

Abstract
Surface diffusion of postsynaptic receptors shapes synaptic transmission. Presynaptic receptors also influence transmission, but the relevance of their mobility for synaptic function is unknown. Using single-particle tracking with quantum dots, we show that calcium-permeable α7-containing nicotinic acetylcholine receptors (α7–nAChRs), capable of promoting transmitter release, are mobile on presynaptic terminals but constrained in synaptic space on rat hippocampal neurons in culture. Additional immobilization of presynaptic α7-nAChRs by antibody crosslinking increases glutamate release capacity as seen in the frequency of spontaneous miniature postsynaptic currents and the size of the readily releasable pool of transmitter. Conversely, blocking glutamate release by targeting tetanus toxin to individual synapses increases α7–nAChR dwell time at presynaptic sites. The effects on release require functional α7–nAChRs and may to depend on CAST/ELKS (calpastatin/glutamine, leucine, lysine, and serine-rich protein), which an unbiased proteomic screen yielded. The results support a new homeostatic regulatory mechanism in which α7–nAChR restrain may be adjusted as needed at presynaptic sites via active zone proteins to maintain transmitter release capability.
Introduction
Positioning of ligand-gated ion channels at postsynaptic sites is critical for achieving fast chemical transmission. Perhaps best characterized in this respect are AMPA-type glutamate receptors that are recruited to postsynaptic sites by mechanisms depending on lateral diffusion of surface receptors for both synaptic maintenance and synaptic plasticity (Tardin et al., 2003; Adesnik et al., 2005; Ashby et al., 2006; Heine et al., 2008; Makino and Malinow, 2009; Hoze et al., 2012). Numerous types of ligand-gated ion channels are also located presynaptically in which they can modulate transmitter release (Dudel and Kuffler, 1961; Eccles, 1964; Khakh and Henderson, 2000; Duguid and Smart, 2009; Larsen et al., 2011). Although their precise location is important for synaptic function, little is known about the mobility of presynaptic ligand-gated ion channels or how this may influence synaptic transmission.
Nicotinic acetylcholine receptors containing the α7-gene product (α7–nAChRs) are located both presynaptically and postsynaptically at many types of synapses in the CNS (Fabian-Fine et al., 2001; Dajas-Bailador and Wonnacott, 2004; Albuquerque et al., 2009). Because of their high relative permeability to calcium (Bertrand et al., 1993; Séguéla et al., 1993), α7–nAChRs can influence a variety of calcium-dependent events, including neuronal development and gene transcription (Jones et al., 1999; Chang and Berg, 2001; Hu et al., 2002; Dajas-Bailador and Wonnacott, 2004; Liu et al., 2006; Albuquerque et al., 2009; Campbell et al., 2010; Miwa et al., 2011; Lozada et al., 2012). A prominent feature of presynaptic α7–nAChRs is their ability to enhance transmitter release at both glutamatergic and GABAergic synapses (McGehee et al., 1995; Gray et al., 1996; Alkondon and Albuquerque, 2001; Dickinson et al., 2008; Zhong et al., 2008; Albuquerque et al., 2009; Gu and Yakel, 2011). However, the relationship between the lateral mobility of presynaptic α7–nAChRs and receptor control of transmitter release is unknown.
A strategy used recently to access the lateral mobility of α7–nAChRs at postsynaptic sites has been single-particle tracking (SPT) with quantum dots (QDs; Fernandes et al., 2010; Gómez-Varela et al., 2012). Here we use QDs to show that α7–nAChRs on presynaptic terminals of hippocampal neurons are mobile but constrained in synaptic space. Immobilizing and clustering presynaptic α7–nAChRs by antibody (Ab) crosslinking increases the frequency of spontaneous miniature EPSCs (mEPSCs) and increases the size of the readily releasable pool (RRP) of vesicles. This represents an increase in the capacity for transmitter release. Conversely, silencing transmitter release in a synapse-specific way by using genetically targeted expression of tetanus toxin induces additional constraint of α7–nAChRs at presynaptic sites. A proteomics screen suggests that the presynaptic scaffold protein CAST/ELKS (calpastatin/glutamine, leucine, lysine, and serine-rich protein) may be associated with α7–nAChRs and could mediate the α7–nAChR effects on transmitter release. The results demonstrate for the first time that the mobility of presynaptic receptors on the cell surface is a critical variable determining transmitter release capability. Control of receptor mobility by the nerve terminal may represent both a homeostatic mechanism to sustain release and a novel intrinsic regulatory mechanism to adjust synaptic strength in response to changing demands.
Materials and Methods
Mass spectrometry of α7–nAChR complexes from rat brain.
Purification of α7-nAChR complexes, analysis of the samples by mass spectrometry, and database searches were performed as described previously (Gómez-Varela et al., 2012).
DNA and RNA interference constructs.
Synaptophysin–green fluorescent protein (Sph–GFP) and tetanus toxin light chain (TeT) downstream from Sph–GFP (TeT/Sph–GFP) vectors were obtained from Michael Ehlers (Duke University Medical Center, Durham, NC; Ehlers et al., 2007). α7–HA was obtained from Stephen F. Heinemann (Salk Institute for Biological Studies, La Jolla, CA; Xu et al., 2006). To generate an RNA interference (RNAi) targeting ELKS1, we used the sequence aaggagagcaaattaagttct generated by a Genscript algorithm. The scrambled sequence as control was generated using the Genscript sequence scrambler and was shown not to be homologous to any identified sequence in the rat and mouse genomes. Sequences were inserted into a lentiviral vector (FG12; Addgene plasmid 14884) under an H1 promoter along with GFP under an ubiquitin promoter and were linked to their reverse complement by the loop sequence TCTCTTGAA to form a short-hairpin RNA. Viral particles were obtained by standard techniques. Viral infection was performed by incubating 7-d-old hippocampal cultures with 2 × 106 viral particles for 12 h at 37°C. Effectiveness of the knockdown was assessed by ELKS immunostaining in 14-d-old hippocampal cultures.
Hippocampal cultures.
Hippocampal cultures were prepared from 18- to 19-d-old Sprague Dawley male and female rat embryos as described previously (Kawai et al., 2002). Briefly, hippocampi were removed rapidly under stereomicroscopic observation, cut into small pieces, and digested with 20 U/ml trypsin (Invitrogen) in HBSS (Invitrogen) at 37°C for 12 min. The tissue segments were then transferred to Neurobasal medium (Invitrogen) with 10% heat-inactivated horse serum (Invitrogen), triturated with a fire-polished Pasteur pipette, and plated at 105 cells per 12 mm glass coverslip coated with poly-d-lysine (>300 kDa; Sigma). Subsequent feeding occurred twice weekly, each time replacing half the volume with fresh Neurobasal media with 2% B-27 (Invitrogen). The cultures were maintained in a humidified tissue culture incubator with 5% CO2 and taken for use after 15–17 d. Cultures were transfected using the calcium–phosphate method (Goetze et al., 2004) on days 7–8. Hippocampal mouse cultures from mice lacking the α7–nAChR gene [α7 knock-outs; (α7KO)] mice were prepared in the same way from P0–P1 animals.
Immunocytochemistry.
For immunostaining of surface GluA1-containing AMPA receptors (GluA1s) and surface HA–α7–nAChRs, hippocampal neurons in culture were incubated with an extracellular GluA1 Ab (1:50; Calbiochem) and anti-HA Ab (1:100; Santa Cruz Biotechnology), respectively, for 30 min at 4°C in PBS plus 5% donkey serum. For immunostaining of intracellular proteins, cultures were fixed with 2% paraformaldehyde (PFA) in PBS for 10 min at room temperature (RT). After three washes with PBS, cells were permeabilized by incubating 10 min with 0.1% Triton X-100 in PBS. To label synapsin, ELKS, and the vesicular glutamate transporter (VGluT), neurons were incubated with an anti-synapsin 1,2 Ab (1:1000; Synaptic Systems), anti-ELKS Ab (1:500; Synaptic Systems), or VGluT Ab (1:500; Synaptic Systems), respectively, for 1 h at RT in PBS containing 5% normal donkey serum. After washing three times in PBS, cells were incubated with appropriate donkey FITC-, Cy3-, or Cy5-conjugated secondary Ab 2 h at RT (1:250; Jackson ImmunoResearch), washed three times with PBS, and mounted on slides for imaging.
To crosslink α7–nAChRs, cells were incubated first with 100 nm biotinylated α-bungarotoxin (Biot-Bgt) for 1 h to bind to the receptors, rinsed and then incubated with anti-biotin Ab (1 μg/μl for 10 min), and then rinsed and incubated with secondary Ab (2.5 μg/μl for 10 min). Controls substituted non-immune IgG for the primary Ab. All incubations were performed in Neurobasal medium at 37°C. To visualize surface α7–nAChRs after crosslinking, an Alexa Fluor 488-labeled Ab was used as the secondary (2.5 μg/μl for 10 min). Controls (no crosslinking) used a Alexa Fluor 488-labeled anti-biotin Ab (1 μg/μl for 10 min) and no secondary Ab.
