

Abstract
Glutamatergic lateral habenula (LHb) output communicates negative motivational valence to ventral tegmental area (VTA) dopamine (DA) neurons via activation of the rostromedial tegmental nucleus (RMTg). However, the LHb also receives a poorly understood DA input from the VTA, which we hypothesized constitutes an important feedback loop regulating DA responses to stimuli. Using whole-cell electrophysiology in rat brain slices, we find that DA initiates a depolarizing inward current (IDAi) and increases spontaneous firing in 32% of LHb neurons. IDAi was also observed upon application of amphetamine or the DA uptake blockers cocaine or GBR12935, indicating involvement of endogenous DA. IDAi was blocked by D4 receptor (D4R) antagonists (L745,870 or L741,742), and mimicked by a selective D4R agonist (A412997). IDAi was associated with increased whole-cell conductance and was blocked by Cs+ or a selective blocker of hyperpolarization-activated cyclic nucleotide-gated (HCN) ion channel, ZD7288. IDAi was also associated with a depolarizing shift in half-activation voltage for the hyperpolarization-activated cation current (Ih) mediated by HCN channels. Recordings from LHb neurons containing fluorescent retrograde tracers revealed that IDAi was observed only in cells projecting to the RMTg and not the VTA. In parallel with direct depolarization, DA also strongly increased synaptic glutamate release and reduced synaptic GABA release onto LHb cells. These results demonstrate that DA can excite glutamatergic LHb output to RMTg via multiple cellular mechanisms. Since the RMTg strongly inhibits midbrain DA neurons, activation of LHb output to RMTg by DA represents a negative feedback loop that may dampen DA neuron output following activation.
Introduction
The lateral habenula (LHb) is a brain structure involved in the control of motivated behavior via signaling the absence of predicted reward, and information regarding aversive stimuli to brain reward areas (Matsumoto and Hikosaka, 2007, 2009; Jhou et al., 2009a; Bromberg-Martin and Hikosaka, 2011). This is supported by anatomical data indicating that the LHb receives input from forebrain nuclei, whereas its efferents terminate in midbrain dopamine (DA) areas, such as the ventral tegmental area (VTA) and substantia nigra, as well as serotonergic brain regions (Herkenham and Nauta, 1979; Araki et al., 1984; Behzadi et al., 1990; Omelchenko et al., 2009). Many studies implicate these monoamine neurons in motivation, reward, and psychiatric illness, suggesting that the LHb may be involved in these behavioral phenomena (Hikosaka et al., 2008). Physiological studies also support this role as there is an inverted relationship between LHb and DA neuron activity, and activation of the LHb strongly inhibits midbrain DA neuron firing (Ji and Shepard, 2007; Matsumoto and Hikosaka, 2007). There is evidence that the LHb, which is largely composed of glutamatergic neurons (Geisler and Trimble, 2008; Omelchenko et al., 2009), sends a direct projection to DA and non-DA neurons in the ventral midbrain (Hikosaka et al., 2008; Omelchenko et al., 2009). However, this is inconsistent with strong inhibition of DA neurons during LHb activation (Christoph et al., 1986; Ji and Shepard, 2007; Hikosaka et al., 2008). Therefore, an intermediary structure is hypothesized that provides inhibitory input to DA neurons (Matsumoto and Hikosaka, 2007; Hikosaka et al., 2008). Recent evidence strongly implicates a collection of GABAergic neurons located in the caudal VTA, termed the rostral medial tegmental nucleus (RMTg), or “tail” of the VTA, as a primary target of LHb efferents (Jhou et al., 2009b; Kaufling et al., 2009), and as the intermediary nucleus that inhibits midbrain DA neurons (Jhou et al., 2009b; Balcita-Pedicino et al., 2011).
In addition to the projection from LHb to the VTA, VTA DA neurons also project to the LHb, suggesting that DA may modulate LHb output. Both tyrosine hydroxylase (TH), the rate-limiting enzyme in DA synthesis, and DA D2 receptors are found in the LHb (Mansour et al., 1990; Bouthenet et al., 1991; Meador-Woodruff et al., 1991; Weiner et al., 1991; Aizawa et al., 2012; Jhou et al., 2013), and single-unit electrophysiology studies show that DA receptor activation alters LHb neuron activity (Dougherty et al., 1990; Kowski et al., 2009). However, the influence of endogenous DA on LHb neurons, the receptors mediating these effects and the mechanism of this modulation have received little attention. Here we describe a population of LHb neurons that are depolarized by DA via D4 receptors (D4Rs). We also identify the mechanism through which this occurs, and we demonstrate that these LHb neurons project to the RMTg, and not the VTA. We propose that these LHb neurons may exist within a negative feedback circuit in which VTA DA neuron activation is terminated by RMTg neurons receiving input from these DA-depolarized LHb neurons.
Materials and Methods
Animals.
Male Sprague Dawley rats, 15–40 d old (Charles River Laboratories) were used for all experiments. All protocols were conducted under National Institutes of Health (NIH) guidelines using the NIH handbook Animals in Research and were approved by the National Institute on Drug Abuse (NIDA) Intramural Research Program Animal Care and Use Committee.
Brain slice preparation and recording.
Animals were decapitated using a guillotine, and their brains were removed and transferred to a beaker containing the following oxygenated (95% O2/5% CO2), ice-cold cutting solution (in mm): sucrose 194, NaCl 30, KCl 4.5, MgCl2 1, NaH2PO4 1.2, glucose 10, NaHCO3 26. The brain was then trimmed with a razor blade, and a block of tissue containing the LHb was glued onto the cutting stage of a vibrating tissue slicer (VT1000, Leica). The brain tissue was immediately submersed in ice-cold, oxygenated cutting solution. Four sagittal, or three coronal, slices (280 μm) containing the LHb were obtained from each rat. The slices were transferred to an oxygenated holding chamber filled with normal artificial CSF (aCSF; in mm: NaCl 126, KCl 3, MgCl2 1.5, CaCl2 2.4, NaH2PO4 1.2, glucose 11, NaHCO3 26) at 31°C for 20–30 min, then the holding chamber was permitted to reach room temperature and was maintained there for the rest of the incubation period.
One brain slice was transferred to a heated chamber (31–33°C) and perfused with normal aCSF (2 ml/min) that was identical to that used for slice storage. Visualization of LHb neurons was performed with an upright microscope modified to provide a gradient contrast image with infrared illumination (Axioskop, Zeiss), or a microscope equipped for epifluorescence and differential interference contrast microscopy (BX51WI, Olympus). Unless otherwise stated, recording electrodes (∼5 MΩ) were filled with a standard internal solution (in mm): K-gluconate 140, KCl 5, HEPES 10, EGTA 0.2, MgCl2 2, Mg-ATP 4, Na2-GTP 0.3, Na2-phosphocreatine 10, pH 7.2, with KOH.
Whole-cell voltage or current-clamp recordings were performed using an Axopatch 200B (Molecular Devices). Voltage steps and ramps were delivered using the Strathclyde electrophysiology software package (WCP, courtesy of Dr. John Dempster, Strathclyde University, Glasgow, UK; http://spider.science.strath.ac.uk/sipbs/software_ses.htm) or WinLTP (WinLTP Ltd, The University of Bristol, Bristol, UK), and an analog-to-digital board (ITC-18, Instrutech Corp.; or PCI-6251, National Instruments) residing in a personal computer.
In some neurons, IPSCs were isolated by including the glutamate antagonists AP-5 (40 μm) and 2,3-dihydroxy-6-nitro-7-sulfonyl-benzo[f]quinoxaline (NBQX; 5–10 μm) in the aCSF, and using recording electrodes (∼5 MΩ) filled with the following (in mm): KCl 145, HEPES 10, EGTA 0.2, MgCl2 2, Mg-ATP 4, Na2-GTP 0.3, Na2-phosphocreatine 10, pH 7.2, with KOH. Under these conditions, spontaneous GABAA receptor-mediated IPSCs were inward at the approximately −70 mV holding potential used in these experiments. Holding potentials are corrected for a liquid junction potential, calculated to be −12.2 mV based on the intracellular and extracellular ion concentrations. Spontaneous EPSCs were measured in some cells during the blockade of IPSCs with picrotoxin (100 μm) added to the extracellular perfusate. Analysis of spontaneous synaptic currents was performed with the MiniAnalysis program (Synaptosoft). For hyperpolarization-activated cation current (Ih) measurements, cells were voltage-clamped at −52 mV, and 2 s hyperpolarizing steps were delivered in 10 mV increments to −122 mV. Three steps were performed at each potential and current responses averaged for each cell. To identify the Ih contribution to the total membrane current, the family of currents obtained in Cs+ or ZD7288 was subtracted from those obtained in control extracellular media or from those obtained during the peak inward DA current (IDAi). Peak Ih was then normalized to the largest current and a Boltzmann function fit to these data using the following equation:
where V1/2 is the membrane potential in which Ih is half-maximal, and k is the slope factor.
Reversal potentials for Ih were measured in aCSF containing TTX (500 nm) and NiCl2 (1 mm) to block voltage-dependent sodium and calcium channels, respectively. Neuron membrane potentials were initially held at −52 mV then stepped to −122 mV for 2 s. The membrane was then stepped from −122 to −52 mV in 10 mV increments for 250 ms. Tail current relaxations measured during these 250 ms steps were then plotted against the membrane step potential, and the Ih reversal potential extrapolated using a linear regression were fit to these points (Aponte et al., 2006).
Stereotactic retrograde tracer injections.
Male Sprague Dawley rats weighing 95–115 g were anesthetized with 80 mg/kg ketamine (80 mg/ml)/xylazine (12 mg/ml; Sigma-Aldrich) and placed in a stereotaxic frame (Kopf Instruments). Two holes were drilled above either the VTA (anterior/posterior, −4.6 mm; mediolateral, 0.9 mm; dorsoventral, −7.8 mm) or the RMTg (anterior/posterior, −5.0 mm; mediolateral, 0.9 mm; dorsoventral, −8 mm). A sharp glass pipette (outside diameter, 1 mm; inside diameter, 0.25 mm) was manufactured using a vertical pipette puller (Narashige International). The fine tip of the pipette was then broken to a diameter of 25–50 μm, measured under microscopic observation, and filled with either cholera toxin subunit B-Alexa Fluor 594 conjugate (CTB; 5 μg/μl in PBS; Invitrogen) or Neuro-DiI (7% in ethanol; Biotium). The pipette tip was slowly lowered into the target nucleus, and pneumatic pressure (Picospritzer III, General Valve) was used to eject ∼150 nl of Neuro-DiI, or iontophoresis (BAB-501, Kation Scientific) was used for injection of CTB. For CTB injections, a positive current (+5 μA) was delivered to the pipette with a 7 s on/7 s off cycle for 10 min. Pipettes were left in place for 5 min before being slowly withdrawn 300 μm, and then were left in place for an additional 3–5 min before being withdrawn from the brain. Animals were allowed to recover 3–5 d before brain slice preparation for electrophysiological recording.
Tyrosine hydroxylase immunohistochemistry.
Male Sprague Dawley rats weighing 95–115 g were perfused transcardially with 4% paraformaldehyde (PFA) and the brains post-fixed in 4% PFA for 24 h. The brains were then transferred to an 18% sucrose solution for 48 h and frozen on powdered dry ice. The brains were blocked, mounted in a cryostat, sectioned at 40 μm, and collected in 0.1 m PBS. The slices were then processed using the following protocol.
