

Abstract
Prediction of clinical efficacy, toxicity, and drug-drug interactions may be improved by accounting for the intracellular unbound drug concentration (Cunbound) in vitro and in vivo. Furthermore, subcellular drug distribution may aid in predicting efficacy, toxicity, and risk assessment. The present study was designed to quantify the intracellular Cunbound and subcellular localization of drugs in rat sandwich-cultured hepatocytes (SCH) compared with rat isolated perfused liver (IPL) tissue. Probe drugs with distinct mechanisms of hepatocellular uptake and accumulation were selected for investigation. Following drug treatment, SCH and IPL tissues were homogenized and fractionated by differential centrifugation to enrich for subcellular compartments. Binding in crude lysate and cytosol was determined by equilibrium dialysis; the Cunbound and intracellular-to-extracellular Cunbound ratio (Kpu,u) were used to describe accumulation of unbound drug. Total accumulation (Kpobserved) in whole tissue was well predicted by the SCH model (within 2- to 3-fold) for the selected drugs. Ritonavir (Kpu,u ∼1) was evenly distributed among cellular compartments, but highly bound, which explained the observed accumulation within liver tissue. Rosuvastatin was recovered primarily in the cytosolic fraction, but did not exhibit extensive binding, resulting in a Kpu,u >1 in liver tissue and SCH, consistent with efficient hepatic uptake. Despite extensive binding and sequestration of furamidine within liver tissue, a significant portion of cellular accumulation was attributed to unbound drug (Kpu,u >16), as expected for a charged, hepatically derived metabolite. Data demonstrate the utility of SCH to predict quantitatively total tissue accumulation and elucidate mechanisms of hepatocellular drug accumulation such as active uptake versus binding/sequestration.
Introduction
The impact of drugs on intracellular targets of efficacy and/or toxicity, and susceptibility to elimination, is driven by local unbound concentrations (Cunbound) according to the “free drug hypothesis” (Smith et al., 2010). Prediction of clinical efficacy, toxicity, and drug-drug interactions (DDIs) could be improved by accounting for Cunbound in vitro and in vivo (Zhou et al., 2011; Zhang et al., 2012). Furthermore, subcellular drug distribution may aid prediction and/or correlation of efficacy and/or toxicity and risk assessment for drugs (Reasor and Kacew, 2001; Gunawan and Kaplowitz, 2007; Labbe et al., 2008). Measurement of blood or plasma Cunbound is convenient and may correlate with tissue concentrations for compounds with sufficient passive permeability. However, for hydrophilic compounds (highly polar or ionized at physiologic pH) that rely on uptake/efflux transporters for distribution and do not readily cross membrane barriers, the intracellular and extracellular (blood or plasma) Cunbound may be highly disparate.
The ratio of intracellular and extracellular Cunbound (Kpu,u) has been used extensively in central nervous system pharmacokinetics-pharmacodynamics to elucidate the role of blood-brain barrier penetration and transport (Gupta et al., 2006) and to correlate in vivo effects with in vitro potency (Hammarlund-Udenaes et al., 2008; Liu et al., 2008). Figure 1 illustrates the concept of Kpu,u as it applies to hepatobiliary drug disposition. Compounds that are taken up efficiently into hepatocytes may exhibit a Kpu,u >>1 (Zhou et al., 2011; Shitara et al., 2013). The important role of the organic anion–transporting polypeptides (OATPs) has been recognized in recent years, highlighting the need to elucidate the role of active uptake in the hepatic accumulation of drugs (Giacomini et al., 2010; Shitara et al., 2013). Indeed, any situation in which the net rate of appearance exceeds elimination under steady-state conditions will result in a Kpu,u >>1; this includes formation of metabolites, which are often polar and poorly permeable, relying on active transport for elimination. A Kpu,u <<1 in the liver indicates that the net effects of elimination (efflux, metabolism) outweigh appearance in the tissue/cellular compartment (low passive permeability and/or rate-limited influx). The role of metabolic clearance and transporter-enzyme interplay in complicating the prediction of intracellular concentrations in hepatocyte systems has been emphasized (Parker and Houston, 2008; Brown et al., 2010).
Fig. 1.

The tissue-to-plasma unbound concentration ratio (Kpu,u) applied to hepatobiliary drug disposition.