To label the recycling pool of synaptic vesicles, neuronal cultures were incubated for 45 s with 5 μm FM4-64FX [fixable version of N-(3-triethylammoniumpropyl)-4-(4-(dibutylamino)styryl) pyridinium dibromide; Invitrogen] in HEPES buffer containing 60 mm KCl. After washing three times with HEPES buffer containing 4 mm KCl, neurons were fixed with 2% PFA for 10 min at RT. To label the lumenal domain of synaptotagmin (Syn–Lum), cells were incubated with anti-synaptotagmin Ab (1:100; Synaptic Systems) for 5 min at 37°C in culture media as described previously (Biederer and Scheiffele, 2007). Neurons were fixed and permeabilized as above. Destaining of FM4-64FX dye was achieved by perfusing labeled neurons with PBS containing 40 mm K+ while imaging at 5 Hz (see below).
Confocal images were acquired in sequential mode using a Leica SP5 confocal microscope with settings that did not saturate the fluorescence signals and that fulfilled Nyquist sampling criteria. NIH ImageJ software was used for quantifying the number of specific protein clusters. Four regions of interest (ROIs) of 20 μm in length were randomly selected per field of view along the dendrites of neurons. ROIs were binarized automatically using the mean of the background value inside the dendrite plus three times the SD as an intensity threshold value for defining a cluster or puncta in each image. Clusters (puncta) within an ROI were counted if they had at least 3 × 3 pixels above threshold (pixel diameter, 80 nm). Data are expressed as the mean ± SEM per dendritic length of 20 μm and represent at least 10 images from each of three or more cultures.
Postsynaptic HA–α7–nAChRs were identified in transfected α7KO neuronal cultures as those receptors located on a major dendrite emanating from a nearby transfected cell body. Presynaptic HA–α7–nAChRs were identified as those receptors present on thin neurites (putative axons) far from transfected cell bodies and often found to cross untransfected dendrites as visualized with bright-field microscopy.
Calcium imaging.
Crosslinking of α7–nAChRs was performed as described above. Calcium imaging was performed as described previously (Gómez-Varela et al., 2012).
SPT.
Labeling of surface α7–nAChRs with QD nanocrystals, acquisition, and analysis of the trajectories were performed as described previously (Gómez-Varela et al., 2012). Briefly, labeling of surface GluA1s with QD nanocrystals was achieved by first incubating neuronal cultures with 0.5 μg/ml anti-GluA1 rabbit Ab (Calbiochem; PC246; recognizes the extracellular N-terminus domain) in PBS plus 0.1% BSA for 10 min at 37°C. The neurons were then rinsed five times with PBS and incubated with 500 pm QD 605 goat F(ab′)2 anti-rabbit IgG conjugate (heavy and light chain; Invitrogen) in PBS plus 0.1% BSA for 1 min at RT. The neurons were then rinsed five more times with PBS before imaging. Negative controls included substituting nonspecific IgG for the GluA1 Ab or incubating the neurons directly with QD 605 goat F(ab′)2 anti-rabbit IgG conjugate. No QD signal was detected in these negative controls. All trajectories analyzed in the SPT experiments were obtained from at least four independent hippocampal culture platings.
Electrophysiology.
Hippocampal cultures were immersed in artificial CSF (aCSF; in mm: 150 NaCl2, 5 KCl, 2 CaCl2, 1 MgCl2, 10 glucose, and 10 HEPES) and placed in a recording chamber mounted on a Zeiss Axiovert. Patch pipettes (2.0–4.0 MΩ) were visually guided to the surface of individual neurons. Patch pipettes contained the following (in mm): 125 K-gluconate, 15 KCl, 8 NaCl2, 2 EGTA, 10 Na2-phosphocreatine, 4 Mg-ATP, 0.3 Na-GTP, and 10 HEPES, pH 7.25. Voltage-clamp recordings were obtained with an Axopatch 200A and Clampex 8.2 software (Molecular Devices). Data were acquired at 5 kHz and filtered at 2 kHz. Pipette capacitance was canceled, whole-cell capacitance was neutralized, and series resistance was compensated by 80% in all recordings. All activity was recorded at −70 mV.
To record glutamatergic mEPSCs, 1 μm tetrodotoxin (TTX) and 20 μm gabazine were included in the recording medium. Mini Analysis software was used to detect the mEPSCs, with an amplitude threshold of four times the SD of the mean background noise level. All detected events were verified by visual inspection.
To assay the size of the RRP, neurons patch clamped in the presence of 1 μm TTX were subjected to hyperosmotic shock (Rosenmund and Stevens, 1996). Briefly, a 4 s application of aCSF containing 0.5 m sucrose and 1 μm TTX was ejected from a multibarrel pipette positioned 30 μm away from the cell body of the patched neuron. The sucrose-containing solution was applied from the central barrel, which was surrounded by barrels ejecting extracellular media at the same flux speed. The time needed to change between barrels was 5–7 ms. This configuration ensured a rapid application and washing of the sucrose solution. To study the refill properties of the RRP, a 4 s pulse of sucrose was applied a second and third time at 30 s intervals. Total charge was calculated by integrating the area of the response after each application using Clampfit 8.2 software.
Results
Presynaptic α7–nAChRs are constrained in synaptic space
To examine the mobility of presynaptic α7–nAChRs, we used SPT with QDs. Hippocampal neurons in culture were incubated first with Biot-Bgt to label α7–nAChRs and then with streptavidin–QD to attach the fluorescent nanocrystal to the bound Biot-Bgt. To distinguish QD-tagged presynaptic α7–nAChRs, axons were marked by sparsely transfecting the cultures 7–9 d earlier with Sph–GFP. Incubating with a low concentration of a rhodamine-derivative mitochondrial marker (Mitotracker) indicated synaptic sites along the axon (Fig. 1A). This enabled us to select QD-labeled α7–nAChRs confined to axons and distinguish those moving into and out of synaptic spaces (Tardin et al., 2003; Groc et al., 2004; Ehlers et al., 2007). The dynamics of such QD-labeled α7–nAChRs were followed over a 60 s period with ∼50 nm accuracy (see Materials and Methods). The instantaneous diffusion coefficient (Di), calculated as a function of time, showed that the receptors displayed either low or high mobility, depending on their being in either synaptic or extrasynaptic space, respectively (Fig. 1B). The mean square displacement (MSD) of the trajectory was consistent with Brownian diffusion outside synapses but indicated confined movement within presynaptic terminals (Fig. 1C). Cumulative frequency plots display the consistent differences in Di values depending on receptor location (Fig. 1D). Notably, the SPT analysis indicates that extrasynaptic α7–nAChRs on axons diffuse more rapidly than on dendrites [median ± 25–75% interquartile range (IQR), 0.21 ± 0.08–0.26 vs 0.165 ± 0.095–0.20 for presynaptic vs postsynaptic α7–nAChR–QD complexes; p < 0.001, Mann–Whitney test; n = 149, 60 trajectories; Gómez-Varela et al., 2012]. The results demonstrate that surface α7–nAChRs are mobile on axon terminals but constrained within synaptic space.
Figure 1.

QD tracking indicates that presynaptic α7–nAChRs are constrained within synaptic space. A, Left, Hippocampal neurons in culture stained with Mitotracker (green) and transfected with Sph–GFP (blue). Neuronal contours (white boundaries) and transfected axonal trajectories were visualized by saturating the Mitotracker and Sph–GFP images, respectively. Right, Higher magnification of the left inset (white box) showing an α7–nAChR–QD complex (red) diffusing (white trajectory) on the membrane of an axon transfected with Sph–GFP (blue) and accessing synapses labeled with Mitotracker (green). Scale bar, 1 μm. B, Representative example showing the variation in Di for the α7–nAChR–QD in A as a function of time. Horizontal line indicates time in synaptic (green) and extrasynaptic (black) spaces. C, Representative example of MSD versus lag time for α7–nAChR–QD complexes in extrasynaptic (Extra) and synaptic (Syn) locations. The linear increase in MSD with time indicates free diffusion outside synapses, whereas the curved MSD with time inside synapses indicates confined diffusion. D, Cumulative distribution of Di values for synaptic and extrasynaptic α7–nAChR–QD (n = 118 and 149 for synaptic and extrasynaptic trajectories, respectively, from ≥3 independent culture sets for each condition; Mann–Whitney test, ***p < 0.001).
Decreasing the mobility of presynaptic α7–nAChRs increases glutamate release
To test the functional relevance of presynaptic α7–nAChR lateral mobility, we measured transmitter release after additional restraining α7–nAChR movement using Biot-Bgt/Ab crosslinking (Heine et al., 2008; see Materials and Methods). As expected, the crosslinking procedure increased the mean size of α7–nAChR clusters on the dendritic surface (Fig. 2A) and decreased α7–nAChR mobility (Fig. 2B). We assessed effects on glutamate release first by patch-clamp recording of mEPSCs in the presence of TTX to block action potentials and gabazine to block GABAergic responses. Remarkably, the Ab crosslinking of α7–nAChRs significantly increased the frequency of mEPSCs more than twofold (α7X in Fig. 2C,D). Neither the mean amplitude nor decay kinetics of the mEPSCs were changed by the crosslinking. These findings are compatible with greater release at preexisting synapses or possibly with an increase in the number of synapses. To exclude the latter possibility, we quantified the number of synaptic sites as revealed by colocalization of the presynaptic marker synapsin-1,2 with the postsynaptic marker GluA1. No increases were seen in either the colocalization of GluA1 with synapsin (Fig. 2E) or their puncta number (data not shown) when α7–nAChRs were crosslinked. This argues against the increase in mEPSC frequency representing an increase in synapse number.
