

Abstract
The loss of dopaminergic neurons in the substantia nigra pars compacta (SNc) and consequent depletion of striatal dopamine are known to underlie the motor deficits observed in Parkinson's disease (PD). Adaptive changes in dopaminergic terminals and in postsynaptic striatal neurons can compensate for significant losses of striatal dopamine, resulting in preservation of motor behavior. In addition, compensatory changes independent of striatal dopamine have been proposed based on PD therapies that modulate nondopaminergic circuits within the basal ganglia. We used a genetic strategy to selectively destroy dopaminergic neurons in mice during development to determine the necessity of these neurons for the maintenance of normal motor behavior in adult and aged mice. We find that loss of 90% of SNc dopaminergic neurons and consequent depletion of >95% of striatal dopamine does not result in changes in motor behavior in young-adult or aged mice as evaluated by an extensive array of motor behavior tests. Treatment of aged mutant mice with the dopamine receptor antagonist haloperidol precipitated motor behavior deficits in aged mutant mice, indicating that <5% of striatal dopamine is sufficient to maintain motor function in these mice. We also found that mutant mice exhibit an exaggerated response to l-DOPA compared with control mice, suggesting that preservation of motor function involves sensitization of striatal dopamine receptors. Our results indicate that congenital loss of dopaminergic neurons induces remarkable adaptions in the nigrostriatal system where limited amounts of dopamine in the dorsal striatum can maintain normal motor function.
Introduction
Parkinson's disease (PD) is the most common neurodegenerative movement disorder, affecting 1% of the population >60 years of age. Progressive degeneration of dopaminergic neurons in the substantia nigra pars compacta (SNc) and resultant depletion of dopamine from the striatum underlie the motor behavior deficits observed in PD patients and in animals that have sustained damage to the nigrostriatal pathway (Dauer and Przedborski, 2003). Although the central role of midbrain dopaminergic neurons in the pathogenesis of PD is well established, efforts to rescue degenerating SNc dopaminergic neurons in PD patients have been unsuccessful. Alternative strategies that bypass dopaminergic neuron function by altering activity of basal ganglia circuits independently of dopamine, such as deep-brain stimulation of the subthalamic nucleus or globus palidus (Ponce and Lozano, 2010) or antagonism of A2 adenosine receptors (Hickey and Stacy, 2012), have shown efficacy in relieving the motor symptoms of PD. These findings suggest that basal ganglia function can be restored independently of SNc dopaminergic neurons.
Motor behavior changes resulting from damage to dopaminergic neurons during development are of interest for several reasons. First, 6-hydroxydopamine (6-OHDA) administered to neonatal rats results in near complete depletion of striatal dopamine but produces less severe deficits in motor behavior compared with adult animals sustaining damage of the same magnitude (Breese et al., 2005). The mechanism underlying this behavior sparing is reportedly independent of dopamine (Bruno et al., 1985; Duncan et al., 1987). Thus, animal models in which dopamine is severely depleted during development provide an opportunity to study potential nondopaminergic mechanisms of plasticity in developing dopaminergic circuits. Animal models in which developmental damage to dopaminergic neurons is moderate are also of significant interest. Partial damage to the nigrostriatal pathway resulting from prenatal or early postnatal exposure to toxins renders mice more vulnerable to dopaminergic neuron damage later in life and has been proposed to predispose humans to PD (Carvey et al., 2003; Bakos et al., 2004; Barlow et al., 2007; Muthian et al., 2010). These studies have provided important insights into the effects of developmental damage of dopaminergic neurons on motor behavior and on increased vulnerability of dopaminergic neurons to a second toxin treatment in adulthood. However, the use of exogenous toxins to destroy dopaminergic neurons during development is limited by a number of factors, including uncertain specificity of the toxins and dosing limitations necessitated by the potential for systemic toxicity to pregnant animals. Models that avoid these confounds have the potential to provide new insights into the consequences of developmental dopaminergic neuron damage and into the mechanisms underlying plasticity in the developing nigrostriatal system.
Here we expressed diphtheria toxin in dopaminergic neurons to create a model of developmental damage that is specific for dopaminergic neurons. A broad spectrum of motor behavior tests demonstrated that, surprisingly, near complete loss of striatal dopamine produced no detectable motor behavior deficits in adult or aged mutant mice. Transient blockade of dopaminergic transmission using haloperidol precipitated motor dysfunction in mutant mice indistinguishable from that seen in control mice, indicating that the small amount of residual dopamine present in the striatum of these mice is sufficient to maintain normal motor function. In addition, mutant mice show increased locomotor behavior in response to l-DOPA at a dose that does not alter locomotor activity in control mice, suggesting that the mechanism by which motor behavior is preserved in DAT-DTA (Dopamine transporterCre-RosafloxSTOPfloxdta) mice involves functional sensitization of striatal dopamine receptors.
Materials and Methods
Animal generation.
Institutionally approved protocols were followed for all animal studies. To generate mice with conditional expression of attenuated diphtheria toxin (DTA) in dopaminergic neurons, mice expressing DTA from the Rosa locus (RosafloxSTOPfloxdta/+) (Wu et al., 2006) were crossed with heterozygous mice expressing Cre from the dopamine transporter (DAT) locus (DATCre/+; Zhuang et al., 2005; Fig. 1A). These experimental mice are hereafter referred to as DAT-DTA mice (RosafloxSTOPfloxdta/+:DATCre/+). Control mice used in this study are littermates or age-matched mice that have one DTA allele but no Cre allele (RosafloxSTOPfloxdta/+:DAT+/+). Previous studies have found no difference in locomotor activity in DAT+/− and wild-type mice (Giros et al., 1996).
Figure 1.

Dramatic loss of SNc and VTA neurons in DAT-DTA mice. A, Schematic illustrating breeding strategy to produce DAT-DTA mice. B, C, Weight of male and female DAT-DTA and control mice. Female DAT-DTA mice weigh significantly less than control mice at 3–5 and 10–21 months. Male DAT-DTA mice weigh significantly less than control mice at 10–21 months (mean ± SEM, Student's t test, control vs DAT-DTA at each time point; *p < 0.05, **p < 0.01, ***p < 0.001). D, Loss of TH-positive neurons in the ventral midbrain of DAT-DTA mice. The number of TH-positive neurons is significantly decreased in the SNc and VTA of DAT-DTA mice compared with control mice at all ages studied (mean ± SEM, Student's t test, control vs DAT-DTA at each time point; *p < 0.05, **p < 0.01, ***p < 0.001). P0, N = 3 of each genotype; 1.5 months, N = 2 of each genotype; 5 months, N = 4 of each genotype; 12 months, N = 3 of each genotype. E, F, Nissl-labeled coronal brain sections from 5-month-old mice showing large neurons in the SNc of control mice (E, arrows). These large neurons are absent in the SNc of DAT-DTA mice (F, arrows).
Behavior testing.
Behavior testing was performed using male and female mice from 1.5 to 24 months of age. The experimenter was blind to the genotypes of the mice and to drug treatment during data acquisition.
Gross motor function (Rotarod).
An accelerating Rotarod (Ugo Basile) was used to evaluate motor coordination and balance. Five consecutive acceleration trials were performed with a 5 min interval between trials as described previously (Montana et al., 2009).
Sensorimotor battery.
Tests in the sensorimotor battery were used as previously described to evaluate balance, strength, coordination, and skilled forelimb use (Jain et al., 2006; Meredith and Kang, 2006). In addition, we evaluated movement initiation by measuring the time it took a mouse to exit an 18 × 18 cm square (all four paws outside the square) marked on a flat horizontal surface.
Locomotor activity (open field).
Locomotor activity was measured in an open field using a VersaMax Animal Activity Monitoring System (AccuScan Instruments). Mice were habituated to the test room in their cages for 1 h before testing. Locomotor activity was assessed by recording photo beam breaks in a 42 (length) × 42 (width) × 30 (height) cm chamber for 60 min. Total distance traveled, time spent moving, and the number of beam breaks (horizontal activity) were calculated for the entire chamber.
Gait analysis.
Mice were trained to walk down a narrow walkway (1 m × 6.5 cm) leading to a dark box. Following training, the walkway was covered with plain white paper and ink was applied to the hindpaws by pressing the mouse's foot onto an ink pad. Toe spread (TS), intertoe spread (ITS), stride width (SW), and stride length (SL) were measured as previously described (Taylor et al., 2001). For each mouse, a minimum of two footprints (TS and ITS) or two sets of prints (SW and SL) were measured for each parameter.
Haloperidol treatment.
Twelve-month-old control mice and DAT-DTA mice were given a single intraperitoneal injection of haloperidol (Sigma-Aldrich) dissolved in DMSO at a dose of 1 mg/kg. Ten minutes after administration of haloperidol, movement initiation was evaluated as described above. Catalepsy was evaluated by measuring the time required for the mouse to step down after its front or hindpaws were placed on a 3 cm block (Weihmuller and Bruno, 1989; Mohajjel Nayebi, 2010). Following these two tests (∼15 min after haloperidol administration), mice were tested in the open field (as above). Following the measurement of open-field activity (∼80 min after haloperidol administration) movement initiation and catalepsy were again evaluated.
Assessment of possible hypersensitivity to dopamine.
Six-to-seven-month-old control and DAT-DTA mice were given a single intraperitoneal injection of l-DOPA (25 mg/kg) and carbidopa (10 mg/kg; both drugs obtained from Sigma-Aldrich) dissolved in PBS containing 0.25% ascorbic acid. Ten minutes after administration of l-DOPA, movement initiation was evaluated as described above. Following this test (∼15 min after l-DOPA administration) mice were tested in the open field (as above). After the measurement of open-field activity (∼80 min after l-DOPA administration) movement initiation was again evaluated.
Quantitative real-time PCR.