On day 1, they were incubated in 1% sodium borohydrate in 0.1 m PBS for 15 min, followed by six rinses in 0.1 m PBS for 2 min each. Sections were then incubated in PBS with 0.3% Triton X-100 (PBST; pH 7.4) overnight with a primary monoclonal mouse anti-TH antibody (1/1000; catalog #MAB318, Millipore Bioscience Research Reagents). On day 2, after six rinses in 0.3% PBST (2 min each), the sections were incubated for 2 h in biotinylated donkey antimouse-IgG (1/1000; Jackson ImmunoResearch Laboratories). They were rinsed six times in 0.3% PBST, then incubated for 90 min in avidin–biotin–peroxidase complex (1/1000; Vector Laboratories). The sections were next rinsed once in 0.3% PBST and twice in PBS, and then reacted with the peroxidase kit VectorSG SK-4700 (Vector Laboratories) to produce a dark blue stain. The sections were kept overnight in PBS with sodium azide. On day 3, sections were mounted on subbed slides and dried overnight in a slide warmer. On day 4, the sections were counterstained with methyl green (Vector Laboratories); dehydrated in alcohols using the sequence 30%, 60%, 90%, twice in 95%, and twice in 100%; placed twice in Histoclear; and coverslipped with Permount.
Statistical analysis.
Data in the text and figures are reported as the mean ± SEM. Statistical analysis was performed using either a two-tailed Student's t test or a one-way ANOVA for multiple comparisons unless otherwise noted. The Kolmogorov–Smirnov (KS) test was used to analyze differences in the cumulative probability distributions. Statistical analyses and graphical representation of the data were performed using GraphPad Prism version 6.00 for Windows.
Drugs.
Picrotoxin, eticlopride, prazosin, forskolin, SKF38393, and dopamine were purchased from Sigma-Aldrich. Amphetamine was obtained from the NIDA Drug Supply Program. ZD7288, L741,742, L745,870, SCH23390, A412997, KN-62, KN-93, and quinpirole were obtained from Tocris Bioscience. The D3 receptor antagonist PG619 was generously provided by Dr. Amy Newman (NIDA Intramural Research Program). The drugs were applied through bath superfusion using calibrated syringe pumps (Razel Scientific Instruments) or were included in the aCSF.
Results
Dopamine innervation of the LHb
To determine the areas of the habenula demonstrating the highest levels of DA innervation we used TH immunohistochemistry (Fig. 1). TH staining was strong along the medial aspect of the LHb at most anterior–posterior levels, and was not observed at high levels in the medial habenula nucleus (MHb) proper (Fig. 1). These data generally agree with a previous anatomical study, which described the strongest TH staining of the LHb to be within the parvocellular and central subnuclei of the LHb (Geisler et al., 2003). We hypothesized that DA signaling would be strongest in these regions that we collectively refer to as the medial LHb (mLHb). Therefore, all of the electrophysiological recordings we present were confined to this mLHb area.
Figure 1.
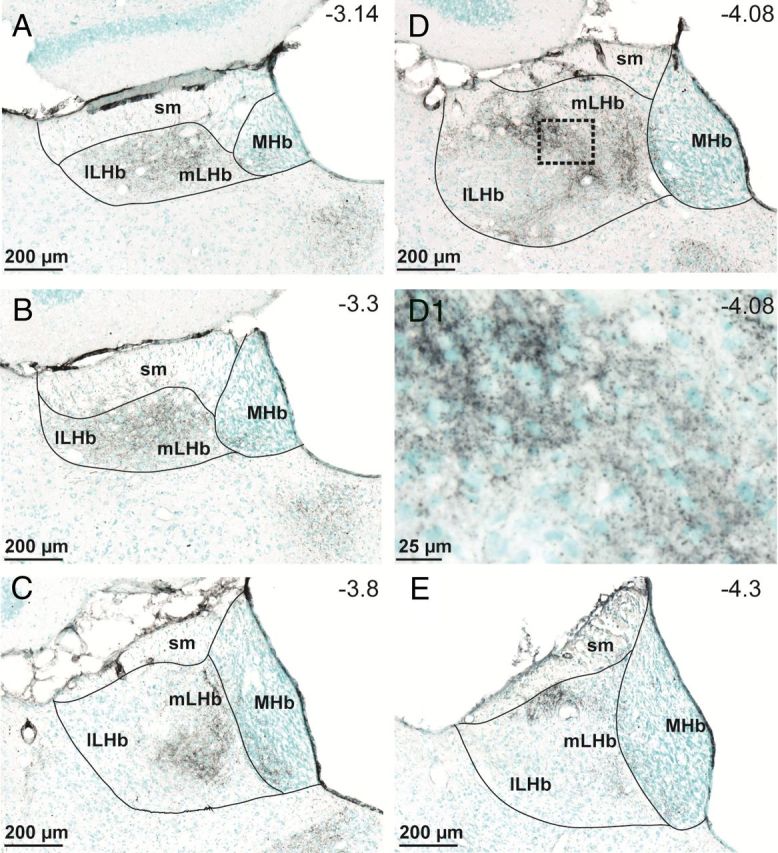
TH fiber innervation of the LHb. TH-immunopositive fibers are stained in black, whereas cell bodies are labeled with methyl-green. A–E, TH fiber density is highest in an area that we designate the medial lateral habenula (mLHb) (A–E), and our recordings were made in this area as represented in C and D. D1, Higher-resolution image from the area designated by the dashed box in D, showing TH varicosities surrounding LHb neuron somata. Anterior–posterior coordinates from bregma (−3.14 to −4.3 mm) are estimated from the stereotaxic atlas of Paxinos and Watson (1998). sm, Stria medullaris; lLHb, lateral LHb; MHb, medial habenula.
Dopamine depolarizes a subpopulation of LHb neurons
To determine whether neurons in the parvocellular and central subnuclei of the mLHb respond to DA, we briefly bath applied DA (2–20 μm) to a total of 349 cells during voltage- or current-clamp whole-cell recordings. Of these neurons, 112 of 349 (32%) were depolarized or exhibited inward currents and 87 of 349 (25%) were hyperpolarized or exhibited outward currents upon DA application. Also, 150 of 349 mLHb neurons (43%) demonstrated no response to DA application. As we previously reported that the DA-mediated hyperpolarization was mediated by D2Rs (Jhou et al., 2013), the present study was designed to investigate the mechanisms involved in the depolarizing response of mLHb neurons to DA. In a further current-clamp analysis, from a subgroup of mLHb neurons we observed that the cells could be divided into two groups; those exhibiting high rates (>5 Hz) of spontaneous action potentials (12.86 ± 1.48 Hz, n = 16; Fig. 2B,C), and those exhibiting low rates (<2 Hz) of spontaneous action potentials (1.4 ± 0.51 Hz, n = 17; Fig. 2A,C). DA caused a membrane depolarization that led to large reversible increases in action potential frequency in the relatively silent neurons (13.32 ± 1.79 Hz, n = 8; F(3,43) = 25.83, p < 0.001; Fig. 2A,C), but did not alter the rates of spontaneous activity in mLHb neurons that exhibited high baseline action potential frequencies (n = 16, p > 0.05; Fig. 2B,C). Further analysis revealed that neurons depolarized by DA exhibited resting membrane potentials (RMPs) that were significantly more negative than cells that were insensitive (DA-depolarized RMP: −64.39 ± 1.43 mV, n = 17; DA-insensitive RMP: −55.80 ± 1.36 mV, n = 14; p = 0.0002, t test; Fig. 2D).
Figure 2.
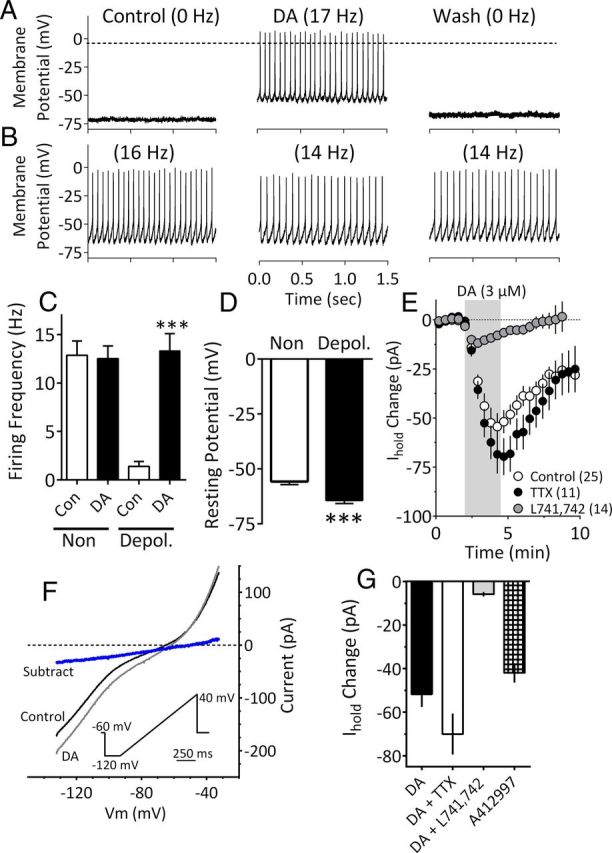
DA depolarizes a population of mLHb neurons. A, Current-clamp recording from an mLHb neuron demonstrating membrane depolarization and an increase in spontaneous action potential frequency caused by bath application of DA (3 μm). B, Absence of DA effect on a mLHb neuron. C, Mean (± SEM) effects of DA (3 μm) on mLHb neuron firing rates. Note that a significant increase in cell firing was observed only in LHb neurons that demonstrated low action potential frequencies before DA application (Depol.). D, Mean RMPs of mLHb neurons that were either depolarized by (Depol.) or insensitive to (Non) DA. Neurons that were depolarized by DA exhibited significantly larger mean RMP. E, Time course of IDAi under control conditions or following application of TTX (500 nm) or the DA D4R antagonist L741,742 (200 nm). The effect of TTX was not significant, whereas that of L741,742 was significantly different from that observed with DA alone. DA application is noted by the vertical shaded bar. F, I–V relationships generated during peak IDAi demonstrate that whole-cell conductance was increased at membrane voltages negative to approximately −45 mV and that IDAi (blue line) had a mean reversal potential of −51.0 mV (95% CI = −50.9 mV to −51.6 mV; linear regression, R2 = 0.99; n = 9 neurons). I–V curves were obtained by slowly changing the membrane potential from −120 to −40 mV using the indicated voltage ramp protocol (inset). The subtracted current was obtained by subtracting the curve obtained during DA from that obtained during the control condition. G, The effects of DA (3 μm) on Ihold, alone, or in the presence of TTX (500 nm) or L741,742 (200 nm) compared with those of the selective D4R agonist A412997 (1 μm; n = 6). DA alone, and in TTX, and A412997 significantly increased Ihold (F(3,45) = 16.44, p < 0.0001, one-way ANOVA). However, DA did not significantly affect Ihold after L741,742 treatment.
In voltage-clamp experiments, the depolarizations could be observed as inward currents upon DA application (IDAi) (Fig. 2E). To determine whether this was direct, the DA effect was reversed by washing and then was re-examined after treatment with TTX (1 μm). The response to DA persisted in the presence of TTX and was not significantly different from that seen in its absence (DA, −54.40 ± 4.16 pA; TTX + DA, −67.06 ± 5.19 pA; p = 0.09; Fig. 2E). We next asked whether IDAi was associated with the opening or closing of ion channels by examining current–voltage relationships initiated by slowly changing the membrane potential from −120 to −40 mV during the peak DA response (Fig. 2F). The DA-induced inward currents were associated with an increase in whole-cell conductance (Fig. 2F), and the current obtained by subtracting the control curve from that obtained in DA had a reversal potential of −51.0 mV (95% CI = −50.9 to −51.6 mV; Fig. 2F). These data suggest that IDAi resulted from the opening of ion channels.
