

Abstract
The complex effects of stress on learning and memory are mediated, in part, by stress-induced changes in the composition and structure of excitatory synapses. In the hippocampus, the effects of stress involve several factors including glucocorticoids and the stress-released neuropeptide corticotropin-releasing hormone (CRH), which influence the integrity of dendritic spines and the structure and function of the excitatory synapses they carry. CRH, at nanomolar, presumed-stress levels, rapidly abolishes short-term synaptic plasticity and destroys dendritic spines, yet the mechanisms for these effects are not fully understood. Here we tested the hypothesis that glutamate receptor-mediated processes, which shape synaptic structure and function, are engaged by CRH and contribute to spine destabilization. In cultured rat hippocampal neurons, CRH application reduced dendritic spine density in a time- and dose-dependent manner, and this action depended on the CRH receptor type 1. CRH-mediated spine loss required network activity and the activation of NMDA, but not of AMPA receptors; indeed GluR1-containing dendritic spines were resistant to CRH. Downstream of NMDA receptors, the calcium-dependent enzyme, calpain, was recruited, resulting in the breakdown of spine actin-interacting proteins including spectrin. Pharmacological approaches demonstrated that calpain recruitment contributed critically to CRH-induced spine loss. In conclusion, the stress hormone CRH co-opts mechanisms that contribute to the plasticity and integrity of excitatory synapses, leading to selective loss of dendritic spines. This spine loss might function as an adaptive mechanism preventing the consequences of adverse memories associated with severe stress.
Introduction
Molecular and cellular correlates of learning and memory are generally considered to take place at excitatory synapses (Larson and Lynch, 1986; Martin et al., 2000; Neves et al., 2008), by influencing synaptic function (Bear et al., 1987; Malenka et al., 1988; Barria and Malinow, 2002). These processes commonly involve changes in the number, composition, and function of glutamate receptors at the postsynaptic density (Baudry and Lynch, 1979; Scannevin and Huganir, 2000; Derkach et al., 2007) and structural changes of synapses. The postsynaptic component of excitatory hippocampal synapses is located on dendritic spines (Hering and Sheng, 2001; Segal, 2005). Indeed, memory-related changes are associated with changes in spine size and shape (Yuste and Bonhoeffer, 2001; Fukazawa et al., 2003; Park et al., 2006; Penzes et al., 2011). Classically, memory-related synaptic plasticity involves enlargement of dendritic spines (Hering and Sheng, 2001; Chen et al., 2007; Bourne and Harris, 2008; Lynch et al., 2008; Holtmaat and Svoboda, 2009), whereas processes associated with memory loss involve spine shrinkage or loss (Tada and Sheng, 2006; Collingridge et al., 2010; Kasai et al., 2010).
Stress influences memory (Conrad et al., 1999; Kim and Diamond, 2002; Joëls and Baram, 2009; Gray et al., 2013) with concomitant changes in synapse and spine integrity (Shors, 2001; Kole et al., 2004; Stewart et al., 2005; Chen et al., 2008, 2010; Jafari et al., 2012). Much work exists on role of the archetypical stress hormones, corticosteroids (Chen et al., 2007; Alberini and Chen, 2012), which activate both glucocorticoid (De Kloet, 2004; Ulrich-Lai and Herman, 2009; Liston and Gan, 2011; McEwen and Gianaros, 2011) and mineralocorticoid receptors (Joëls and Baram, 2009; Wang et al., 2013a,b).
More recently, corticotropin-releasing hormone (CRH) has been implicated in deficits of hippocampus-dependent memory and long-term potentiation (LTP) resulting from chronic and short (hours-long) stress (Diamond and Rose, 1994; Garcia et al., 1997; Pawlak et al., 2003; Chen et al., 2008, 2010; Ivy et al., 2010). CRH is synthesized by pyramidal cell layer interneurons (Chen et al., 2001), and released during stress (Chen et al., 2004, 2010). Hippocampal pyramidal neurons express CRH receptor type 1 (Chen et al., 2000; Refojo et al., 2011) within the postsynaptic density on dendritic spine heads (Chen et al., 2004). The use of live, two-photon imaging has demonstrated that CRH provokes retraction of existing spines rather than reduction of spine formation (Chen et al., 2008, 2013). Thus, CRH is a candidate molecular mediator of the structural effects of stress in hippocampus. During minutes-long stress, CRH enhances LTP (Blank et al., 2002) whereas longer exposures reduce synaptic function and spine density in CA1 and CA3: behavioral deficits and spine loss induced by short stress can be largely prevented by CRHR1 antagonists (Chen et al., 2010), suggesting that CRH contributes to stress-related memory deficits. However, how CRH elicits spine loss is largely unknown. Because activation of ionotropic glutamate receptors by network activity can rapidly and dynamically change synapse size and spine structure, we tested the hypothesis that CRH-induced spine loss involves the co-option of fundamental mechanisms that influence synapse and spine dynamics.
Materials and Methods
Experiments conformed to National Institutes of Health guidelines and were approved by the Institutional Animal Care and Use Committee of the University of California-Irvine (UCI).
Hippocampal neuron cultures
Timed-pregnant Sprague Dawley rat dams gave birth in the UCI vivarium. Hippocampal neuron cultures were prepared on the day of birth (P0) from pups of either sex as previously described (Noam et al., 2010). Briefly, hippocampi were dissected and incubated in dissection solution (137 mm NaCl, 5.4 mm KCl, 0.17 mm Na2PO4, 0.22 mm KH2PO4, 33.3 mm d-glucose, and 43.8 mm sucrose in 9.9 mm HEPES, pH 7.4) with 10 U/ml papain (Worthington). After removal of papain, cells were triturated and plated at a density of 400–600 cell/mm2 on 12 mm coverslips (Thermo Fisher) or 60 mm CellBIND dishes (Corning) precoated with poly-d-lysine (Sigma). Cultures were initially maintained in Neurobasal Medium (NBM) with B27 (Invitrogen) at 36°C and 5% CO2. After 3–4 h, half the culture medium was replaced with NBM preconditioned for 24 h over 1- to 3-week-old glia cell cultures (conditioned medium). Cultures were treated with 1 μm arabinoside-cytosine (Sigma) 3 days in vitro (3 DIV) to inhibit glial proliferation and refreshed twice a week with conditioned medium. Neurons were used for experiments on 17–21 DIV. At this age, mature synapses are generally present and the presence of postsynaptic structures was verified using postsynaptic density protein 95 (PSD95) immunocytochemistry (ICC).
Visualization of dendritic spines using three independent methods
Several independent measures were used to visualize dendritic spines. (1) The postsynaptic density located on spine heads was visualized using ICC for PSD95. Quantification of PSD95 puncta is a reliable marker of mature synapses. In addition, it enables visualization of small, thin spines that have PSD95 but might be missed by other methods (see below). Because PSD95 might rarely occur on dendritic shaft (Arnold and Clapham, 1999), additional methods were used. (2) Neurons were infected with green fluorescent protein (GFP)-expressing lentiviruses. This led to migration of GFP to all neuronal compartments including dendritic spines; spines were visualized using anti-GFP ICC. Very small, thin spines might not be fully filled; therefore, the method was complemented by the others. (3) Because the core components of dendritic spines consist of polymerized actin (Lynch et al., 2007), low doses of phalloidin conjugated to Alexa Fluor568 were used to label filamentous (F), polymerized actin. Phalloidin was diluted to a final concentration of 165 nm in PBS and incubated with the cultures for 1 h at room temperature. When phalloidin labeling was combined with ICC, phalloidin was added to the blocking buffer along with secondary antibody. Phalloidin labeling was used primarily for qualitative colocalization experiments. Phalloidin interacts preferentially with polymerized actin (Levitsky et al., 2008), and can be used to visualize enlarging spines (Lin et al., 2005). However, imaging resolution might not allow all spines to be visualized, resulting in lower spine densities.
Experimental design and pharmacological manipulations
CRH (Bachem) was maintained in a stock solution of 100 μm prepared in sterile water and then diluted to 100 nm in NBM + B27 just before use. Dissociated neurons on glass coverslips were treated in 24-well plates with a minimum volume of 0.5 ml at 36°C for 30–60 min. The selective blocker of CRH receptor type 1 (CRHR1), 3-[6-(dimethylamino-4-methyl-pyrid-3-yl]-2,5-dimethyl-N,N-dipropyl-pyrazolo[2,3-a]pyrimindin-7-amine (NBI30775), was a gift from D.E. Grigoriadis (Neurocrine Biosciences). The CRHR1 antagonist was dissolved in sterile water and used at a final concentration of 100 nm. NBI30775 was applied alone for 5 min to allow the compound to bind CRHR1 receptors, and this was followed by application of a solution containing both NBI30775 and CRH.
Tetrodotoxin (TTX; Abcam) was stored as a 1 mm stock solution in sterile water and freshly diluted to a final concentration of 1 μm. The selective blockers of ionotropic glutamate receptors, APV, CNQX, MK-801, and NBQX (Sigma) were dissolved in sterile water. Neurons were treated with NMDA and AMPA glutamate receptor antagonists together or each blocker separately for 60 min at the following final concentrations: 100 μm APV, 50 μm CNQX, 10 μm MK-801, and 10 μm NBQX.
The calpain inhibitor III (Calbiochem), an antagonist of both calpain I and II, was dissolved in cell culture grade dimethylsulfoxide (DMSO; Sigma). Neurons were treated with a final concentration of 100 nm calpain inhibitor III 5 min before incubation with CRH. After 60 min, neurons from all of the experimental groups were fixed for ICC or rapidly processed for Western blot analysis.
ICC of fixed cells and F-actin labeling
Coverslips with cultured neurons were placed immediately into ice slush (0°C). Neurons were fixed with ice-cold 4% paraformaldehyde (PFA) in PBS, pH 7.4, for 20 min. All antibodies were diluted in blocking buffer (3% bovine serum albumin, 0.1% Triton-X in PBS, pH 7.4) overnight at 4°C. The following antibodies were used: mouse anti-PSD95 1:4000 (Thermo Fisher), goat anti-CRHR1 1:2000 (directed against the N terminus of the receptor; Everest Biotech), mouse anti-GFP 1:1000 (Sigma), rabbit anti-calpain-1 1:1000 (Abcam), rabbit anti-GluR1 1:1000 (Millipore), and rabbit anti-NR2A 1:500 (Invitrogen). The next day, coverslips were washed and incubated in the appropriate secondary antibodies conjugated to Alexa Fluor 488, Alexa Fluor 568, or Alexa Fluor 633 at a concentration of 1:400 (Invitrogen) at room temperature for 1 h.
F-actin was visualized using phalloidin conjugated to Alexa Fluor 568 (Invitrogen). Neurons were fixed in 4% PFA in PBS, pH 7.4, in an ice slush for 20 min. Phalloidin (final concentration of 165 nm in PBS) was applied for 1 h at room temperature. When phalloidin labeling was combined with ICC it was added along with the secondary antibody. Neurons were processed for confocal imaging with Fluoromount G (Southern Biotech) mounting medium to protect against bleaching.
ICC of nonpermeabilized neurons.
Coverslips containing cultured neurons were briefly fixed with cold 4% PFA for 10 min on ice, and washed gently three times for 1 min each in PBS without detergents. These nonpermeabilized neurons were incubated in anti-GluR1 1:100 (directed against the extracellular N terminus domain of the receptor; Calbiochem) for 48 h or anti-CRHR1 1:50 (directed against the extracellular domain of the receptor; Everest Biotech) overnight at 4°C. Antibodies against the surface receptor were diluted in 3% fetal bovine serum (FBS) in PBS (lacking detergent). The next day, visualization of surface receptor was performed by diluting the appropriate secondary antibody 1:400 in 3% FBS in PBS. Coverslips were washed three times in PBS. To enable detection of internal antigens, neurons were incubated with antibodies against PSD95 and internal CRHR1 diluted in blocking buffer (containing detergents; see method for fixed cells) for 1 h at room temperature. Secondary antibodies for internal proteins were diluted 1:400 in blocking buffer, and coverslips were incubated for 1 h at room temperature.
GFP lentiviral infection
Recombinant lentiviruses expressing GFP under the H1 promoter were produced by transient transfection in HEK293T cells as previously described (Regev et al., 2011). Supernatant was collected from transfected HEK293T cells and virus particles were titered to 2.5 × 105 particles per microliter. Lentiviral infections were performed on 13 DIV and neurons were used on 17–21 DIV. All GFP-expressing neurons were processed for anti-GFP ICC.
