorial boards of Brain, Annals of Neurology, Human Brain Mapping, and Journal of Cognitive Neuroscience; receives royalties from the publication of Principles of Behavioral and Cognitive Neurology (Oxford University Press, 2000); and receives research support from the NIH. T. Bird receives licensing fees from Athena Diagnostics, Inc. C. White III serves as a scientific advisor for the Michael J. Fox Foundation for Parkinson's Research and receives research support from the NIH, the Winspear Family Center for Research on the Neuropathology of Alzheimer's Disease, and the McCune Foundation. A. Kertesz serves on a scientific advisory board for Pfizer Inc.; serves on the editorial boards of Cognitive and Behavioral Neurology and Aphasiology; receives royalties from the publication of The Western Aphasia Battery (Grune and Stratton, 1982); and has received support from the Lawson Research Institute, the American Neurological Society, and the Whitaker professorship. D. Geschwind serves on scientific advisory boards for Autism Speaks, the Alzheimer Research Forum, and the March of Dimes Birth Defects Foundation; serves on the editorial boards of Neuron, Neurogenetics, Neurobiology of Disease, Current Genomics, Biological Psychiatry, Autism Research, Encyclopedia of Autism and Related Disorders, and Biomed Central; may accrue revenue on patents re: peripheral gene expression biomarkers for autism and full biomarkers in Friedreich ataxia; and receives research support from NIH grant R01 AG026938, The Simons Foundation, and the Consortium for Frontotemporal Dementia Research, and has received institutional support from Alzheimer's Disease Research Center of California grant 03-7527. V. Van Deerlin reports no disclosures. R. Petersen chairs a Data Monitoring Committee for Pfizer, Inc. and Janssen Alzheimer Immunotherapy, and is a consultant for GE Healthcare and Elan Pharmaceuticals. He receives royalties from Oxford University Press for Mild Cognitive Impairment. G. Binetti reports no disclosures. B. Miller serves on a scientific advisory board for the Alzheimer's Disease Clinical Study, serves as an Editor for Neurocase, and as an Associate Editor of ADAD; receives royalties from the publication of Behavioral Neurology of Dementia (Cambridge, 2009), Handbook of Neurology (Elsevier, 2009), and The Human Frontal Lobes (Guilford, 2008); serves as board member on the John Douglas French Alzheimer's Foundation and Larry L. Hillblom Foundation; serves as a consultant for Lundbeck Inc., Allon Therapeutics, Inc., TauRx, Ltd., the Tau Consortium, the Consortium for Frontotemporal Research, and Novartis; and receives research support from Novartis, NIH grants P50 AG023501, P01 AG019724, and P50 AG1657303, and the State of California Alzheimer's Center. L. Petrucelli has received support from the Mayo Clinic Foundation, NIH/NIA (R01 AG026251), NIH/NINDS (R01 NS063964-01, R01 NS077402, ES20395-01), Amyotrophic Lateral Sclerosis Association, and the Department of Defense (W81XWH-10-1-0512-1 and W81XWH-09-1-0315AL093108). Z. Wszolek serves as Co-Editor-in-Chief of Parkinsonism and Related Disorders, Regional Editor of the European Journal of Neurology, and on the editorial boards of Neurologia i Neurochirurgia Polska, Advances in Rehabilitation, Medical Journal of the Rzeszow University, and Clinical and Experimental Medical Letters; holds and has contractual rights for receipt of future royalty payments from patents re: a novel polynucleotide involved in heritable Parkinson disease; receives royalties from publishing Parkinsonism and Related Disorders (Elsevier, 2010, 2011, 2012) and the European Journal of Neurology (Wiley-Blackwell, 2010, 2011, 2012); and receives educational grant support from Allergan, Inc., and research support from the NIH/NINDS P50 NS072187, the Mayo Clinic Center for Regenerative Medicine, and the Dystonia Medical Research Foundation. K. Boylan receives research support from the ALS Association, Biogen Idec, Cytokinetics Inc., and Mayo Foundation, and has received research support from Neuraltus Pharmaceuticals, Avanir Pharmaceuticals, and Synapse Biomedical. N. Graff-Radford serves as board member of The Neurologist, serves as a consultant for Codman, and receives research support from Medivation, Janssen, Allon, Forest, Pfizer, and NIA. I. Mackenzie serves on the