RNA was purified from dorsal striatum from 12-month-old DAT-DTA (N = 9) and control (N = 7) mice. RNA (1 μg) was reverse transcribed using M-MLV Reverse Transcriptase (Invitrogen). Quantitative real-time PCR (qRT-PCR) was performed using Fast SYBR Green Master Mix (Applied Biosystems) and reactions were run in an Applied Biosystems 7900HT Real Time Thermal Cycler. All reactions were run in triplicate and relative fold changes in RNA amount were calculated by the ΔΔCT method using GAPDH to normalize samples (Livak and Schmittgen, 2001). qRT-PCR primers were designed by Primer Express software (Applied Biosystems). Primer sequences are as follows (5′→3′): D1: forward-GCCATTCTGCATTGATTCCA, reverse-ATTCGCCCAGCCAAACC; D2: forward-CGTTATCATGAAGTCTAATGGGAGTTT, reverse-GGCGGGCAGCATCCA.
Immunohistochemistry.
At the conclusion of behavior analysis, mice were deeply anesthetized and perfused through the heart with PBS followed by 4% paraformaldehyde in PBS. Brains were removed immediately after perfusion, postfixed overnight in paraformaldehyde, cryoprotected with 30% sucrose, and rapidly frozen on dry ice. Brains from postnatal day 0 (P0) mice were sectioned in the coronal plane at 20 μm and were collected in series on slides. For 1.5–12-month-old mice, 40 μm coronal brain sections were collected in PBS. Free-floating (1.5–12-month-old mice) coronal sections or slides with coronal sections (P0 mice) of midbrain and striatum were labeled with an antibody to tyrosine hydroxylase (TH; Millipore Bioscience Research Reagents) as previously described (Jain et al., 2006). TH-labeled neurons in the SNc and the ventral tegmental area (VTA) were counted in every fourth section by an investigator blinded to genotype. Profiles identified as neurons were TH-positive profiles that contained a nucleus and at least one process. The total number of neurons was determined by multiplying the number of neurons counted by four. The entire SNc and VTA were included. For SNc counts, all sections from Plate 52-63 (Franklin and Paxinos, 1997) were included. For VTA counts, all sections from Plate 55-63 were included. The border between the SNc and VTA was determined in each section according to the borders outlined for these structures in the Franklin and Paxinos Brain Atlas (Franklin and Paxinos, 1997). TH-positive neurons were also counted in all of the nuclei of the hypothalamus, in the dorsal thalamus, and in the periaqueductal gray of the same 5-month-old control and DAT-DTA mice used for SNc and VTA counts. Neurons were identified as described above and were counted in every fourth section (40 μm). The total neuron number for each nucleus was obtained by multiplying the neuron count for each nucleus by four. Individual hypothalamic nuclei were identified based on borders outlined for these structures in the Franklin and Paxinos Brain Atlas. Sections corresponding to Plate 27-54 were included in these counts (Franklin and Paxinos, 1997).
Using an unbiased stereological method for determining the areal fraction occupied by an object (in this case TH-positive fibers) as previously described (Gundersen et al., 1988; Jain et al., 2006), TH-positive fibers were quantified in the dorsal and ventral striatum of 5-month-old DAT-DTA and control mice. For fiber counts in the dorsal striatum, three 40 μm coronal sections, corresponding to Plates 23, 26, and 29 (Franklin and Paxinos, 1997) were sampled. Two images from each section were used in the analysis for a total of six images per brain. For the ventral striatum, three 40 μm coronal sections corresponding to Plates 21, 23, and 26 were sampled using two images per section for a total of six images per brain. The sections of striatum used for this analysis were obtained from the same brains that provided midbrain sections for 5-month-old SNc and VTA neuron counts.
Evaluation of dopaminergic neurons in the gut was performed on whole-mount peeled sections of the submucosa from terminal ileum using TH immunohistochemistry as previously described (Li et al., 2004). Briefly, intestines were harvested from mice perfused with 4% paraformaldehyde, fixed flat overnight, rinsed with PBS, peeled, and prepared for TH immunohistochemistry using diaminobenzidine for visualization.
Microscopy.
All images were captured using an upright microscope equipped for epifluorescence microscopy (Nikon 80i; CoolSnapES camera). Images were processed using MetaMorph or Adobe Photoshop software using global adjustments in brightness and contrast.
Biochemistry.
Levels of dopamine and its metabolites homovanillic acid (HVA) and 3,4-dihydroxyphenylacetic acid (DOPAC) were determined in samples of dorsal striatum using HPLC with electrochemical detection as previously described (Pehek et al., 1992). Mice were killed by cervical dislocation. The brain was rapidly removed, frozen on dry ice, and cut into 1 mm slices using a precooled mouse brain matrix (Stoelting). The dorsal striatum was dissected from slices corresponding to Plate 23–31 in the Franklin and Paxinos Mouse Brain Atlas (Franklin and Paxinos, 1997).
Statistical analysis.
Data are reported as the mean ± SEM. Statistically significant differences between multiple groups were determined using two-way ANOVA and Bonferroni post-test. Comparisons between two groups were determined using the Student's t test.
Results
At all ages examined, DAT-DTA mice appear healthy and are indistinguishable from littermate control mice. DAT-DTA mice were weaned successfully with littermates and did not exhibit any gross deficits in drinking or feeding behavior. However, at 3–5 months of age, female DAT-DTA mice weighed on average 10% less than female control mice and at 10–21 months of age both male and female DAT-DTA mice weighed ∼20% less than age-matched and sex-matched control mice (Fig. 1B,C). DAT-DTA mice survived to 24 months (at which time they were killed), similar to control mice.
Expression of DTA from the DAT locus results in rapid, progressive loss of dopaminergic neurons in the SNc and VTA
Loss of TH-positive neurons in the SNc was readily apparent by P0 in DAT-DTA mice (Figs. 1D, 2A–D). Nissl staining of sections adjacent to those used for TH labeling show that the loss of TH-positive neurons is due to cell loss in the SNc and VTA and not to loss of TH expression (Fig. 1E,F). Quantification of the number of TH-positive cells revealed a significant 73% decrease in the number of TH-positive neurons in the SNc of DAT-DTA mice compared with control mice at P0 (Fig. 1D). The remaining TH-positive cells appeared abnormal compared with TH-positive cells in the SNc of control mice at this stage (Fig. 2E,F,H). Remaining neurons had neurites with large varicose swellings (Fig. 2F). In addition, a large number of cells were swollen and appeared to be disintegrating, suggesting active loss of neurons at this time point (Fig. 2H). By 1.5 months of age, TH-positive cell loss in the SNc of DAT-DTA mice was 90% compared with control mice (Fig. 1D). No additional cell loss relative to control mice was observed in the SNc through 12 months of age. Loss of TH-positive cells was less in the VTA than in the SNc of DAT-DTA mice at all ages analyzed as has been reported in PD patients and in several animal models of PD (Dauer and Przedborski, 2003). This pattern of cell loss in DAT-DTA mice is consistent with reported lower levels of DAT expression in the VTA compared with the SNc (Storch et al., 2004). Cell loss in the VTA was also progressive. In the VTA of DAT-DTA mice, the number of TH-positive cells was significantly decreased by 50% at P0 (Figs. 1D, 2A–D). By 1.5 months of age, 72% of TH-positive neurons were lost in the VTA of DAT-DTA mice. The number of TH-positive neurons in the VTA of DAT-DTA mice further declined by 85% at 12 months of age (Figs. 1D, 2I,J). At all ages examined, remaining TH-positive neurons in the SNc and VTA exhibited an abnormal appearance, including nonsmooth, irregular profiles and distended neurites with large varicose swellings (Fig. 2F,H). As expected, degeneration was specific to dopaminergic neurons. TH-positive noradrenergic neurons in the locus ceruleus of DAT-DTA mice have a normal morphology (Fig. 2G) and do not appear to be decreased in number compared with control mice, demonstrating that DTA expression is restricted to dopaminergic (DAT-positive) neurons.
Figure 2.
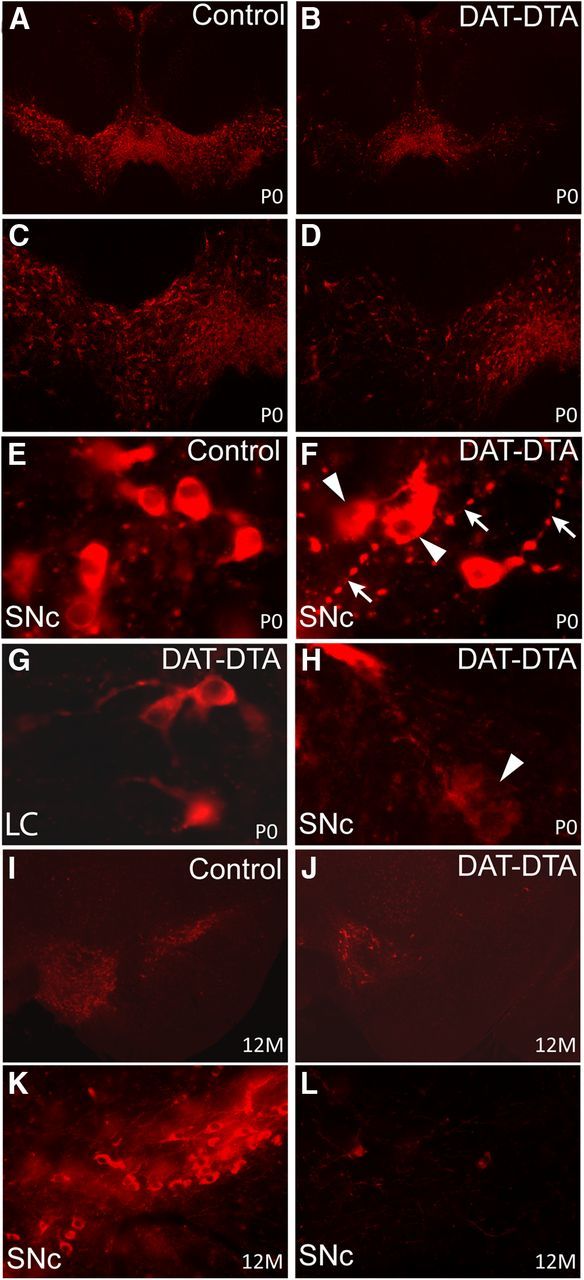
Loss of TH-positive neurons in the ventral midbrain of DAT-DTA mice. A–D, Fewer TH-positive neurons are observed in the SNc and VTA of DAT-DTA mice at P0 compared with control mice. TH-positive neuron loss is less severe in the VTA at this age. E, F, H, At P0, TH-positive neurons in the SNc of DAT-DTA mice (F, H) appear abnormal compared with neurons in the SNc of control mice (E). TH-positive neurons in the SNc of DAT-DTA mice (F, H) appear to be actively degenerating (arrowheads) and have neurites with large varicose swellings (F, arrows). G, TH-positive neurons in the locus ceruleus (LC) of DAT-DTA mice have a normal appearance. I–L, At 12 months of age, fewer TH-positive neurons are observed in the SNc and VTA of DAT-DTA mice compared with control mice. TH-positive neuron loss appears to be more severe at 12 months compared with P0 in both the SNc and VTA of DAT-DTA mice.