Spectrin breakdown product Western blots and analyses
Presence of activated calpain was determined using Western blot analysis for the presence of a cleaved substrate of calpain, spectrin. Specifically, spectrin breakdown products (SBDP) were examined using an antiserum to spectrin, and looking for low molecular weight moieties of the expected size. The expected molecular weight of full-length brain spectrin is 240 kDa, whereas the SBDP is 140 kDa (Siman et al., 1984). Neurons were cultured in 60 mm CellBIND dishes coated with PDL, and exposed to CRH at a final concentration of 100 nm CRH, with or without 100 nm calpain inhibitor for 1 h at 36°C. To determine whether CRH-mediated NMDA receptor activation was responsible for the increase in SBDP, neurons were exposed to 100 nm CRH with or without 100 μm APV. To distinguish CRH-induced SBDP from constitutive calpain activity, all cultures were pre-incubated with 500 nm calpain inhibitor for 3 h and washed with ice-cold sterile PBS before vehicle or CRH exposure. Dishes were immediately chilled on an ice slush, carefully washed twice with sterile PBS, and neurons were lifted off with a 1% Triton X-100 lysis buffer (50 mm Tris-HCl, 150 mm NaCl, 2 mm EDTA, 1% Triton) with a protease inhibitor cocktail. Lysate was centrifuged at 16,000 rpm for 20 min twice, both times discarding the pellet. Following a Bradford assay, protein samples were boiled in sample buffer (6× loading buffer; 375 mm Tris-HCl, 6% SDS, 9% β-mercaptoethanol, 0.03% brilliant blue, 48% glycerol) and loaded on a 4–12% PAGE gel (Lonza; VWR) run at 125 V for 90 min. Samples were transferred to a PVDF membrane (GE Healthcare) and blocked for 1 h in 10% milk in PBS-Tween. The membrane was incubated in 1:20,000 anti-α-spectrin (also called α-fodrin; Abcam) overnight at 4°C in 5% milk. This was followed by incubating the membrane in secondary conjugated to horse radish peroxidase at 1:10,000 at room temperature in 5% milk and developed using the ECL detection kit (Thermo Fisher). Optical densities from Western blot analysis were quantified using ImageJ. Individual experiments were combined by assigning a value of 1 to the optical densities of each experiment's control group. Other groups were compared with control using one-way ANOVA with post hoc Bonferroni's multiple-comparisons test.
Systematic analyses and statistical considerations
Each experiment included 3–4 sister coverslips per treatment group, and neurons were sampled equally from each coverslip for imaging. For PSD95 quantification, a total of at least 12 dendrites from six neurons per treatment group were analyzed. Each experiment was repeated at least twice; therefore, a minimum of 24 dendrites per treatment group were included in the analyses. For GFP spine quantification, six neurons per treatment group were analyzed, and each experiment was repeated twice for a total of 12 dendrites.
All imaging and quantification was done without knowledge of treatment group. To ensure appropriate comparisons and address quantitative differences between imaging studies, dendritic spines were systematically sampled among treatment groups. Images were scaled for distance per pixel length, and the distance from the soma was measured and divided into 20 μm segments using ImageJ. A total of 4800–6000 μm of dendritic length, derived from at least 12 dendrites per treatment group, was analyzed. Spine counts were pooled from two to three experiments. For PSD95 quantification, each individual puncta was considered a separate spine and counts were not adjusted for puncta size. For GFP-expressing neurons, spines were defined by a clear neck and head protruding from the dendrite. Images for analysis were generated using confocal microscopy, Zeiss LSM 510. Images (40×) were generated to show the whole neuron using an oil-immersion objective (NA 1.3). The 3 μm z-series (0.5 μm steps) images were captured from dendrites that were distinct from other dendrites and dendritic crossings and extended at least 100 μm from the soma at 63× (NA 1.4) using an oil-immersion objective. Analysis of all treatment groups across the distance of the dendrite was accomplished using two-way repeated-measures (RM)-ANOVA. Control and CRH treatment groups were combined if there was no significant interaction. All RM-ANOVAs were followed by Tukey's post hoc multiple-comparisons test. Significance levels were set at 0.05 and data are presented as the mean ± SEM. Data were analyzed using GraphPad Prism 5.0 Software.
Results
CRH, at nanomolar concentrations, reduces dendritic spine density in 17–21 DIV hippocampal neurons
Severe, hours-long stress reduces spine density in apical dendrites of CA3 and CA1 hippocampal pyramidal cells, and this is significantly abrogated by application of a CRHR1 blocker directly into the brain. This finding suggests that the effects of stress are mediated, at least in part, by the actions of endogenous, hippocampal CRH (Chen et al., 2010). In accord, we have previously found that CRH application to organotypic or acute hippocampal slices can cause loss of dendritic spines in a distribution similar to the effects of stress (Chen et al., 2008, 2013). In the current study, we used 17–21 DIV hippocampal neurons in culture as an accessible and controllable system that enables a better understanding of the mechanisms. We first examined if CRH-induced dendritic spine loss could be replicated in these neurons by exposing them to CRH levels that may reflect levels present in hippocampus during severe stress (100 nm) (Khan et al., 2004; Chen et al., 2010), followed by two independent methods of analysis. First, we conducted ICC for the integral postsynaptic density protein PSD95 (Fig. 1A,B), followed by sampling and quantification of the number of PSD95-immunoreactive (ir) puncta (Fig. 1C–F). Spine density varies considerably with branch order, as does the vulnerability of dendritic spines to stress (Chen et al., 2010) and to CRH (Chen et al., 2008) Therefore, we quantified PSD95-ir puncta density by branch order. CRH did not change the shape, size, or general appearance of neurons (Fig. 1A,B). Exposure to CRH reduced the density of PSD95-ir puncta along dendritic branches (F(1,66) = 3.81, p = 0.006), and this reduction became more apparent in the third and fourth order branches (p < 0.05; Fig. 1E). The inhomogeneous reduction in dendritic spine density along the dendrite in vitro was reminiscent of the predilection of short stress–and of CRH in acute hippocampal slices–to preferentially affect third- and fourth-order dendritic spines in the stratum radiatum, where they contact terminals of the commissural projection/associational fibers (Chen et al., 2008).
Figure 1.
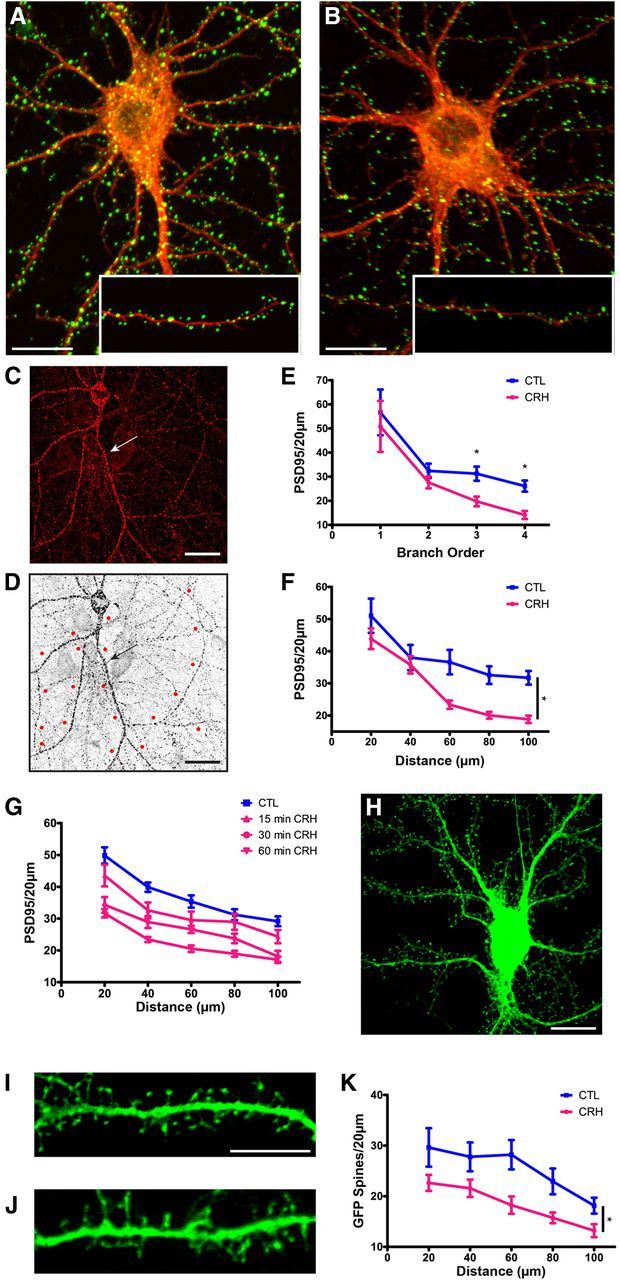
The stress neuropeptide CRH causes a loss of PSD95-ir puncta and dendritic spines in cultured rat hippocampal neurons. Exposure to 100 nm CRH leads to a significant reduction in PSD95-ir puncta, an indication of dendritic spine loss. A, B, Control neuron and (B) neuron exposed to 100 nm CRH at 36°C for 30 min and processed for ICC for PSD95 (green) and for F-actin (red). C, Confocal images were used to quantify PSD95-ir puncta and dendritic spines from GFP-expressing neurons. Neurons used for quantification were clearly demarcated and devoid of dendritic crossings from other neurons that could confound counts. D, Example of a confocal image processed for quantification with 20 μm segments measured out from the soma. E, Exposure to CRH reduced the density of PSD95-ir puncta along dendritic branches (F(1,66) = 3.81, p = 0.006), and this effect became more apparent in third-order (p = 0.004) and fourth-order (p < 0.001) branches (n = 12). Similar results were obtained by quantifying PSD95 by distance from the soma. F, Graph quantifying PSD95-ir puncta per 20 μm segment in cultures incubated in the presence or absence of CRH (F(1,88) = 7.09, p = 0.014; n = 12). G, CRH reduced PSD95 puncta in a time-dependent manner (F(3,80) = 17.66, p < 0.001; n = 6). H, Lentiviral infection of neurons did not change the size or shape of the soma, and enabled direct visualization of spines. I, J, An example of a control GFP-filled dendrite and (J) a dendrite after exposure to 100 nm CRH at 36°C for 60 min. K, Graph quantifying GFP-filled spines per 20 μm segment with and without exposure to CRH (F(1,48) = 7.55, p = 0.017; n = 12). The values for spine density were approximately half of those found using PSD95-ir puncta because the GFP analysis is limited to spines perpendicular to the dendrite. Scale bars: A–D, H, 20 μm; I, J, 5 μm.
We also measured the effects of CRH on spine density as a function of distance from the soma and found that spine density depended on this distance (F(4,88) = 50.28, p < 0.0001; Fig. 1F). CRH reduced the density of PSD95-ir puncta along dendrites (F(1,88) = 7.09, p = 0.014), and post hoc comparisons showed that the peptide reduced PSD95 puncta density as compared with controls at 40–100 μm from the soma (p < 0.05). In view of the comparable results using branch order or distance from soma, we elected to use a single type of analysis. We chose to analyze by distance from soma (0–100 μm, using 20 μm segments) to avoid the possibility that, in the high-magnification microscope images (63×) obtained for each treatment group, the numbers of third- and fourth-order branches might be small or unequal among groups.
At 30 nm, the effects of CRH on spine density were not significant (F(1,50) = 2.31, p = 0.135). Peptide concentrations of 60 nm provoked reduction of spine density (F(1,50) = 6.86, p = 0.012), as did 100 nm (F(1,50) = 4.14, p = 0.047; Chen et al., 2013). CRH-induced loss of PSD95-ir puncta occurred in a time-dependent manner (F(3,80) = 17.66, p < 0.001; Fig. 1G). The significant reduction of synapses by 30–60 min was in line with the actions of the peptide in hippocampal slices (Chen et al., 2008).
The postsynaptic density, labeled with anti-PSD95, represents an intact synaptic structure at the head of dendritic spines. However, it remains unclear if the correlation of PDS95 puncta and the integrity of dendritic spines is absolute. Therefore, we examined directly the effects of CRH on spine density using an independent visualization of dendritic spines expressing a fluorescent protein (Fig. 1H–K). Neurons (13 DIV) were infected with a lentivirus to induce expression of GFP, rendering the whole neuron, including dendritic spines, visible. Four to six days later (17–19 DIV), a subset of cultures was exposed to 100 nm CRH for 60 min. Initial experiments with the GFP-filled neurons used the same 30 min exposure duration that reduced the density of PSD95-immunoreactive puncta. However, there was only a modest reduction in the density of GFP-filled dendritic spines after this exposure duration (data not shown), suggesting that loss of visible PSD95 aggregates might precede spine breakdown. For this reason, the majority of experiments used both methods and consisted of a 60 min exposure to 100 nm CRH. Using GFP-infected neurons, spines, defined as dendritic protrusions with a clear head and distinct neck, were quantified (Fig. 1I,J). Similar to the results found using PSD95 immunolabeling, the number of dendritic spines decreased with increased distance from the soma (F(4,48) = 17.37, p < 0.0001; Fig. 1K). CRH significantly reduced dendritic spine density (F(1,48) = 7.55, p < 0.018), and post hoc comparisons showed that CRH significantly reduced dendritic spine density at 60–100 μm from the cell body (p < 0.05; Fig. 1K). Spine density was approximately half of that observed using PSD95-ir puncta because analysis was limited to spines perpendicular to the dendrite in GFP-expressing neurons. Together, these data, using two independent and systematic quantification measures of dendritic spines, indicated that the effects of CRH on spine density in cultured neurons recapitulate the results observed following short stress in vivo, and following CRH application to the intact hippocampal slice.