Figure 2.
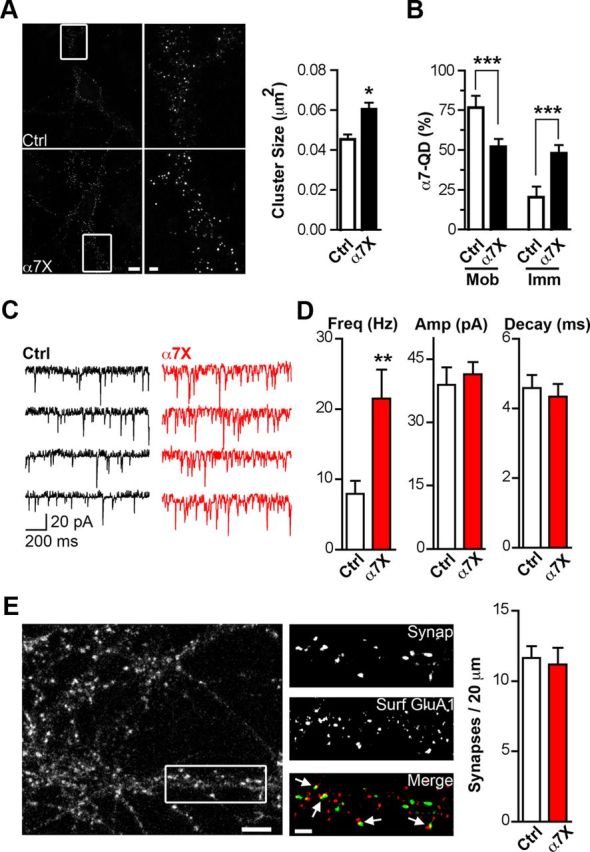
Reducing α7–nAChR mobility increases synaptic transmission. A, Left, Representative images showing α7–nAChR staining in control (Ctrl) or crosslinked (α7X) conditions. Middle, Higher magnification of the left insets (white boxes). Scale bars, 10 and 2 μm. Right, Quantification of α7–nAChR cluster size for Ctrl and α7X (n = 22 neurons, 4 culture sets; mean ± SEM; Student's t test, *p < 0.05). B, Quantification of mobile and immobile α7–nAChR–QD complexes for Ctrl and α7X (n = 18 and 26 images from 3 culture sets for Ctrl and α7X; mean ± SEM; Student's t test, ***p < 0.001). C, Sample traces of mEPSCs in Ctrl (black) and α7X (red) conditions. D, Quantitative analysis of the frequency (Freq), amplitude (Amp), and decay time of mEPSCs for Ctrl and α7X (n = 16 neurons, 4 culture sets; mean ± SEM; Student's t test, **p < 0.01). E, Left, Image shows synapsin immunostaining in hippocampal cultures. Middle, Higher magnification of the box from the left showing synapsin-1,2 staining (Synap, top), surface GluA1 staining (Surf GluA1, middle), and overlay of the two (Merge, bottom; Synap, green; GluA1, red). Arrows indicate examples of colocalization of the two, representing likely synapses. Scale bars, 5 and 2 μm. Right, Quantification of synaptic number (colocalized Synap and GluA1 puncta) for Ctrl and α7X (n = 19 neurons, 4 culture sets for both Ctrl and α7X; mean ± SEM; Student's t test, p > 0.05).
In contrast, crosslinking did affect release capability as monitored by uptake of the dye FM4-64FX, which can be used to label recycling vesicle pools (Pyle et al., 2000; Südhof, 2000). Stimulation of FM4-64FX-labeled terminals depletes the accumulated dye, consistent with its having been in recycling vesicles (Fig. 3A,B). Crosslinking α7–nAChRs increased the size of FM4-64FX-labeled clusters in terminals after potassium-induced vesicle release and re-endocytosis (Fig. 3C). The same result was obtained using a Syn–Lum Ab (Fig. 3C) that recognizes an intravesicular epitope (Biederer and Scheiffele, 2007). Importantly, the effect of crosslinking α7–nAChRs on transmitter release appears to require functional α7–nAChRs. This was found by repeating the α7–nAChR crosslinking experiment in the presence of the α7–nAChR-specific antagonist methyllycaconitine (MLA) during both the Ab incubation and the potassium pulse used for FM4-64FX labeling. MLA prevented the increase in labeling otherwise seen after crosslinking presynaptic α7–nAChRs (Fig. 3C). Direct evidence that a fraction of the α7–nAChRs remain functional after crosslinking was obtained by challenging them with choline while using a calcium fluor to report depolarization-induced calcium increases (Fig. 3D) as described previously (Gómez-Varela et al., 2012). Receptor crosslinking did not increase the number of FM4-64FX-labeled terminals (data not shown), and MLA treatment in the absence of crosslinking had no effect on transmitter release (FM4-64FX cluster size, 0.10 ± 0.02 vs 0.12 ± 0.03 μm2 for control and MLA, respectively). The results are consistent with the crosslinking causing an increase in the capacity for synaptic vesicle release at existing synapses. The effect was specific for α7–nAChRs; Ab crosslinking of mobile presynaptic cannabinoid 1b receptors (CB1; Mikasova et al., 2008) did not change FM4-64FX uptake compared with controls (FM4-64FX cluster size, 0.13 ± 0.03 vs 0.11 ± 0.02 μm2 for control and CB1 crosslink, respectively).
Figure 3.

Reducing the mobility of α7–nAChRs increases the pool size of recycling vesicles. A, Representative image of FM4-64FX staining in hippocampal cultures. Scale bar, 5 μm. B, Left, Representative image of a presynaptic terminal at various times after stimulation with KCl. Right, Trace showing a representative destaining time course following a sigmoidal kinetic (solid line) in arbitrary units (au). C, Left, Representative images of FM4-64FX and Syn–Lum staining in control (Ctrl) and crosslinked (α7X) conditions. Right, Quantification of FM4-64FX cluster size in Ctrl, α7X, and α7X plus MLA, or Syt–Lum cluster size in Ctrl and α7X (n = 21–24 neurons from 4–5 culture sets for each condition; mean ± SEM; one-way ANOVA, **p < 0.01). D, Top, Representative fluorescent images of a dendrite loaded with the calcium fluor Fluo-4 captured before (Basal) and after (Choline) puffing on choline. Bottom trace shows a representative time course for the decay of the fluorescent signal after α7–nAChR activation by choline.
To confirm that presynaptic, and not postsynaptic, α7–nAChRs were responsible for the increased transmitter release, we repeated the experiments with dissociated hippocampal cell cultures prepared from α7KOs. The α7KO cells were sparsely transfected in culture with a construct encoding HA-tagged α7–nAChR, enabling the receptors to be labeled and crosslinked using an anti-HA Ab to an extracellular epitope. Crosslinking presynaptic HA–α7–nAChRs in this manner did increase release, as monitored by increased labeling by Syn–Lum (Fig. 4A,C). Crosslinking postsynaptic HA–α7–nAChRs had no effect (Fig. 4B,C). Control experiments were performed to show that mouse hippocampal cultures behaved like the rat cultures described above with respect to α7–nAChR crosslinking, causing increased Syn–Lum labeling (Fig. 4C). The results demonstrate that reducing the mobility of presynaptic α7–nAChRs enhances spontaneous transmitter release by increasing release at existing synapses.
Figure 4.