In contrast to other models of dopaminergic neuron damage (Breese and Traylor, 1972; Zhou and Palmiter, 1995; Kobayashi et al., 2004), we did not observe dramatic weight loss and wasting in DAT-DTA mice (Fig. 1B,C). Wasting in some of these prior studies has been attributed to abnormalities in the hypothalamus and/or damage to dopaminergic neurons in the gastrointestinal (GI) tract (Kobayashi et al., 2004). In DAT-DTA mice, expression of the DTA toxin is dependent on DAT-Cre expression. DAT is expressed at significantly lower levels in dopaminergic neurons of the hypothalamus compared with dopaminergic neurons in the midbrain (Storch et al., 2004). We counted TH-positive neurons in the nuclei of the hypothalamus. We found no significant difference in the number of TH-positive neurons in the hypothalamus of 5-month-old DAT-DTA mice compared with control mice (Fig. 3A). In addition, the morphology of TH-positive neurons is similar in the hypothalamus of DAT-DTA and control mice (Fig. 3B,C).
Figure 3.
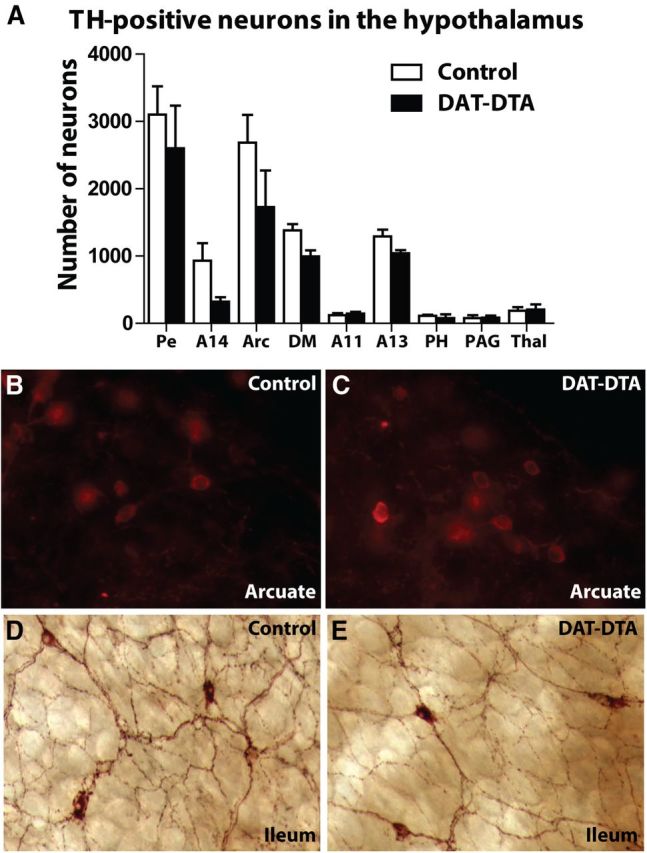
Dopaminergic neurons are normal in the hypothalamus and ileum of DAT-DTA mice. A, Counts of TH-positive neurons in the nuclei of the hypothalamus reveal no significant differences in neuron number between 5-month-old control mice and DAT-DTA mice. Pe, Periventricular and paraventricular nucleus; Arc, arcuate nucleus; DM, dorsomedial nucleus; PH, posterior hypothalamus; PAG, periaqueductal gray; Thal, dorsal thalamus (mean ± SEM, Student's t test, p > 0.05; N = 3 control mice, N = 3 DAT-DTA mice). B, C, TH-positive neurons in the arcuate nucleus of 5-month-old control mice (B) and DAT-DTA mice (C) appear similar. D, E, TH-positive neurons in the ileum of control mice (D) and DAT-DTA mice (E) appear similar in morphology and density. Representative images are from one of three control and one of three DAT-DTA mice examined.
DAT expression is also reportedly lower in the gut compared with the brain (Li et al., 2004). We examined TH-labeled intrinsic dopaminergic neurons in the terminal ileum of DAT-DTA and control mice. We found that both the apparent density and morphological appearance of TH-labeled dopaminergic neurons in the terminal ileum was similar in DAT-DTA mice and control mice (Fig. 3D,E).
Locomotor activity is not impaired in DAT-DTA mice
Locomotor activity in an open field has been frequently used as a measure of motor impairment following neurotoxin-induced damage to midbrain dopaminergic neurons and in rodent genetic models of PD (Dunnett and Lelos, 2010; Taylor et al., 2010). Surprisingly, open-field activity was not different in DAT-DTA mice compared with control mice at 1.5, 5, 12, or 18–24 months of age. The total number of horizontal movements, distance traveled, and time spent moving were not different during the 1 h testing period in DAT-DTA mice compared with age-matched or littermate control mice (Fig. 4A–C). We did observe an age-dependent decrease in all three measures of spontaneous locomotor activity in control mice as reported previously (Gage et al., 1984; Da Silva Costa et al., 2009) and in DAT-DTA mice.
Figure 4.

DAT-DTA mice do not show motor behavior deficits compared with control mice. A–C, Open-field test: DAT-DTA mice are not significantly different from control mice at any age studied (mean ± SEM, 2-Way ANOVA, Bonferroni post-test, p > 0.05). Control mice at 1.5 months exhibit significantly greater locomotor activity compared with 12-month-old control mice (distance, time, activity, p < 0.05). DAT-DTA mice an 1.5 months exhibit significantly greater locomotor activity compared with 12-month-old DAT-DTA mice (distance, time, p < 0.001; activity, p < 0.01). Control mice at 1.5 months exhibit significantly greater locomotor activity compared with 18–24-month-old control mice (activity, p < 0.05). DAT-DTA mice at 1.5 months exhibit significantly greater locomotor activity compared with 18–24-month-old DAT-DTA mice (distance, p < 0.01; time, p < 0.001; activity, p < 0.05). Control mice at 5 months exhibit significantly greater locomotor activity compared with 12-month-old control mice (distance, time, activity, p < 0.001). DAT-DTA mice at 5 months exhibit significantly greater locomotor activity compared with 12-month-old DAT-DTA mice (time, p < 0.05). Control mice at 5 months exhibit significantly greater locomotor activity compared with 18–24-month-old control mice (distance, time, activity, p < 0.001). Two-way ANOVA, Bonferroni post-test was used for all the above comparisons. Five months, N = 9 control mice, 8 DAT-DTA mice; 12 months: N = 9 control mice, 12 DAT-DTA mice; 18–24 months: N = 8 control mice, 6 DAT-DTA mice. D–H, Rotarod test: two-way ANOVA, Bonferroni post-test was used for all the following comparisons. Performance on the Rotarod is not different for DAT-DTA mice compared with control mice at 5 or 12 months (D, E, p > 0.05). At 18–24 months, the performance of DAT-DTA mice is significantly less than control mice in Trials 2–5 (F, *p < 0.05, **p < 0.01, ***p < 0.001). At 18–24 months, the performance of control mice is significantly better in Trials 3–5 compared with Trial 1 (F, Trial 1 vs 3, p < 0.01; Trial 1 vs 4, p < 0.001; Trial 1 vs 5, p < 0.01). The performance of 18–24-month-old DAT-DTA mice is not significantly different between any of the five trials (F, p > 0.05). Five-month-old control mice perform significantly better than 12 or 18–24 month control mice. Five month DAT-DTA mice perform significantly better than 12 or 18–24 month DAT-DTA mice (G, H, *p < 0.05, **p < 0.01, ***p < 0.001). Five months: N = 13 control mice, 12 DAT-DTA mice; 12 months: N = 9 control mice, 12 DAT-DTA mice; 18–24 months: N = 11 control mice, 6 DAT-DTA mice.
Performance on the Rotarod is not impaired in DAT-DTA mice
At 5 (Fig. 4D) and 12 (Fig. 4E) months of age, the time spent on an accelerating Rotarod was not different in each of five consecutive trials for DAT-DTA mice compared with control mice. However, both control mice (Fig. 4G) and DAT-DTA mice (Fig. 4H) exhibited an age-dependent decrease in the amount of time on the accelerating Rotarod between 5 and 12 months and between 5 and 18–24 months. At 18–24 months of age, performance of DAT-DTA mice in Trial 1 on the Rotarod was not different from control mice, suggesting equivalent motor ability in the two groups (Fig. 4F). However, 18–24-month-old DAT-DTA mice were not able to remain on the accelerating rod for as long as control mice in the second through fifth trials. The performance of control mice improved between Trial 1 and Trials 3–5 while the performance of DAT-DTA mice did not improve after Trial 1. This result suggests the possibility that the differences between DAT-DTA and control mice in Trials 2–5 may be due to a learning impairment in aged DAT-DTA mice. Learning deficits have been observed in rats depleted of dopamine in the neonatal period (Shaywitz et al., 1976; Whishaw et al., 1987; Archer et al., 1988).
Sensorimotor function is not impaired in DAT-DTA mice
To determine whether the extensive loss of SNc dopaminergic neurons alters sensorimotor behavior of adult or aged DAT-DTA mice, we performed a battery of sensorimotor tests. The pole test is used to evaluate performance of a complex motor task that requires skilled forelimb use, strength, and balance (Meredith and Kang, 2006). At all ages studied, DAT-DTA mice were not different from control mice either in the time required to turn around and orient downward on a vertical pole (Fig. 5A) or the time required to climb down from the pole (Fig. 5B). We did observe an age-related increase in the time to orient downward and the time to climb down in both control and DAT-DTA mice.