CRH-induced spine loss requires activation of CRHR1
Of the two types of CRH receptors, rat hippocampal pyramidal neurons express primarily CRHR1 (Eghbal-Ahmadi et al., 1998; Sánchez et al., 1999; Chen et al., 2000; Van Pett et al., 2000; Lim et al., 2005; Refojo et al., 2011). Colocalization and electron microscopy studies have shown that CRHR1 immunoreactivity is present throughout the neuron, including dendritic spines and within the postsynaptic density (Chen et al., 2004). Here, we examined specifically the localization of surface CRHR1, because cell-membrane located G-protein-coupled receptors (GPCRs) are those activated by their respective ligands. We conducted ICC under conditions where the cell membrane was either permeabilized or not (Fig. 2A), a method enabling distinction between surface CRHR1 and internal pools of the receptor. In line with previous work, CRHR1 immunoreactivity was apparent in the soma and dendritic shafts of permeabilized hippocampal neurons (red), consistent with intracellular synthesis and trafficking. Interestingly, the majority of surface CRHR1 (green) was found on apparent dendritic spines, away from the shaft, and likely within the synaptic components on spine heads. These data are consistent with a role for CRHR1 activation in CRH-induced spine loss.
Figure 2.
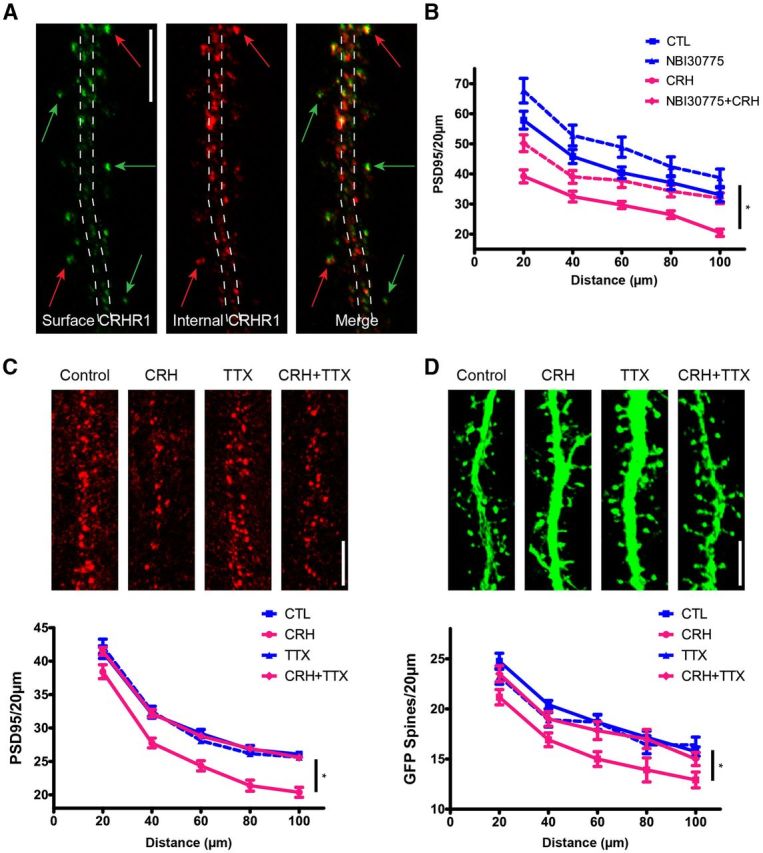
CRH-induced spine loss involves CRHR1. Cultured hippocampal neurons express CRHR1. Nonpermeable and permeabilized ICC conditions discriminate surface versus internal pools of CRHR1. A, Surface CRHR1 (green) was immunolabeled under nonpermeable conditions. After permeabilization, the neuron was processed for ICC for internal CRHR1 (red). The dashed lines approximate the contour of the dendrite. Green arrows point to surface CRHR1, the majority of which are located away from the dendrite and likely on dendritic spines. Red arrows point to external CRHR1 away from the dendritic shaft that colocalize with internal pools of CRHR1. These colocalized puncta are likely spines carrying the external receptor within the postsynaptic density as well as internal pools of CRHR1. B, Graph depicting that the CRHR1-selective antagonist (NBI30775; 100 nm) prevents CRH-induced loss of PSD95 puncta (p > 0.05; n = 12). C, D, CRH-induced spine loss requires neuronal activity. In the presence of TTX, CRH no longer reduces PSD95 puncta (F(3,368) = 20.31, p < 0.001; n = 12; C) or GFP-filled spines (F(3,176) = 6.29, p = 0.001; n = 12; D). Scale bars: A, C, D, 5 μm.
To test directly whether activation of CRHR1 was required for CRH-induced spine loss, neurons were treated with CRH in the presence of the CRHR1-specific blocker, NBI30775. We chose short incubation periods and modest blocker doses, because cultured hippocampal neurons contain populations of interneurons that release CRH, and long incubation of organotypic slice cultures in the presence of CRHR1 blockers increased the numbers of dendritic spines (and eventually dendritic branching), likely by interfering with the endogenous peptide (Chen et al., 2004). Previous studies identified 60 nm CRH as the lowest effective dose to cause a significant loss of PSD95-ir puncta (Chen et al., 2013), and we used this dose here for 30 min in the presence or absence of 100 nm NBI30775 (Fig. 2B). Under these conditions, dendritic spine density in the presence of NBI30775 did not differ significantly from that in controls (p > 0.05). CRH significantly altered dendritic spine density compared with the untreated control groups (F(3,128) = 17.90, p < 0.001), and post hoc comparisons revealed that CRH reduced PSD95-ir elements compared with controls at all distance points along the dendrites (p < 0.05). The CRHR1 blocker restored numbers of PSD95-ir puncta to control values (all distance points p > 0.05). These data indicate that activation of the GPCR, CRHR1, contributes to the rapid CRH-induced destabilization and loss of dendritic spines.
CRH-induced loss of dendritic spines requires neuronal activity
How might CRH-CRHR1 signaling lead to loss of dendritic spines? Considering the rapid action of this neuropeptide and the location of the CRHR1 within the PSD, we reasoned that canonical mechanisms that influence spine integrity and size (Tada and Sheng, 2006; Chen et al., 2007; Anggono and Huganir, 2012) might be exploited by CRH. Because afferent stimulation of neurons is a robust signal that leads to rapid functional and structural changes in synapses and spines, we used TTX to examine if such stimulation was required for CRH-induced spines and synapse loss. In the presence of TTX, CRH no longer reduced the density of PSD95-ir puncta along neuronal dendrites (F(3,368) = 20.31, p < 0.001; Fig. 2C). TTX also protected GFP-filled dendritic spines from CRH-induced spine loss (F(3,176) = 6.29, p = 0.001; Fig. 2D). These findings indicate that neurotransmission is required for CRH-induced spine loss in vitro, and suggest that during stress, concurrent neurotransmitter and CRH release functions to rapidly destroy excitatory synapses.
Potential role of ionotropic glutamate receptors in CRH-induced loss of dendritic spines
In view of the requirement for axon-potential firing for CRH-induced loss of dendritic spines, we next studied if activation of glutamate receptors, molecules that contribute crucially to activity-dependent dendritic spine dynamics, was required for the underlying mechanisms. We first examined the colocalization of CRHR1 with AMPA- and NMDA-type glutamate receptors. Using triple-labeled ICC, a subset of GFP-expressing dendritic spines coexpressed the GluR1 subunit of AMPA receptors and CRHR1 (Fig. 3A). Similarly, CRHR1 and the NR2A subunit of the NMDA-type glutamate receptor co-resided on dendritic spines (Fig. 3B). Quantification of the triple-labeled ICCs revealed that 46.7% of spines were positive for GluR1, 59% of spines were positive for NR2A, and 39.5% of spines contained CRHR1 receptors. Of dual-labeled spines sampled (n = 446), 31.9% contained both GluR1 and CRHR1. When dual-labeled CRHR1/NR2A spines were evaluated (n = 369), 35% contained both NR2A and CRHR1. More spines expressed NR2A compared with GluR1, consistent with the typical location of the latter selectively on large, mushroom-type spine heads. This observation is consistent with several electron microscopic studies showing that not all synapses have GluR1/AMPA receptors (Takumi et al., 1999; Nusser, 2000; Petralia et al., 2005) and GluR1/AMPA receptor content increases as the synapse or spine increases in size. We visualized the mature neuronal NR2A subunit (Williams et al., 1993; Tovar and Westbrook, 1999) rather than NR1, the obligate subunit of the receptor, because of technical difficulties with several NR1 antisera, and in view of literature supporting that NR2A and NR1 have similar ICC patterns (Petralia et al., 1994). Together, these data suggest that the CRH receptor and ionotropic glutamate receptors reside in close physical proximity, providing support for the notion that they might interact to mediate CRH-induced spines loss.
Figure 3.
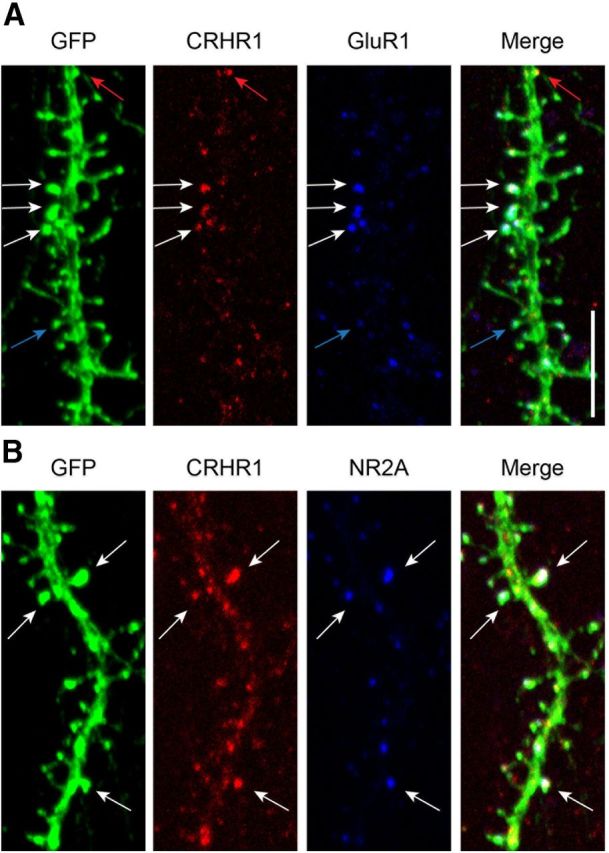
CRHR1 colocalizes with ionotropic glutamate receptors on dendritic spines. A, GFP-expressing neurons were immunostained for CRHR1 (red) and the GluR1 subunit of AMPA receptors (blue). White arrows point to spines that contain both CRHR1 and GluR1, red arrows point to spines that have CRHR1 without GluR1, and blue arrows point to spines that have GluR1 but lack CRHR1. B, GFP-expressing neurons immunostained for CRHR1 (red) and the NR2A subunit of NMDA receptors (blue). White arrows point to spines that contain both CRHR1 and NR2A. Scale bar, 5 μm.
Exposure to CRH selectively eliminates GluR1-lacking dendritic spines
To probe the interaction between CRH and ionotropic glutamate receptors, we tested if dendritic spines impacted by CRH-CRHR1 signaling contained specific types of ionotropic glutamate receptors. ICC analyses of surface AMPA receptor subunit GluR1 (sGluR1) together with PSD95 (Fig. 4A), indicated that the total number of PSD95-ir puncta was significantly reduced after exposure to CRH (F(1,88) = 20.11, p = 0.0002; Fig. 4A,B, solid lines). Remarkably, the number of PSD95-ir puncta that also expressed sGluR1 (PSD95-sGluR1; Fig. 4B, dotted lines) was not significantly affected by CRH (F(1,88) = 1.43, p = 0.244). This observation can be also expressed as a ratio of double-labeled, GluR1-positive PSD95-ir puncta (sGluR1-PSD95) over the total number of PSD95-ir elements (Fig. 4C). On dendritic segments 40–100 μm from the soma, ∼40% of the PSD95 colocalized with sGluR1. In CRH-exposed neurons, the number of PSD95-ir puncta decreased, and 50–60% of the remaining spines expressed sGluR1 (F(1,88) = 35.27, p < 0.001) as compared with controls. Together, these data indicate that GluR1-lacking synapses and spines are more vulnerable to CRH compared with those containing GluR1.
Figure 4.

CRH selectively eliminates GluR1-lacking dendritic spines. A, Neurons were exposed to CRH or a control medium for 30 min. ICC for surface GluR1 was performed under nonpermeabilized conditions (see Materials and Methods). In the control condition, ∼40% of PSD95-ir puncta (red) colocalized with sGluR1 (green). CRH reduced the number of PSD95-ir puncta, and of those remaining, the majority colocalized with surface GluR1. These findings indicate that GluR1-negative spines are more vulnerable to CRH. B, Graph showing the effects of CRH on sGluR1-positive PSD95-ir puncta (dotted lines; F(1,88) = 1.43, p = 0.244; n = 12) compared with total, PSD95-ir puncta (solid lines). These differed among groups (F(1,88) = 20.11, p = 0.0002; n = 12): CRH led to a marked reduction in total PSD95-ir puncta, but did not influence significantly the density of dual-labeled puncta. C, Upon exposure to CRH, the ratio of sGluR1-positive PSD95-ir puncta over the total PSD95-ir puncta was increased throughout the dendrite (F(1,88) = 35.27, p < 0.0001; n = 12), supporting the preferential loss of sGluR1-lacking dendritic spines. Scale bars: A, 5 μm.
The emergence of subpopulation-specific spine vulnerability to CRH is intriguing: large, mushroom-type dendritic spines typically harbor AMPA receptors, in particular sGluR1-containing AMPA receptors. In contrast, thin spines contain very few AMPA receptors, and little to no sGluR1. Our previous work suggested that thin spines are preferentially lost in intact hippocampus exposed to CRH, in accord with the current data. These findings are important, because thin spines are thought to carry the population of potentiation-ready excitatory synapses (Bourne and Harris, 2007). Hence, preferential loss of sGluR1-lacking spines may result in a paucity of synapses amenable to potentiation, with effects on learning and memory that are disproportionate to the total number of spines lost.