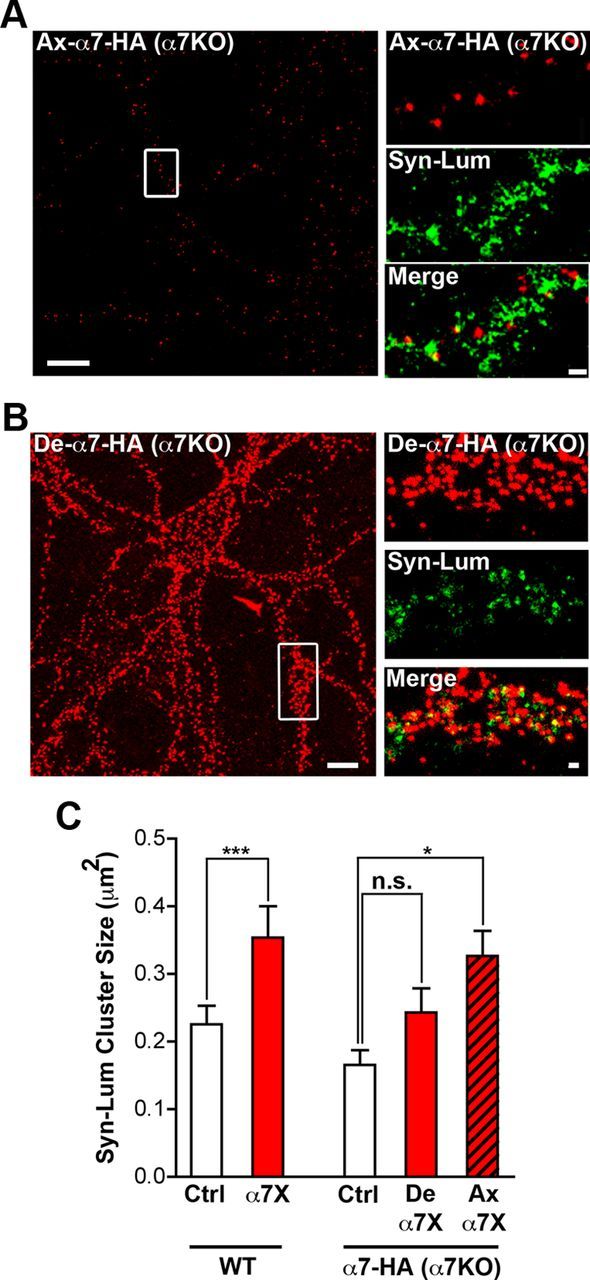
Crosslinking presynaptic α7–nAChRs increases synaptic transmission. A, Left, Representative image of a transfected α7KO hippocampal neuron in culture expressing HA–α7–nAChR on an axon (Ax-α7-HA; boxed region). Scale bar, 10 μm. Right, Magnified images of the boxed region showing HA–α7–nAChR staining (red, top), Syn–Lum staining (green, middle), and the two merged (Merge, bottom). Scale bar, 1 μm. B, Left, Representative image of a transfected α7KO neuron expressing HA–α7–nAChR on dendrites (De-α7-HA). Scale bar, 10 μm. Right, Magnified image of the boxed region showing HA–α7–nAChR, Syn–Lum, and the two merged as in A. Scale bar, 1 μm. C, Left, Quantification of Syn–Lum puncta size in control (Ctrl) and crosslinked (α7X) conditions in wild-type mouse hippocampal neurons in culture (n = 14 neurons from 3 culture sets for both conditions; mean ± SEM; Student's t test, ***p < 0.001). Right, Syn–Lum puncta size in α7KO mouse hippocampal neurons in controls (Ctrl), after crosslinking dendritic HA–α7–nAChRs (De α7X), or after crosslinking axonal HA–α7–nAChRs (Ax α7X; n = 12 neurons from 3 culture sets for each condition; mean ± SEM; one-way ANOVA, *p < 0.05).
Immobilizing presynaptic α7–nAChRs enhances transmitter release by increasing the RRP
The increase in mEPSC frequency, together with the increase in size of both FM4-64FX puncta and Syn–Lum puncta, suggests that immobilizing presynaptic α7–nAChRs by crosslinking increases the release capability of presynaptic terminals. This could result from either an increase in the size of the RRP or an increase in the kinetics of synaptic vesicle release and recycling. To resolve this, we quantified the RRP by stimulating terminals with a 4 s application of hypertonic sucrose while recording the total evoked EPSC in postsynaptic neurons. Crosslinking α7–nAChRs increased the initial phase of the response (0–2 s), which is proportional to RRP size (Fig. 5A). It had no effect on the second phase of the response (2–4 s), which reflects ongoing exocytosis of vesicles and RRP refilling (Fig. 5A; Rosenmund and Stevens, 1996; Deng et al., 2011). No change occurred in the kinetics of sucrose-induced release analyzed by quantifying cumulative charge transfer (Fig. 5B). The refilling dynamics of the RRP was also unaffected. This was determined by measuring the response (total charge transfer in the postsynaptic cell) to three successive applications of hypertonic sucrose 30 s apart (Fig. 5C; Deng et al., 2011). The fact that crosslinking of α7–nAChRs increased RRP size without affecting either the rate of release or the refilling of the RRP suggests a role for specific active zone scaffold proteins linking α7–nAChRs to latent transmitter release sites.
Figure 5.

Effects of α7–nAChR crosslink on RRP capacity and kinetics. A, Sample traces (left) and summary charge-transfer graphs (right) of EPSCs induced by hypertonic sucrose (0.5 m) in control (Ctrl, black trace) and crosslinked α7–nAChR (α7X, red trace) conditions. Hypertonic sucrose was puffed using a multibarrel pipette (see Materials and Methods) onto the patched neuron for 4 s in the presence of 1 μm TTX and 50 μm gabazine. Charge transfer during the initial (Charge 0–2 s) and steady-state responses (Charge 2–4 s) were quantified to estimate the RRP size (n = 17 neurons, 4 culture sets for both conditions; mean ± SEM; Student's t test, *p < 0.05). B, Cumulative charge transfer as a function of time (left) and the corresponding kinetic analyses (right) of sucrose-induced EPSCs in control (Ctrl, black) and crosslinked (α7X, red) conditions. Dotted gray line shows the scaled control (Ctrl scaled) for comparison. The integrated charge transfer was fitted with a double-exponential function to determine kinetic parameters for the fast and slow components (τfast and τslow, respectively) and for their amplitudes (Afast and Aslow). C, Representative EPSC traces (left) and summary graphs (right) showing absolute and normalized data for RRP recovery over three sucrose applications at 30 s interpuff intervals in control (black) and crosslinked (red) conditions. RRP recovery was calculated by dividing the second and third sucrose-induced charge transfer by the value of the first and then normalized to the first current amplitude (n = 19 neurons, 4 culture sets for both conditions; mean ± SEM; Student's t test, *p < 0.05).
The degree of α7–nAChR mobility has downstream consequences for AMPA trafficking
Silencing of transmitter release increases the mobility of postsynaptic AMPA receptors (Ehlers et al., 2007); increasing the amount of release can have the opposite effect (Heine et al., 2008; Opazo et al., 2010). To determine whether the increases in transmitter release caused by reducing α7–nAChR mobility was likely to be physiologically significant, we examined a potential downstream effect, namely the effect on postsynaptic AMPA receptor mobility. SPT with QDs was used to track the mobility of endogenous GluA1s on neurons in hippocampal cultures. Crosslinking of α7–nAChRs clearly decreased the proportion of GluA1s that was mobile and, correspondingly, increased the proportion that scored as immobile (Fig. 6A). No change was seen in the rates of diffusion for the remaining mobile GluA1s after α7–nAChR crosslinking (Fig. 6B), but there was a significant increase in the dwell time of the receptors in synaptic space (Fig. 6C). The results are consistent with immobilization of presynaptic α7–nAChRs causing enhanced transmitter release sufficient to exert transynaptic effects, in this case altering the trafficking of postsynaptic AMPA receptors. Therefore, presynaptic α7–nAChR mobility may not only have local effects on transmitter release but may also exert more extensive changes through downstream signaling.
Figure 6.
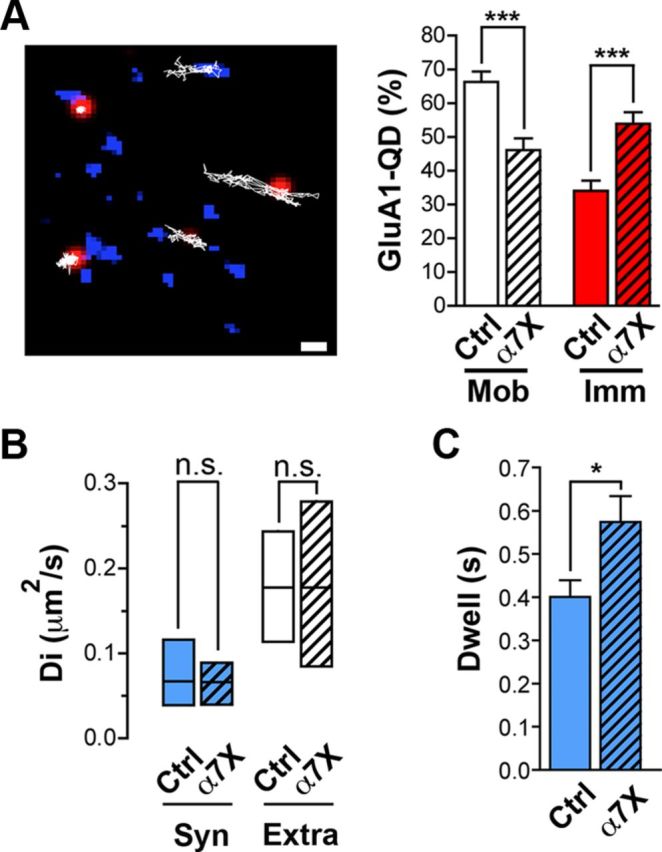
AMPA mobility is reduced after α7–nAChR crosslinking. A, Left, Image shows synapses of hippocampal neurons in culture stained with Mitotracker (blue) and the trajectories (white lines) of mobile and immobile postsynaptic GluA1–QD complexes (red). Scale bar, 1 μm. Right, Quantification of mobile and immobile GluA1–QD complexes in control (Ctrl) and crosslinked α7–nAChR (α7X) conditions (n = 21 and 36 images, 4 culture sets for both conditions; mean ± SEM; Student's t test, ***p < 0.001). B, Median diffusion ± 25–75% IQR of GluA1–QD complexes in synaptic (Syn) and extrasynaptic (Extra) spaces in control and crosslinked conditions (n = 78, 138 and 72, 63 for synaptic and extrasynaptic trajectories of Ctrl and α7X conditions, respectively, from 3 culture sets for each; Mann–Whitney test, p > 0.05). C, Synaptic dwell time of GluA1–QD complexes in control and crosslinked conditions (n = 22 and 16 QDs, 4 culture sets for each; mean ± SEM; Student's t test, *p < 0.05).