Hepatic intracellular Cunbound, Kpu,u, and subcellular localization information has improved predictions and/or explained seemingly discrepant pharmacokinetic-pharmacodynamic relationships with a variety of endpoints involving efficacy (Dollery, 2013; Shitara et al., 2013), toxicity (Chen et al., 2008), and drug disposition, including transport (Kudo et al., 2007), metabolism (Obach, 1996, 1999; Deshmukh and Harsch, 2011), and related DDIs (Yamano et al., 1999; Chen et al., 2008; Sato et al., 2010; Pfeifer et al., 2013). Intracellular Cunbound and subcellular localization of drugs are challenging to measure accurately, and our understanding of hepatocellular drug disposition remains rudimentary (Chu et al., 2013). A versatile and reliable method to determine these parameters in a relevant hepatic in vitro model would be of value to differentiate the contribution of active uptake versus binding/sequestration as mechanisms of hepatocellular accumulation, and to assess routes and rates of elimination (e.g., metabolism and biliary excretion) (Giacomini et al., 2010; Zhou et al., 2011; Chu et al., 2013).
Isolated perfused livers (IPLs) are considered the gold standard for studying the hepatobiliary system in isolation, without the influence of extrahepatic components (Brouwer and Thurman, 1996). Unfortunately, the IPL system has a number of drawbacks, including a limited experimental period (≤2 hours), the time- and labor-intensive nature of the procedure, and limited applicability to nonrodent species including humans. Sandwich-cultured hepatocytes (SCH) are a common in vitro model used to assess hepatic uptake and excretory function, including DDIs and transporter-metabolism interplay (Swift et al., 2010). Recapitulation of functional bile networks in SCH provides a key advantage for predicting cellular accumulation of drugs that are eliminated from hepatocytes via biliary excretion. Expression of transport proteins and metabolizing enzymes in SCH may be altered over days in culture compared with whole tissue (Li et al., 2009; Tchaparian et al., 2011); however, the use of medium additives can modulate expression levels to more closely mimic the in vivo situation (Swift et al., 2010). Methods to estimate the cellular unbound fraction (fu) and resulting Kpu,u in hepatocyte models in vitro include the use of temperature, inhibitors, and/or pharmacokinetic modeling (Parker and Houston, 2008; Yabe et al., 2011; Jones et al., 2012; Shitara et al., 2013), but direct measurement of Kpu,u in SCH has not been reported.
The objective of this work was to determine the intracellular Cunbound and subcellular localization of drugs in SCH compared with IPL tissue. A set of probe drugs with distinct mechanisms of hepatocellular uptake and accumulation was selected for investigation. Ritonavir inhibits drug transport and metabolism in vitro and in vivo and accumulates in liver tissue (Denissen et al., 1997). Surprisingly, the contribution of active transport to the cellular uptake and accumulation of ritonavir has not been characterized definitively, and reports of hepatic Kpu,u and intracellular Cunbound for prediction of ritonavir DDIs are inconsistent (Parker and Houston, 2008; Griffin et al., 2011; Mateus et al., 2013; Pfeifer et al., 2013). Rosuvastatin, a 3-hydroxy-3-methylglutaryl–coenzyme A reductase inhibitor with a primary site of action in hepatocytes, is taken up efficiently by OATPs and accumulates within hepatocytes, with a Kpu,u >1 (Nezasa et al., 2002a, 2003; Yabe et al., 2011; Shitara et al., 2013). Furamidine is formed in the liver via sequential metabolism from the prodrug pafuramidine; furamidine is poorly permeable and must be excreted from hepatocytes into the systemic circulation to exert antiparasitic activity (Paine et al., 2010; Yan et al., 2011). This set of drugs provided a useful range of tissue accumulation, hepatic fu, subcellular distribution, and Kpu,u values for comparison of methods to determine hepatocellular Cunbound.
Materials and Methods
All chemicals were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise stated. Rosuvastatin (Nezasa et al., 2002b) and the deuterated internal standard (d6-rosuvastatin), as well as ritonavir (Denissen et al., 1997), were purchased from Toronto Research Chemicals (Toronto, ON, Canada). Furamidine, the prodrug pafuramidine, and internal standard (deuterium-labeled furamdine) were synthesized in the laboratory of Dr. David W. Boykin (Georgia State University, Atlanta, GA), as reported previously (Yan et al., 2011).
Tissue Accumulation.
Male Wistar rats (Charles River Laboratories, Wilmington, MA) were used for IPL studies. Rats were allowed free access to water and food and acclimated for a minimum of 1 week prior to experimentation. All animal procedures complied with the guidelines of the Institutional Animal Care and Use Committee (University of North Carolina, Chapel Hill, NC). All procedures were performed under full anesthesia with ketamine/xylazine (140/8 mg/kg i.p.). Livers were perfused in a single-pass manner (30 ml/min continually oxygenated Krebs-Ringer bicarbonate buffer in the presence of 5 μM taurocholate to maintain bile flow); following a 15-minute equilibration period, ritonavir (1 μM) or rosuvastatin (1 μM) was included in the perfusate for 30 minutes, and then the livers were flushed briefly with blank buffer. Pafuramidine (10 μM) was perfused in a recirculating IPL system with 20% blood-containing perfusate for 120 minutes as reported previously (Yan et al., 2011). Livers were harvested and stored whole at −80°C until homogenization and analysis.