Ionotropic glutamate receptor activation is required for CRH-induced spine loss
The differential effects of CRH on dendritic spines based on their glutamate receptor complement suggested that activation of ionotropic glutamate receptors might contribute to CRH-induced spine destabilization. To test this idea, neurons were exposed to CRH in the presence or absence of selective blockers of the NMDA- and AMPA-type glutamate receptors: APV and CNQX, respectively (Fig. 5A,B), and analyzed using the two independent measures of GFP-visible spines and PSD95-ir puncta. CRH reduced the number of PSD95 puncta as compared with controls (treatment, F(2,180) = 64.22, p < 0.0001), and the combined use of glutamate receptor antagonists restored numbers of PSD95-ir elements to control levels (at all distance points p < 0.001). Densities of GFP-labeled dendritic spines were not different between the control and APV + CNQX treatment groups (F(1,96) = 0.12, p = 0.728). Similar to the PSD95 analyses, there was a significant effect of exposure to CRH (F(2,196) = 25.46, p < 0.0001) and post hoc analyses showed that the combined treatment with ionotropic glutamate receptor antagonists and CRH was significantly different from CRH exposure alone (all distance points p < 0.01).
Figure 5.
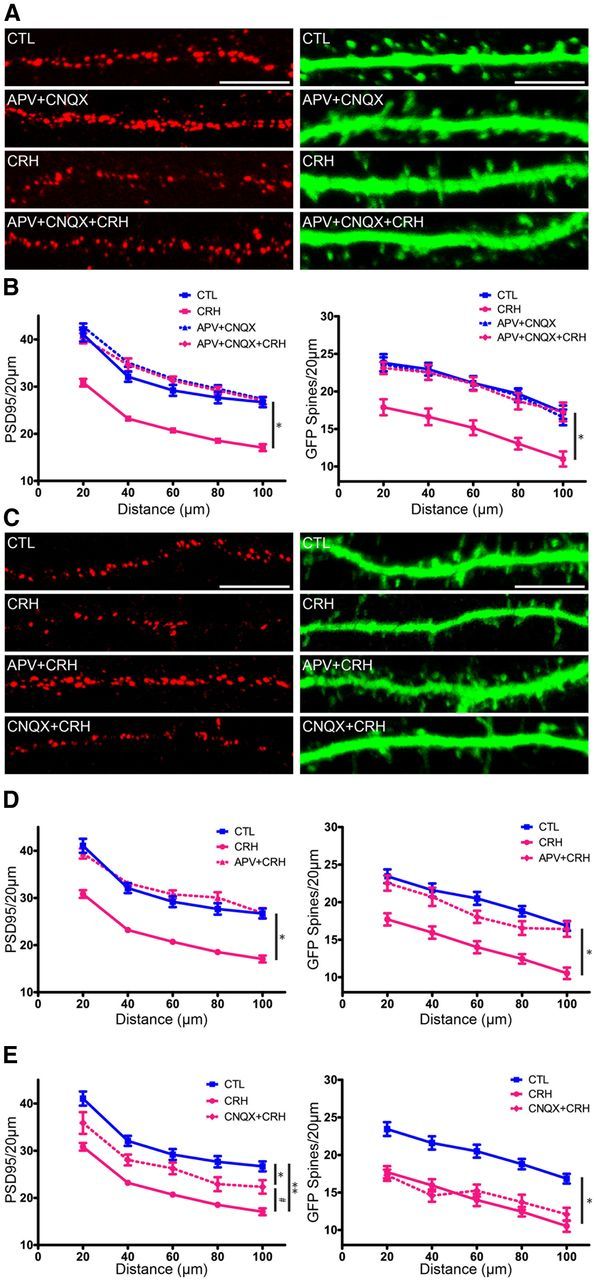
CRH-induced spine loss requires NMDA receptor activation, but not AMPA receptor activation. To distinguish the roles of NMDA- and AMPA-type receptors, neurons were exposed to CRH in the presence of selective blockers, APV and CNQX, respectively. A, Example of dendrites exposed to CRH in the presence of APV + CNQX. The combined antagonists prevented CRH-induced spine loss. B, Graph quantifying CRH spine loss in the presence of both glutamate receptor antagonists (F(2,180) = 64.22, p < 0.0001; n = 12). C, Exposing neurons to CRH in the presence of APV or of CNQX revealed that NMDA receptor activation was required for CRH-induced spine loss. D, PSD95 quantification demonstrates that the AMPA receptor blocker CNQX (purple) partially ameliorated the effects of CRH on PSD95-ir puncta (F(2,180) = 38.79, p < 0.0001; post hoc CRH vs CNQX + CRH p > 0.05 at all distance points; n = 12), but did not protect from the effects of CRH on GFP-filled spines (F(2,240) = 43.05, p < 0.0001 n = 12). E, In contrast, NMDA receptor blockade abolished CRH-induced reduction in PSD95 puncta (F(2,180) = 62.78, p < 0.0001; n = 12) and GFP-filled spines (F(2,240) = 34.65, p < 0.0001; n = 12). Scale bars: A, C, 5 μm.
CRH-induced spine loss requires activation of NMDA receptors
To distinguish whether the mechanism of CRH-induced spine loss required AMPA or NMDA receptor activation (or both); we blocked the receptors individually and tested the effects of exposure to nanomolar levels of CRH (Fig. 5C–E). In the studies quantifying PSD95 puncta, neurons incubated with APV + CRH were indistinguishable from controls (Fig. 5E; treatment, F(2,180) = 62.78, p < 0.0001; interaction, F(8,180) = 2.36, p = 0.019), and differed significantly from those treated with CRH alone (at all distance points p < 0.001). These results were confirmed using GFP-expressing neurons: neurons exposed to APV + CRH had similar numbers of spines as did controls (at all distance points, p > 0.05). In addition, spine counts from the APV + CRH treatment group were significantly different from those of neurons treated with CRH alone (F(2,240) = 34.65, p < 0.0001; at all distance points p < 0.01). These results suggest that the CRH-mediated spine loss involves NMDA receptor activation.
Indeed, studies blocking AMPA receptors selectively with CNQX and assessing PSD95 puncta density confirmed the predominant role of NMDA receptors. There was a significant effect of exposure to CRH (Fig. 5D; F(2,180) = 38.79, p < 0.0001; post hoc analysis: reduction of PSD95-ir puncta at all distance points p < 0.001), and no protective effect of concurrent treatment with CNQX (significant difference of PSD95-ir puncta in CRH-CNQX vs controls at all distance points). Effects of CNQX + CRH exposure were significantly different from those of CRH treatment alone (p > 0.05 at all distance points), suggesting a modest effect of blocking AMPA receptors. The use of GFP-expressing dendritic spines yielded largely analogous results: CRH reduced the number of dendritic spines (F(2,240) = 43.05, p < 0.0001) and CNQX did not block this spine loss. Here, spine counts did not differ between neurons exposed to CNQX + CRH and cells treated with CRH alone. Together, these data indicate that NMDA rather than AMPA receptor activation is largely responsible for CRH-provoked dendritic spine loss. The partial rescue of PSD95-ir puncta compared with the lack of effect of CNQX on dendritic spines assessed structurally was intriguing, and might be attributed to the previously observed incomplete correlation of PSD95-ir puncta and structural spines (Woods et al., 2011). These complex relationships support the need for several independent methods for assessing dendritic spine density.
A potential explanation for the failure of CNQX to protect from the effects of CRH might derive from pharmacological properties unique to this compound (Menuz et al., 2007; Milstein and Nicoll, 2008). To test this possibility, we used structurally distinct blockers for both AMPA and NMDA receptors. Neurons expressing GFP were exposed to 100 nm CRH in the presence or absence of the noncompetitive NMDA antagonist MK-801. As found for APV, CRH reduced dendritic spine density compared with controls (all distance points p < 0.01), and MK-801 restored spine numbers to control levels at all distance points (treatment, F(2,84) = 23.52, p < 0.0001; interaction, F(8,84) = 1.51, p = 0.166). When a second competitive antagonist of the AMPA-type glutamate receptor, NBQX, was used in the same manner, CRH still reduced dendritic spine density and NBQX failed to protect from this effect (treatment, F(2,84) = 27.27, p < 0.001; interaction, F(8,84) = 1.13, p = 0.349). Together, these data suggest that CRH-mediated spine loss requires the activation of NMDA receptor-mediated signaling pathways.
The NMDA receptor-activated, calcium-dependent enzyme, calpain, contributes to CRH-induced spine loss
What mechanisms downstream from NMDA receptor activation mediate CRH-induced spine loss? Spine disintegration involves the breakdown of the spine's actin cytoskeleton (Halpain, 2000; Hering and Sheng, 2001; Yuste and Bonhoeffer, 2001; Chen et al., 2007; Holtmaat and Svoboda, 2009; Kramár et al., 2009; Kasai et al., 2010; Penzes et al., 2011; Chen et al., 2013). NMDA receptor stimulation and the subsequent influx of calcium activate the enzyme calpain (Vanderklish et al., 2000), which is expressed within dendritic spines (Perlmutter et al., 1990). Calpain substrates include spectrin (also known as fodrin in the brain), and homologous proteins (e.g., actinin), which cross-link and stabilize actin filaments. Spectrin cleavage disrupts the spine cytoskeleton as well as the organization of the postsynaptic density (Dosemeci and Reese, 1995). Previous work has shown that calpain activation in hippocampal neurons depends on NMDA receptor activation (Adamec et al., 1998) and that antagonists to NMDA receptors prevent the activation of calpain (del Cerro et al., 1994).
Cultured rat hippocampal neurons expressed calpain throughout the soma and dendrites (Fig. 6A). To test the role of calpain in CRH-induced loss of dendritic spines, we initially tested if CRH-CRHR1 signaling activated this enzyme. To distinguish CRH-induced SBDP from constitutively active calpain, neurons were incubated with 500 nm calpain inhibitor III for 3 h before the onset of the experiment. Following an exposure to CRH (100 nm for 1 h), Western blot analysis revealed the presence of calpain-cleaved SBDP (Fig. 6B). Whereas full-length α-spectrin (MW ∼240 kDa) and the principal breakdown product (MW∼140 kDa) were detected in both control and CRH-exposed cultures, the ratio of the breakdown product to intact spectrin was significantly increased in cultures exposed to CRH. This breakdown product was largely eliminated by treating the cultures with 100 nm calpain inhibitor III (Fig. 6B,C), and this was apparent both as the ratio of SBDP to full-length α-spectrin (F(4,13) = 5.592, p = 0.008) and as the ratio of the SBDP to its actin loading control (F(4,13) = 9.356, p = 0.001). CRH significantly increased the ratio of SBDP to intact spectrin (post hoc Bonferroni's multiple-comparison test, p < 0.05). These data indicate that CRH increases the activation of endogenous hippocampal calpain. CRH-induced calpain activation required NMDA receptor function, because it was abrogated by the NMDA receptor blocker APV (Fig. 6D,E): CRH increased the ratio of SBDP to full-length α-spectrin (F(3,12) = 7.758, p = 0.004), and increased the ratio of SBDP to its actin loading control (F(3,12) = 15.28, p = 0.001). This effect was prevented by adding APV to the medium, whereas APV alone had little effect.
Figure 6.
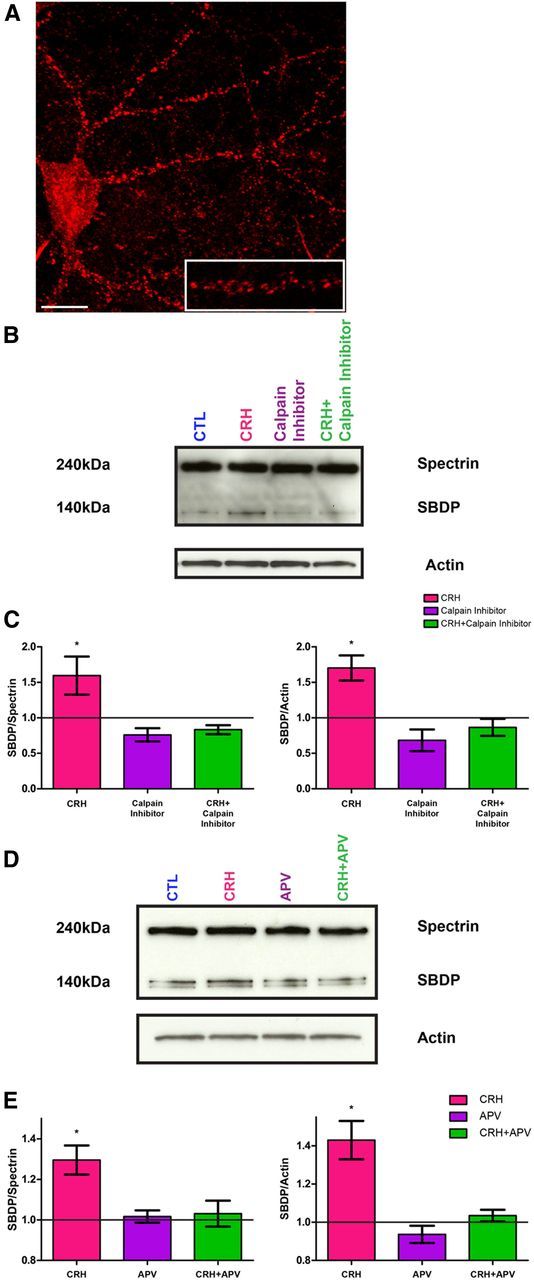
Exposure to CRH increases NMDA receptor-dependent calpain activity. A, Cultured hippocampal neurons express calpain throughout the soma and dendrites. The punctate appearance of calpain-1 is consistent with its presence in dendritic spines. B, To distinguish CRH-induced calpain activation from constitutively active calpain, all of the groups were exposed to 500 nm calpain inhibitor III for 3 h before the onset of the experiment. The amount of SBDP (140 kDa) increased with exposure to CRH (pink column). The increase in the SBDP was abolished by calpain inhibitor III (purple and green columns). C, Graph showing optical density analysis of the ratio of SBDP/full-length spectrin for each treatment group (F(4,13) = 5.592, p = 0.001) and of the ratio of the SBDP/actin loading control (F(4,13) = 9.356, p = 0.001); results were from two to four experiments. D, Representative gel, showing that the NMDA receptor blocker, APV, prevented CRH-induced increase in calpain activation. E, Quantitative graph derived from two experiments. Optical densities of SBDP/full-length spectrin and of SBDP/actin loading control were increased by CRH, and this effect was blocked by APV. Scale bar, A, 7 μm.