Synapse-specific blockade of transmitter release further constrains presynaptic α7–nAChRs
If constraining presynaptic α7–nAChRs at synaptic sites enables nerve terminals to increase transmitter release, one might predict that blockade of release would induce a homeostatic response in the terminal that further restrained α7–nAChRs at that site in an attempt to restore release capability. To test this, we transfected neurons with TeT/Sph–GFP. In neurons expressing the construct, TeT cleaves vesicle-associated membrane protein 2, thereby blocking transmitter release (Harms et al., 2005; Ehlers et al., 2007). Di values calculated from MSD plots for α7–nAChRs on neurons showed that TeT/Sph–GFP expression lowered the mobility of axonal α7–nAChRs specifically at synaptic sites and not at extrasynaptic sites (Fig. 7A). TeT/Sph–GFP expression also significantly increased the dwell time of α7–nAChRs on the axon that enter synaptic space (Fig. 7B). The results demonstrate that the constraint of surface α7–nAChRs within synaptic space is increased when transmitter release is compromised by TeT.
Figure 7.

Silencing of transmitter release reduces α7–nAChR mobility inside presynaptic terminals. The mobility of α7–nAChRs on hippocampal neurons in culture was assessed with QDs as in Figure 1. Mobility on neurons with normal synaptic activity (transfected with Sph–GFP) was compared with those on neurons in which transmitter release was silenced (transfected with TeT/Sph–GFP). A, Median Di ± 25–75% IQR of α7–nAChR–QD complexes in synaptic (Syn) and extrasynaptic (Extra) spaces on neurons transfected with Sph–GFP (Ctrl) or TeT/Sph–GFP (TeT). Silencing of transmitter release significantly decreased the mobility of synaptic receptors specifically inside terminals (n = 50, 55 and 118, 149 for synaptic and extrasynaptic trajectories of Ctrl and TeT conditions, 3 culture sets for each; Mann–Whitney test, ***p < 0.001). B, Synaptic silencing reduces the dwell time of α7–nAChR–QDs inside terminals (n = 12 and 22 QDs from 3 culture sets for Ctrl and TeT, respectively; mean ± SEM; Student's t test, ***p < 0.001).
CAST/ELKS is a candidate mediator of α7–nAChR mobility effects on glutamate release
To enhance RRP size, presynaptic α7–nAChRs presumably must be linked to the presynaptic scaffold, positioning the receptors to augment release. To identify candidate proteins tethering α7–nAChRs to presynaptic release machinery, we performed an unbiased proteomic screen from rat brain extracts. Bgt linked to Actigel beads was used to purify α7–nAChRs and associated proteins (Gómez-Varela et al., 2012). Negative controls included unlinked beads, inclusion of the competitor nicotine along with the Bgt beads, and soluble Bgt with the Bgt beads, as described previously (Gómez-Varela et al., 2012). Proteins specifically bound to α7–nAChRs were identified by comparative liquid chromatography–tandem mass spectrometry analysis of the eluates to determine which were uniquely associated with Bgt beads. Interestingly, the procedure revealed three specific peptides belonging to CAST/ELKS in eluates from Bgt beads (data not shown). ELKS—also known as Rab6-interacting proteins, CASTs, and ERCs—are central components of the presynaptic active zone with proposed roles in transmitter release (Kaeser et al., 2009; Südhof, 2012). The “bait protein” α7–nAChR was found only in the Bgt-bead eluate (data not shown), consistent with specificity.
Because rat brain extracts include α7–nAChRs from multiple sources, we felt it necessary to target directly the presynaptic population. Presynaptic α7–nAChRs cannot presently be uniquely distinguished by immunostaining. Accordingly, to visualize them specifically, we transfected α7KO cultures with HA–α7–nAChR as above and confirmed that the receptors colocalize with ELKS on presynaptic terminals identified by an Ab against VGluT (Fig. 8A). The α7KO cultures, with or without transfection, had VGluT and ELKS puncta equivalent in numbers to those found in wild-type cultures (29.7 ± 1.9 vs 30.9 ± 2.3 for VGluT and 37.3 ± 3 vs 32.6 ± 3.4 for ELKS on wild types and α7KOs, respectively), suggesting no deficit in the total number of glutamatergic synapses on α7KO neurons in culture under these conditions. Notably, crosslinking α7–nAChRs increased the size of ELKS clusters and decreased the size of synaptophysin clusters on axons, consistent with the presynaptic α7–nAChRs being closely positioned near release machinery (Fig. 8B).
Figure 8.
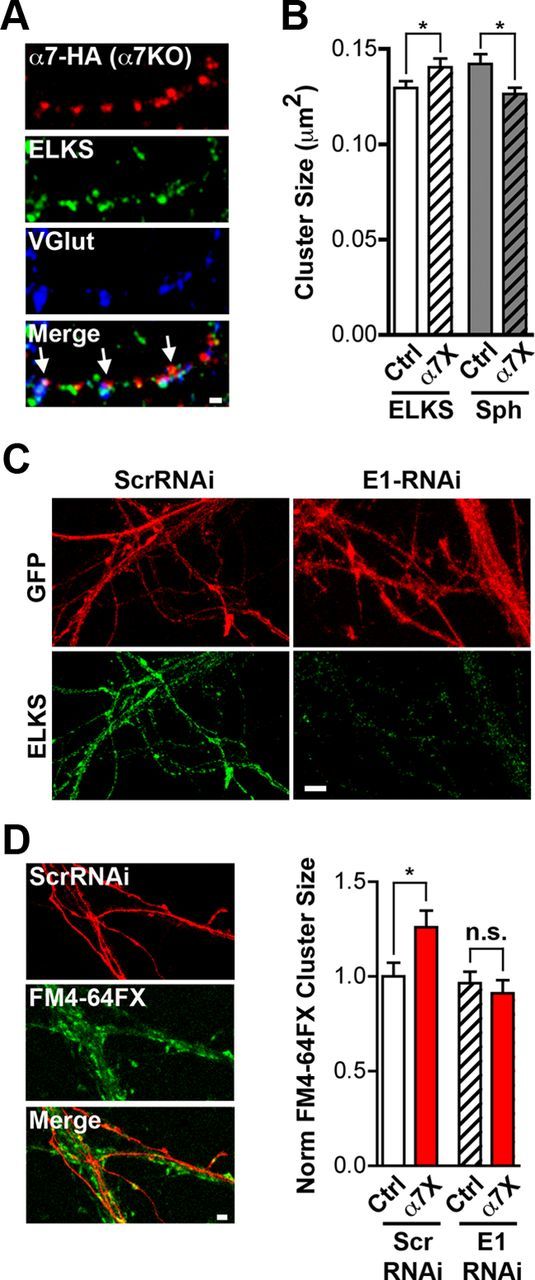
Functional proteomics suggests ELKS as a possible mediator of α7–nAChR crosslinking effects on release capacity. A, Representative images of an axon from a transfected neuron expressing HA–α7–nAChR (red), ELKS (green), and VGluT immunostaining (blue) and the three merged (Merge) with arrows indicating colocalization characteristic of presynaptic terminals. Scale bar, 1 μm. B, Quantification of ELKS and synaptophysin (Sph) cluster size on rat hippocampal neurons in culture under control (Ctrl) and crosslinked α7–nAChR (α7X) conditions (n = 27 neurons, 4 culture sets per condition; mean ± SEM; Student's t test, *p < 0.05). C, Representative images of neurons expressing GFP (red) and ELKS (green) after being infected with either control Scr–RNAi or E1–RNAi virus. Scale bar, 5 μm. D, Left, Representative images of cells expressing control Scr–RNAi (red), stained for FM4-64FX (green), and the images merged (Merge). Scale bar, 2 μm. Right, Quantification of FM4-64FX puncta size in Ctrl and α7X cultures infected with either Scr–RNAi virus as control or E1–RNAi virus (n = 25 neurons, 4 culture sets per condition; mean ± SEM; Student's t test, *p < 0.05).
RNAi was used to knockdown ELKS1 levels in neurons by transfecting them with a short-hairpin RNA directed against ELKS1 mRNA (E1–RNAi; Fig. 8C). The E1–RNAi construct, but not a scrambled control (Scr–RNAi), prevented the FM4-64FX increase in puncta size otherwise induced by presynaptic α7–nAChR crosslinking (Fig. 8D). ELKS1 knockdown did not alter basal uptake by the terminal. The results are consistent with CAST2/ELKS1 being a contributor to the effects of presynaptic α7–nAChRs on glutamate release.
Discussion
The results demonstrate that calcium-permeable α7–nAChRs are mobile on the axon surface but are restrained at presynaptic release sites in which they can act to enhance transmitter release. Immobilizing the receptors further, such that they display increased dwell time in synaptic space, increases the release capability of the synapse. Conversely, the synaptic restraint is increased at the site when transmitter release is compromised. The mobility-dependent effects of α7–nAChRs on release represent changes in RRP size and may be mediated in part by ELKS1. The results suggest a regulatory mechanism in which α7–nAChRs are recruited to and restrained at transmitter release sites as needed to promote transmitter release capability (Fig. 9).
Figure 9.