Figure 5.
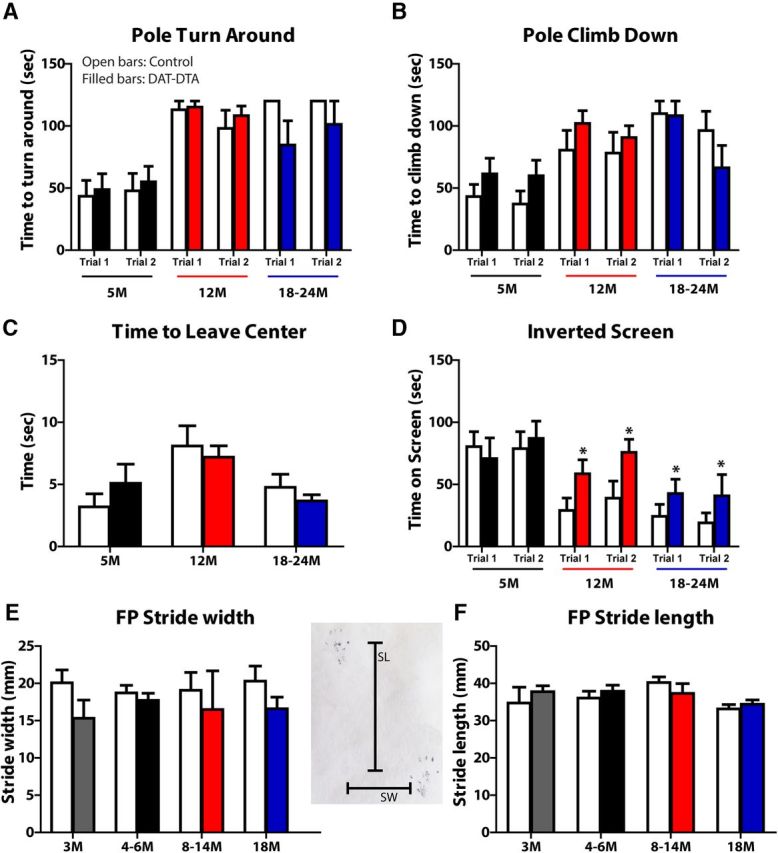
Motor behavior and gait are not impaired in DAT-DTA mice compared with control mice. A, B, Pole test: performance in the pole test is not different in DAT-DTA mice compared with control mice at any age studied (mean ± SEM. Student's t test). There is an age-dependent decrease in performance on the pole test in both DAT-DTA and control mice (2-Way ANOVA, Bonferroni post-test). Pole turn around (A): control mice: 5 vs 12 months, Trial 1: p < 0.001; Trial 2: p < 0.05; 5 vs 18–24 months, Trial 1: p < 0.001; Trial 2: p < 0.001. DAT-DTA mice: 5 vs 12 months, Trial 1: p < 0.001; Trial 2: p < 0.01; 5 vs 18–24 months, Trial 1: p > 0.05; Trial 2: p < 0.05. Pole climb down (B): control mice: 5 vs 12 months, Trial 1: p > 0.05; Trial 2: p > 0.05; 5 vs 18–24 months, Trial 1: p < 0.001; Trial 2: p < 0.01. DAT-DTA mice: 5 vs 12 months, Trial 1: p < 0.05; Trial 2: p > 0.05; 5 vs 18–24 months, Trial 1: p < 0.05; Trial 2: p > 0.05. Five months: N = 13 control mice, N = 12 DAT-DTA mice; 12 months: N = 9 control mice, N = 12 DAT-DTA mice; 18–24 months: N = 8 control mice, N = 6 DAT-DTA mice. C, Movement-initiation test: movement initiation is not significantly different in DAT-DTA mice compared with control mice at any age studied (mean ± SEM, Student's t test, p > 0.05). Five months: N = 9 control mice, N = 7 DAT-DTA mice; 12 months: N = 9 control mice, N = 12 DAT-DTA mice; 18–24 months: N = 8 control mice, N = 6 DAT-DTA mice. D, Inverted screen test: at 5 months, time on an inverted screen is not different in DAT-DTA mice compared with control mice (mean ± SEM, Student's t test, p > 0.05). At 12 and 18–24 months DAT-DTA mice remain on an inverted screen significantly longer than control mice (mean ± SEM, Student's t test, *p < 0.05). There is an age-dependent decrease in performance on the inverted screen test in control mice: Trial 1: 5 vs 12 months, p < 0.01; 5 vs 18–24 months, p < 0.01; Trial 2: 5 vs 12 months, p > 0.05; 5 vs 18–24 months, p < 0.01 (mean ± SEM, 2-Way ANOVA, Bonferroni post-test). Five months: N = 9 control mice, N = 7 DAT-DTA mice; 12 months: N = 9 control mice, N = 12 DAT-DTA mice; 18–24 months, N = 7 control mice, N = 6 DAT-DTA mice. E, F, Footprint test: there is no difference in SW or SL between DAT-DTA mice and control mice at any age studied (mean ± SEM, Student's t test, p > 0.05). Three months: N = 5 control mice, N = 4 DAT-DTA mice; 4–6 months: N = 8 control mice, N = 8 DAT-DTA mice; 8–14 months: N = 4 control mice, N = 3 DAT-DTA mice; 18 months: N = 6 control mice, N = 3 DAT-DTA mice.
The platform test evaluates balance and the ledge test evaluates balance and strength. The performance of DAT-DTA mice (N = 13 control, 12 DAT-DTA) in the platform and ledge tests was not different from age-matched control mice at any age studied (data not shown). Tests designed to evaluate the initiation and speed of movement, including measurement of the time required to leave the center of a square (18 × 18 cm; Fig. 5C) or the time required to reach the top of a screen inclined at 60 or 90° from the horizontal (data not shown) revealed no significant differences in performance between DAT-DTA mice and control mice at any age studied. PD patients exhibit decreased muscle strength that may be a secondary consequence of decreased activity or a direct result of dopamine loss (Cano-de-la-Cuerda et al., 2010). We used the inverted screen test to evaluate muscle strength in DAT-DTA mice (Tillerson et al., 2002; Fig. 5D). At 5 months of age, DAT-DTA mice remained on the inverted screen for the same amount of time as control mice. At 12 and 18–24 months, DAT-DTA mice performed better than control mice, remaining on the screen twice as long as control mice. This result suggests that DAT-DTA mice do not exhibit decreased muscle strength. We also observed an age-dependent decrease in the amount of time on the inverted screen for both control and DAT-DTA mice.
Gait analysis
Gait analysis has been used previously to document motor impairment in a number of rodent models, including models of nigrostriatal dysfunction (Tillerson et al., 2002; Byler et al., 2009; Taylor et al., 2010). Our analysis revealed no differences in SW (Fig. 5E) or SL (Fig. 5F) in DAT-DTA mice compared with age-matched control mice at any age studied. In addition, measures of paw length, TS, and ITS were not different between DAT-DTA mice and control mice at any age studied (data not shown).
Marked reduction in TH-positive fiber density in the dorsal and ventral striatum of DAT-DTA mice
The absence of motor behavior deficits in DAT-DTA mice, despite near complete loss of SNc dopaminergic neurons, prompted us to evaluate dopaminergic innervation of the striatum. We examined TH-positive fibers in the dorsal and ventral striatum of DAT-DTA mice to determine whether dopaminergic cell loss in the SNc and VTA is accompanied by loss of striatal dopaminergic afferents. At P0, TH-positive striatal afferents are greatly reduced in the dorsal striatum of DAT-DTA mice compared with control mice (Fig. 6A,B). At this age, fiber density is also reduced in the ventral striatum of DAT-DTA mice, but the reduction in the ventral striatum appears less severe than the reduction in the dorsal striatum (Fig. 6B, arrow). TH-positive fiber density appears to be further decreased in the dorsal striatum of DAT-DTA mice at 5 (Fig. 6D,E) and 12 months of age (data not shown), which is consistent with the increased loss of dopaminergic neurons from the SNc after P0 (Fig. 1D). We quantified the areal fraction of TH-positive fibers in the dorsal and ventral striatum of 5-month-old DAT-DTA and control mice using an unbiased stereological method as previously described (Gundersen et al., 1988; Jain et al., 2006). At 5 months of age, TH-positive fiber density is decreased by ∼93% in the dorsal striatum of DAT-DTA mice compared with control mice (Fig. 6C–E). At this age, TH-positive fiber density is decreased by ∼58% in the ventral striatum of DAT-DTA mice compared with control mice (Fig. 6C,F,G). The more severe fiber loss in the dorsal striatum compared with the ventral striatum (p < 0.001) is consistent with the greater magnitude of SNc cell loss compared with VTA cell loss at this age (Fig. 1D).
Figure 6.
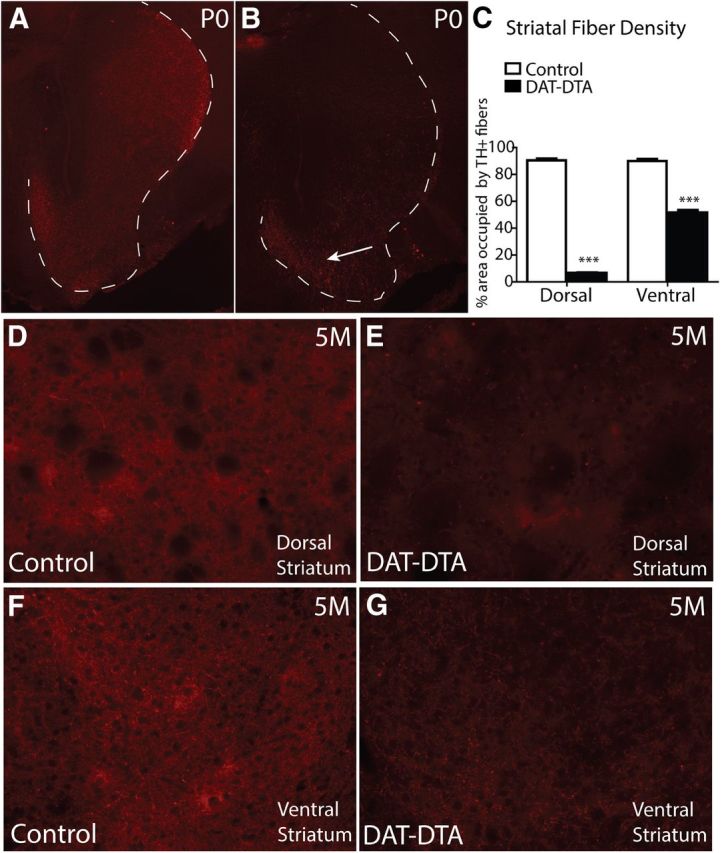
Loss of TH-positive fibers in the dorsal and ventral striatum of DAT-DTA mice. A, B, At P0, TH-positive fibers are decreased in the dorsal striatum of DAT-DTA mice compared with control mice. TH-positive fiber density is less affected in the ventral striatum of DAT-DTA mice (B, arrow). The white dotted line in A and B outlines the area of the dorsal and ventral striatum. C, Quantification of TH-positive fiber density in the striatum of 5-month-old mice reveals a significant decrease in fiber density in both the dorsal and ventral striatum of DAT-DTA mice compared with control mice (mean ± SEM, Student's t test, ***p < 0.001). N = 3 control mice, N = 3 DAT-DTA mice. D–G, Representative images used for quantifying fiber density showing decreased TH-positive fiber density in both the dorsal and ventral striatum of DAT-DTA mice compared with control mice.