editorial boards of Journal of Neuroinflammation, Clinical Neuropathology, Acta Neuropathologica, American Journal of Neurodegenerative Disease, and Neurobiology of Aging; and receives research support from the CIHR, the Pacific Alzheimer Research Fund, and Michael Smith Foundation for Health Research. B. Boeve receives royalties from the publication of Behavioral Neurology of Dementia (Cambridge University Press, 2009) and receives research support from Cephalon, Inc., the NIH, and the Alzheimer's Association. D. Dickson serves on the editorial boards of the American Journal of Pathology, Journal of Neuropathology and Experimental Neurology, Brain Pathology, Neurobiology of Aging, Journal of Neurology, Neurosurgery, and Psychiatry, Annals of Neurology, and Neuropathology; is supported by NIH grants (P50 AG16574, P50 NS72187, and P01 AG03949), the Mangurian Foundation, CurePSP, and the Robert E. Jacoby Professorship for Alzheimer's Research. R. Rademakers receives research support from the NIH (R01 NS080882, R01 NS065782, R01 AG026251, P50 NS072187, and P50 AG16574), the ALS Therapy Alliance, and the Consortium for Frontotemporal Degeneration Research. Dr. Rademakers further received honoraria for lectures or educational activities not funded by industry; she serves on the medical advisory board of the Association for Frontotemporal Degeneration, on the board of directors of the International Society for Frontotemporal Dementia, and holds a patent on methods to screen for the hexanucleotide repeat expansion in the C9ORF72 gene. Go to Neurology.org for full disclosures.
REFERENCES
- 1.DeJesus-Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011;72:245–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Renton AE, Majounie E, Waite A, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011;72:257–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rademakers R, Neumann M, Mackenzie IR. Advances in understanding the molecular basis of frontotemporal dementia. Nat Rev Neurol 2012;8:423–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Blitterswijk M, van Es MA, Hennekam EA, et al. Evidence for an oligogenic basis of amyotrophic lateral sclerosis. Hum Mol Genet 2012;21:3776–3784 [DOI] [PubMed] [Google Scholar]
- 5.van Blitterswijk M, Dejesus-Hernandez M, Rademakers R. How do C9ORF72 repeat expansions cause amyotrophic lateral sclerosis and frontotemporal dementia: can we learn from other noncoding repeat expansion disorders? Curr Opin Neurol 2012;25:689–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.King A, Al-Sarraj S, Troakes C, et al. Mixed tau, TDP-43 and p62 pathology in FTLD associated with a C9ORF72 repeat expansion and p.Ala239Thr MAPT (tau) variant. Acta Neuropathol 2013;125:303–310 [DOI] [PubMed] [Google Scholar]
- 7.Kaivorinne AL, Moilanen V, Kervinen M, et al. Novel TARDBP sequence variant and C9ORF72 repeat expansion in a family with frontotemporal dementia. Alzheimer Dis Assoc Disord Epub 2012 Aug 12. [DOI] [PMC free article] [PubMed]
- 8.Zhang YJ, Xu YF, Cook C, et al. Aberrant cleavage of TDP-43 enhances aggregation and cellular toxicity. Proc Natl Acad Sci USA 2009;106:7607–7612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ash PE, Bieniek KF, Gendron TF, et al. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 2013;77:639–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mackenzie IR, Neumann M, Baborie A, et al. A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol 2011;122:111–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mackenzie IR, Baker M, Pickering-Brown S, et al. The neuropathology of frontotemporal lobar degeneration caused by mutations in the progranulin gene. Brain 2006;129:3081–3090 [DOI] [PubMed] [Google Scholar]
- 12.Al-Sarraj S, King A, Troakes C, et al. p62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MND/ALS. Acta Neuropathol 2011;122:691–702 [DOI] [PubMed] [Google Scholar]
- 13.Mori K, Weng SM, Arzberger T, et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 2013;339:1335–1338 [DOI] [PubMed] [Google Scholar]
- 14.Beck J, Poulter M, Hensman D, et al. Large C9orf72 hexanucleotide repeat expansions are seen in multiple neurodegenerative syndromes and are more frequent than expected in the UK population. Am J Hum Genet 2013;92:345–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Daoud H, Noreau A, Rochefort D, et al. Investigation of C9orf72 repeat expansions in Parkinson's disease. Neurobiol Aging 2013;34:1710.e7–1710.e9 [DOI] [PubMed] [Google Scholar]