Model of crosstalk between α7–nAChR lateral mobility and transmitter release. A, α7–nAChRs are highly mobile at the axonal surface, exchanging between extrasynaptic and synaptic spaces. TeT impairs spontaneous release and reduces the mobility of α7–nAChRs specifically inside terminals. B, Conversely, reducing the mobility of α7–nAChRs by extracellular crosslinking increases transmitter release, possibly via scaffold proteins such as ELKS1, reduces the mobility of GluA1s.
SPT with QDs has been an effective technique for defining the mobility of postsynaptic membrane proteins (Triller and Choquet, 2008; Czöndör et al., 2012), including α7–nAChRs at both nicotinic (Fernandes et al., 2010) and glutamatergic (Gómez-Varela et al., 2012) synapses. Although numerous classes of ligand-gated ion channels are located presynaptically in which they can influence transmitter release (Dudel and Kuffler, 1961; Eccles, 1964; Khakh and Henderson, 2000; Langer, 2008; Duguid and Smart, 2009; Larsen et al., 2011), their diffusion properties have been less well characterized (Mikasova et al., 2008; Mercer et al., 2011). Here we show for the first time that α7–nAChRs are mobile on axon surfaces but are constrained in synaptic space. The mean Di value obtained here for α7–nAChRs traversing synaptic space on the presynaptic side is similar to the values reported for α7–nAChRs in synaptic space on the postsynaptic side on both ciliary ganglion neurons and hippocampal interneuron dendrites (Fernandes et al., 2010; Gómez-Varela et al., 2012). This reduced mobility indicates the existence of molecular constraints on nicotinic receptors at the active zone.
For all three cases of α7–nAChR mobility examined, the shape of the MSD versus lag-time plot indicates restrained movement of the receptors when in synaptic space (Fig. 1C; Fernandes et al., 2010; Gómez-Varela et al., 2012). For postsynaptic α7–nAChRs, the constraint has been shown to depend primarily on interactions with scaffold proteins, e.g., PSD-95 or F-actin (Fernandes et al., 2010; Gómez-Varela et al., 2012). For presynaptic α7–nAChRs, active zone-associated components may be key players. Proteomics here identified ELKS as a candidate for being associated with α7–nAChRs. Because ELKS bind to Rab3a-interacting molecules (RIMs) and liprins (Wang et al., 2002; Ko et al., 2003), the ELKS1 link could embed α7–nAChRs in the scaffold, linking them to presynaptic sites. Additional analysis is required to determine whether ELKS1 is actually associated with α7–nAChRs and mediates the effects of α7–nAChR crosslinking on RRP size, as suggested by the RNAi experiments.
A major finding here is that crosslinking presynaptic α7–nAChRs produces a substantial increase in transmitter release capability as measured by electrophysiology, FM4-64FX labeling, and Syn–Lum immunostaining. Several lines of evidence indicate that the increased capacity represents an increase in RRP size rather than a change in the steady-state rate of RRP refilling or other parameters. Crosslinking produced no change in the second phase of transmitter release triggered by sucrose, no change in the kinetics of release, and no change in the refilling rate of the RRP. It did increase the initial release elicited by sucrose, which is an indication of RRP size, and it did increase mEPSC frequency with no change in mEPSC amplitude. That, together with the lack of change in number of synapses detectable by immunostaining, suggests that crosslinking α7–nAChRs may increase release capability at preexisting sites rather than creating more synapses de novo.
Long-term stimulation of α7–nAChRs can increase the number of glutamatergic synapses identified by immunostaining, mEPSC frequency, and ultrastructural analysis (Lozada et al., 2012). An increase in synapse number was not the basis for the increased release seen here because only relatively short time periods of α7–nAChR crosslinking were required. In addition, changes in synapse number depended on α7–nAChRs on the postsynaptic neuron (Lozada et al., 2012), whereas the effects seen here on release required presynaptic α7–nAChRs.
How might immobilization of presynaptic α7–nAChRs affect transmitter release? The possibility of presynaptic α7–nAChR association with active zone components suggests two mechanisms. ELKS1, for example, links to RIMs (Wang et al., 2002; Ko et al., 2003; Südhof, 2012) and could position α7–nAChRs near N-P/Q calcium channels and sites of transmitter release (Kaeser et al., 2012), optimizing the impact of an α7–nAChR calcium signal. It is well documented that presynaptic α7–nAChRs can promote transmitter release by virtue of their high relative permeability to calcium and activation of calcium release from internal stores (Bertrand et al., 1993; Séguéla et al., 1993; McGehee et al., 1995; Gray et al., 1996; Dickinson et al., 2008; Zhong et al., 2008; Albuquerque et al., 2009; Gu and Yakel, 2011). The size of the effective RRP can be influenced by calcium (Thanawala and Regehr, 2013). Furthermore, the efficacy of transmitter release is known to depend critically on the submicrometer location of calcium-permeable channels in the active zone (Augustine et al., 1991; Meinrenken et al., 2003; Schneggenburger and Neher, 2005).
A second possibility is that the interaction between α7–nAChRs and active zone components may increase RRP size by rearranging the presynaptic scaffold during the crosslinking rather than providing an acute effect on calcium-dependent transmitter release. The crosslinking of α7–nAChRs did, in fact, rearrange proteins comprising the active zone (Fig. 7B). This could, for example, help constrain the brake on release thought to be imposed by ELKS (Kaeser et al., 2009). However, if ELKS is involved, yet to be explained is how synaptic silencing by TeT immobilizes α7–nAChRs given that a global reduction in activity is thought to reduce ELKS at presynaptic terminals (Gundelfinger and Fejtova, 2012).
In either case, functional α7–nAChRs appear to be required for the enhancement of RRP by receptor crosslinking. This was inferred from the observation that MLA prevented the crosslinking from increasing RRP when added along with the Ab, whereas MLA in the absence of crosslinking had no effect on RRP. Although the Biot-Bgt used in the crosslinking of α7–nAChRs is a competitive antagonist of the receptors, only a single Biot-Bgt need be bound for Ab tethering of a receptor to form a cluster. Unoccupied sites remaining on the homopentameric α7–nAChR would have permitted functionality (Williams et al., 2011). Activation of the receptors in culture could have resulted either from choline in the medium (Alkondon et al., 1997, 1999) or by ACh released from cholinergic neurons in the cultures (Frotscher et al., 1986, 2000). Indeed, application of choline was able to activate the crosslinked receptors (Fig. 3D).
Crosslinking α7–nAChRs likely did more than simply immobilize the receptors. It should have increased receptor number in clusters such as those at presynaptic sites. This is expected because individual Biot-Bgt-bound receptors would remain mobile until within range of a receptor cluster for linkage. Reduced diffusion and increased dwell time of α7–nAChRs in presynaptic space is certainly consistent with elevated receptor density at such sites, comprising a cluster that could serve as a seed for crosslinking. Consistent with this, crosslinking increased the mean size of α7–nAChR clusters (Fig. 2A). When methods become available to image presynaptic α7–nAChRs directly, it will be interesting to determine how the number of clustered α7–nAChRs at presynaptic terminals varies with different physiological conditions.
The SPT approach allowed us to label and study specifically the dynamics of α7–nAChRs on axons. The differences seen in axonal versus dendritic α7–nAChR mobilities (Gómez-Varela et al., 2012) confirm that different populations can be distinguished in this way. An important challenge for the future will be identifying the molecular mechanisms responsible for reversibly recruiting α7–nAChRs to release sites as a function of regulatory need. Although the RNAi experiments initially implicate ELKS1, it is not clear how it participates or whether other components act as the primary sensors linking α7–nAChR mobility to the need for transmitter release. Additional analysis of complexes containing ELKS and α7–nAChRs will be needed to identify the key players.
The relationship between α7–nAChR immobility and transmitter release suggests a homeostatic mechanism that sustains an appropriate capacity for transmitter release. The range over which the mechanism operates appears to be physiological. The observed increase in transmitter release resulting from crosslinking α7–nAChRs correlates with a decrease in the mobility of postsynaptic AMPA receptors, consistent with a downstream effect. Although the manipulation could not distinguish presynaptic versus postsynaptic α7–nAChR contributions to the effect, the change in AMPA receptor mobility is consistent with previous results for the inverse situation, namely the finding that decreasing glutamate release increases AMPA receptor mobility at postsynaptic sites (Ehlers et al., 2007). Although glutamate release was the primary focus here, presynaptic α7–nAChRs are found on many kinds of terminals throughout the CNS (McGehee et al., 1995; Gray et al., 1996; Alkondon and Albuquerque, 2001; Fabian-Fine et al., 2001; Dajas-Bailador and Wonnacott, 2004; Dickinson et al., 2008; Zhong et al., 2008; Gu and Yakel, 2011). That, and the pervasiveness of cholinergic innervation (Umbriaco et al., 1995; Descarries et al., 1997; Bansal et al., 2000; Hanson and Landmesser, 2003; Witten et al., 2010), suggest that the regulation seen here may be widespread in the nervous system.
Footnotes
This work was supported by National Institutes of Health Grants NS012601, NS034569, and DA034216 and Tobacco-Related Disease Research Program Grants 17FT-0053 and 19XT-0072. We thank Xiao-Yun Wang and Jeff Schoellerman for expert technical assistance and Massimo Scanziani for comments on this manuscript.