Biochemical analysis reveals a marked reduction of striatal dopamine in DAT-DTA mice
Compensatory mechanisms that maximize dopaminergic transmission have been identified following damage to the nigrostriatal dopaminergic pathway (Zigmond et al., 1984, 1990). These include increased dopamine synthesis and increased dopamine release (suggested by an increase in the ratio of dopamine metabolites to dopamine). We used HPLC to quantify dopamine and the dopamine metabolites HVA and DOPAC in the dorsal striatum of DAT-DTA and control mice. Dopamine, DOPAC, and HVA are significantly decreased in the dorsal striatum of DAT-DTA mice compared with control mice at 2, 5–7, and 12–24 months of age (Fig. 7A). The ratio of DOPAC to dopamine was not different in DAT-DTA mice compared with control mice at any age studied (Fig. 7B). At 2 months of age, the ratio of HVA to dopamine was not significantly different in DAT-DTA mice compared with control mice (Fig. 7C). However, at 5–7 and 12–24 months of age, the ratio of HVA to dopamine was significantly increased in DAT-DTA mice compared with control mice.
Figure 7.
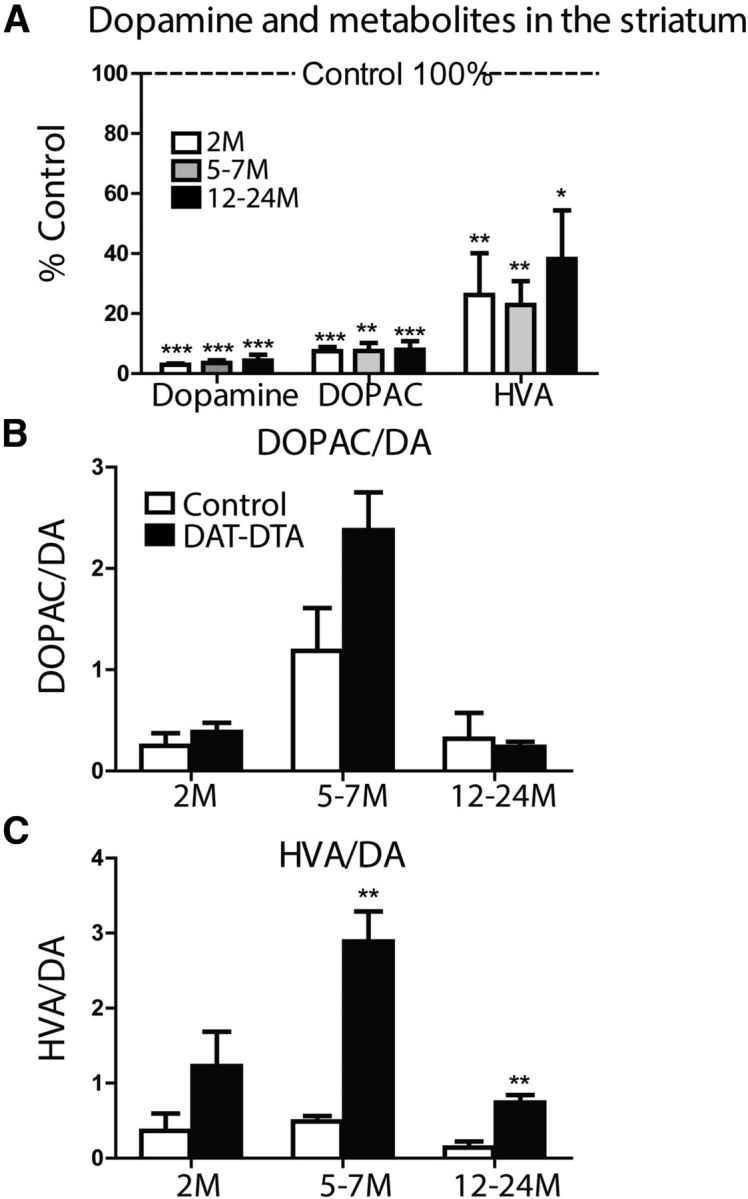
Dopamine, DOPAC, and HVA are depleted in the dorsal striatum of DAT-DTA mice. A, Dopamine, DOPAC, and HVA are decreased in the dorsal striatum of DAT-DTA mice compared with control mice at all ages studied (mean ± SEM, Student's t test, control vs DAT-DTA at each time point; *p < 0.05, **p < 0.01, ***p < 0.001). B, The ratio of DOPAC to dopamine is not significantly different between DAT-DTA mice and control mice at any age studied (mean ± SEM, Student's t test, p > 0.05). C, The ratio of HVA to dopamine is significantly increased in DAT-DTA mice compared with control mice at 5–7 and 12–24 months of age (mean ± SEM, Student's t test, **p < 0.01). Two months: N = 5 of each genotype; 5–7 months: N = 3 of each genotype; 12–24 months: N = 5 control mice, 4 DAT-DTA mice.
Dopamine depletion in the dorsal striatum of DAT-DTA mice is nearly complete (97%) at 2 months, suggesting that dopamine-independent mechanisms could be responsible for normal motor behavior in DAT-DTA mice. To determine whether the normal motor behavior of DAT-DTA mice is dependent on residual dopamine, we treated 12-month-old male and female DAT-DTA and control mice with the dopamine receptor antagonist haloperidol and evaluated motor behavior. As expected, haloperidol treatment resulted in a significant decrease in the locomotor activity of 12-month-old control mice in the open-field test compared with vehicle (DMSO)-treated control mice (Fig. 8A). Haloperidol-treated control mice were similarly impaired in the movement-initiation test (Fig. 8A) and the catalepsy test (data not shown) compared with vehicle-treated control mice. The performance of 12-month-old haloperidol-treated DAT-DTA mice was not different from haloperidol-treated littermate control mice in the open-field test (Fig. 8A), the movement-initiation test (Fig. 8A), or the catalepsy test (data not shown). These results indicate that maintenance of normal motor behavior in DAT-DTA mice is dependent on the small amount of remaining striatal dopamine (3% of control; Fig. 7A) in DAT-DTA mice.
Figure 8.

DAT-DTA mice are sensitive to haloperidol and hypersensitive to l-DOPA. A, Haloperidol (1 mg/kg) significantly decreases locomotor activity in both DAT-DTA mice and control mice at 12 months of age. Locomotor activity is not significantly different in haloperidol-treated DAT-DTA mice compared with haloperidol-treated control mice. The time required to exit an 18 × 18 cm square is significantly increased in haloperidol-treated DAT-DTA mice and haloperidol-treated control mice compared with vehicle-treated control mice. Exit time is not significantly different in haloperidol-treated DAT-DTA mice compared with haloperidol-treated control mice. Mean ± SEM, Student's t test, control vs DAT-DTA, *p < 0.05, **p < 0.01, ***p < 0.001. Control mice/DMSO, N = 2; control mice/haloperidol, N = 7; DAT-DTA mice/haloperidol, N = 7. B, The locomotor activity of l-DOPA-treated control mice is not significantly different from vehicle-treated control mice. The locomotor activity of l-DOPA-treated DAT-DTA mice is significantly increased compared with l-DOPA-treated control mice. The time required to exit an 18 × 18 cm square is not significantly different in l-DOPA-treated DAT-DTA mice compared with l-DOPA-treated control mice. Mean ± SEM, Student's t test, control vs DAT-DTA, *p ≤ 0.05, **p < 0.01, ***p < 0.001. Control mice/vehicle, N = 4; control mice/l-DOPA, N = 5; DAT-DTA mice/l-DOPA, N = 7.
Previous studies have shown that dopamine receptor agonists induce an exaggerated increase in locomotor activity in animals with nigrostriatal damage. The increased activity in response to dopamine agonists observed in injured animals is reportedly due to sensitization of striatal dopamine receptors (Zigmond and Stricker, 1980; Breese et al., 1985; Brotchie and Fitzer-Attas, 2009). To gain insight into the mechanism by which motor behavior is preserved in DAT-DTA mice, we treated 6–7-month-old DAT-DTA mice and control mice with l-DOPA (25 mg/kg) and measured locomotor activity. This dose of l-DOPA did not alter the locomotor activity of control mice in the open-field test compared with vehicle-treated control mice (Fig. 8B). The locomotor activity of l-DOPA-treated DAT-DTA mice was significantly increased compared with l-DOPA-treated control mice (Fig. 8B). The observed increase in locomotor activity in DAT-DTA mice compared with control mice in response to l-DOPA suggests that striatal dopamine receptors are functionally sensitized in DAT-DTA mice. This sensitization likely contributes to the preservation of motor behavior observed in these mice. Movement initiation was not altered in either DAT-DTA mice or control mice in response to l-DOPA (Fig. 8B).