- 16.Lesage S, Le Ber I, Condroyer C, et al. C9orf72 repeat expansions are a rare genetic cause of parkinsonism. Brain 2013;136:385–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Akimoto C, Forsgren L, Linder J, et al. No GGGGCC-hexanucleotide repeat expansion in C9ORF72 in parkinsonism patients in Sweden. Amyotroph Lateral Scler Frontotemporal Degener 2013;14:26–29 [DOI] [PubMed] [Google Scholar]
- 18.Takada LT, Pimentel ML, Dejesus-Hernandez M, et al. Frontotemporal dementia in a Brazilian kindred with the c9orf72 mutation. Arch Neurol 2012;69:1149–1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ishiura H, Takahashi Y, Mitsui J, et al. C9ORF72 repeat expansion in amyotrophic lateral sclerosis in the Kii peninsula of Japan. Arch Neurol 2012;69:1154–1158 [DOI] [PubMed] [Google Scholar]
- 20.Byrne S, Elamin M, Bede P, et al. Cognitive and clinical characteristics of patients with amyotrophic lateral sclerosis carrying a C9orf72 repeat expansion: a population-based cohort study. Lancet Neurol 2012;11:232–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Levine TP, Daniels RD, Gatta AT, Wong LH, Hayes MJ. The product of C9orf72, a gene strongly implicated in neurodegeneration, is structurally related to DENN Rab-GEFs. Bioinformatics 2013;29:499–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang D, Iyer LM, He F, Aravind L. Discovery of novel DENN proteins: implications for the evolution of eukaryotic intracellular membrane structures and human disease. Front Genet 2012;3:283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sieben A, Van Langenhove T, Engelborghs S, et al. The genetics and neuropathology of frontotemporal lobar degeneration. Acta Neuropathol 2012;124:353–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ferrari R, Mok K, Moreno JH, et al. Screening for C9ORF72 repeat expansion in FTLD. Neurobiol Aging 2012;33:1850.e1–1850.e11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bieniek KF, Murray ME, Rutherford NJ, et al. Tau pathology in frontotemporal lobar degeneration with C9ORF72 hexanucleotide repeat expansion. Acta Neuropathol 2013;125:289–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Van Langenhove T, van der Zee J, Gijselinck I, et al. Distinct clinical characteristics of C9orf72 expansion carriers compared with GRN, MAPT, and nonmutation carriers in a Flanders-Belgian FTLD cohort. JAMA Neurol 2013;70:365–373 [DOI] [PubMed] [Google Scholar]
- 27.Le Ber I, Camuzat A, Guillot-Noel L, et al. C9ORF72 repeat expansions in the frontotemporal dementias spectrum of diseases: a flow-chart for genetic testing. J Alzheimers Dis 2013;34:485–499 [DOI] [PubMed] [Google Scholar]
- 28.Whitwell JL, Weigand SD, Boeve BF, et al. Neuroimaging signatures of frontotemporal dementia genetics: C9ORF72, tau, progranulin and sporadics. Brain 2012;135:794–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boeve BF, Boylan KB, Graff-Radford NR, et al. Characterization of frontotemporal dementia and/or amyotrophic lateral sclerosis associated with the GGGGCC repeat expansion in C9ORF72. Brain 2012;135:765–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Simon-Sanchez J, Dopper EG, Cohn-Hokke PE, et al. The clinical and pathological phenotype of C9ORF72 hexanucleotide repeat expansions. Brain 2012;135:723–735 [DOI] [PubMed] [Google Scholar]
- 31.Majounie E, Renton AE, Mok K, et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol 2012;11:323–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mok KY, Koutsis G, Schottlaender LV, Polke J, Panas M, Houlden H. High frequency of the expanded C9ORF72 hexanucleotide repeat in familial and sporadic Greek ALS patients. Neurobiol Aging 2012;33:1851.e1–1851.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gass J, Cannon A, Mackenzie IR, et al. Mutations in progranulin are a major cause of ubiquitin-positive frontotemporal lobar degeneration. Hum Mol Genet 2006;15:2988–3001 [DOI] [PubMed] [Google Scholar]
- 34.Cohn-Hokke PE, Elting MW, Pijnenburg YA, van Swieten JC. Genetics of dementia: update and guidelines for the clinician. Am J Med Genet B Neuropsychiatr Genet 2012;159B:628–643 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.