DA depolarization of mLHb neurons occurs through D4 receptors
To identify the receptor mediating the depolarizing effect of DA, IDAi was measured in mLHb neurons and reversed by washing. Then, antagonists for D1R (SCH23390, 1 μm), D2R (eticlopride, 200 nm), D3R (PG619, 10 nm), or D4R (741,742, 200 nm; l-745,870, 200 nm), or α1 adrenergic receptors (prazosin, 10 μm) were applied to the brain slices for 10 min, followed by DA reapplication. Also tested for their ability to generate inward currents in these neurons were the D2 agonist quinpirole (1 μm), and the D1 agonist SKF38393 (20 μm). We found that only the D4R antagonists l-741,742 (n = 14, 200 nm) and l-745,870 (n = 7, 200 nm) significantly inhibited the DA-induced inward currents in mLHb neurons (Fig. 2E), and that the D1 and D2 agonists did not alter holding currents in these cells (data not shown). We next examined the effect of the selective D4R agonist A412997 (1 μm; Moreland et al., 2005) on mLHb neuron holding currents. Similar to DA, A412997 activated inward currents in 35% of the neurons (6 of 17 neurons; Fig. 2G), providing further evidence that IDAi was mediated by the activation of D4Rs.
Since DA did not alter action potential frequency in the subpopulation of neurons that exhibited high resting rates of activity (Fig. 2B), we tested the hypothesis that these neurons were tonically excited by endogenous DA acting at D4Rs. We found that the D4R antagonist L741,742 (200 nm) did not significantly alter the basal firing rates of these cells (control, 6.1 ± 1.3 Hz; L741,742, 7.5 ± 1.6 Hz; p > 0.05, paired t test, n = 4), suggesting that they were not tonically excited by DA acting at these receptors.
Depolarization of mLHb neurons by endogenous DA
Immunohistochemical detection of TH suggested that the mLHb receives substantial innervation from midbrain DA neurons (Beckstead et al., 1979; Phillipson and Pycock, 1982; Reisine et al., 1984; Skagerberg et al., 1984; Gruber et al., 2007). Therefore, we evaluated whether endogenous DA could also depolarize mLHb neurons. Amphetamine is a potent psychostimulant and a well known releaser of DA (Sulzer, 2011). After establishing that IDAi could be observed with DA (3 μm) application, its effect was reversed by washing, and then amphetamine (3 μm) applied for 2.5 min. In four of four mLHb neurons that previously demonstrated inward currents with DA application, amphetamine generated a depolarizing inward current (Fig. 3B). Inward currents were also observed in mLHb neurons with cocaine (10 μm, n = 6) and the selective DA transporter (DAT) inhibitor GBR12935 (1 μm, n = 7; Fig. 3), indicating that DAT inhibition was sufficient to depolarize these mLHb neurons.
Figure 3.
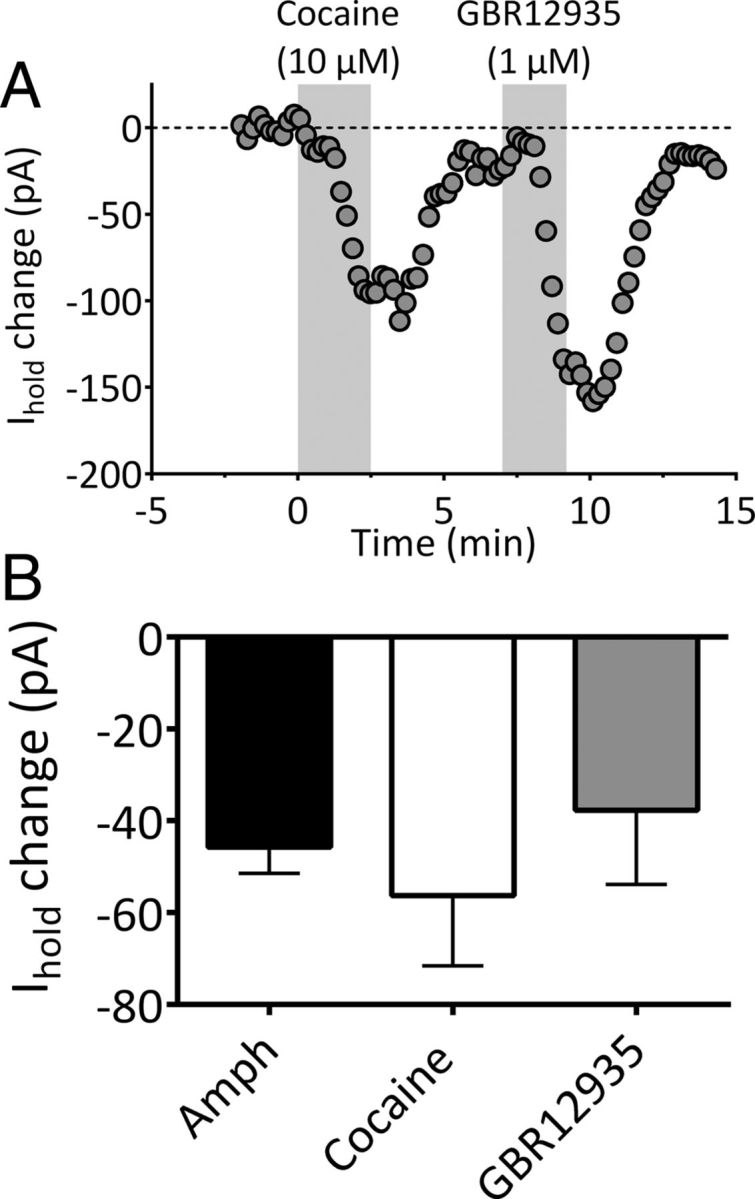
Endogenous DA generates depolarizing inward currents in mLHb neurons. A, Inward currents generated by the DA transport inhibitors cocaine and GBR12935 in the same mLHb neuron. The time of drug application is indicated with the vertical shaded bar. B, Mean inward current (±SEM) generated by in LHb neurons by amphetamine (Amph, n = 4), cocaine (n = 6), and GBR12935 (n = 7).
Mechanism of DA depolarization of mLHb neurons
The mean current–voltage (I–V) relationship obtained during the peak of IDAi indicated an increase in whole-cell current at membrane potentials greater than approximately −45 mV, and a reversal potential for this subtracted current of −51.0 mV (Fig. 2F). This is consistent with the activation of nonselective ion channels, such as those that mediate Ih. These currents result from the opening of hyperpolarization-activated and cyclic nucleotide-gated nonselective ion channels (HCNs) that are found in many neurons in the CNS. HCN2, HCN3, and HCN4 subunit markers are expressed in mLHb neurons (Notomi and Shigemoto, 2004; Poller et al., 2011), where they are primarily associated with dendrites (Poller et al., 2011). We therefore examined the potential role of these ion channels in mediating IDAi in mLHb neurons. Ih can be identified by its time-dependent contribution to the whole-cell current, described as an inward “sag” in voltage-clamp when the membrane voltage is stepped to more hyperpolarized values from the RMP (Maccaferri and McBain, 1996; Lupica et al., 2001). However, recent evidence also indicates that Ih can be present despite the absence of an inward sag, identified when either of the Ih channel blockers Cs+ or ZD7288 are applied (Aponte et al., 2006) or when inwardly rectifying K+ leak channels are blocked (Rateau and Ropert, 2006).
Bath application of Cs+ (3 mm; Fig. 4A) or ZD7288 (50 μm; Fig. 4B) at the peak of IDAi significantly reversed the inward current. Similarly, the inward current produced by the selective D4R agonist A412997 (1 μm) was rapidly reversed by 3 mm Cs+ (n = 4; Fig. 4C). The effect of Cs+ on IDAi was more rapid than that of ZD7288 (Fig. 4A,B), which may be attributed to their differing sites of action on HCN channels (Harris and Constanti, 1995). The absence of inward current sag on voltage steps near the membrane holding potential (Vhold) used in these experiments (approximately −70 mV; Fig. 4D1,E) could indicate that Ih is not active in this voltage range. However, prior studies have shown that Ih can be present in the absence of an overt current sag and revealed via blockade of HCN channels with ZD7288 (Aponte et al., 2006). If this is true of mLHb neurons, then application of ZD7288 should result in an outward current due to the blockade of a tonic inward conductance mediated by Ih. Therefore, we examined the effect of ZD7288 on the holding current (Ihold) necessary to set the voltage-clamp potential at −70 mV. ZD7288 (50 μm, n = 6) caused a significant outward shift in Ihold (control, −58 ± 22 pA; ZD7288, −34 ± 10 pA; p = 0.03, paired t test), consistent with membrane hyperpolarization, and indicating that Ih is active at this membrane potential. Also consistent with the possibility that Ih is active in this voltage range, and in agreement with the calculated IDAi reversal potential (Fig. 2F), its mean reversal potential (see Materials and Methods) was −49.8 mV (n = 7; 95% CI = −62.5 to −20.5 mV).
Figure 4.
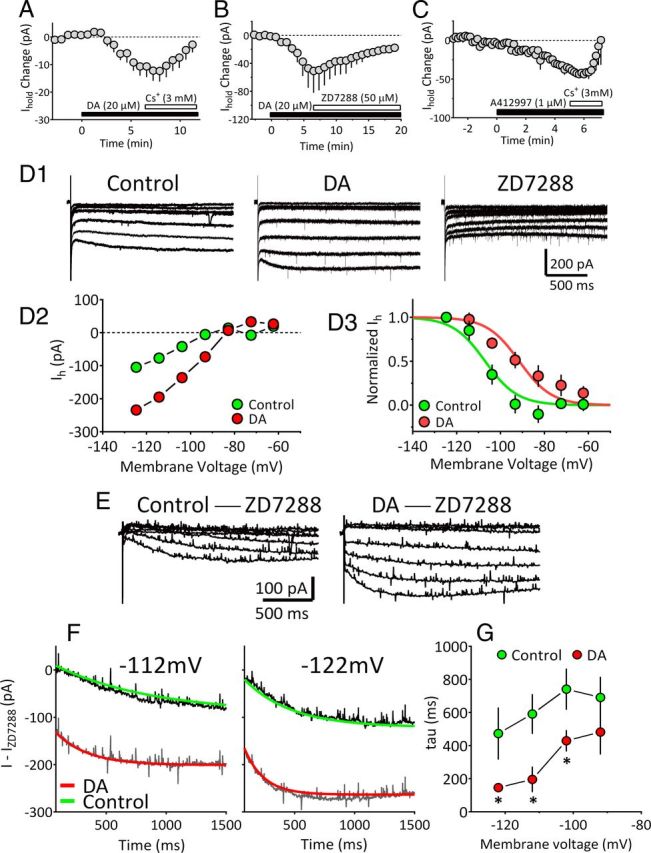
DA enhancement of hyperpolarization-activated cation current in mLHb neurons. A, Mean time course of reversal of inward currents produced by DA (IDAi; 20 μm) by cesium (Cs+; 3 mM; n = 5 neurons). B, Mean time course of the reversal of IDAi by the HCN channel blocker ZD 7288 (50 μm; n = 3 neurons). C, Mean time course of the inward current generated by the selective D4R agonist A412997 (1 μm) in mLHb neurons (n = 4), and the rapid reversal of the current by Cs+ (Vhold = −70 mV). D1, Current traces from an mLHb neuron in which 2 s, 10 mV hyperpolarizing pulses were applied from Vhold = −52 mV to activate Ih. Steps were initiated during the peak IDAi, and again during the peak ZD7288 effect. D2, Mean I–V relationship obtained by measuring current during the last 50 ms of each voltage step demonstrates the voltage-dependent enhancement of Ih by DA. D3, Normalized Ih current amplitude versus membrane potential during the control period and during peak IDAi. Boltzmann functions were fit to data points for control (green symbols; V1/2 = −107 mV) and DA conditions (red symbols; V1/2 = −91.5 mV). Statistical comparison of the curves indicated a significant shift in the V1/2 by DA (extra sum-of-squares F test, F(1,11) = 21.12, p = 0.0008). E, Representative Ih traces obtained by subtracting currents obtained during ZD7288 application from those obtained during the control period (control −ZD7288) or during the peak IDAi (DA −ZD7288). This isolated Ih shows enhancement during DA application. F, Traces, isolated as described in E, in which monoexponential curves have been fit to the activation phase of Ih, measured at −122 and − 112 mV, during control (green lines) and DA application conditions (red lines). DA decreased the time constant for Ih activation at these membrane potentials (−112 mV: control, 815.7 ms; DA, 267.4 ms; −122 mV; control, 321.6 ms; DA, 161.6 ms). G, Summary of the effect of DA on ZD7288-isolated Ih activation time constants for all mLHb neurons included in this analysis (n = 6). DA significantly decreased the time constant for Ih activation (repeated-measures ANOVA, F(1,12) = 9.2, p = 0.04) at all membrane potentials greater than −92 mV. *p < 0.05, Tukey's post hoc comparison).