We then examined if calpain activation was required for CRH-induced dendritic spine loss by exposing neurons to CRH in the absence or presence of 100 nm calpain inhibitor. Quantification of PSD95-ir puncta showed that CRH significantly reduced their numbers (Fig. 7A–C; F(5,264) = 37.21, p < 0.0001; post hoc analysis, CRH vs control, p < 0.001 at all distances). Inhibition of calpain fully prevented this CRH-induced loss (post hoc analysis at all distance points, p > 0.05). Visualization of GFP-expressing spines confirmed these results: CRH reduced spine density, and blocking calpain activity during CRH exposure prevented CRH-induced reduction of spine density (F(5,264) = 65.56, p < 0.0001; post hoc analysis at all distance points p > 0.05). Together, these results indicate that the mechanism through which CRH disrupts dendritic spines involves NMDA-mediated activation of calpain, resulting in cleavage of actin-associated proteins and destabilization of the spine's actin cytoskeleton.
Figure 7.
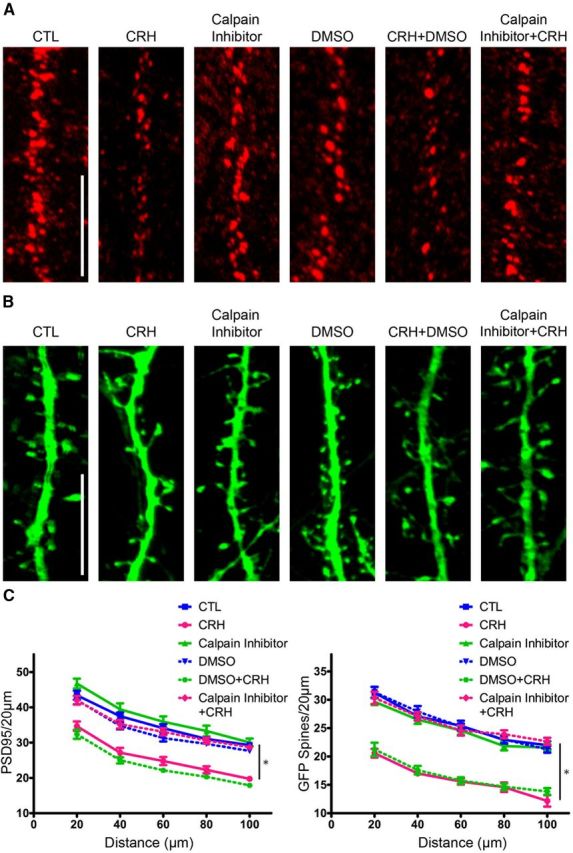
The calpain inhibitor prevents dendritic spine loss induced by CRH. A, The 100 nm calpain inhibitor prevented CRH-induced reduction of PSD95-ir puncta. B, The calpain inhibitor abolished the loss of GFP-filled spines. C, Graph showing quantification of PSD95-ir puncta (F(5,264) = 37.21, p < 0.0001; n = 12) and GFP-filled spines (F(5,264) = 65.56, p < 0.0001; n = 12). Scale bars: A, B, 5 μm.
Discussion
This series of experiments demonstrates how fundamental mechanisms of dendritic spine dynamics are co-opted by a stress hormone to enable rapid loss of hippocampal dendritic spines. Because this CRH-mediated loss of dendritic spines results in loss of excitatory synapses and contributes to stress-provoked memory defects, it might represent an adaptive mechanism to reduce pathological memories associated with short, yet severe, stress.
In essence we found that 60–100 nm CRH, concentrations expected during severe stress (Khan et al., 2004; Chen et al., 2012), reduced dendritic spine density. The levels of CRH bathing hippocampal synapses during stress are difficult to estimate. Microdialysis studies in the amygdala and hypothalamus have reported that stress-induced increases in CRH occur within 20 min, and are in the 100–200 nm range (Richter et al., 1995; Cook, 2004; Merali et al., 2004; Maras and Baram, 2012). Microdialysis studies in the hippocampus are not available, so basal and stress levels of CRH in this area of the brain remain speculative. Indirect observations suggest that severe stress and increased network activity (e.g., seizures) may lead to peptide levels as high as 200 nm (Khan et al., 2004; Tringali et al., 2009). The loss of spines by the presumed-stress levels of CRH required the binding of the peptide to the CRHR1 receptor located at the surface of dendritic spine heads in the presence of neuronal activity, followed by the selective activation of NMDA-type glutamate receptors. Downstream of NMDA receptors, the calcium-dependent enzyme calpain contributed crucially to degradation of the spine actin-skeleton, resulting in destabilization and loss of GluR1-lacking (typically thin) dendritic spines.
Dendritic spines, memory processes, and stress
Dendritic spines are specialized structures that are crucial for synaptic function and plasticity (Augustine et al., 2003; Yuste and Bonhoeffer, 2004). Changes in the number and shape of spines may be critical components of mechanisms of synaptic plasticity (Zhou et al., 2004; Segal, 2005; Chen et al., 2007), and are regulated by factors including neurotransmitters, growth factors, and hormones that, in turn, are governed by environmental signals including stress (Calabrese and Halpain, 2005; Segal, 2005). Both the formation (Engert and Bonhoeffer, 1999; Maletic-Savatic et al., 1999) and retraction (Fu et al., 2007; Biou et al., 2008) of spines (“spine dynamics”) are regulated, and are considered crucial elements of synaptic plasticity in both developing and mature neurons (Nägerl et al., 2004; Zhou et al., 2004). Thus, a derangement of spine dynamics that favors loss of spines is a plausible candidate mechanism for stress-induced synaptic dysfunction and memory problems. Whereas earlier work regarding the effects of stress on dendritic spines and dendrites themselves focused on chronic stress, i.e., the time frame of days and weeks (Luine et al., 1994; Margariños and McEwen, 1995; Conrad et al., 1996; Krugers et al., 1997; Pavlides et al., 2002; Wilson et al., 2007), more recently, hours-long stress has been found to influence dendritic spine integrity in adult hippocampus (de Quervain et al., 1998; Conrad et al., 1999; Pawlak et al., 2003; Diamond et al., 2006; Chen et al., 2010, 2013). Together, this body of work suggests that while acute stress (lasting seconds) enhances memory, hours-long stress (similar to chronic stress) might impair memory processes, at least in part via loss of dendritic spines and their excitatory synapses (Conrad et al., 1999; de Kloet et al., 2005; Joëls and Baram, 2009). Whereas a key role for glucocorticoids and their receptors has been established (Liston and Gan, 2011), the mechanisms for the relatively rapid effects of hours-long stress on hippocampus-dependent memory processes are not fully understood.
Glutamate receptors and the regulation of dendritic spine dynamics
Mechanisms of dendritic spine dynamics in the time frame of minutes to hours are often initiated by afferent stimulation of the neuron and involve glutamate receptors. The major forces that influence dendritic spine growth, shrinkage, and collapse take place through glutamate receptor activation. A large body of work has focused on the mechanisms involved (Halpain, 2000; Tada and Sheng, 2006; O'Donnell et al., 2011; Penzes et al., 2011). Afferent input to the postsynaptic density and the activation of glutamate receptors leads to a number of molecular and structural cascades. The recruitment of AMPA receptors containing GluR1 subunits into the postsynaptic density within spines is associated with spine enlargement and LTP (Shi et al., 1999; Boehm et al., 2006; Ehlers et al., 2007; Fedulov et al., 2007; Kopec et al., 2007), whereas internalization and trafficking of the receptors away from the postsynaptic density is a fundamental mechanism for the rapid spine remodeling during LTD (Barria and Malinow, 2002; Bredt and Nicoll, 2003; Brown et al., 2005; Biou et al., 2008). In view of the established and potent role of ionotropic glutamate receptors in dendritic spine dynamics, we queried if the rapid actions of CRH, leading to spine loss within minutes, involved co-option of these mechanisms of synaptic plasticity.
How might CRH receptor occupancy interact with glutamate receptor function?
The CRHR1 receptor is a member of the B subfamily of GPCRs (Perrin and Vale, 1999; Holmes et al., 2006). However, unlike the majority of GPCRs, which are extrasynaptic, surface CRHR1 localizes to the postsynaptic density, in close proximity to glutamate receptors (Chen et al., 2004; Fig. 3). The current studies demonstrate that NMDA receptor function was required for CRH-mediated spine loss. CRH-CRHR1 signaling might influence NMDA receptor function at several potential molecular steps. CRHR1 occupancy might influence the activation of NMDA via phosphorylation (as found, for example, for interleukin-1β). Alternatively, CRHR1 internalization (Holmes et al. (2006); Reyes et al., 2006, 2008) might influence the anchoring or trafficking of closely located NMDA receptors within the postsynaptic density. Here we found that CRH enhanced the NMDA-dependent (Adamec et al., 1998) activity of calpain, which was required for spine loss. However, it is feasible that other mechanisms exist, including, for example, augmentation or inhibition of NMDA receptor-mediated changes of actin-regulating Rho GTPases (Swinny and Valentino, 2006; Chen et al., 2013).
Indeed, dendritic spine integrity depends on the presence of a stable, F-actin “skeleton” (Dent et al., 2011; Penzes et al., 2011; Penzes and Rafalovich, 2012; Chen et al., 2013). Actin polymerization and disintegration (“dynamics”) are tightly regulated by a large number of proteins including scaffolding proteins and enzymes (Hotulainen and Hoogenraad, 2010; Moutin et al., 2012). Activation of specific glutamate receptors phosphorylates (activates) or dephosphorylates (deactivates) families of GTPases that influence actin polymerization directly or through additional molecular interactions (Rao and Craig, 2000; Hering and Sheng, 2001; Penzes et al., 2003; Ethell and Pasquale, 2005). The growing repertoire of actin-interacting proteins that govern the formation and stability of the polymerized F-actin include Rac1, RhoA, Cdc42, GEF-H1 (Lfc), Kalirin-7, and others (Nakayama et al., 2000; Xie et al., 2007; Kang et al., 2009). We have previously found that RhoA was involved in CRH-induced spine loss in adult hippocampus. Notably, RhoA activation is also required for increases in spine F-actin associated with the induction of LTP (Rex et al., 2009). However, the degree, time frame, and balance of activated actin-regulating factors are important to their effect on spine integrity: modest activation of RhoA enabled F-actin branching and growth, whereas runaway CRH-induced activation provoked F-actin degradation. As discussed below, a similar principle (U-shaped curve) might obtain activation of calpain (Lynch and Seubert, 1989).
NMDA receptor-mediated calpain activation is necessary for CRH-induced spine loss
NMDA receptors play complex roles in spine dynamics and integrity during learning and memory processes (Shi et al., 1999; Barria and Malinow, 2002, Brown et al., 2005; Gao et al., 2011). NMDA receptor activation as a result of network activity, and the resulting calcium influx, recruit the enzyme calpain in dendritic spines (Perlmutter et al., 1990; Vanderklish et al., 2000). NMDA-mediated activation of calpain is necessary for LTP (Vanderklish et al., 1996), and calpain-dependent reorganization of actin is required for spine (and likely synapse) expansion. Presumably, this involves moderate, regulated action of the enzyme, because significant breakdown of calpain substrates including spectrin disrupts spine actin-backbone (Dosemeci and Reese, 1995; Lu et al., 2000). Here, CRH-induced NMDA receptor-dependent calpain activation contributed to dendritic spine loss. This might result from a quantitatively higher (“runaway”) enzyme activity, from activation of calpain in atypical dendritic spine compartments, or in the context of distinct calcium signals produced by different types of stimulation and leading to distinct biochemical cascades.