To determine whether increased expression of dopamine receptors contributes to functional sensitization, we evaluated the expression of dopamine receptors in 12-month-old DAT-DTA mice using qRT-PCR. Our analysis shows no change in the expression of either D1 or D2 dopamine receptors in the dorsal striatum of DAT-DTA mice compared with control mice (data not shown). The observed functional sensitization in response to l-DOPA may be a result of mechanisms that regulate the translation of dopamine receptors, the post-translational modification of receptors, or receptor trafficking. Additional studies will be required to evaluate the contribution of these potential changes to sensitization.
Discussion
DTA expressed from the DAT locus produces a substantial and reproducible loss of SNc dopaminergic neurons and striatal dopamine in DAT-DTA mice without the systemic toxicity, dosing limitations, or lack of specificity inherent in models using neurotoxins to destroy dopaminergic neurons (Blandini and Armentero, 2012). Despite loss of 90% of SNc dopaminergic neurons, dysfunction of surviving neurons (as inferred from degenerative morphological changes), and depletion of 97% of striatal dopamine, we were not able to detect motor behavior defects in DAT-DTA mice using behavior tests that have been used previously to demonstrate motor dysfunction in other models of nigrostriatal damage (Meredith and Kang, 2006; Byler et al., 2009; Taylor et al., 2010). Importantly, we were able to detect an age-dependent decline in motor performance in both control and DAT-DTA mice, demonstrating that our testing methods are sufficient to detect motor dysfunction. Our results indicate that preserved motor function in DAT-DTA mice is dependent on residual dopamine. Other, nondopaminergic mechanisms may also contribute to the maintenance of motor function in DAT-DTA mice, but such mechanisms are insufficient to maintain normal motor behavior when dopaminergic transmission is blocked by haloperidol. Previous studies in which dopaminergic neurons have been destroyed by administration of toxins to adult animals have found motor behavior deficits with far less severe SNc neuron loss and striatal dopamine depletion than those seen in DAT-DTA mice (Bové and Perier, 2012). However, the majority of SNc dopaminergic neurons (>70%) are lost in DAT-DTA mice during prenatal development. Our results show that the nigrostriatal dopaminergic pathway has a significant capacity to compensate for damage sustained during prenatal development. The remarkable preservation of motor function in DAT-DTA mice that occurs in the absence of growth retardation and nonspecific damage to nondopaminergic pathways suggests that additional mechanisms of plasticity may be induced when damage occurs in developing animals. Identification of the molecular mechanisms that underlie motor behavior sparing in DAT-DTA mice may suggest novel strategies for treating PD or for improving current therapies.
Studies in which prenatal exposure to a neurotoxin was used to damage the nigrostriatal system have found that less severe damage, compared with that seen in DAT-DTA mice, results in defects in motor performance. For example, prenatal administration of MPTP reduces striatal dopamine by ∼14% and results in decreased spontaneous locomotion (Muthian et al., 2010). Prenatal administration of lipopolysaccharide produces a moderate decrease in SNc dopaminergic neurons (∼30%; Ling et al., 2002, 2009; Carvey et al., 2003) and results in decreased locomotion in the open field and an increased number of skips in the beam-walking test (Bakos et al., 2004). These findings are consistent with the idea that damage to dopaminergic neurons during prenatal development can result in motor behavior defects in adult animals. A much greater percentage of SNc dopaminergic neurons are destroyed during prenatal development in DAT-DTA mice. A greater magnitude of neuron loss may be required to induce mechanisms of compensation. It is also possible that toxicity to the mother or other unknown effects of the toxin in the fetus could affect motor behavior in animals treated prenatally with toxins.
Moderate feeding and motor behavior deficits occur in rats in which 6-OHDA is used to damage dopaminergic neurons during early postnatal development (Breese et al., 2005). Adult rats sustaining >95% depletion of striatal dopamine following intracerebralventricular or intranigral administration of 6-OHDA exhibit adipsia, aphagia, and akinesia (Ungerstedt, 1971; Zigmond and Stricker, 1973; Marshall et al., 1974). Although rats treated with intracerebralventricular 6-OHDA as neonates (P3) sustain comparable depletion of striatal dopamine as adult-treated rats, they exhibit much less severe feeding and motor behavior defects. Locomotor behavior is reportedly increased (Shaywitz et al., 1976; Erinoff et al., 1979, 1984; Archer et al., 1988) or decreased (Smith et al., 1973) in adult rats treated with 6-OHDA as neonates. In addition, defects in skilled forearm and tongue use have been observed in these rats (Whishaw et al., 1987). In contrast to neonatal-treated 6-OHDA rats that have some motor behavior deficits, we were not able to detect any deficits in motor behavior in DAT-DTA mice that sustained comparable depletion of striatal dopamine. This difference may be related to the time course of SNc cell damage in the two models, to species differences, or to other effects of the toxin. In addition to dopaminergic neurons, 6-OHDA also damages noradrenergic neurons (Ungerstedt, 1968). Damage to noradrenergic neurons alone increases locomotor behavior (Smith et al., 1973). Although, desmethylimipramine (DMI) is routinely administered with 6-OHDA to minimize damage to noradrenergic neurons, DMI is not completely effective in preventing damage to noradrenergic neurons (Breese and Traylor, 1971; Castañeda et al., 1990). In addition, mice treated with 6-OHDA at early postnatal stages show marked growth retardation, which could impact motor behavior (Breese and Traylor, 1972). Our data show normal motor behavior in a mouse model in which substantial damage specifically to dopaminergic neurons is sustained during prenatal development in the absence of adverse effects on general health.
Rats treated with 6-OHDA as neonates are resistant to haloperidol treatment in adulthood, suggesting that nondopaminergic systems are compensating for the loss of dopamine to maintain normal motor behavior in these rats (Bruno et al., 1985; Duncan et al., 1987; Weihmuller and Bruno, 1989). In contrast, our results indicate that with comparable depletion of striatal dopamine, residual dopamine is responsible for the maintenance of normal motor behavior in DAT-DTA mice. It is surprising that normal motor behavior in DAT-DTA mice is dependent on residual dopamine. TH-positive fibers are virtually absent in the striatum of adult DAT-DTA mice and striatal dopamine is decreased by 97%, indicating nearly complete depletion of dopamine. Preservation of motor behavior following partial damage to the nigrostriatal system is thought to be due to adaptations in residual dopaminergic terminals, such as increased dopamine synthesis and release and attenuated dopamine reuptake (Zigmond et al., 1984, 1990). In addition, changes in postsynaptic striatal neurons can also maximize dopaminergic transmission when dopamine levels are severely depleted.
Increased dopamine release is reflected by an increase in the ratio of dopamine metabolites to dopamine, particularly the HVA/dopamine ratio. DAT-DTA mice have a significantly greater HVA/dopamine ratio, which is consistent with the hypothesis that maintenance of motor function in DAT-DTA mice is dependent upon mechanisms that maximize the efficiency of residual dopamine. However, dopamine-dependent mechanisms that preserve normal motor behavior in animals with partial nigrostriatal damage are reportedly limited. Previous studies indicate that these mechanisms are only effective when ≥5–10% of dopaminergic terminals remain in the striatum (Reader and Dewar, 1999). In addition, the nigrostriatal system is more susceptible to toxin-induced damage in aged animals and compensatory mechanisms that maximize dopamine function are attenuated in aged animals (Ovadia et al., 1995; Thiruchelvam et al., 2003; Collier et al., 2007, 2011; Peng et al., 2007). It is therefore surprising that no motor defects are observed in DAT-DTA mice, even at 18–24 months of age, and that aged DAT-DTA mice are sensitive to haloperidol, suggesting that compensatory mechanisms that rely on residual dopamine are intact even in aged DAT-DTA mice. Our results further indicate that the mechanism underlying preservation of motor behavior in DAT-DTA mice involves functional sensitization of dopamine receptors.
Interestingly, developmental elimination of dopaminergic transmission by deletion of TH or D1 and D2 dopamine receptors results in more severe behavior deficits than destruction of dopaminergic neurons in DAT-DTA mice. Dopamine-deficient mice are hypoactive, exhibit severe growth retardation, and die during the third postnatal week (Zhou and Palmiter, 1995). Mice in which both the D1 and D2 dopamine receptors are deleted have a similar phenotype to dopamine-deficient mice and also die in the third postnatal week (Kobayashi et al., 2004). Premature death of D1/D2-null mice is thought to be due to peripheral GI defects that result in a failure to thrive and severe growth retardation. In contrast, DAT-DTA mice are generally healthy and appear to grow and develop normally. The absence of severe growth retardation in DAT-DTA mice may be due to lower DAT expression and thus lower DAT-CRE activity in peripheral dopaminergic neurons, which results in less DTA-mediated cell damage in the GI system (Li et al., 2004). Our observation that intrinsic dopaminergic neurons in the terminal ileum appear similar in DAT-DTA mice and control mice is consistent with this possibility. In addition, we observed no significant loss of dopaminergic neurons in the hypothalamus of DAT-DTA mice compared with control mice. Since DAT expression is also reported to be significantly lower in hypothalamic dopaminergic neurons compared with midbrain dopaminergic neurons (Storch et al., 2004), this observation also supports the hypothesis that expression of the DTA toxin is lower in dopaminergic neurons with low DAT expression. The olfactory bulbs contain a large number of dopaminergic neurons in which DAT expression is also reportedly low (Cerruti et al., 1993; Fujita et al., 1993). It will be interesting to determine whether these neurons are altered in DAT-DTA mice. It is also possible that dopaminergic transmission is more significantly impaired in dopamine-deficient and D1/D2-null mice compared with DAT-DTA mice. Studies using microdialysis show that the decrease in extracellular dopamine concentration is not as severe as the decrease in tissue dopamine following neonatal 6-OHDA treatment (Castañeda et al., 1990). Therefore, although striatal dopamine is decreased by 97% in DAT-DTA mice, extracellular dopamine may not be entirely depleted in these mice.