To determine the properties of Ih in these cells, we examined membrane currents in response to a series of 2 s hyperpolarizing voltage steps from −52 to −122 mV (Fig. 4D1). Although the inward sag indicative of Ih was modest at the largest voltage step, DA increased the size of this current, and application of ZD7288 revealed that Ih contributed substantially to the whole-cell conductance (Fig. 4D1). To identify the Ih contribution to the total membrane current activated by voltage steps, the family of currents obtained in ZD7288 was subtracted from those obtained in control extracellular media, or from those obtained during the peak IDAi (Fig. 4D2,E). Thus, only the ZD7288-sensitive Ih component of the response that was present under control and DA conditions was observed using this approach. When peak Ih was plotted, a large increase was observed during DA application (Fig. 4D2,D3,E). Boltzmann functions fit to the normalized Ih currents revealed a significant shift in the V1/2 of Ih from −107.6 ± 2.7 mV in control to −97.5 ± 3.14 mV in the presence of DA (extra sum-of-squares F test, F(1,11) = 21.12, p = 0.0008, Fig. 4D3). DA also significantly affected the kinetics of Ih by decreasing the time constant (τ) of its activation (Fig. 4F,G). Together, these data suggest that DA acts to increase mLHb neuron excitability by shifting the activation curve of Ih to more depolarized membrane potentials.
Potential mechanisms of Ih modulation by DA
The data suggest that DA enhances Ih activation in LHb neurons. Since cyclic nucleotides can act directly on HCN channels to shift their activation curve to more depolarized potentials (Ingram and Williams, 1994; DiFrancesco, 1999; Lüthi and McCormick, 1999), we sought to determine whether Ih in mLHb neurons was sensitive to forskolin-stimulated cAMP accumulation, and whether this altered the D4R modulation of IDAi. Forskolin (25 μm) alone generated inward currents (−56.2 ± 10.2 pA, n = 4) in mLHb neurons, and caused a significant depolarizing shift in the Ih activation curve (control, −107.6 ± 2.7 mV; forskolin, −94.05 ± 3.1 mV; n = 4; data not shown), as reported previously (Ingram and Williams, 1994). However, the inward current generated by DA (4 μm) application in the presence of forskolin was not significantly different from that produced by DA alone (n = 5; Fig. 5C; one-way ANOVA, F(3,18) = 0.6561, p = 0.5896). This suggests that whereas HCN channels in mLHb neurons are sensitive to cAMP, modulation of these channels by D4Rs is unaffected by increased levels of this cyclic nucleotide. Since D4Rs on prefrontal cortex (PFC) neurons have also been linked to regulation of AMPA receptor trafficking via α-calcium/calmodulin-dependent protein kinase II (CaMKII)-control of microtubules (Yuen and Yan, 2011), and since CaMKII has been implicated in the activity-dependent insertion of HCN1 channels into hippocampal neuron membranes (Fan et al., 2005), we also evaluated whether the inhibition of CaMKII would reduce IDAi. However, we found that intracellular loading of mLHb neurons with the CaMKII inhibitors KN-62 (5 μm, n = 5) or KN-93 (10 μm, n = 5) did not significantly reduce the inward currents produced by DA (Fig. 5B,C; one-way ANOVA, F(3,18) = 0.6561, p = 0.5896).
Figure 5.
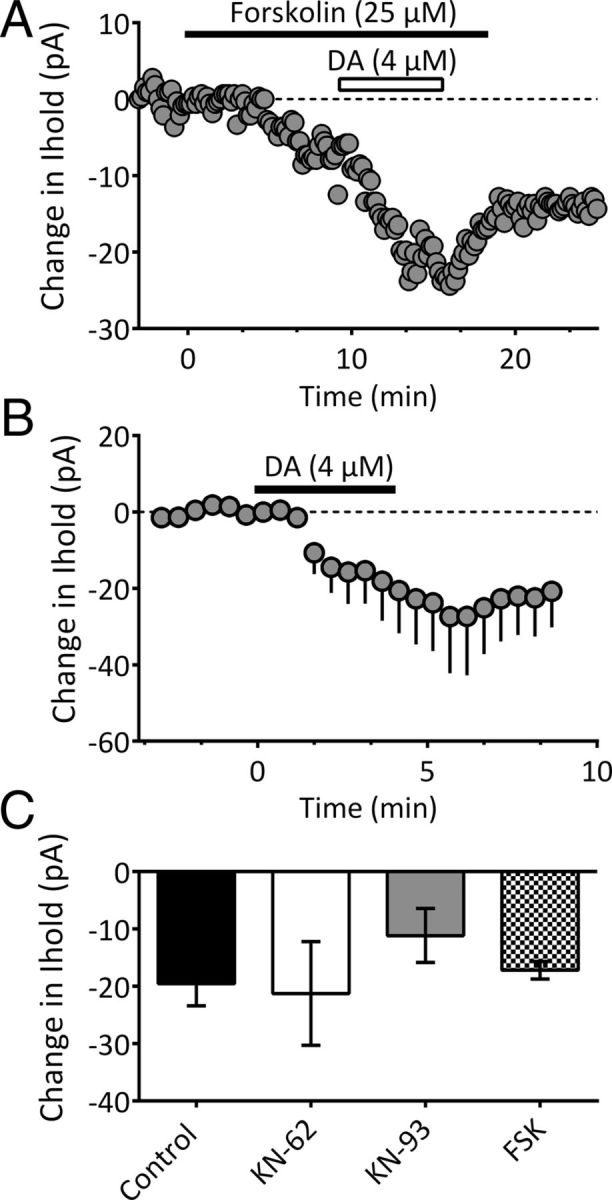
Effects of cAMP activation and CaMKII inhibition on inward currents produced by DA. A, Time course from a single neuron (Vhold = −70mV). Application of forskolin (25 μm) resulted in an inward current. Subsequent application of DA (4 μm) generated an additional inward current. B, Mean time course of the effect of DA (4 μm) during intracellular application of the CaMKII inhibitor KN-62 (5 μm; n = 5). C, Summary of inward currents in untreated cells (Control, n = 7), and in cells treated with intracellular KN-62 (n = 5), intracellular KN-93 (10 μm, n = 5), or forskolin (FSK, n = 5). None of the treatments significantly altered IDAi relative to control (one-way ANOVA, F(3,18) = 0.6561, p = 0.5896).
DA modulation of synaptic neurotransmitter release contributes to the excitation of mLHB neurons
Increase synaptic glutamate release in DA-depolarized mLHb neurons
Robust spontaneous inward synaptic currents were also observed in mLHb neurons exhibiting IDAi. These currents were blocked by the AMPA/kainate receptor antagonist NBQX (10 μm; Fig. 6A1), indicating that they were synaptic spontaneous EPSCs (sEPSCs) mediated by glutamate. In mLHb neurons exhibiting robust IDAi, there was a concomitant large increase in the frequency of sEPSCs during DA treatment (KS test, p < 0.0001; Fig. 6A2) and no change in the amplitudes of these events (KS test, p > 0.05; Fig. 6A2). An increase in sEPSC frequency was not observed in mLHb neurons that did not display IDAi (data not shown). To determine whether the increase in sEPSCs occurred presynaptically, we measured quantal synaptic currents during TTX application. These miniature EPSCs (mEPSCs) were also sensitive to DA, as their frequency was significantly increased (KS test, p < 0.0001; Fig. 6A3). However, as with sEPSCs, the amplitudes of mEPSCs were not affected by DA. These data strongly suggest that DA acted at axon terminals to increase glutamate release onto these mLHb neurons.
Figure 6.
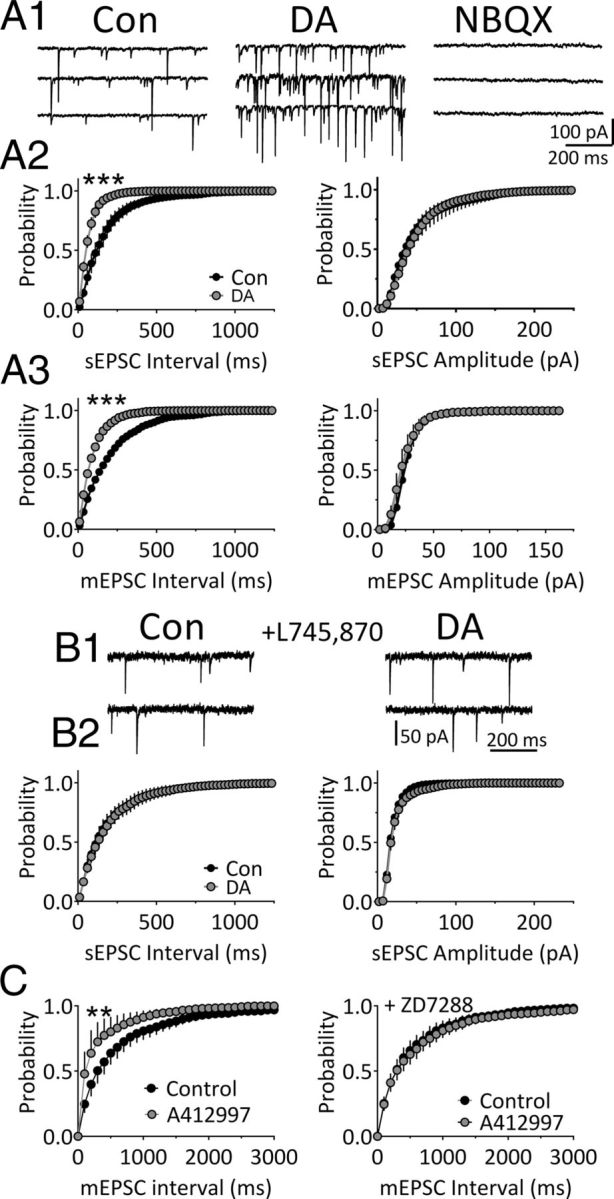
DA increases quantal glutamate release via D4Rs in mLHb neurons. A1, Traces showing sEPSCs recorded during baseline (Con) and during application of DA (3 μm) in an mLHb neuron. The AMPA/kainate receptor antagonist NBQX (10 μm) completely blocked the sEPSCs, indicating that they were mediated by glutamate receptors. A2, Mean cumulative probability histograms showing the effect of DA on sEPSC frequency (interval) and amplitude in mLHb neurons (n = 6). A3, Mean cumulative probability histograms showing the effects of DA on mEPSC frequency and amplitude in LHb neurons preincubated in TTX (1 μm). sEPSC and mEPSC intervals were significantly shortened (i.e., frequencies were increased) by DA (***p < 0.001, **p < 0.01, KS test). B1, Traces of sEPSCs recorded during baseline and DA application following preincubation in the DA D4R antagonist L745,870 (200 nm). B2, Mean cumulative probability histograms showing the lack of effect of DA on sEPSC frequency (interval) and amplitude in mLHb neurons preincubated in L745,870 (n = 6). C, The selective D4R agonist A412997 (1 μm) increases the frequency of mEPSCs (left, n = 8), and this is blocked by ZD7288 (50 μm; right, n = 9), suggesting that D4Rs increase glutamate release via augmentation of Ih in axon terminals.