In summary, the current studies probed the mechanisms by which CRH contributes to loss of hippocampal dendritic spines, an important basis of memory problems that arise after short severe stress (Fig. 8). We found that CRH-induced spine loss required action potentials, and the activation of NMDA receptors but not of AMPA receptors. CRH receptor occupancy together with the activation of NMDA receptors recruited the calcium-dependent enzyme, calpain, and the breakdown of the spine-actin interacting protein, spectrin. Pharmacological approaches demonstrated that calpain recruitment contributed critically to CRH-induced spine loss. Thus, the stress hormone CRH co-opts mechanisms that contribute to the plasticity and integrity of excitatory synapses, leading to selective loss of dendritic spines. Whereas other mechanisms for CRH effects on dendritic spines exist (Wang et al., 2013), and the effects of stress or CRH on inhibitory synapses have not yet been studied, this spine loss described here might function as an adaptive mechanism preventing the consequences of adverse memories associated with severe stress.
Figure 8.
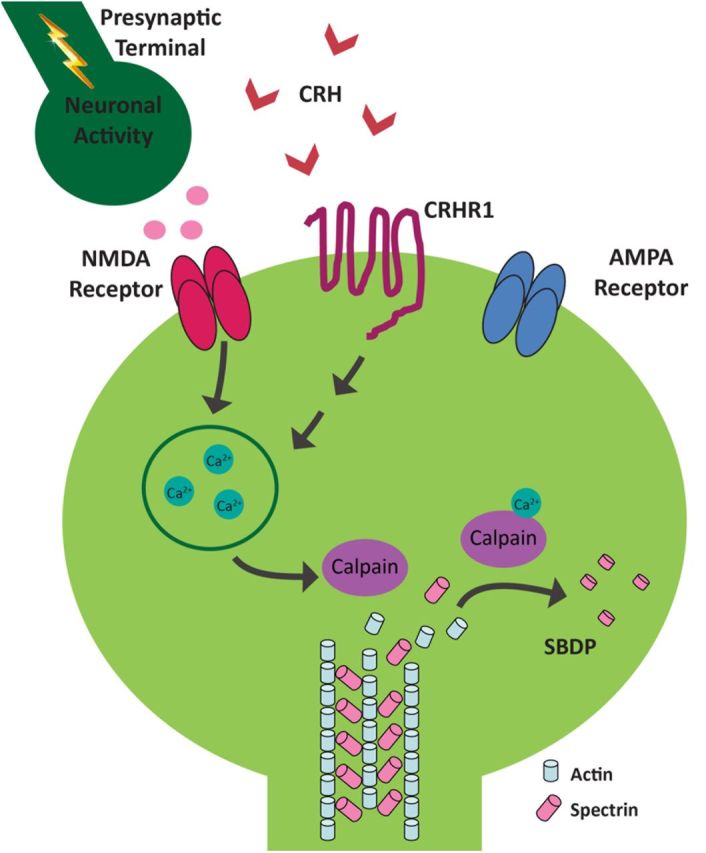
Schematic of the proposed molecular signaling involved in CRH-induced dendritic spine loss. The CRH receptor, CRHR1, is located on dendritic spine heads, within the postsynaptic density and in close proximity to NMDA- and AMPA-type ionotropic glutamate receptors. When CRH (released during stress from hippocampal interneurons), binds CRHR1 in the presence of network activity, this triggers an NMDA receptor-dependent signaling cascade that culminates in spine loss. Specifically, the influx of calcium ions through NMDA receptors activates calpain. Calpain cleaves actin-associated scaffolding proteins, such as spectrin, leading to the breakdown of the spine cytoskeleton and spine loss. The presence of GluR1 may protect subsets of mature spines from the actions of CRH.
Footnotes
This research was supported by National Institutes of Health grants NS28912, NS45260, and MH73136. The authors thank Drs. Pamela M. Maras and Alex H. Babayan for helpful discussions and support.
The authors declare no competing financial interests.
References
- Adamec E, Beermann ML, Nixon RA. Calpain I activation in rat hippocampal neurons in culture is NMDA receptor selective and not essential for excitotoxic cell death. Brain Res Mol Brain Res. 1998;54:35–48. doi: 10.1016/S0169-328X(97)00304-5. [DOI] [PubMed] [Google Scholar]
- Alberini CM, Chen DY. Memory enhancement: consolidation, reconsolidation and insulin-like growth factor 2. Trends Neurosci. 2012;35:274–283. doi: 10.1016/j.tins.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anggono V, Huganir RL. Regulation of AMPA receptor trafficking and synaptic plasticity. Curr Opin Neurobiol. 2012;22:461–469. doi: 10.1016/j.conb.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold DB, Clapham DE. Molecular determinants for subcellular localization of PSD-95 with an interacting K+ channel. Neuron. 1999;23:149–157. doi: 10.1016/S0896-6273(00)80761-8. [DOI] [PubMed] [Google Scholar]
- Augustine GJ, Santamaria F, Tanaka K. Local calcium signaling in neurons. Neuron. 2003;40:331–346. doi: 10.1016/S0896-6273(03)00639-1. [DOI] [PubMed] [Google Scholar]
- Barria A, Malinow R. Subunit-specific NMDA receptor trafficking to synapses. Neuron. 2002;35:345–353. doi: 10.1016/S0896-6273(02)00776-6. [DOI] [PubMed] [Google Scholar]
- Baudry M, Lynch G. Regulation of glutamate receptors by cations. Nature. 1979;282:748–750. doi: 10.1038/282748a0. [DOI] [PubMed] [Google Scholar]
- Bear MF, Cooper LN, Ebner FF. A physiological basis for a theory of synapse modification. Science. 1987;237:42–48. doi: 10.1126/science.3037696. [DOI] [PubMed] [Google Scholar]
- Biou V, Bhattacharyya S, Malenka RC. Endocytosis and recycling of AMPA receptors lacking GluR2/3. Proc Natl Acad Sci U S A. 2008;105:1038–1043. doi: 10.1073/pnas.0711412105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blank T, Nijholt I, Eckart K, Spiess J. Priming of long-term potentiation in mouse hippocampus by corticotropin-releasing factor and acute stress: implications for hippocampus-dependent learning. J Neurosci. 2002;22:3788–3794. doi: 10.1523/JNEUROSCI.22-09-03788.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm J, Kang MG, Johnson RC, Esteban J, Huganir RL, Malinow R. Synaptic incorporation of AMPA receptors during LTP is controlled by a PKC phosphorylation site on GluR1. Neuron. 2006;51:213–225. doi: 10.1016/j.neuron.2006.06.013. [DOI] [PubMed] [Google Scholar]
- Bourne J, Harris KM. Do thin spines learn to be mushroom spines that remember? Curr Opin Neurobiol. 2007;17:381–386. doi: 10.1016/j.conb.2007.04.009. [DOI] [PubMed] [Google Scholar]
- Bourne JN, Harris KM. Balancing structure and function at hippocampal dendritic spines. Annu Rev Neurosci. 2008;31:47–67. doi: 10.1146/annurev.neuro.31.060407.125646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredt DS, Nicoll RA. AMPA receptor trafficking at excitatory synapses. Neuron. 2003;40:361–379. doi: 10.1016/S0896-6273(03)00640-8. [DOI] [PubMed] [Google Scholar]
- Brown TC, Tran IC, Backos DS, Esteban JA. NMDA receptor-dependent activation of the small GTPase Rab5 drives the removal of synaptic AMPA receptors during hippocampal LTD. Neuron. 2005;45:81–94. doi: 10.1016/j.neuron.2004.12.023. [DOI] [PubMed] [Google Scholar]
- Calabrese B, Halpain S. Essential role for the PKC target MARCKS in maintaining dendritic spine morphology. Neuron. 2005;48:77–90. doi: 10.1016/j.neuron.2005.08.027. [DOI] [PubMed] [Google Scholar]
- Chen LY, Rex CS, Casale MS, Gall CM, Lynch G. Changes in synaptic morphology accompany actin signaling during LTP. J Neurosci. 2007;27:5363–5372. doi: 10.1523/JNEUROSCI.0164-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Brunson KL, Müller MB, Cariaga W, Baram TZ. Immunocytochemical distribution of corticotropin-releasing hormone receptor type-1 (CRF(1))-like immunoreactivity in the mouse brain: light microscopy analysis using an antibody directed against the C-terminus. J Comp Neurol. 2000;420:305–323. doi: 10.1002/(SICI)1096-9861(20000508)420:3<305::AID-CNE3>3.0.CO%3B2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Bender RA, Frotscher M, Baram TZ. Novel and transient populations of corticotropin-releasing hormone-expressing neurons in developing hippocampus suggest unique functional roles: a quantitative spatiotemporal analysis. J Neurosci. 2001;21:7171–7181. doi: 10.1523/JNEUROSCI.21-18-07171.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Brunson KL, Adelmann G, Bender RA, Frotscher M, Baram TZ. Hippocampal corticotropin releasing hormone: pre- and postsynaptic location and release by stress. Neuroscience. 2004;126:533–540. doi: 10.1016/j.neuroscience.2004.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Dubé CM, Rice CJ, Baram TZ. Rapid loss of dendritic spines after stress involves derangement of spine dynamics by corticotropin-releasing hormone. J Neurosci. 2008;28:2903–2911. doi: 10.1523/JNEUROSCI.0225-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Rex CS, Rice CJ, Dubé CM, Gall CM, Lynch G, Baram TZ. Correlated memory defects and hippocampal dendritic spine loss after acute stress involve corticotropin-releasing hormone signaling. Proc Natl Acad Sci U S A. 2010;107:13123–13128. doi: 10.1073/pnas.1003825107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Andres AL, Frotscher M, Baram TZ. Tuning synaptic transmission in the hippocampus by stress: the CRH system. Front Cell Neurosci. 2012;6:13. doi: 10.3389/fncel.2012.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Kramár EA, Chen LY, Babayan AH, Andres AL, Gall CM, Lynch G, Baram TZ. Impairment of synaptic plasticity by the stress mediator CRH involves selective destruction of thin dendritic spines via RhoA signaling. Mol Psychiatry. 2013;18:485–496. doi: 10.1038/mp.2012.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collingridge GL, Peineau S, Howland JG, Wang YT. Long-term depression in the CNS. Nat Rev Neurosci. 2010;11:459–473. doi: 10.1038/nrn2867. [DOI] [PubMed] [Google Scholar]
- Conrad CD, Galea LA, Kuroda Y, McEwen BS. Chronic stress impairs rat spatial memory on the Y maze, and this effect is blocked by tianeptine pretreatment. Behav Neurosci. 1996;110:1321–1334. doi: 10.1037/0735-7044.110.6.1321. [DOI] [PubMed] [Google Scholar]
- Conrad CD, LeDoux JE, Magariños AM, McEwen BS. Repeated restraint stress facilitates fear conditioning independently of causing hippocampal CA3 dendritic atrophy. Behav Neurosci. 1999;113:902–913. doi: 10.1037/0735-7044.113.5.902. [DOI] [PubMed] [Google Scholar]
- Cook CJ. Stress induces CRF release in the paraventricular nucleus, and both CRF and GABA release in the amygdala. Physiol Behav. 2004;82:751–762. doi: 10.1016/j.physbeh.2004.06.013. [DOI] [PubMed] [Google Scholar]
- De Kloet ER. Hormones and the stressed brain. Ann N Y Acad Sci. 2004;1018:1–15. doi: 10.1196/annals.1296.001. [DOI] [PubMed] [Google Scholar]
- de Kloet ER, Joëls M, Holsboer F. Stress and the brain: from adaptation to disease. Nat Rev Neurosci. 2005;6:463–475. doi: 10.1038/nrn1683. [DOI] [PubMed] [Google Scholar]
- del Cerro S, Arai A, Kessler M, Bahr BA, Vanderklish P, Rivera S, Lynch G. Stimulation of NMDA receptors activates calpain in cultured hippocampal slices. Neurosci Lett. 1994;167:149–152. doi: 10.1016/0304-3940(94)91049-9. [DOI] [PubMed] [Google Scholar]
- Dent EW, Merriam EB, Hu X. The dynamic cytoskeleton: backbone of dendritic spine plasticity. Curr Opin Neurobiol. 2011;21:175–181. doi: 10.1016/j.conb.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Quervain DJ, Roozendaal B, McGaugh JL. Stress and glucocorticoids impair retrieval of long-term spatial memory. Nature. 1998;394:787–790. doi: 10.1038/29542. [DOI] [PubMed] [Google Scholar]
- Derkach VA, Oh MC, Guire ES, Soderling TR. Regulatory mechanisms of AMPA receptors in synaptic plasticity. Nat Rev Neurosci. 2007;8:101–113. doi: 10.1038/nrn2055. [DOI] [PubMed] [Google Scholar]
- Diamond DM, Rose GM. Stress impairs LTP and hippocampal-dependent memory. Ann N Y Acad Sci. 1994;746:411–414. doi: 10.1111/j.1749-6632.1994.tb39271.x. [DOI] [PubMed] [Google Scholar]
- Diamond DM, Campbell AM, Park CR, Woodson JC, Conrad CD, Bachstetter AD, Mervis RF. Influence of predator stress on the consolidation versus retrieval of long-term spatial memory and hippocampal spinogenesis. Hippocampus. 2006;16:571–576. doi: 10.1002/hipo.20188. [DOI] [PubMed] [Google Scholar]
- Dosemeci A, Reese TS. Effect of calpain on the composition and structure of postsynaptic densities. Synapse. 1995;20:91–97. doi: 10.1002/syn.890200113. [DOI] [PubMed] [Google Scholar]
- Eghbal-Ahmadi M, Hatalski CG, Lovenberg TW, Avishai-Eliner S, Chalmers DT, Baram TZ. The developmental profile of the corticotropin releasing factor receptor (CRF2) in rat brain predicts distinct age-specific functions. Brain Res Dev Brain Res. 1998;107:81–90. doi: 10.1016/S0165-3806(98)00002-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers MD, Heine M, Groc L, Lee MC, Choquet D. Diffusional trapping of GluR1 AMPA receptors by input-specific synaptic activity. Neuron. 2007;54:447–460. doi: 10.1016/j.neuron.2007.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engert F, Bonhoeffer T. Dendritic spine changes associated with hippocampal long-term synaptic plasticity. Nature. 1999;399:66–70. doi: 10.1038/19978. [DOI] [PubMed] [Google Scholar]