Our studies reveal remarkable preservation of motor behavior in mice with severe loss of SNc dopaminergic neurons and nearly complete depletion of striatal dopamine. The observation that preserved motor behavior depends on mechanisms that require intact dopaminergic transmission in aged DAT-DTA mice suggests even greater plasticity of the nigrostriatal dopaminergic pathway than previously thought. The DAT-DTA mouse is a novel model for studying the behavioral effects of dopamine depletion in the absence of motor impairment or growth retardation. For example, studies examining the role of dopaminergic pathways in addiction and in the regulation of circadian rhythm use behavioral assays, such as conditioned place preference and voluntary wheel running, that depend on intact motor function. Interpretation of these assays is complicated by impaired motor performance in neurotoxin models of dopaminergic neuron damage. The DAT-DTA mouse provides an opportunity to evaluate the effects of damage to dopaminergic pathways in the absence of motor dysfunction.
Footnotes
This work was supported by National Institutes of Health (NIH) Grants R21NS059566 (J.P.G.); DK081644 and DK082531 (S.J.); AGO13370 (E.M.J.); and NS048602 (R.W.G.). This work was supported by NIH Neuroscience Blueprint Interdisciplinary Center Core Grant P30 NS057105 and George M. O'Brien Center for Kidney Disease Research Grant P30-DK079333 to Washington University. The Michael J. Fox Foundation for Parkinson's Research provided salary support for J.P.G. We thank Angela Lluka, Patricia Osbourne, and Sherri Vogt for providing excellent technical assistance and animal care. We thank Daniel O'Brien for many useful comments and suggestions. We are grateful to Dr. Jeff Milbrandt for providing initial support and resources for these studies. We are grateful to Dr. Xiaoxi Zhuang, University of Chicago, and Dr. Mario Capecchi, University of Utah, for their generosity in providing the DAT-Cre and RosaDTA mice, respectively.
The authors declare no competing financial interests.
References
- Archer T, Danysz W, Fredriksson A, Jonsson G, Luthman J, Sundström E, Teiling A. Neonatal 6-hydroxydopamine-induced dopamine depletions: motor activity and performance in maze learning. Pharmacology, biochemistry, and behavior. 1988;31:357–364. doi: 10.1016/0091-3057(88)90358-9. [DOI] [PubMed] [Google Scholar]
- Bakos J, Duncko R, Makatsori A, Pirnik Z, Kiss A, Jezova D. Prenatal immune challenge affects growth, behavior, and brain dopamine in offspring. Ann N Y Acad Sci. 2004;1018:281–287. doi: 10.1196/annals.1296.033. [DOI] [PubMed] [Google Scholar]
- Barlow BK, Cory-Slechta DA, Richfield EK, Thiruchelvam M. The gestational environment and Parkinson's disease: evidence for neurodevelopmental origins of a neurodegenerative disorder. Reprod Toxicol. 2007;23:457–470. doi: 10.1016/j.reprotox.2007.01.007. [DOI] [PubMed] [Google Scholar]
- Blandini F, Armentero MT. Animal models of Parkinson's disease. FEBS J. 2012;279:1156–1166. doi: 10.1111/j.1742-4658.2012.08491.x. [DOI] [PubMed] [Google Scholar]
- Bové J, Perier C. Neurotoxin-based models of Parkinson's disease. Neuroscience. 2012;211:51–76. doi: 10.1016/j.neuroscience.2011.10.057. [DOI] [PubMed] [Google Scholar]
- Breese GR, Traylor TD. Depletion of brain noradrenaline and dopamine by 6-hydroxydopamine. Br J Pharmacol. 1971;42:88–99. doi: 10.1111/j.1476-5381.1971.tb07089.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breese GR, Traylor TD. Developmental characteristics of brain catecholamines and tyrosine hydroxylase in the rat: effects of 6-hydroxydopamine. Br J Pharmacol. 1972;44:210–222. doi: 10.1111/j.1476-5381.1972.tb07257.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breese GR, Napier TC, Mueller RA. Dopamine agonist-induced locomotor activity in rats treated with 6-hydroxydopamine at differing ages: functional supersensitivity of D-1 dopamine receptors in neonatally lesioned rats. J Pharmacol Exp Ther. 1985;234:447–455. [PubMed] [Google Scholar]
- Breese GR, Knapp DJ, Criswell HE, Moy SS, Papadeas ST, Blake BL. The neonate-6-hydroxydopamine-lesioned rat: a model for clinical neuroscience and neurobiological principles. Brain Res Brain Res Rev. 2005;48:57–73. doi: 10.1016/j.brainresrev.2004.08.004. [DOI] [PubMed] [Google Scholar]
- Brotchie J, Fitzer-Attas C. Mechanisms compensating for dopamine loss in early Parkinson disease. Neurology. 2009;72:S32–S38. doi: 10.1212/WNL.0b013e318198e0e9. [DOI] [PubMed] [Google Scholar]
- Bruno JP, Stricker EM, Zigmond MJ. Rats given dopamine-depleting brain lesions as neonates are subsensitive to dopaminergic antagonists as adults. Behav Neurosci. 1985;99:771–775. doi: 10.1037/0735-7044.99.4.771. [DOI] [PubMed] [Google Scholar]
- Byler SL, Boehm GW, Karp JD, Kohman RA, Tarr AJ, Schallert T, Barth TM. Systemic lipopolysaccharide plus MPTP as a model of dopamine loss and gait instability in C57BL/6J mice. Behav Brain Res. 2009;198:434–439. doi: 10.1016/j.bbr.2008.11.027. [DOI] [PubMed] [Google Scholar]
- Cano-de-la-Cuerda R, Pérez-de-Heredia M, Miangolarra-Page JC, Muñoz-Hellín E, Fernández-de-Las-Peñas C. Is there muscular weakness in Parkinson's disease? Am J Phys Med Rehabil. 2010;89:70–76. doi: 10.1097/PHM.0b013e3181a9ed9b. [DOI] [PubMed] [Google Scholar]
- Carvey PM, Chang Q, Lipton JW, Ling Z. Prenatal exposure to the bacteriotoxin lipopolysaccharide leads to long-term losses of dopamine neurons in offspring: a potential, new model of Parkinson's disease. Front Biosci. 2003;8:s826–837. doi: 10.2741/1158. [DOI] [PubMed] [Google Scholar]
- Castañeda E, Whishaw IQ, Lermer L, Robinson TE. Dopamine depletion in neonatal rats: effects on behavior and striatal dopamine release assessed by intracerebral microdialysis during adulthood. Brain Res. 1990;508:30–39. doi: 10.1016/0006-8993(90)91113-U. [DOI] [PubMed] [Google Scholar]
- Cerruti C, Walther DM, Kuhar MJ, Uhl GR. Dopamine transporter mRNA expression is intense in rat midbrain neurons and modest outside midbrain. Brain Res Mol Brain Res. 1993;18:181–186. doi: 10.1016/0169-328X(93)90187-T. [DOI] [PubMed] [Google Scholar]
- Collier TJ, Lipton J, Daley BF, Palfi S, Chu Y, Sortwell C, Bakay RA, Sladek JR, Jr, Kordower JH. Aging-related changes in the nigrostriatal dopamine system and the response to MPTP in nonhuman primates; diminished compensatory mechanisms as a prelude to parkinsonism. Neurobiol Dis. 2007;26:56–65. doi: 10.1016/j.nbd.2006.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier TJ, Kanaan NM, Kordower JH. Ageing as a primary risk factor for Parkinson's disease: evidence from studies of non-human primates. Nat Rev Neurosci. 2011;12:359–366. doi: 10.1038/nrn3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da Silva Costa V, Duchatelle P, Boulouard M, Dauphin F. Selective 5-HT6 receptor blockade improves spatial recognition memory and reverses age-related deficits in spatial recognition memory in the mouse. Neuropsychopharmacology. 2009;34:488–500. doi: 10.1038/npp.2008.94. [DOI] [PubMed] [Google Scholar]
- Dauer W, Przedborski S. Parkinson's disease: mechanisms and models. Neuron. 2003;39:889–909. doi: 10.1016/S0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- Duncan GE, Criswell HE, McCown TJ, Paul IA, Mueller RA, Breese GR. Behavioral and neurochemical responses to haloperidol and SCH-23390 in rats treated neonatally or as adults with 6-hydroxydopamine. J Pharmacol Exp Ther. 1987;243:1027–1034. [PubMed] [Google Scholar]
- Dunnett SB, Lelos M. Behavioral analysis of motor and non-motor symptoms in rodent models of Parkinson's disease. Prog Brain Res. 2010;184:35–51. doi: 10.1016/S0079-6123(10)84003-8. [DOI] [PubMed] [Google Scholar]
- Erinoff L, MacPhail RC, Heller A, Seiden LS. Age-dependent effects of 6-hydroxydopamine on locomotor activity in the rat. Brain Res. 1979;164:195–205. doi: 10.1016/0006-8993(79)90015-5. [DOI] [PubMed] [Google Scholar]
- Erinoff L, Kelly PH, Basura M, Snodgrass SR. Six-hydroxydopamine induced hyperactivity: neither sex differences nor caffeine stimulation are found. Pharmacol Biochem Behav. 1984;20:707–713. doi: 10.1016/0091-3057(84)90189-8. [DOI] [PubMed] [Google Scholar]
- Franklin BJ, Paxinos GT. The mouse brain in stereotaxic coordinates. Academic: New York; 1997. [Google Scholar]
- Fujita M, Shimada S, Nishimura T, Uhl GR, Tohyama M. Ontogeny of dopamine transporter mRNA expression in the rat brain. Brain Res Mol Brain Res. 1993;19:222–226. doi: 10.1016/0169-328X(93)90031-J. [DOI] [PubMed] [Google Scholar]
- Gage FH, Dunnett SB, Björklund A. Spatial learning and motor deficits in aged rats. Neurobiol Aging. 1984;5:43–48. doi: 10.1016/0197-4580(84)90084-8. [DOI] [PubMed] [Google Scholar]
- Giros B, Jaber M, Jones SR, Wightman RM, Caron MG. Hyperlocomotion and indifference to cocaine and amphetamine in mice lacking the dopamine transporter. Nature. 1996;379:606–612. doi: 10.1038/379606a0. [DOI] [PubMed] [Google Scholar]