We next determined whether, like IDAi, the increase in quantal synaptic glutamate release also occurred through D4R activation. Each mLHb neuron used in these experiments initially demonstrated IDAi and an increase in mEPSC frequency upon DA (3 μm) application. Then, following DA washout, brain slices were treated with the D4 antagonist L745,870 (200 nm) for 10 min, and DA was reapplied in its presence. The effects of DA on both glutamate release and Ihold were blocked by the D4 antagonist (KS test, p > 0.05; Fig. 6B1,B2). Next, we examined the effects of the selective D4R agonist A412997 (1 μm) on mEPSCs in mLHb neurons. Similar to the effect of DA, A412997 significantly increased the frequency of mEPSCs (Fig. 6C, left; KS test, p < 0.01, n = 8) without altering mEPSC amplitudes (data not shown). To determine whether the increase in synaptic glutamate release was mediated by Ih, we measured the effect of the D4R agonist on the frequency of mEPSCs in a separate group of mLHb neurons that were pretreated with ZD7288 (50 μm, n = 9). The D4R agonist had no effect on mEPSCs after ZD7288 treatment (Fig. 6C, right; p > 0.05, KS test), suggesting that the D4R-mediated increase in mEPSCs occurs via activation of HCN ion channels. To determine whether endogenous DA could also increase sEPSCs, we examined the effect of amphetamine. As with the inward currents, amphetamine significantly increased the sEPSC frequency (i.e., decreased the inter-sEPSC interval; KS test, p < 0.0001; data not shown), without altering sEPSC amplitudes (KS test, p > 0.05).
Finally, to determine whether IDAi is mediated by glutamate acting at AMPA/kainate receptors we examined the effects of NBQX on this DA-activated current. We found that NBQX (10 μm) did not reverse IDAi (n = 4; DA, −22 ± 6 pA; DA + NBQX, −27 ± 7 pA; p = 0.1344, paired t test), suggesting that AMPA/kainate receptors did not contribute to the DA depolarizing current.
Decreased synaptic GABA release onto DA-depolarized mLHb neurons
To more fully determine the scope of DA actions on mLHb neurons, we measured spontaneous GABA release and its activation of synaptic GABAA receptors. In the absence of TTX, robust spontaneous IPSCs (sIPSCs) were observed both in mLHb neurons that exhibited IDAi and in those that did not. Neurons that exhibited IDAi demonstrated significant reductions in sIPSC frequency, whereas DA did not affect sIPSCs in mLHb neurons that did not exhibit this current (Fig. 7A,B). We next examined the quantal properties of TTX-insensitive mIPSCs in mLHb neurons. In the absence of DA, miniature IPSCs (mIPSCs), recorded in TTX (500 nM), occurred at a wide range of frequencies across neurons (range, 0.65–10.5 Hz; mean, 2.9 ± 0.94 Hz; n = 13; Fig. 7E). Similarly, mean mIPSC amplitudes were widely distributed in these same cells (29.7–133.3 pA; mean = 54.7 ± 7.3 pA; n = 13; Fig. 7E), and individual amplitude histograms for several mLHb neurons demonstrated clear multimodal peaks when fitted with Gaussian distributions (Fig. 7C). Together, these data indicate a high probability of multiquantal GABA release in the absence of action potentials in a subgroup of mLHb neurons.
Figure 7.
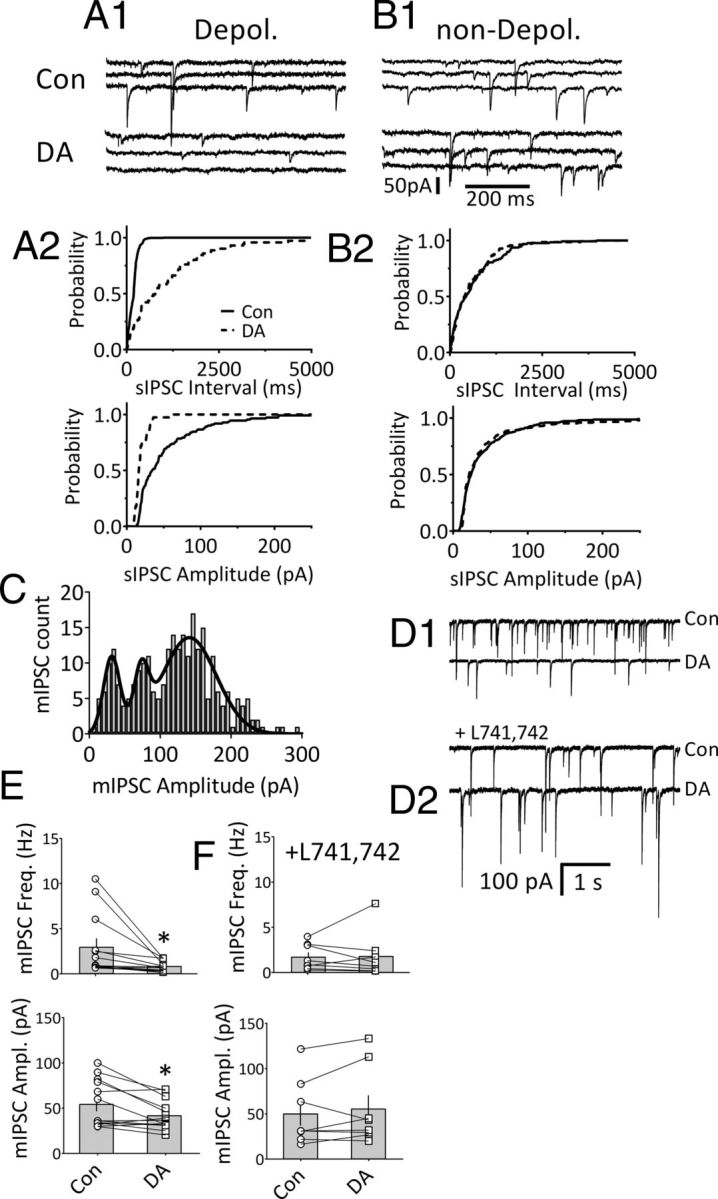
DA reduces GABAergic synaptic transmission via D4Rs in mLHb neurons. A1, sIPSC traces from an mLHb neuron that was also depolarized (Depol.) by DA (3 μm). A2, Cumulative probability distributions of inter-sIPSC interval and sIPSC amplitude (same cell as in A1) reveal a significant increase in sIPSC interval and decrease in sIPSC amplitude during DA (p < 0.001, KS test). B1, sIPSCs in an mLHb neuron that was not depolarized by DA before (top) and during (bottom) DA application. B2, Cumulative probability distributions showing no effect of DA on sIPSC interval or amplitude in the same cell shown in B1. C, TTX-insensitive mIPSC event histogram (bin width = 7 pA) demonstrating multimodal distribution of mIPSC amplitudes in a representative mLHb neuron. The distribution was fitted with three Gaussian curves (r2 = 0.855), and the means for each curve were statistically different, as indicated by non-overlapping 95% confidence intervals (mean, 95% CI: curve 1 = 31.59 pA, 27.69–35.48 pA; curve 2 = 73.20 pA, 67.85–78.55 pA; curve 3 = 141.1 pA, 134.2–147.9 pA). D1, D2, mIPSCs are inhibited by DA (3 μm) in mLHb neurons, and this is prevented by the D4 antagonist L741,742 (200 nm). D1, mIPSC traces showing DA inhibition of mIPSCs in an mLHb neuron. D2, mIPSC traces showing no effect of DA in a neuron preincubated in the D4 antagonist L741,742. E, mIPSC frequency (top) and amplitudes (bottom) from all mLHb neurons (open symbols) superimposed on group means (shaded bars ± SEM) in cells that were not pretreated with L741,742 (n = 13). DA significantly decreased mean mIPSC frequency (p = 0.02, paired t test) and amplitude (p = 0.01, paired t test) in these groups. F, Effects of DA on mIPSC frequency and amplitude in mLHb neurons preincubated with the D4 antagonist L741,742. DA did not significantly affect mIPSCs in these cells (n = 8; p < 0.05). *p < 0.05, paired Student's t test.
DA (3 μm) significantly reduced mIPSC frequency (control, 2.93 ± 0.94 Hz; DA, 0.81 ± 0.16 Hz; p = 0.02, paired Student's t test; n = 13; Fig. 7D1,E) and mIPSC amplitude (control, 54.66 ± 7.22; DA, 42.12 ± 4.62 Hz; p = 0.01, paired t test; n = 13; Fig. 7E). However, in a separate group of LHb neurons that were treated with the D4R antagonist L741,742 (200 nm), DA did not significantly affect mIPSC frequency or amplitude (mIPSC frequency: control, 1.70 ± 0.50 Hz; DA + L741,742, 1.78 ± 0.88 Hz; mIPSC amplitude: control, 49.90 ± 13.0 pA; DA + L741,742, 55.35 ± 15.20 pA; n = 8; p = 0.90 and p = 0.33, respectively, paired t test; Fig. 7D2,F).
Depolarizing neurons project to the RMTg and not the VTA
Anatomical evidence suggests that LHb neurons project to both the RMTg and the rostral VTA in the ventral midbrain. To determine whether the mLHb neurons that are depolarized by DA project to these areas, injections of the retrograde tracers CTB or Neuro-DiI were targeted to either the VTA or the RMTg region in different groups of rats. Three to 5 d after these injections, sagittal slices containing the LHb, VTA, and RMTg were cut. The injections were first visually confirmed to be located in either the RMTg or the rostral VTA (Fig. 8A) before commencing electrophysiological recordings from the fluorescence-containing neurons in the mLHb. The effects of DA were tested on 80 fluorescently labeled neurons projecting to the VTA. Surprisingly, none of these neurons were depolarized by DA. Instead, 41 of 80 neurons (51.3%) demonstrated a D2R-mediated hyperpolarization, whereas 37 cells (46.2%) showed no response to DA (Fig. 8B). Two neurons demonstrated a biphasic response marked by an initial hyperpolarization (outward current), followed by a delayed depolarization (inward current) that we have previously described in the LHb (Jhou et al., 2013). We then recorded from nonretrogradely labeled neurons in the vicinity of fluorescent VTA-projecting cells and found two neurons exhibiting a depolarizing response to DA that was blocked with the D4 antagonist L745,870 (100 nm; data not shown).
Figure 8.