- Ethell IM, Pasquale EB. Molecular mechanisms of dendritic spine development and remodeling. Prog Neurobiol. 2005;75:161–205. doi: 10.1016/j.pneurobio.2005.02.003. [DOI] [PubMed] [Google Scholar]
- Fedulov V, Rex CS, Simmons DA, Palmer L, Gall CM, Lynch G. Evidence that long-term potentiation occurs within individual hippocampal synapses during learning. J Neurosci. 2007;27:8031–8039. doi: 10.1523/JNEUROSCI.2003-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Z, Lee SH, Simonetta A, Hansen J, Sheng M, Pak DT. Differential roles of Rap1 and Rap2 small GTPases in neurite retraction and synapse elimination in hippocampal spiny neurons. J Neurochem. 2007;100:118–131. doi: 10.1111/j.1471-4159.2006.04195.x. [DOI] [PubMed] [Google Scholar]
- Fukazawa Y, Saitoh Y, Ozawa F, Ohta Y, Mizuno K, Inokuchi K. Hippocampal LTP is accompanied by enhanced F-actin content within the dendritic spine that is essential for late LTP maintenance in vivo. Neuron. 2003;38:447–460. doi: 10.1016/S0896-6273(03)00206-X. [DOI] [PubMed] [Google Scholar]
- Gao C, Frausto SF, Guedea AL, Tronson NC, Jovasevic V, Leaderbrand K, Corcoran KA, Guzmán YF, Swanson GT, Radulovic J. IQGAP1 regulates NR2A signaling, spine density, and cognitive processes. J Neurosci. 2011;31:8533–8542. doi: 10.1523/JNEUROSCI.1300-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia R, Musleh W, Tocco G, Thompson RF, Baudry M. Time-dependent blockade of STP and LTP in hippocampal slices following acute stress in mice. Neurosci Lett. 1997;233:41–44. doi: 10.1016/S0304-3940(97)00621-6. [DOI] [PubMed] [Google Scholar]
- Gray JD, Milner TA, McEwen BS. Dynamic plasticity: the role of glucocorticoids, brain-derived neurotrophic factor and other trophic factors. Neuroscience. 2013;239:214–227. doi: 10.1016/j.neuroscience.2012.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halpain S. Actin and the agile spine: how and why do dendritic spines dance? Trends Neurosci. 2000;23:141–146. doi: 10.1016/S0166-2236(00)01576-9. [DOI] [PubMed] [Google Scholar]
- Hering H, Sheng M. Dendritic spines: structure, dynamics and regulation. Nat Rev Neurosci. 2001;2:880–888. doi: 10.1038/35104061. [DOI] [PubMed] [Google Scholar]
- Holmes KD, Babwah AV, Dale LB, Poulter MO, Ferguson SS. Differential regulation of corticotropin releasing factor 1alpha receptor endocytosis and trafficking by beta-arrestins and Rab GTPases. J Neurochem. 2006;96:934–949. doi: 10.1111/j.1471-4159.2005.03603.x. [DOI] [PubMed] [Google Scholar]
- Holtmaat A, Svoboda K. Experience-dependent structural synaptic plasticity in the mammalian brain. Nat Rev Neurosci. 2009;10:647–658. doi: 10.1038/nrn2699. [DOI] [PubMed] [Google Scholar]
- Hotulainen P, Hoogenraad CC. Actin in dendritic spines: connecting dynamics to function. J Cell Biol. 2010;189:619–629. doi: 10.1083/jcb.201003008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivy AS, Rex CS, Chen Y, Dubé C, Maras PM, Grigoriadis DE, Gall CM, Lynch G, Baram TZ. Hippocampal dysfunction and cognitive impairments provoked by chronic early-life stress involve excessive activation of CRH receptors. J Neurosci. 2010;30:13005–13015. doi: 10.1523/JNEUROSCI.1784-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jafari M, Seese RR, Babayan AH, Gall CM, Lauterborn JC. Glucocorticoid receptors are localized to dendritic spines and influence local actin signaling. Mol Neurobiol. 2012;46:304–315. doi: 10.1007/s12035-012-8288-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joëls M, Baram TZ. The neuro-symphony of stress. Nat Rev Neurosci. 2009;10:459–466. doi: 10.1038/nrn2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang MG, Guo Y, Huganir RL. AMPA receptor and GEF-H1/Lfc complex regulates dendritic spine development through RhoA signaling cascade. Proc Natl Acad Sci U S A. 2009;106:3549–3554. doi: 10.1073/pnas.0812861106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasai H, Fukuda M, Watanabe S, Hayashi-Takagi A, Noguchi J. Structural dynamics of dendritic spines in memory and cognition. Trends Neurosci. 2010;33:121–129. doi: 10.1016/j.tins.2010.01.001. [DOI] [PubMed] [Google Scholar]
- Khan S, Milot M, Lecompte-Collin J, Plamondon H. Time-dependent changes in CRH concentrations and release in discrete brain regions following global ischemia: effects of MK-801 pretreatment. Brain Res. 2004;1016:48–57. doi: 10.1016/j.brainres.2004.04.062. [DOI] [PubMed] [Google Scholar]
- Kim JJ, Diamond DM. The stressed hippocampus, synaptic plasticity and lost memories. Nat Rev Neurosci. 2002;3:453–462. doi: 10.1038/nrm832. [DOI] [PubMed] [Google Scholar]
- Kole MH, Costoli T, Koolhaas JM, Fuchs E. Bidirectional shift in the cornu ammonis 3 pyramidal dendritic organization following brief stress. Neuroscience. 2004;125:337–347. doi: 10.1016/j.neuroscience.2004.02.014. [DOI] [PubMed] [Google Scholar]
- Kopec CD, Real E, Kessels HW, Malinow R. GluR1 links structural and functional plasticity at excitatory synapses. J Neurosci. 2007;27:13706–13718. doi: 10.1523/JNEUROSCI.3503-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramár EA, Chen LY, Rex CS, Gall CM, Lynch G. Estrogen's place in the family of synaptic modulators. Mol Cell Pharmacol. 2009;1:258–262. [PMC free article] [PubMed] [Google Scholar]
- Krugers HJ, Douma BR, Andringa G, Bohus B, Korf J, Luiten PG. Exposure to chronic psychosocial stress and corticosterone in the rat: effects on spatial discrimination learning and hippocampal protein kinase Cgamma immunoreactivity. Hippocampus. 1997;7:427–436. doi: 10.1002/(SICI)1098-1063(1997)7:4<427::AID-HIPO8>3.0.CO%3B2-F. [DOI] [PubMed] [Google Scholar]
- Larson J, Lynch G. Induction of synaptic potentiation in hippocampus by patterned stimulation involves two events. Science. 1986;232:985–988. doi: 10.1126/science.3704635. [DOI] [PubMed] [Google Scholar]
- Levitsky DI, Pivovarova AV, Mikhailova VV, Nikolaeva OP. Thermal unfolding and aggregation of actin. FEBS J. 2008;275:4280–4295. doi: 10.1111/j.1742-4658.2008.06569.x. [DOI] [PubMed] [Google Scholar]
- Lim CS, Jin DQ, Mok H, Oh SJ, Lee JU, Hwang JK, Ha I, Han JS. Antioxidant and antiinflammatory activities of xanthorrhizol in hippocampal neurons and primary cultured microglia. J Neurosci Res. 2005;82:831–838. doi: 10.1002/jnr.20692. [DOI] [PubMed] [Google Scholar]
- Lin B, Kramár EA, Bi X, Brucher FA, Gall CM, Lynch G. Theta stimulation polymerizes actin in dendritic spines of hippocampus. J Neurosci. 2005;25:2062–2069. doi: 10.1523/JNEUROSCI.4283-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liston C, Gan WB. Glucocorticoids are critical regulators of dendritic spine development and plasticity in vivo. Proc Natl Acad Sci U S A. 2011;108:16074–16079. doi: 10.1073/pnas.1110444108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Rong Y, Baudry M. Calpain-mediated degradation of PSD-95 in developing and adult rat brain. Neurosci Lett. 2000;286:149–153. doi: 10.1016/s0304-3940(00)01101-0. [DOI] [PubMed] [Google Scholar]
- Luine V, Villegas M, Martinez C, McEwen BS. Repeated stress causes reversible impairments of spatial memory performance. Brain Res. 1994;639:167–170. doi: 10.1016/0006-8993(94)91778-7. [DOI] [PubMed] [Google Scholar]
- Lynch G, Seubert P. Links between long-term potentiation and neuropathology. An hypothesis involving calcium-activated proteases. Ann N Y Acad Sci. 1989;568:171–180. doi: 10.1111/j.1749-6632.1989.tb12505.x. [DOI] [PubMed] [Google Scholar]
- Lynch G, Rex CS, Gall CM. LTP consolidation: substrates, explanatory power, and functional significance. Neuropharmacology. 2007;52:12–23. doi: 10.1016/j.neuropharm.2006.07.027. [DOI] [PubMed] [Google Scholar]
- Lynch G, Rex CS, Chen LY, Gall CM. The substrates of memory: defects, treatments, and enhancement. Eur J Pharmacol. 2008;585:2–13. doi: 10.1016/j.ejphar.2007.11.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Kauer JA, Zucker RS, Nicoll RA. Postsynaptic calcium is sufficient for potentiation of hippocampal synaptic transmission. Science. 1988;242:81–84. doi: 10.1126/science.2845577. [DOI] [PubMed] [Google Scholar]
- Maletic-Savatic M, Malinow R, Svoboda K. Rapid dendritic morphogenesis in CA1 hippocampal dendrites induced by synaptic activity. Science. 1999;283:1923–1927. doi: 10.1126/science.283.5409.1923. [DOI] [PubMed] [Google Scholar]
- Maras PM, Baram TZ. Sculpting the hippocampus from within: stress, spines, and CRH. Trends Neurosci. 2012;35:315–324. doi: 10.1016/j.tins.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margariños AM, McEwen BS. Stress-induced atrophy of apical dendrites of hippocampal neurons: comparison of stressors. Neuroscience. 1995;69:83–88. doi: 10.1016/0306-4522(95)00256-I. [DOI] [PubMed] [Google Scholar]
- Martin SJ, Grimwood PD, Morris RG. Synaptic plasticity and memory: an evaluation of the hypothesis. Annu Rev Neurosci. 2000;23:649–711. doi: 10.1146/annurev.neuro.23.1.649. [DOI] [PubMed] [Google Scholar]
- McEwen BS, Gianaros PJ. Stress- and allostasis-induced brain plasticity. Annu Rev Med. 2011;62:431–445. doi: 10.1146/annurev-med-052209-100430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menuz K, Stroud RM, Nicoll RA, Hays FA. TARP auxiliary subunits switch AMPA receptor antagonists into partial agonists. Science. 2007;318:815–817. doi: 10.1126/science.1146317. [DOI] [PubMed] [Google Scholar]
- Merali Z, Khan S, Michaud DS, Shippy SA, Anisman H. Does amygdaloid corticotropin-releasing hormone (CRH) mediate anxiety-like behaviors? Dissociation of anxiogenic effects and CRH release. Eur J Neurosci. 2004;20:229–239. doi: 10.1111/j.1460-9568.2004.03468.x. [DOI] [PubMed] [Google Scholar]
- Milstein AD, Nicoll RA. Regulation of AMPA receptor gating and pharmacology by TARP auxiliary subunits. Trends Pharmacol Sci. 2008;29:333–339. doi: 10.1016/j.tips.2008.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moutin E, Raynaud F, Roger J, Pellegrino E, Homburger V, Bertaso F, Ollendorff V, Bockaert J, Fagni L, Perroy J. Dynamic remodeling of scaffold interactions in dendritic spines controls synaptic excitability. J Cell Biol. 2012;198:251–263. doi: 10.1083/jcb.201110101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nägerl UV, Eberhorn N, Cambridge SB, Bonhoeffer T. Bidirectional activity-dependent morphological plasticity in hippocampal neurons. Neuron. 2004;44:759–767. doi: 10.1016/j.neuron.2004.11.016. [DOI] [PubMed] [Google Scholar]
- Nakayama AY, Harms MB, Luo L. Small GTPases Rac and Rho in the maintenance of dendritic spines and branches in hippocampal pyramidal neurons. J Neurosci. 2000;20:5329–5338. doi: 10.1523/JNEUROSCI.20-14-05329.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neves G, Cooke SF, Bliss TV. Synaptic plasticity, memory and the hippocampus: a neural network approach to causality. Nat Rev Neurosci. 2008;9:65–75. doi: 10.1038/nrn2303. [DOI] [PubMed] [Google Scholar]