The authors declare no competing financial interests.
References
- Adesnik H, Nicoll RA, England PM. Photoinactivation of native AMPA receptors reveals their real-time trafficking. Neuron. 2005;48:977–985. doi: 10.1016/j.neuron.2005.11.030. [DOI] [PubMed] [Google Scholar]
- Albuquerque EX, Pereira EF, Alkondon M, Rogers SW. Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol Rev. 2009;89:73–120. doi: 10.1152/physrev.00015.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkondon M, Albuquerque EX. Nicotinic acetylcholine receptor alpha7 and alpha4beta2 subtypes differentially control GABAergic input to CA1 neurons in rat hippocampus. J Neurophysiol. 2001;86:3043–3055. doi: 10.1152/jn.2001.86.6.3043. [DOI] [PubMed] [Google Scholar]
- Alkondon M, Pereira EF, Cortes WS, Maelicke A, Albuquerque EX. Choline is a selective agonist of alpha7 nicotinic acetylcholine receptors in the rat brain neurons. Eur J Neurosci. 1997;9:2734–2742. doi: 10.1111/j.1460-9568.1997.tb01702.x. [DOI] [PubMed] [Google Scholar]
- Alkondon M, Pereira EF, Eisenberg HM, Albuquerque EX. Choline and selective antagonists identify two subtypes of nicotinic acetylcholine receptors that modulate GABA release from CA1 interneurons in rat hippocampal slices. J Neurosci. 1999;19:2693–2705. doi: 10.1523/JNEUROSCI.19-07-02693.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashby MC, Maier SR, Nishimune A, Henley JM. Lateral diffusion drives øconstitutive exchange of AMPA receptors at dendritic spines and is regulated by spine morphology. J Neurosci. 2006;26:7046–7055. doi: 10.1523/JNEUROSCI.1235-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustine GJ, Adler EM, Charlton MP. The calcium signal for transmitter secretion from presynaptic nerve terminals. Ann NY Acad Sci. 1991;635:365–381. doi: 10.1111/j.1749-6632.1991.tb36505.x. [DOI] [PubMed] [Google Scholar]
- Bansal A, Singer JH, Hwang BJ, Xu W, Beaudet A, Feller MB. Mice lacking specific nicotinic acetylcholine receptor subunits exhibit dramatically altered spontaneous activity patterns and reveal a limited role for retinal waves in forming ON and OFF circuits in the inner retina. J Neurosci. 2000;20:7672–7681. doi: 10.1523/JNEUROSCI.20-20-07672.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand D, Galzi JL, Devillers-Thiéry A, Bertrand S, Changeux JP. Mutations at two distinct sites within the channel domain M2 alter calcium permeability of neuronal alpha 7 nicotinic receptor. Proc Natl Acad Sci U S A. 1993;90:6971–6975. doi: 10.1073/pnas.90.15.6971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biederer T, Scheiffele P. Mixed-culture assays for analyzing neuronal synapse formation. Nat Protoc. 2007;2:670–676. doi: 10.1038/nprot.2007.92. [DOI] [PubMed] [Google Scholar]
- Campbell NR, Fernandes CC, Halff AW, Berg DK. Endogenous signaling through α7-containing nicotinic receptors promotes maturation and integration of adult-born neurons in the hippocampus. J Neurosci. 2010;30:8734–8744. doi: 10.1523/JNEUROSCI.0931-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang KT, Berg DK. Voltage-gated channels block nicotinic regulation of CREB phosphorylation and gene expression in neurons. Neuron. 2001;32:855–865. doi: 10.1016/S0896-6273(01)00516-5. [DOI] [PubMed] [Google Scholar]
- Czöndör K, Mondin M, Garcia M, Heine M, Frischknecht R, Choquet D, Sibarita JB, Thoumine OR. Unified quantitative model of AMPA receptor trafficking at synapses. Proc Natl Acad Sci U S A. 2012;109:3522–3527. doi: 10.1073/pnas.1109818109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dajas-Bailador F, Wonnacott S. Nicotinic acetylcholine receptors and the regulation of neuronal signalling. Trends Pharmacol Sci. 2004;25:317–324. doi: 10.1016/j.tips.2004.04.006. [DOI] [PubMed] [Google Scholar]
- Deng L, Kaeser PS, Xu W, Südhof TC. RIM proteins activate vesicle priming by reversing autoinhibitory homodimerization of Munc13. Neuron. 2011;69:317–331. doi: 10.1016/j.neuron.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Descarries L, Gisiger V, Steriade M. Diffuse transmission by acetylcholine in the CNS. Prog Neurobiol. 1997;53:603–625. doi: 10.1016/S0301-0082(97)00050-6. [DOI] [PubMed] [Google Scholar]
- Dickinson JA, Kew JN, Wonnacott S. Presynaptic alpha 7- and beta 2-containing nicotinic acetylcholine receptors modulate excitatory amino acid release from rat prefrontal cortex nerve terminals via distinct cellular mechanisms. Mol Pharmacol. 2008;74:348–359. doi: 10.1124/mol.108.046623. [DOI] [PubMed] [Google Scholar]
- Dudel J, Kuffler SW. The quantal nature of transmission and spontaneous miniature potentials at the crayfish neuromuscular junction. J Physiol. 1961;155:514–529. doi: 10.1113/jphysiol.1961.sp006644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duguid IC, Smart TG. Presynaptic NMDA receptors. In: Van Dongen AM, editor. Biology of the NMDA receptor. Boca Raton, FL: CRC; 2009. Ch 14. [PubMed] [Google Scholar]
- Eccles JC. Presynaptic inhibition in the spinal cord. Prog Brain Res. 1964;12:65–91. doi: 10.1016/S0079-6123(08)60618-4. [DOI] [PubMed] [Google Scholar]
- Ehlers MD, Heine M, Groc L, Lee MC, Choquet D. Diffusional trapping of GluR1 AMPA receptors by input-specific synaptic activity. Neuron. 2007;54:447–460. doi: 10.1016/j.neuron.2007.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabian-Fine R, Skehel P, Errington ML, Davies HA, Sher E, Stewart MG, Fine A. Ultrastructural distribution of the α7 nicotinic acetylcholine receptor subunit in rat hippocampus. J Neurosci. 2001;21:7993–8003. doi: 10.1523/JNEUROSCI.21-20-07993.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes CC, Berg DK, Gómez-Varela D. Lateral mobility of nicotinic acetylcholine receptors on neurons is determined by receptor composition, local domain, and cell type. J Neurosci. 2010;30:8841–8851. doi: 10.1523/JNEUROSCI.6236-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frotscher M, Schlander M, Léránth C. Cholinergic neurons in the hippocampus a combined light- and electron-microscopic immunocytochemical study in the rat. Cell Tissue Res. 1986;246:293–301. doi: 10.1007/BF00215891. [DOI] [PubMed] [Google Scholar]
- Frotscher M, Vida I, Bender R. Evidence for the existence of non-GABAergic, cholinergic interneurons in the rodent hippocampus. Neuroscience. 2000;96:27–31. doi: 10.1016/S0306-4522(99)00525-4. [DOI] [PubMed] [Google Scholar]
- Goetze B, Grunewald B, Baldassa S, Kiebler M. Chemically controlled formation of a DNA/calcium phosphate coprecipitate: application for transfection of mature hippocampal neurons. J Neurobiol. 2004;60:517–525. doi: 10.1002/neu.20073. [DOI] [PubMed] [Google Scholar]
- Gómez-Varela D, Schmidt M, Schoellerman J, Peters EC, Berg DK. PMCA2 via PSD-95 controls calcium signaling by α7-containing nicotinic acetylcholine receptors on aspiny interneurons. J Neurosci. 2012;32:6894–6905. doi: 10.1523/JNEUROSCI.5972-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray R, Rajan AS, Radcliffe KA, Yakehiro M, Dani JA. Hippocampal synaptic transmission enhanced by low concentrations of nicotine. Nature. 1996;383:713–716. doi: 10.1038/383713a0. [DOI] [PubMed] [Google Scholar]
- Groc L, Heine M, Cognet L, Brickley K, Stephenson FA, Lounis B, Choquet D. Differential activity-dependent regulation of the lateral mobilities of AMPA and NMDA receptors. Nat Neurosci. 2004;7:695–696. doi: 10.1038/nn1270. [DOI] [PubMed] [Google Scholar]
- Gu Z, Yakel JL. Timing-dependent septal cholinergic induction of dynamic hippocampal synaptic plasticity. Neuron. 2011;71:155–165. doi: 10.1016/j.neuron.2011.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundelfinger ED, Fejtova A. Molecular organization and plasticity of the cytomatrix at the active zone. Curr Opin Neurobiol. 2012;22:423–430. doi: 10.1016/j.conb.2011.10.005. [DOI] [PubMed] [Google Scholar]