- Gundersen HJ, Bendtsen TF, Korbo L, Marcussen N, Møller A, Nielsen K, Nyengaard JR, Pakkenberg B, Sørensen FB, Vesterby A. Some new, simple and efficient stereological methods and their use in pathological research and diagnosis. APMIS. 1988;96:379–394. doi: 10.1111/j.1699-0463.1988.tb05320.x. [DOI] [PubMed] [Google Scholar]
- Hickey P, Stacy M. Adenosine A2A antagonists in Parkinson's disease: what's next? Curr Neurol Neurosci Rep. 2012;12:376–385. doi: 10.1007/s11910-012-0279-2. [DOI] [PubMed] [Google Scholar]
- Jain S, Golden JP, Wozniak D, Pehek E, Johnson EM, Jr, Milbrandt J. RET is dispensable for maintenance of midbrain dopaminergic neurons in adult mice. J Neurosci. 2006;26:11230–11238. doi: 10.1523/JNEUROSCI.1876-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi M, Iaccarino C, Saiardi A, Heidt V, Bozzi Y, Picetti R, Vitale C, Westphal H, Drago J, Borrelli E. Simultaneous absence of dopamine D1 and D2 receptor-mediated signaling is lethal in mice. Proc Natl Acad Sci U S A. 2004;101:11465–11470. doi: 10.1073/pnas.0402028101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li ZS, Pham TD, Tamir H, Chen JJ, Gershon MD. Enteric dopaminergic neurons: definition, developmental lineage, and effects of extrinsic denervation. J Neurosci. 2004;24:1330–1339. doi: 10.1523/JNEUROSCI.3982-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling Z, Gayle DA, Ma SY, Lipton JW, Tong CW, Hong JS, Carvey PM. In utero bacterial endotoxin exposure causes loss of tyrosine hydroxylase neurons in the postnatal rat midbrain. Mov Disorder. 2002;17:116–124. doi: 10.1002/mds.10078. [DOI] [PubMed] [Google Scholar]
- Ling Z, Zhu Y, Tong CW, Snyder JA, Lipton JW, Carvey PM. Prenatal lipopolysaccharide does not accelerate progressive dopamine neuron loss in the rat as a result of normal aging. Exp Neurol. 2009;216:312–320. doi: 10.1016/j.expneurol.2008.12.004. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Marshall JF, Richardson JS, Teitelbaum P. Nigrostriatal bundle damage and the lateral hypothalamic syndrome. J Comp Physiol Psychol. 1974;87:808–830. doi: 10.1037/h0037223. [DOI] [PubMed] [Google Scholar]
- Meredith GE, Kang UJ. Behavioral models of Parkinson's disease in rodents: a new look at an old problem. Mov Disord. 2006;21:1595–1606. doi: 10.1002/mds.21010. [DOI] [PubMed] [Google Scholar]
- Mohajjel Nayebi AA, Sheidaei H. Buspirone improves haloperidol-induced Parkinson disease in mice through 5-HT1A receptors. DARU. 2010;18:41–45. [PMC free article] [PubMed] [Google Scholar]
- Montana MC, Cavallone LF, Stubbert KK, Stefanescu AD, Kharasch ED, Gereau RW., 4th The metabotropic glutamate receptor subtype 5 antagonist fenobam is analgesic and has improved in vivo selectivity compared with the prototypical antagonist 2-methyl-6-(phenylethynyl)-pyridine. J Pharmacol Exp Ther. 2009;330:834–843. doi: 10.1124/jpet.109.154138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muthian G, Mackey V, King J, Charlton CG. Modeling a sensitization stage and a precipitation stage for Parkinson's disease using prenatal and postnatal 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine administration. Neuroscience. 2010;169:1085–1093. doi: 10.1016/j.neuroscience.2010.04.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovadia A, Zhang Z, Gash DM. Increased susceptibility to MPTP toxicity in middle-aged rhesus monkeys. Neurobiol Aging. 1995;16:931–937. doi: 10.1016/0197-4580(95)02012-8. [DOI] [PubMed] [Google Scholar]
- Pehek EA, Crock R, Yamamoto BK. Selective subregional dopamine depletions in the rat caudate-putamen following nigrostriatal lesions. Synapse. 1992;10:317–325. doi: 10.1002/syn.890100406. [DOI] [PubMed] [Google Scholar]
- Peng J, Peng L, Stevenson FF, Doctrow SR, Andersen JK. Iron and paraquat as synergistic environmental risk factors in sporadic Parkinson's disease accelerate age-related neurodegeneration. J Neurosci. 2007;27:6914–6922. doi: 10.1523/JNEUROSCI.1569-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponce FA, Lozano AM. Deep brain stimulation state of the art and novel stimulation targets. Prog Brain Res. 2010;184:311–324. doi: 10.1016/S0079-6123(10)84016-6. [DOI] [PubMed] [Google Scholar]
- Reader TA, Dewar KM. Effects of denervation and hyperinnervation on dopamine and serotonin systems in the rat neostriatum: implications for human Parkinson's disease. Neurochem Int. 1999;34:1–21. doi: 10.1016/S0197-0186(98)00048-5. [DOI] [PubMed] [Google Scholar]
- Shaywitz BA, Yager RD, Klopper JH. Selective brain dopamine depletion in developing rats: an experimental model of minimal brain dysfunction. Science. 1976;191:305–308. doi: 10.1126/science.942800. [DOI] [PubMed] [Google Scholar]
- Smith RD, Cooper BR, Breese GR. Growth and behavioral changes in developing rats treated intracisternally with 6-hydroxydopamine: evidence for involvement of brain dopamine. J Pharmacol Exp Ther. 1973;185:609–619. [PubMed] [Google Scholar]
- Storch A, Ludolph AC, Schwarz J. Dopamine transporter: involvement in selective dopaminergic neurotoxicity and degeneration. J Neural Transm. 2004;111:1267–1286. doi: 10.1007/s00702-004-0203-2. [DOI] [PubMed] [Google Scholar]
- Taylor MD, Vancura R, Patterson CL, Williams JM, Riekhof JT, Wright DE. Postnatal regulation of limb proprioception by muscle-derived neurotrophin-3. J Comp Neurol. 2001;432:244–258. doi: 10.1002/cne.1100. [DOI] [PubMed] [Google Scholar]
- Taylor TN, Greene JG, Miller GW. Behavioral phenotyping of mouse models of Parkinson's disease. Behav Brain Res. 2010;211:1–10. doi: 10.1016/j.bbr.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiruchelvam M, McCormack A, Richfield EK, Baggs RB, Tank AW, Di Monte DA, Cory-Slechta DA. Age-related irreversible progressive nigrostriatal dopaminergic neurotoxicity in the paraquat and maneb model of the Parkinson's disease phenotype. Eur J Neurosci. 2003;18:589–600. doi: 10.1046/j.1460-9568.2003.02781.x. [DOI] [PubMed] [Google Scholar]
- Tillerson JL, Caudle WM, Reverón ME, Miller GW. Detection of behavioral impairments correlated to neurochemical deficits in mice treated with moderate doses of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Exp Neurol. 2002;178:80–90. doi: 10.1006/exnr.2002.8021. [DOI] [PubMed] [Google Scholar]
- Ungerstedt U. 6-Hydroxy-dopamine induced degeneration of central monoamine neurons. Eur J Pharmacol. 1968;5:107–110. doi: 10.1016/0014-2999(68)90164-7. [DOI] [PubMed] [Google Scholar]
- Ungerstedt U. Adipsia and aphagia after 6-hydroxydopamine induced degeneration of the nigro-striatal dopamine system. Acta Physiol Scand Suppl. 1971;367:95–122. doi: 10.1111/j.1365-201x.1971.tb11001.x. [DOI] [PubMed] [Google Scholar]
- Weihmuller FB, Bruno JP. Drinking behavior and motor function in rat pups depleted of brain dopamine during development. Dev Psychobiol. 1989;22:101–113. doi: 10.1002/dev.420220202. [DOI] [PubMed] [Google Scholar]
- Whishaw IQ, Funk DR, Hawryluk SJ, Karbashewski ED. Absence of sparing of spatial navigation, skilled forelimb and tongue use and limb posture in the rat after neonatal dopamine depletion. Physiol Behav. 1987;40:247–253. doi: 10.1016/0031-9384(87)90215-0. [DOI] [PubMed] [Google Scholar]
- Wu S, Wu Y, Capecchi MR. Motoneurons and oligodendrocytes are sequentially generated from neural stem cells but do not appear to share common lineage-restricted progenitors in vivo. Development. 2006;133:581–590. doi: 10.1242/dev.02236. [DOI] [PubMed] [Google Scholar]
- Zhou QY, Palmiter RD. Dopamine-deficient mice are severely hypoactive, adipsic, and aphagic. Cell. 1995;83:1197–1209. doi: 10.1016/0092-8674(95)90145-0. [DOI] [PubMed] [Google Scholar]
- Zhuang X, Masson J, Gingrich JA, Rayport S, Hen R. Targeted gene expression in dopamine and serotonin neurons of the mouse brain. J Neurosci Methods. 2005;143:27–32. doi: 10.1016/j.jneumeth.2004.09.020. [DOI] [PubMed] [Google Scholar]
- Zigmond MJ, Stricker EM. Recovery of feeding and drinking by rats after intraventricular 6-hydroxydopamine or lateral hypothalamic lesions. Science. 1973;182:717–720. doi: 10.1126/science.182.4113.717. [DOI] [PubMed] [Google Scholar]
- Zigmond MJ, Stricker EM. Supersensitivity after intraventricular 6-hydroxydopamine: relation to dopamine depletion. Experientia. 1980;36:436–438. doi: 10.1007/BF01975133. [DOI] [PubMed] [Google Scholar]
- Zigmond MJ, Acheson AL, Stachowiak MK, Stricker EM. Neurochemical compensation after nigrostriatal bundle injury in an animal model of preclinical parkinsonism. Arch Neurol. 1984;41:856–861. doi: 10.1001/archneur.1984.04050190062015. [DOI] [PubMed] [Google Scholar]
- Zigmond MJ, Abercrombie ED, Berger TW, Grace AA, Stricker EM. Compensations after lesions of central dopaminergic neurons: some clinical and basic implications. Trends Neurosci. 1990;13:290–296. doi: 10.1016/0166-2236(90)90112-N. [DOI] [PubMed] [Google Scholar]