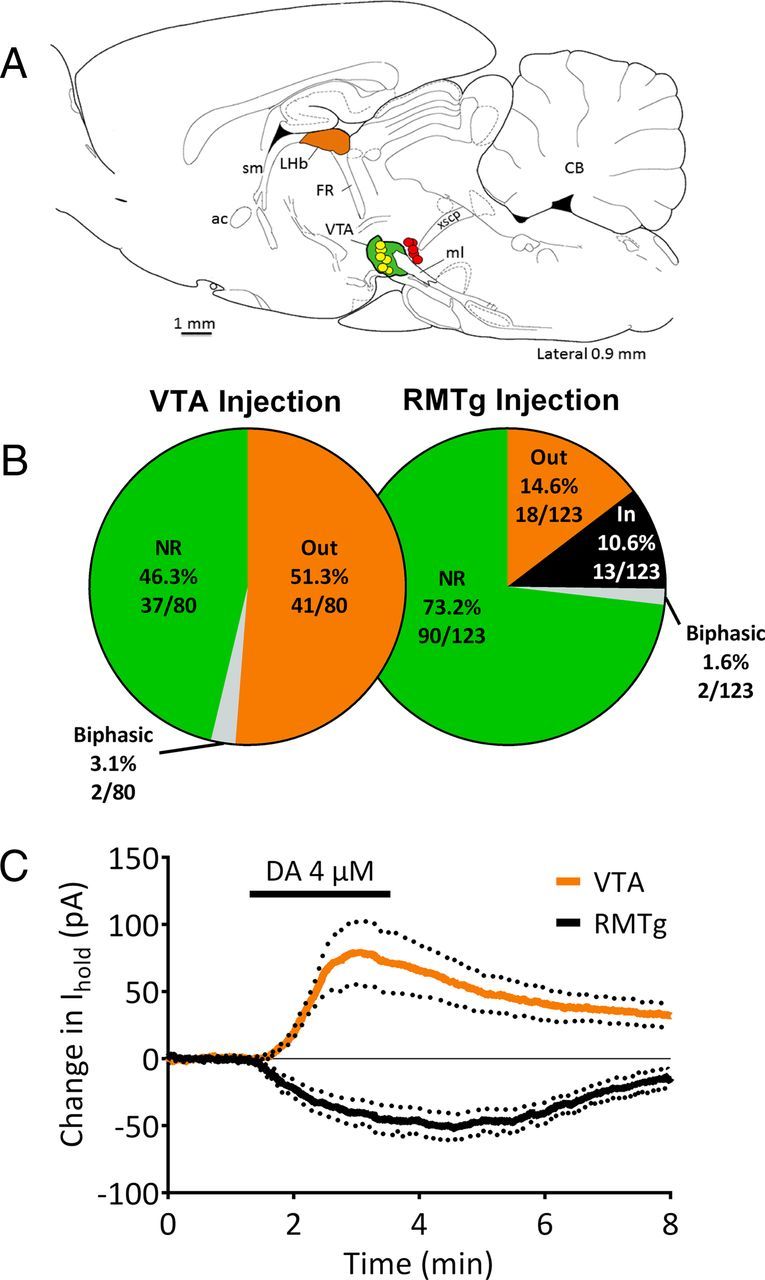
Responses to DA in mLHb neurons labeled with retrograde fluorescent markers. A, Composite diagram indicating the locations of representative Neuro-DiI or CTB retrograde tracer injections into the VTA or RMTg regions (caudal to VTA) of the ventral midbrain (see Materials and Methods for VTA and RMTg injection coordinates). The locations of tracer found upon histological examination for injections targeting the VTA are shown in yellow (n = 5 rats), and those targeting the RMTg are shown in red (n = 4 rats). The VTA region is colored in green. Whole-cell recordings were then made in tracer-containing neurons located in the medial portion of the LHb (orange) using epifluorescence microscopy in brain slices. The figure is modified from the electronic rat brain atlas of Paxinos and Watson, (1998). B, The distribution of responses to DA in retrogradely labeled mLHb neurons following VTA or RMTg injections of fluorescent dyes. NR, No response to DA; Out, outward hyperpolarizing current; In, inward depolarizing current; biphasic, cells demonstrating initial outward currents followed by inward currents. C, Mean outward currents recorded in mLHb neurons (n = 14) labeled by VTA dye injections (orange line), and mean inward currents recorded in mLHb neurons labeled by RMTg (n = 12) dye injections (black line) during DA application in subpopulation of responsive LHb neurons. DA (4 μm) was applied as indicated by the black bar, and dotted lines indicate the SEM. sm, Stria medullaris; ac, anterior commissure; FR, fasciculus retroflexus; ml, medial lemniscus; xscp, decussation of the superior cerebellar peduncle; CB, cerebellum.
Anatomical studies have shown that RMTg-projecting LHb neurons are found in both lateral and medial segments of the LHb (Gonçalves et al., 2012), and those in the mLHb are intermingled with projections to the VTA (Li et al., 2011). Consistent with these reports, we found many mLHb neurons that were retrogradely labeled following targeting of tracers to the RMTg, and we tested the effect of DA on 123 of these fluorescently labeled neurons. We found that 13 of 123 of the mLHb neurons (10.6%) exhibited depolarizing inward currents with DA application (Fig. 8C). In addition, 18 neurons (14.6%) were hyperpolarized by DA, and 90 neurons (73.2%) demonstrated no response to DA. Two additional neurons showed a biphasic response consisting of depolarization followed by hyperpolarization (Jhou et al., 2013). These data suggest that at least some of the DA-depolarized neurons project to the RMTg, and not to the VTA, whereas mLHb neurons projecting to the VTA were largely inhibited by DA acting at D2Rs.
Discussion
Our study demonstrates that DA strongly excites a subgroup of mLHb neurons through multiple mechanisms. First, mLHb neurons are depolarized through a shift in the voltage dependence of Ih. Second, DA increases synaptic glutamate release onto these same mLHb neurons. Third, GABA release onto GABAA receptors on mLHb neurons is suppressed, consistent with disinhibitory excitation. We also provide data to suggest that mLHb neurons that are activated by DA project to the RMTg, a region that strongly inhibits the majority of midbrain DA neurons (Jhou et al., 2009b). As the mLHb output neurons are thought to be glutamatergic (Geisler and Trimble, 2008; Omelchenko et al., 2009), this effect of DA would increase RMTg output to midbrain DA neurons, resulting in their inhibition. We therefore propose that the excitatory actions of DA on mLHb neurons constitute a negative feedback loop in which the duration of midbrain DA neuron activity is limited via RMTg activation.
There is strong evidence that LHb function is influenced by DA. The LHb receives DA innervation from the midbrain (Gruber et al., 2007), and DA receptors are found in LHb (Mansour et al., 1990; Bouthenet et al., 1991; Meador-Woodruff et al., 1991; Weiner et al., 1991; Aizawa et al., 2012; Jhou et al., 2013). We confirm the presence of TH-positive fibers within the medial aspect of the LHb, where we performed electrophysiological recordings. In addition, we find that cocaine, GBR 12935, or amphetamine all mimic the depolarizing effect of exogenous DA, suggesting that endogenous DA is released in functionally relevant concentrations in the LHb in vitro.
Functional data also suggest that DA modulates LHb activity. Expression of the immediate early gene c-fos, indicative of heightened neuronal activity, is increased by DA agonists, or cocaine treatment in vivo (Wirtshafter et al., 1994; Jhou et al., 2009a; Zahm et al., 2010). Also, LHb neuron activity increases following intravenous administration of DA ligands, application of cocaine (Dougherty et al., 1990; Kowski et al., 2009; Zuo et al., 2013), or electrical stimulation of DA neurons (Shen et al., 2012). In contrast, cocaine also inhibits LHb activity in vivo (Dougherty et al., 1990), consistent with our observation that some LHb neurons are hyperpolarized via D2 receptors during cocaine or DA exposure (Jhou et al., 2013). Also, in vivo exposure to cocaine increases AMPA receptor expression and long-term potentiation, specifically in LHb neurons projecting to RMTg (Maroteaux and Mameli, 2012).
Among other nuclei, LHb neurons project to the VTA and RMTg, and some of the heterogeneity of LHb neuron responses to DA may relate to the targets of these cells. We demonstrate that ∼10% of mLHb neurons retrogradely labeled by RMTg-targeted tracer injections exhibit depolarizing inward currents, whereas neurons projecting to VTA exhibit only outward hyperpolarizing currents. This suggests that the DA innervation arising in the VTA excites some RMTg-projecting LHb neurons, but uniformly inhibits LHb neurons projecting to VTA. Since the proportion of randomly selected nonlabeled neurons that are depolarized by DA is higher (∼30%) than those in the retrograde RMTg-labeling study, it is possible that some DA-depolarized mLHb neurons project to brain regions other than the RMTg or VTA, such as the raphe nucleus or laterodorsal tegmentum (Geisler and Trimble, 2008; Bernard and Veh, 2012), or that the labeling of neurons was incomplete.
The depolarization and alterations in synaptic transmission produced by DA in mLHb neurons is blocked by D4R antagonists, and mimicked by the selective D4R agonist A412997. Although a recent study implicated D2 receptors in the excitation of LHb neurons, involvement of D4Rs was not specifically addressed (Zuo et al., 2013). D4Rs have been previously identified in several other brain areas (Callier et al., 2003; Lauzon and Laviolette, 2010). To our knowledge, this is the first report of their presence in the LHb, where we provide evidence for multiple cellular locations for D4Rs. First, as IDAi is observed when action potentials are blocked by TTX, it is likely that D4Rs are located on mLHb neuron somata and/or dendrites. Second, DA and A412997 increase synaptic glutamate release, and DA decreases synaptic GABA release onto mLHb neurons. These data suggest that D4Rs are also likely located on GABAergic and glutamatergic axon terminals in the LHb.
Prior studies show that D4Rs can inhibit both hyperpolarizing outward currents evoked by GABA application in rodent globus pallidus neurons through a postsynaptic mechanism (Shin et al., 2003), and synaptic GABA release onto thalamic and dorsolateral septal neurons through a presynaptic mechanism via Gi/o coupling (Asaumi et al., 2006; Gasca-Martinez et al., 2010). D4Rs are also coupled to postsynaptic inhibition of glutamate EPSCs at subthalamopallidal synapses (Hernández et al., 2006) and NMDA receptor currents in lateral amygdala neurons (Martina and Bergeron, 2008). D4Rs on PFC neurons are linked to the regulation of AMPA receptor trafficking via CaMKII-control of microtubules (Yuen and Yan, 2011). CaMKII is also implicated in the activity-dependent insertion of HCN1 channels into hippocampal neuron membranes (Fan et al., 2005), and disruption of microtubule-dependent cycling of HCN1 subunits by AMPARs facilitates this process (Noam et al., 2010) through a CaMKII-dependent mechanism (Shin and Chetkovich, 2007). However, we found that intracellular CaMKII inhibitors did not block the inward currents produced by DA. Similarly, although numerous studies have shown that cAMP produces a depolarizing shift in the voltage sensitivity of HCN channels (Ingram and Williams, 1994; DiFrancesco, 1999; Lüthi and McCormick, 1999), we found that stimulation of cAMP accumulation with forskolin did not block IDAi in mLHb neurons. Therefore, it does not appear that D4Rs are coupled to HCN channels via adenylyl cyclase- or CaMKII-dependent mechanisms in these mLHb neurons, and future experiments will focus on identifying the nature of this interaction.