- Hanson MG, Landmesser LT. Characterization of the circuits that generate spontaneous episodes of activity in the early embryonic mouse spinal cord. J Neurosci. 2003;23:587–600. doi: 10.1523/JNEUROSCI.23-02-00587.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harms KJ, Tovar KR, Craig AM. Synapse-specific regulation of AMPA receptor subunit composition by activity. J Neurosci. 2005;25:6379–6388. doi: 10.1523/JNEUROSCI.0302-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heine M, Groc L, Frischknecht R, Béïque JC, Lounis B, Rumbaugh G, Huganir RL, Cognet L, Choquet D. Surface mobility of postsynaptic AMPARs tunes synaptic transmission. Science. 2008;320:201–205. doi: 10.1126/science.1152089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoze N, Nair D, Hosy E, Sieben C, Manley S, Herrmann A, Sibarita JB, Choquet D, Holcman D. Heterogeneity of AMPA receptor trafficking and molecular interactions revealed by superresolution analysis of live cell imaging. Proc Natl Acad Sci U S A. 2012;109:17052–17057. doi: 10.1073/pnas.1204589109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu M, Liu QS, Chang KT, Berg DK. Nicotinic regulation of CREB activation in hippocampal neurons by glutamatergic and nonglutamatergic pathways. Mol Cell Neurosci. 2002;21:616–625. doi: 10.1006/mcne.2002.1202. [DOI] [PubMed] [Google Scholar]
- Jones S, Sudweeks S, Yakel JL. Nicotinic receptors in the brain: correlating physiology with function. Trends Neurosci. 1999;22:555–561. doi: 10.1016/S0166-2236(99)01471-X. [DOI] [PubMed] [Google Scholar]
- Kaeser PS, Deng L, Chávez AE, Liu X, Castillo PE, Südhof TC. ELKS2alpha/CAST deletion selectively increases neurotransmitter release at inhibitory synapses. Neuron. 2009;64:227–239. doi: 10.1016/j.neuron.2009.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeser PS, Deng L, Fan M, Südhof TC. RIM genes differentially contribute to organizing presynaptic release sites. Proc Natl Acad Sci U S A. 2012;109:11830–11835. doi: 10.1073/pnas.1209318109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai H, Zago W, Berg DK. Nicotinic α7 receptor clusters on hippocampal GABAergic neurons: regulation by synaptic activity and neurotrophins. J Neurosci. 2002;22:7903–7912. doi: 10.1523/JNEUROSCI.22-18-07903.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khakh BS, Henderson G. Modulation of fast synaptic transmission by presynaptic ligand-gated cation channels. J Auton Nerv Syst. 2000;81:110–121. doi: 10.1016/S0165-1838(00)00111-9. [DOI] [PubMed] [Google Scholar]
- Ko J, Na M, Kim S, Lee JR, Kim E. Interaction of the ERC family of RIM-binding proteins with the liprin-alpha family of multidomain proteins. J Biol Chem. 2003;278:42377–42385. doi: 10.1074/jbc.M307561200. [DOI] [PubMed] [Google Scholar]
- Langer SZ. Presynaptic autoreceptors regulating transmitter release. Neurochem Int. 2008;52:26–30. doi: 10.1016/j.neuint.2007.04.031. [DOI] [PubMed] [Google Scholar]
- Larsen RS, Corlew RJ, Henson MA, Roberts AC, Mishina M, Watanabe M, Lipton SA, Nakanishi N, Pérez-Otaño I, Weinberg RJ, Philpot BD. NR3A-containing NMDARs promote neurotransmitter release and spike timing-dependent plasticity. Nat Neurosci. 2011;14:338–344. doi: 10.1038/nn.2750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Neff RA, Berg DK. Sequential interplay of nicotinic and GABAergic signaling guides neuronal development. Science. 2006;314:1610–1613. doi: 10.1126/science.1134246. [DOI] [PubMed] [Google Scholar]
- Lozada AF, Wang X, Gounko NV, Massey KA, Duan J, Liu Z, Berg DK. Glutamatergic synapse formation is promoted by α7-containing nicotinic acetylcholine receptors. J Neurosci. 2012;32:7651–7661. doi: 10.1523/JNEUROSCI.6246-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino H, Malinow R. AMPA receptor incorporation into synapses during LTP: the role of lateral movement and exocytosis. Neuron. 2009;64:381–390. doi: 10.1016/j.neuron.2009.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGehee DS, Heath MJ, Gelber S, Devay P, Role LW. Nicotine enhancement of fast excitatory synaptic transmission in CNS by presynaptic receptors. Science. 1995;269:1692–1696. doi: 10.1126/science.7569895. [DOI] [PubMed] [Google Scholar]
- Meinrenken CJ, Borst JG, Sakmann B. Local routes revisited: the space and time dependence of the Ca2+ signal for phasic transmitter release at the rat calyx of Held. J Physiol. 2003;547:665–689. doi: 10.1113/jphysiol.2002.032714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer AJ, Chen M, Thoreson WB. Lateral mobility of presynaptic L-type calcium channels at photoreceptor ribbon synapses. J Neurosci. 2011;31:4397–4406. doi: 10.1523/JNEUROSCI.5921-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikasova L, Groc L, Choquet D, Manzoni OJ. Altered surface trafficking of presynaptic cannabinoid type 1 receptor in and out synaptic terminals parallels receptor desensitization. Proc Natl Acad Sci U S A. 2008;105:18596–18601. doi: 10.1073/pnas.0805959105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miwa JM, Freedman R, Lester HA. Neural systems governed by nicotinic acetylcholine receptors: emerging hypotheses. Neuron. 2011;70:20–33. doi: 10.1016/j.neuron.2011.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opazo P, Labrecque S, Tigaret CM, Frouin A, Wiseman PW, De Koninck P, Choquet D. CaMKII triggers the diffusional trapping of surface AMPARs through phosphorylation of stargazin. Neuron. 2010;67:239–252. doi: 10.1016/j.neuron.2010.06.007. [DOI] [PubMed] [Google Scholar]
- Pyle JL, Kavalali ET, Piedras-Rentería ES, Tsien RW. Rapid reuse of readily releasable pool vesicles at hippocampal synapses. Neuron. 2000;28:221–231. doi: 10.1016/S0896-6273(00)00098-2. [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Stevens CF. Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron. 1996;16:1197–1207. doi: 10.1016/S0896-6273(00)80146-4. [DOI] [PubMed] [Google Scholar]
- Schneggenburger R, Neher E. Presynaptic calcium and control of vesicle fusion. Curr Opin Neurobiol. 2005;15:266–274. doi: 10.1016/j.conb.2005.05.006. [DOI] [PubMed] [Google Scholar]
- Séguéla P, Wadiche J, Dineley-Miller K, Dani JA, Patrick JW. Molecular cloning, functional properties, and distribution of rat brain α7: a nicotinic cation channel highly permeable to calcium. J Neurosci. 1993;13:596–604. doi: 10.1523/JNEUROSCI.13-02-00596.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof TC. The synaptic vesicle cycle revisited. Neuron. 2000;28:317–320. doi: 10.1016/S0896-6273(00)00109-4. [DOI] [PubMed] [Google Scholar]
- Südhof TC. The presynaptic active zone. Neuron. 2012;75:11–25. doi: 10.1016/j.neuron.2012.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tardin C, Cognet L, Bats C, Lounis B, Choquet D. Direct imaging of lateral movements of AMPA receptors inside synapses. EMBO J. 2003;22:4656–4665. doi: 10.1093/emboj/cdg463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thanawala MS, Regehr WG. Presynaptic calcium influx controls neurotransmitter release in part by regulating the effective size of the readily releasable pool. J Neurosci. 2013;33:4625–4633. doi: 10.1523/JNEUROSCI.4031-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triller A, Choquet D. New concepts in synaptic biology derived from single-molecule imaging. Neuron. 2008;59:359–374. doi: 10.1016/j.neuron.2008.06.022. [DOI] [PubMed] [Google Scholar]
- Umbriaco D, Garcia S, Beaulieu C, Descarries L. Relational features of acetylcholine, noradrenaline, serotonin and GABA axon terminals in the stratum radiatum of adult rat hippocampus (CA1) Hippocampus. 1995;5:605–620. doi: 10.1002/hipo.450050611. [DOI] [PubMed] [Google Scholar]
- Wang Y, Liu X, Biederer T, Südhof TC. A family of RIM-binding proteins regulated by alternative splicing: Implications for the genesis of synaptic active zones. Proc Natl Acad Sci U S A. 2002;99:14464–14469. doi: 10.1073/pnas.182532999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams DK, Stokes C, Horenstein NA, Papke RL. The effective opening of nicotinic acetylcholine receptors with single agonist binding sites. J Gen Physiol. 2011;137:369–384. doi: 10.1085/jgp.201010587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witten IB, Lin SC, Brodsky M, Prakash R, Diester I, Anikeeva P, Gradinaru V, Ramakrishnan C, Deisseroth K. Cholinergic interneurons control local circuit activity and cocaine conditioning. Science. 2010;330:1677–1681. doi: 10.1126/science.1193771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Zhu Y, Heinemann SF. Identification of sequence motifs that target neuronal nicotinic receptors to dendrites and axons. J Neurosci. 2006;26:9780–9793. doi: 10.1523/JNEUROSCI.0840-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong C, Du C, Hancock M, Mertz M, Talmage DA, Role LW. Presynaptic type III neuregulin 1 is required for sustained enhancement of hippocampal transmission by nicotine and for axonal targeting of α7 nicotinic acetylcholine receptors. J Neurosci. 2008;28:9111–9116. doi: 10.1523/JNEUROSCI.0381-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]