- Noam Y, Zha Q, Phan L, Wu RL, Chetkovich DM, Wadman WJ, Baram TZ. Trafficking and surface expression of hyperpolarization-activated cyclic nucleotide-gated channels in hippocampal neurons. J Biol Chem. 2010;285:14724–14736. doi: 10.1074/jbc.M109.070391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusser Z. AMPA and NMDA receptors: similarities and differences in their synaptic distribution. Curr Opin Neurobiol. 2000;10:337–341. doi: 10.1016/S0959-4388(00)00086-6. [DOI] [PubMed] [Google Scholar]
- O'Donnell C, Nolan MF, van Rossum MC. Dendritic spine dynamics regulate the long-term stability of synaptic plasticity. J Neurosci. 2011;31:16142–16156. doi: 10.1523/JNEUROSCI.2520-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park M, Salgado JM, Ostroff L, Helton TD, Robinson CG, Harris KM, Ehlers MD. Plasticity-induced growth of dendritic spines by exocytic trafficking from recycling endosomes. Neuron. 2006;52:817–830. doi: 10.1016/j.neuron.2006.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlides C, Nivón LG, McEwen BS. Effects of chronic stress on hippocampal long-term potentiation. Hippocampus. 2002;12:245–257. doi: 10.1002/hipo.1116. [DOI] [PubMed] [Google Scholar]
- Pawlak R, Magarinos AM, Melchor J, McEwen B, Strickland S. Tissue plasminogen activator in the amygdala is critical for stress-induced anxiety-like behavior. Nat Neurosci. 2003;6:168–174. doi: 10.1038/nn998. [DOI] [PubMed] [Google Scholar]
- Penzes P, Rafalovich I. Regulation of the actin cytoskeleton in dendritic spines. Adv Exp Med Biol. 2012;970:81–95. doi: 10.1007/978-3-7091-0932-8_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penzes P, Beeser A, Chernoff J, Schiller MR, Eipper BA, Mains RE, Huganir RL. Rapid induction of dendritic spine morphogenesis by trans-synaptic ephrinB-EphB receptor activation of the Rho-GEF kalirin. Neuron. 2003;37:263–274. doi: 10.1016/S0896-6273(02)01168-6. [DOI] [PubMed] [Google Scholar]
- Penzes P, Cahill ME, Jones KA, VanLeeuwen JE, Woolfrey KM. Dendritic spine pathology in neuropsychiatric disorders. Nat Neurosci. 2011;14:285–293. doi: 10.1038/nn.2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlmutter LS, Gall C, Baudry M, Lynch G. Distribution of calcium-activated protease calpain in the rat brain. J Comp Neurol. 1990;296:269–276. doi: 10.1002/cne.902960207. [DOI] [PubMed] [Google Scholar]
- Perrin MH, Vale WW. Corticotropin releasing factor receptors and their ligand family. Ann N Y Acad Sci. 1999;885:312–328. doi: 10.1111/j.1749-6632.1999.tb08687.x. [DOI] [PubMed] [Google Scholar]
- Petralia RS, Wang YX, Wenthold RJ. The NMDA receptor subunits NR2A and NR2B show histological and ultrastructural localization patterns similar to those of NR1. J Neurosci. 1994;14:6102–6120. doi: 10.1523/JNEUROSCI.14-10-06102.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petralia RS, Sans N, Wang YX, Wenthold RJ. Ontogeny of postsynaptic density proteins at glutamatergic synapses. Mol Cell Neurosci. 2005;29:436–452. doi: 10.1016/j.mcn.2005.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao A, Craig AM. Signaling between the actin cytoskeleton and the postsynaptic density of dendritic spines. Hippocampus. 2000;10:527–541. doi: 10.1002/1098-1063(2000)10:5<527::AID-HIPO3>3.0.CO%3B2-B. [DOI] [PubMed] [Google Scholar]
- Refojo D, Schweizer M, Kuehne C, Ehrenberg S, Thoeringer C, Vogl AM, Dedic N, Schumacher M, von Wolff G, Avrabos C, Touma C, Engblom D, Schütz G, Nave KA, Eder M, Wotjak CT, Sillaber I, Holsboer F, Wurst W, Deussing JM. Glutamatergic and dopaminergic neurons mediate anxiogenic and anxiolytic effects of CRHR1. Science. 2011;333:1903–1907. doi: 10.1126/science.1202107. [DOI] [PubMed] [Google Scholar]
- Regev L, Neufeld-Cohen A, Tsoory M, Kuperman Y, Getselter D, Gil S, Chen A. Prolonged and site-specific over-expression of corticotropin-releasing factor reveals differential roles for extended amygdala nuclei in emotional regulation. Mol Psychiatry. 2011;16:714–728. doi: 10.1038/mp.2010.64. [DOI] [PubMed] [Google Scholar]
- Rex CS, Chen LY, Sharma A, Liu J, Babayan AH, Gall CM, Lynch G. Different Rho GTPase-dependent signaling pathways initiate sequential steps in the consolidation of long-term potentiation. J Cell Biol. 2009;186:85–97. doi: 10.1083/jcb.200901084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes BA, Fox K, Valentino RJ, Van Bockstaele EJ. Agonist-induced internalization of corticotropin-releasing factor receptors in noradrenergic neurons of the rat locus coeruleus. Eur J Neurosci. 2006;23:2991–2998. doi: 10.1111/j.1460-9568.2006.04820.x. [DOI] [PubMed] [Google Scholar]
- Reyes BA, Valentino RJ, Van Bockstaele EJ. Stress-induced intracellular trafficking of corticotropin-releasing factor receptors in rat locus coeruleus neurons. Endocrinology. 2008;149:122–130. doi: 10.1210/en.2007-0705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter RM, Pich EM, Koob GF, Weiss F. Sensitization of cocaine-stimulated increase in extracellular levels of corticotropin-releasing factor from the rat amygdala after repeated administration as determined by intracranial microdialysis. Neurosci Lett. 1995;187:169–172. doi: 10.1016/0304-3940(95)11365-4. [DOI] [PubMed] [Google Scholar]
- Sánchez MM, Young LJ, Plotsky PM, Insel TR. Autoradiographic and in situ hybridization localization of corticotropin-releasing factor 1 and 2 receptors in nonhuman primate brain. J Comp Neurol. 1999;408:365–377. doi: 10.1002/(SICI)1096-9861(19990607)408:3<365::AID-CNE5>3.3.CO%3B2-E. [DOI] [PubMed] [Google Scholar]
- Scannevin RH, Huganir RL. Postsynaptic organization and regulation of excitatory synapses. Nat Rev Neurosci. 2000;1:133–141. doi: 10.1038/35039075. [DOI] [PubMed] [Google Scholar]
- Segal M. Dendritic spines and long-term plasticity. Nat Rev Neurosci. 2005;6:277–284. doi: 10.1038/nrn1649. [DOI] [PubMed] [Google Scholar]
- Shi SH, Hayashi Y, Petralia RS, Zaman SH, Wenthold RJ, Svoboda K, Malinow R. Rapid spine delivery and redistribution of AMPA receptors after synaptic NMDA receptor activation. Science. 1999;284:1811–1816. doi: 10.1126/science.284.5421.1811. [DOI] [PubMed] [Google Scholar]
- Shors TJ. Acute stress rapidly and persistently enhances memory formation in the male rat. Neurobiol Learn Mem. 2001;75:10–29. doi: 10.1006/nlme.1999.3956. [DOI] [PubMed] [Google Scholar]
- Siman R, Baudry M, Lynch G. Brain fodrin: substrate for calpain I, an endogenous calcium-activated protease. Proc Natl Acad Sci U S A. 1984;81:3572–3576. doi: 10.1073/pnas.81.11.3572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart MG, Davies HA, Sandi C, Kraev IV, Rogachevsky VV, Peddie CJ, Rodriguez JJ, Cordero MI, Donohue HS, Gabbott PL, Popov VI. Stress suppresses and learning induces plasticity in CA3 of rat hippocampus: a three-dimensional ultrastructural study of thorny excrescences and their postsynaptic densities. Neuroscience. 2005;131:43–54. doi: 10.1016/j.neuroscience.2004.10.031. [DOI] [PubMed] [Google Scholar]
- Swinny JD, Valentino RJ. Corticotropin-releasing factor promotes growth of brain norepinephrine neuronal processes through Rho GTPase regulators of the actin cytoskeleton in rat. Eur J Neurosci. 2006;24:2481–2490. doi: 10.1111/j.1460-9568.2006.05129.x. [DOI] [PubMed] [Google Scholar]
- Tada T, Sheng M. Molecular mechanisms of dendritic spine morphogenesis. Curr Opin Neurobiol. 2006;16:95–101. doi: 10.1016/j.conb.2005.12.001. [DOI] [PubMed] [Google Scholar]
- Takumi Y, Ramírez-Leon V, Laake P, Rinvik E, Ottersen OP. Different modes of expression of AMPA and NMDA receptors in hippocampal synapses. Nat Neurosci. 1999;2:618–624. doi: 10.1038/10172. [DOI] [PubMed] [Google Scholar]
- Tovar KR, Westbrook GL. The incorporation of NMDA receptors with a distinct subunit composition at nascent hippocampal synapses in vitro. J Neurosci. 1999;19:4180–4188. doi: 10.1523/JNEUROSCI.19-10-04180.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tringali G, Lisi L, De Simone ML, Aubry JM, Preziosi P, Pozzoli G, Navarra P. Effects of olanzapine and quetiapine on corticotropin-releasing hormone release in the rat brain. Prog Neuropsychopharmacol Biol Psychiatry. 2009;33:1017–1021. doi: 10.1016/j.pnpbp.2009.05.012. [DOI] [PubMed] [Google Scholar]
- Ulrich-Lai YM, Herman JP. Neural regulation of endocrine and autonomic stress responses. Nat Rev Neurosci. 2009;10:397–409. doi: 10.1038/nrn2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Pett K, Viau V, Bittencourt JC, Chan RK, Li HY, Arias C, Prins GS, Perrin M, Vale W, Sawchenko PE. Distribution of mRNAs encoding CRF receptors in brain and pituitary of rat and mouse. J Comp Neurol. 2000;428:191–212. doi: 10.1002/1096-9861(20001211)428:2<191::AID-CNE1>3.0.CO%3B2-U. [DOI] [PubMed] [Google Scholar]
- Vanderklish PW, Krushel LA, Holst BH, Gally JA, Crossin KL, Edelman GM. Marking synaptic activity in dendritic spines with a calpain substrate exhibiting fluorescence resonance energy transfer. Proc Natl Acad Sci U S A. 2000;97:2253–2258. doi: 10.1073/pnas.040565597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderklish P, Bednarski E, Lynch G. Translational suppression of calpain blocks long-term potentiation. Learn Mem. 1996;3:209–217. doi: 10.1101/lm.3.2-3.209. [DOI] [PubMed] [Google Scholar]
- Wang Q, Van Heerikhuize J, Aronica E, Kawata M, Seress L, Joels M, Swaab DF, Lucassen PJ. Glucocorticoid receptor protein expression in human hippocampus; stability with age. Neurobiol Aging. 2013a;34:1662–1673. doi: 10.1016/j.neurobiolaging.2012.11.019. [DOI] [PubMed] [Google Scholar]
- Wang XD, Su YA, Wagner KV, Avrabos C, Scharf SH, Hartmann J, Wolf M, Liebl C, Kühne C, Wurst W, Holsboer F, Eder M, Deussing JM, Müller MB, Schmidt MV. Nectin-3 links CRHR1 signaling to stress-induced memory deficits and spine loss. Nat Neurosci. 2013b;16:706–713. doi: 10.1038/nn.3395. [DOI] [PubMed] [Google Scholar]
- Williams K, Russell SL, Shen YM, Molinoff PB. Developmental switch in the expression of NMDA receptors occurs in vivo and in vitro. Neuron. 1993;10:267–278. doi: 10.1016/0896-6273(93)90317-K. [DOI] [PubMed] [Google Scholar]
- Wilson RS, Schneider JA, Boyle PA, Arnold SE, Tang Y, Bennett DA. Chronic distress and incidence of mild cognitive impairment. Neurology. 2007;68:2085–2092. doi: 10.1212/01.wnl.0000264930.97061.82. [DOI] [PubMed] [Google Scholar]
- Woods GF, Oh WC, Boudewyn LC, Mikula SK, Zito K. Loss of PSD-95 enrichment is not a prerequisite for spine retraction. J Neurosci. 2011;31:12129–12138. doi: 10.1523/JNEUROSCI.6662-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Srivastava DP, Photowala H, Kai L, Cahill ME, Woolfrey KM, Shum CY, Surmeier DJ, Penzes P. Kalirin-7 controls activity-dependent structural and functional plasticity of dendritic spines. Neuron. 2007;56:640–656. doi: 10.1016/j.neuron.2007.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuste R, Bonhoeffer T. Morphological changes in dendritic spines associated with long-term synaptic plasticity. Annu Rev Neurosci. 2001;24:1071–1089. doi: 10.1146/annurev.neuro.24.1.1071. [DOI] [PubMed] [Google Scholar]
- Yuste R, Bonhoeffer T. Genesis of dendritic spines: insights from ultrastructural and imaging studies. Nat Rev Neurosci. 2004;5:24–34. doi: 10.1038/nrn1300. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Homma KJ, Poo MM. Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. Neuron. 2004;44:749–757. doi: 10.1016/j.neuron.2004.11.011. [DOI] [PubMed] [Google Scholar]