Whereas enhancement of Ih represents a novel D4R-dependent transduction mechanism, the inhibition of GABA release onto mLHb neurons is consistent with prior results (Asaumi et al., 2006; Gasca-Martinez et al., 2010). However, facilitation of glutamate release onto mLHb neurons is inconsistent with the actions of other Gi/Go G-protein-coupled receptors on neurotransmitter release. It is possible that increased glutamatergic synaptic activity results from IDAi-associated depolarization of mLHb neurons that are connected via recurrent axons. However, the observed increase in mEPSC frequency is suggestive of a direct effect on axon terminals. In support of this, pretreatment of mLHb neurons with ZD7288 prevented the facilitation of synaptic glutamate release produced by the D4R agonist A412997. This suggests that D4Rs may also shift the voltage dependency of HCN channels in glutamate axon terminals to facilitate neurotransmitter release. The modulation of neurotransmitter release via HCN channels has been reported previously (Beaumont and Zucker, 2000; Yu et al., 2004), and our data suggest that this mechanism may be coupled to D4Rs to enhance glutamate release in the LHb.
The LHb is a central component of the dorsal diencephalic conduction system that channels neural information from the limbic forebrain to ascending midbrain and hindbrain dopaminergic, serotonergic, and cholinergic areas (Geisler and Trimble, 2008; Brinschwitz et al., 2010; Bernard and Veh, 2012). Recently, the LHb has been implicated in regulating midbrain DA neuron activity during reward-based learning (Matsumoto and Hikosaka, 2007). However, since the LHb output is largely glutamatergic, and stimulation of the LHb strongly inhibits VTA DA neurons (Christoph et al., 1986; Ji and Shepard, 2007; Matsumoto and Hikosaka, 2007), an indirect influence of LHb output to DA neurons was proposed (Hikosaka et al., 2008). The intermediary RMTg is now thought to be the target of the excitatory LHb output (Balcita-Pedicino et al., 2011) as it is strongly activated by LHb stimulation and inhibits up to 97% of midbrain DA neurons (Ji and Shepard, 2007). Optogenetic activation of the LHb output to the RMTg promotes behavioral avoidance (Stamatakis and Stuber, 2012), consistent with increased c-fos activity in both the LHb and RMTg following aversive stimuli (Jhou et al., 2009a; Mahler and Aston-Jones, 2012). We recently provided evidence that the LHb plays a prominent role in mediating delayed aversive properties of cocaine via its output to the RMTg, supporting its role in anhedonic opponent processes related to psychostimulant use (Jhou et al., 2013).
The LHb receives input from midbrain DA neurons (Herkenham and Nauta, 1979; Phillipson and Pycock, 1982; Gruber et al., 2007), can suppress their activity in behaviorally relevant ways (Matsumoto and Hikosaka, 2007), and is activated by psychostimulants (Jhou et al., 2009a). We propose that the actions of DA and the involvement of D4Rs that we describe here are relevant to these effects. We further hypothesize that the D4R-mediated excitation of LHb neurons projecting to the RMTg represents an indirect inhibitory feedback pathway that suppresses continued DA neuron activity (Fig. 9) and can be contrasted with a direct inhibitory feedback pathway (Fig. 9) that is controlled by D2 receptors on LHb neurons that also project to midbrain DA neurons (Jhou et al., 2013). Activation of either the indirect or direct pathways by DA results in the inhibition of further DA neuron activity, thereby refining the temporal properties of the DA signal to downstream targets.
Figure 9.

Hypothesized direct and indirect inhibitory feedback pathways from the LHb to midbrain DA neurons distinguished by D4 or D2 receptor control. In the direct negative feedback pathway D2Rs on LHb glutamatergic neurons are activated by DA release from midbrain DA neurons. D2Rs inhibit these LHb neurons, thereby decreasing glutamate release onto midbrain DA neurons, causing inhibition. In the indirect negative feedback pathway, DA release onto D4Rs excites LHb neurons via the following three mechanisms; increased Ih; increased glutamate release; and decreased GABA release. The LHb neurons depolarized by D4R activation project to the GABAergic output neurons of the RMTg, increasing their excitability, thereby strongly inhibiting midbrain DA neurons via GABA release from RMTg cells. In both cases, the net effect is the inhibition of DA neuron activity following initial activation. However, the indirect pathway may have a larger influence on midbrain DA neurons because the LHb projection to RMTg is larger than the direct projection from the LHb to the midbrain.
Footnotes
This work was supported by the National Institutes of Health, National Institute on Drug Abuse Intramural Research Program. We thank Dr. Thomas C. Jhou for critical reading of the manuscript and Daniel D. Brauer for assistance with some experiments.
The authors declare no competing financial interests.
References
- Aizawa H, Kobayashi M, Tanaka S, Fukai T, Okamoto H. Molecular characterization of the subnuclei in rat habenula. J Comp Neurol. 2012;520:4051–4066. doi: 10.1002/cne.23167. [DOI] [PubMed] [Google Scholar]
- Aponte Y, Lien CC, Reisinger E, Jonas P. Hyperpolarization-activated cation channels in fast-spiking interneurons of rat hippocampus. J Physiol. 2006;574:229–243. doi: 10.1113/jphysiol.2005.104042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki M, McGeer PL, McGeer EG. Retrograde HRP tracing combined with a pharmacohistochemical method for GABA transaminase for the identification of presumptive GABAergic projections to the habenula. Brain Res. 1984;304:271–277. doi: 10.1016/0006-8993(84)90330-5. [DOI] [PubMed] [Google Scholar]
- Asaumi Y, Hasuo H, Akasu T. Dopamine presynaptically depresses fast inhibitory synaptic transmission via D4 receptor-protein kinase A pathway in the rat dorsolateral septal nucleus. J Neurophysiol. 2006;96:591–601. doi: 10.1152/jn.00966.2005. [DOI] [PubMed] [Google Scholar]
- Balcita-Pedicino JJ, Omelchenko N, Bell R, Sesack SR. The inhibitory influence of the lateral habenula on midbrain dopamine cells: ultrastructural evidence for indirect mediation via the rostromedial mesopontine tegmental nucleus. J Comp Neurol. 2011;519:1143–1164. doi: 10.1002/cne.22561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaumont V, Zucker RS. Enhancement of synaptic transmission by cyclic AMP modulation of presynaptic Ih channels. Nat Neurosci. 2000;3:133–141. doi: 10.1038/72072. [DOI] [PubMed] [Google Scholar]
- Beckstead RM, Domesick VB, Nauta WJ. Efferent connections of the substantia nigra and ventral tegmental area in the rat. Brain Res. 1979;175:191–217. doi: 10.1016/0006-8993(79)91001-1. [DOI] [PubMed] [Google Scholar]
- Behzadi G, Kalén P, Parvopassu F, Wiklund L. Afferents to the median raphe nucleus of the rat: retrograde cholera toxin and wheat germ-conjugated horseradish peroxidase tracing, and selective D-[3H]aspartate labelling of possible excitatory amino acid inputs. Neuroscience. 1990;37:77–100. doi: 10.1016/0306-4522(90)90194-9. [DOI] [PubMed] [Google Scholar]
- Bernard R, Veh RW. Individual neurons in the rat lateral habenular complex project mostly to the dopaminergic ventral tegmental area or to the serotonergic raphe nuclei. J Comp Neurol. 2012;520:2545–2558. doi: 10.1002/cne.23080. [DOI] [PubMed] [Google Scholar]
- Bouthenet ML, Souil E, Martres MP, Sokoloff P, Giros B, Schwartz JC. Localization of dopamine D3 receptor mRNA in the rat brain using in situ hybridization histochemistry: comparison with dopamine D2 receptor mRNA. Brain Res. 1991;564:203–219. doi: 10.1016/0006-8993(91)91456-B. [DOI] [PubMed] [Google Scholar]
- Brinschwitz K, Dittgen A, Madai VI, Lommel R, Geisler S, Veh RW. Glutamatergic axons from the lateral habenula mainly terminate on GABAergic neurons of the ventral midbrain. Neuroscience. 2010;168:463–476. doi: 10.1016/j.neuroscience.2010.03.050. [DOI] [PubMed] [Google Scholar]
- Bromberg-Martin ES, Hikosaka O. Lateral habenula neurons signal errors in the prediction of reward information. Nat Neurosci. 2011;14:1209–1216. doi: 10.1038/nn.2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callier S, Snapyan M, Le Crom S, Prou D, Vincent JD, Vernier P. Evolution and cell biology of dopamine receptors in vertebrates. Biol Cell. 2003;95:489–502. doi: 10.1016/S0248-4900(03)00089-3. [DOI] [PubMed] [Google Scholar]
- Christoph GR, Leonzio RJ, Wilcox KS. Stimulation of the lateral habenula inhibits dopamine-containing neurons in the substantia nigra and ventral tegmental area of the rat. J Neurosci. 1986;6:613–619. doi: 10.1523/JNEUROSCI.06-03-00613.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiFrancesco D. Dual allosteric modulation of pacemaker (f) channels by cAMP and voltage in rabbit SA node. J Physiol. 1999;515:367–376. doi: 10.1111/j.1469-7793.1999.367ac.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty PM, Qiao JT, Wiggins RC, Dafny N. Microiontophoresis of cocaine, desipramine, sulpiride, methysergide, and naloxone in habenula and parafasciculus. Exp Neurol. 1990;108:241–246. doi: 10.1016/0014-4886(90)90129-G. [DOI] [PubMed] [Google Scholar]
- Fan Y, Fricker D, Brager DH, Chen X, Lu HC, Chitwood RA, Johnston D. Activity-dependent decrease of excitability in rat hippocampal neurons through increases in I(h) Nat Neurosci. 2005;8:1542–1551. doi: 10.1038/nn1568. [DOI] [PubMed] [Google Scholar]
- Gasca-Martinez D, Hernandez A, Sierra A, Valdiosera R, Anaya-Martinez V, Floran B, Erlij D, Aceves J. Dopamine inhibits GABA transmission from the globus pallidus to the thalamic reticular nucleus via presynaptic D4 receptors. Neuroscience. 2010;169:1672–1681. doi: 10.1016/j.neuroscience.2010.05.048. [DOI] [PubMed] [Google Scholar]
- Geisler S, Trimble M. The lateral habenula: no longer neglected. CNS Spectr. 2008;13:484–489. doi: 10.1017/s1092852900016710. [DOI] [PubMed] [Google Scholar]
- Geisler S, Andres KH, Veh RW. Morphologic and cytochemical criteria for the identification and delineation of individual subnuclei within the lateral habenular complex of the rat. J Comp Neurol. 2003;458:78–97. doi: 10.1002/cne.10566. [DOI] [PubMed] [Google Scholar]
- Gonçalves L, Sego C, Metzger M. Differential projections from the lateral habenula to the rostromedial tegmental nucleus and ventral tegmental area in the rat. J Comp Neurol. 2012;520:1278–1300. doi: 10.1002/cne.22787. [DOI] [PubMed] [Google Scholar]
- Gruber C, Kahl A, Lebenheim L, Kowski A, Dittgen A, Veh RW. Dopaminergic projections from the VTA substantially contribute to the mesohabenular pathway in the rat. Neurosci Lett. 2007;427:165–170. doi: 10.1016/j.neulet.2007.09.016. [DOI] [PubMed] [Google Scholar]
- Harris NC, Constanti A. Mechanism of block by ZD 7288 of the hyperpolarization-activated inward rectifying current in guinea pig substantia nigra neurons in vitro. J Neurophysiol. 1995;74:2366–2378. doi: 10.1152/jn.1995.74.6.2366. [DOI] [PubMed] [Google Scholar]
- Herkenham M, Nauta WJ. Efferent connections of the habenular nuclei in the rat. J Comp Neurol. 1979;187:19–47. doi: 10.1002/cne.901870103. [DOI] [PubMed] [Google Scholar]
- Hernández A, Ibáñez-Sandoval O, Sierra A, Valdiosera R, Tapia D, Anaya V, Galarraga E, Bargas J, Aceves J. Control of the subthalamic innervation of the rat globus pallidus by D2/3 and D4 dopamine receptors. J Neurophysiol. 2006;96:2877–2888. doi: 10.1152/jn.00664.2006. [DOI] [PubMed] [Google Scholar]
- Hikosaka O, Sesack SR, Lecourtier L, Shepard PD. Habenula: crossroad between the basal ganglia and the limbic system. J Neurosci. 2008;28:11825–11829. doi: 10.1523/JNEUROSCI.3463-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingram SL, Williams JT. Opioid inhibition of Ih via adenylyl cyclase. Neuron. 1994;13:179–186. doi: 10.1016/0896-6273(94)90468-5. [DOI] [PubMed] [Google Scholar]
- Jhou TC, Fields HL, Baxter MG, Saper CB, Holland PC. The rostromedial tegmental nucleus (RMTg), a GABAergic afferent to midbrain dopamine neurons, encodes aversive stimuli and inhibits motor responses. Neuron. 2009a;61:786–800. doi: 10.1016/j.neuron.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhou TC, Geisler S, Marinelli M, Degarmo BA, Zahm DS. The mesopontine rostromedial tegmental nucleus: a structure targeted by the lateral habenula that projects to the ventral tegmental area of Tsai and substantia nigra compacta. J Comp Neurol. 2009b;513:566–596. doi: 10.1002/cne.21891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhou TC, Good CH, Rowley CS, Xu SP, Wang H, Burnham NW, Hoffman AF, Lupica CR, Ikemoto S. Cocaine drives aversive conditioning via delayed activation of dopamine-responsive habenular and midbrain pathways. J Neurosci. 2013;33:7501–7512. doi: 10.1523/JNEUROSCI.3634-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji H, Shepard PD. Lateral habenula stimulation inhibits rat midbrain dopamine neurons through a GABAA receptor-mediated mechanism. J Neurosci. 2007;27:6923–6930. doi: 10.1523/JNEUROSCI.0958-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufling J, Veinante P, Pawlowski SA, Freund-Mercier MJ, Barrot M. Afferents to the GABAergic tail of the ventral tegmental area in the rat. J Comp Neurol. 2009;513:597–621. doi: 10.1002/cne.21983. [DOI] [PubMed] [Google Scholar]
- Kowski AB, Veh RW, Weiss T. Dopaminergic activation excites rat lateral habenular neurons in vivo. Neuroscience. 2009;161:1154–1165. doi: 10.1016/j.neuroscience.2009.04.026. [DOI] [PubMed] [Google Scholar]
- Lauzon NM, Laviolette SR. Dopamine D4-receptor modulation of cortical neuronal network activity and emotional processing: implications for neuropsychiatric disorders. Behav Brain Res. 2010;208:12–22. doi: 10.1016/j.bbr.2009.11.037. [DOI] [PubMed] [Google Scholar]
- Li B, Piriz J, Mirrione M, Chung C, Proulx CD, Schulz D, Henn F, Malinow R. Synaptic potentiation onto habenula neurons in the learned helplessness model of depression. Nature. 2011;470:535–539. doi: 10.1038/nature09742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupica CR, Bell JA, Hoffman AF, Watson PL. Contribution of the hyperpolarization-activated current (I(h)) to membrane potential and GABA release in hippocampal interneurons. J Neurophysiol. 2001;86:261–268. doi: 10.1152/jn.2001.86.1.261. [DOI] [PubMed] [Google Scholar]
- Lüthi A, McCormick DA. Modulation of a pacemaker current through Ca(2+)-induced stimulation of cAMP production. Nat Neurosci. 1999;2:634–641. doi: 10.1038/10189. [DOI] [PubMed] [Google Scholar]
- Maccaferri G, McBain CJ. The hyperpolarization-activated current (Ih) and its contribution to pacemaker activity in rat CA1 hippocampal stratum oriens-alveus interneurones. J Physiol. 1996;497:119–130. doi: 10.1113/jphysiol.1996.sp021754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahler SV, Aston-Jones GS. Fos activation of selective afferents to ventral tegmental area during cue-induced reinstatement of cocaine seeking in rats. J Neurosci. 2012;32:13309–13326. doi: 10.1523/JNEUROSCI.2277-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansour A, Meador-Woodruff JH, Bunzow JR, Civelli O, Akil H, Watson SJ. Localization of dopamine D2 receptor mRNA and D1 and D2 receptor binding in the rat brain and pituitary: an in situ hybridization-receptor autoradiographic analysis. J Neurosci. 1990;10:2587–2600. doi: 10.1523/JNEUROSCI.10-08-02587.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroteaux M, Mameli M. Cocaine evokes projection-specific synaptic plasticity of lateral habenula neurons. J Neurosci. 2012;32:12641–12646. doi: 10.1523/JNEUROSCI.2405-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martina M, Bergeron R. D1 and D4 dopaminergic receptor interplay mediates coincident G protein-independent and dependent regulation of glutamate NMDA receptors in the lateral amygdala. J Neurochem. 2008;106:2421–2435. doi: 10.1111/j.1471-4159.2008.05584.x. [DOI] [PubMed] [Google Scholar]
- Matsumoto M, Hikosaka O. Lateral habenula as a source of negative reward signals in dopamine neurons. Nature. 2007;447:1111–1115. doi: 10.1038/nature05860. [DOI] [PubMed] [Google Scholar]
- Matsumoto M, Hikosaka O. Representation of negative motivational value in the primate lateral habenula. Nat Neurosci. 2009;12:77–84. doi: 10.1038/nn.2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meador-Woodruff JH, Mansour A, Civelli O, Watson SJ. Distribution of D2 dopamine receptor mRNA in the primate brain. Prog Neuropsychopharmacol Biol Psychiatry. 1991;15:885–893. doi: 10.1016/0278-5846(91)90016-T. [DOI] [PubMed] [Google Scholar]
- Moreland RB, Patel M, Hsieh GC, Wetter JM, Marsh K, Brioni JD. A-412997 is a selective dopamine D4 receptor agonist in rats. Pharmacol Biochem Behav. 2005;82:140–147. doi: 10.1016/j.pbb.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Noam Y, Zha Q, Phan L, Wu RL, Chetkovich DM, Wadman WJ, Baram TZ. Trafficking and surface expression of hyperpolarization-activated cyclic nucleotide-gated channels in hippocampal neurons. J Biol Chem. 2010;285:14724–14736. doi: 10.1074/jbc.M109.070391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notomi T, Shigemoto R. Immunohistochemical localization of Ih channel subunits, HCN1–4, in the rat brain. J Comp Neurol. 2004;471:241–276. doi: 10.1002/cne.11039. [DOI] [PubMed] [Google Scholar]
- Omelchenko N, Bell R, Sesack SR. Lateral habenula projections to dopamine and GABA neurons in the rat ventral tegmental area. Eur J Neurosci. 2009;30:1239–1250. doi: 10.1111/j.1460-9568.2009.06924.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Ed 4. San Diego: Academic; 1998. [DOI] [PubMed] [Google Scholar]
- Phillipson OT, Pycock CJ. Dopamine neurones of the ventral tegmentum project to both medial and lateral habenula. Some implications for habenular function. Exp Brain Res. 1982;45:89–94. doi: 10.1007/BF00235766. [DOI] [PubMed] [Google Scholar]
- Poller WC, Bernard R, Derst C, Weiss T, Madai VI, Veh RW. Lateral habenular neurons projecting to reward-processing monoaminergic nuclei express hyperpolarization-activated cyclic nucleotid-gated cation channels. Neuroscience. 2011;193:205–216. doi: 10.1016/j.neuroscience.2011.07.013. [DOI] [PubMed] [Google Scholar]
- Rateau Y, Ropert N. Expression of a functional hyperpolarization-activated current (Ih) in the mouse nucleus reticularis thalami. J Neurophysiol. 2006;95:3073–3085. doi: 10.1152/jn.00922.2005. [DOI] [PubMed] [Google Scholar]
- Reisine TD, Soubrié P, Ferron A, Blas C, Romo R, Glowinski J. Evidence for a dopaminergic innervation of the cat lateral habenula: its role in controlling serotonin transmission in the basal ganglia. Brain Res. 1984;308:281–288. doi: 10.1016/0006-8993(84)91067-9. [DOI] [PubMed] [Google Scholar]
- Shen X, Ruan X, Zhao H. Stimulation of midbrain dopaminergic structures modifies firing rates of rat lateral habenula neurons. PLoS One. 2012;7:e34323. doi: 10.1371/journal.pone.0034323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin M, Chetkovich DM. Activity-dependent regulation of h channel distribution in hippocampal CA1 pyramidal neurons. J Biol Chem. 2007;282:33168–33180. doi: 10.1074/jbc.M703736200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin RM, Masuda M, Miura M, Sano H, Shirasawa T, Song WJ, Kobayashi K, Aosaki T. Dopamine D4 receptor-induced postsynaptic inhibition of GABAergic currents in mouse globus pallidus neurons. J Neurosci. 2003;23:11662–11672. doi: 10.1523/JNEUROSCI.23-37-11662.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skagerberg G, Lindvall O, Björklund A. Origin, course and termination of the mesohabenular dopamine pathway in the rat. Brain Res. 1984;307:99–108. doi: 10.1016/0006-8993(84)90465-7. [DOI] [PubMed] [Google Scholar]
- Stamatakis AM, Stuber GD. Activation of lateral habenula inputs to the ventral midbrain promotes behavioral avoidance. Nat Neurosci. 2012;15:1105–1107. doi: 10.1038/nn.3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzer D. How addictive drugs disrupt presynaptic dopamine neurotransmission. Neuron. 2011;69:628–649. doi: 10.1016/j.neuron.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner DM, Levey AI, Sunahara RK, Niznik HB, O'Dowd BF, Seeman P, Brann MR. D1 and D2 dopamine receptor mRNA in rat brain. Proc Natl Acad Sci U S A. 1991;88:1859–1863. doi: 10.1073/pnas.88.5.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirtshafter D, Asin KE, Pitzer MR. Dopamine agonists and stress produce different patterns of Fos-like immunoreactivity in the lateral habenula. Brain Res. 1994;633:21–26. doi: 10.1016/0006-8993(94)91517-2. [DOI] [PubMed] [Google Scholar]
- Yu X, Duan KL, Shang CF, Yu HG, Zhou Z. Calcium influx through hyperpolarization-activated cation channels (I(h) channels) contributes to activity-evoked neuronal secretion. Proc Natl Acad Sci U S A. 2004;101:1051–1056. doi: 10.1073/pnas.0305167101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen EY, Yan Z. Cellular mechanisms for dopamine D4 receptor-induced homeostatic regulation of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors. J Biol Chem. 2011;286:24957–24965. doi: 10.1074/jbc.M111.221416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahm DS, Becker ML, Freiman AJ, Strauch S, Degarmo B, Geisler S, Meredith GE, Marinelli M. Fos after single and repeated self-administration of cocaine and saline in the rat: emphasis on the Basal forebrain and recalibration of expression. Neuropsychopharmacology. 2010;35:445–463. doi: 10.1038/npp.2009.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo W, Chen L, Wang L, Ye JH. Cocaine facilitates glutamatergic transmission and activates lateral habenular neurons. Neuropharmacology. 2013;70:180–189. doi: 10.1016/j.neuropharm.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]