Rat hepatocytes were seeded in six-well BioCoat plates (BD Biosciences, San Jose, CA) and overlaid with Matrigel basement membrane matrix (BD Biosciences) in a sandwich-cultured configuration and maintained as described previously (Swift et al., 2010). Day 4 rat SCH were preincubated for 10 minutes in Ca2+-free Hanks’ balanced salt solution (HBSS) B-CLEAR technology; Qualyst Transporter Solutions, Durham, NC) to open tight junctions and prevent accumulation in bile canalicular spaces, and then treated with 1 μM ritonavir or [3H]rosuvastatin (100 nCi/ml; American Radiolabeled Chemicals, Inc., St. Louis, MO) for 10 minutes at 37°C as described previously (Swift et al., 2010). Starting on day 3, pafuramidine was incubated for 24 hours at 10 μM to allow complete formation and equilibration of furamidine, as previously reported (Yan et al., 2011), followed by a 5-minute incubation in Ca2+-free HBSS. In all cases, incubation medium was collected at the end of the incubation period, and cells were washed three times in ice-cold HBSS.
Fractionation.
Liver tissue was homogenized in 3–5 volumes of fractionation buffer [250 mM sucrose, 10 mM HEPES, 10 mM KCl, 1 mM EDTA, 1.5 mM MgCl2, 1 mM dithiothreitol, cOmplete Protease Inhibitor Cocktail (Roche Diagnostics, Indianapolis, IN)] using a Potter-Elvehjem homogenizer. Following treatment with the drug of interest, all wells of the six-well SCH plate were harvested and pooled by scraping each well sequentially into 1 ml fractionation buffer. Collected cells in buffer were homogenized by passing 10 times through a 27 gauge needle, resting 10 minutes on ice, followed by an additional 10 passes. Following homogenization to disrupt cell membranes, the resulting crude lysates (liver and SCH) were sampled and reserved to perform analysis for total and unbound drug concentrations, protein content, and enzyme activity assays. The remaining lysate was subjected to stepwise differential centrifugation as published previously (Ward et al., 2000) to separate the following cellular components: 10 minutes at 600g (nuclei and cellular debris), 10,000g (mitochondria), and 35,000g (lysosomes and other medium-sized membrane-bound bodies), and 60 minutes at 100,000g (microsomes, membrane fraction), with the resulting supernatant representing the cytosolic fraction. All spins <100,000g were repeated after resuspending the pellet in 5 (liver) or 0.2 (SCH) ml of fractionation buffer; resulting supernatants were pooled before moving on to subsequent centrifugation steps. Pellets were resuspended in fractionation buffer [10 ml for liver tissue and 0.3 (600g) or 0.15 (>600g) ml for SCH] for analysis of drug and protein content and enzyme activities.
Protein content of each fraction was determined using the Pierce BCA Protein Assay (Thermo Fisher Scientific, Inc., Rockford, IL). Separation and recovery of subcellular fractions were assessed by measuring lactate dehydrogenase (LDH) activity as a cytosolic marker using a Cytotoxicity Detection kit (Roche Diagnostics) and acid phosphatase activity as a lysosomal marker using an Acid Phosphatase kit (Sigma-Aldrich). Succinate dehydrogenase activity was measured to assess the presence of mitochondria as described previously (Gong et al., 2007), adapted to a microplate format as follows: 5 μl sample volume was combined with 60 μl 10 mM sodium succinate in 50 mM phosphate buffer and incubated for 60 minutes at 37°C, followed by the addition of 20 μl 2.5-mg/ml p-iodonitrotetrazolium and incubation for another 15 minutes. The reaction was quenched by the addition of 0.2 ml of 5:5:1 (v/v/w) ethanol/ethyl acetate/trichloroacetic acid and absorbance was determined at 492 nm on a plate-based spectrophotometer (BioTek PowerWave HT, Winooski, VT). Similarly, glucose 6-phosphatase activity was measured to assess the presence of microsomes as described previously (Ockerman, 1967), adapted to a microplate format as follows: 10 μl sample volume was plated in duplicate and combined with 10 μl 20 mM Tris-HCl, pH 7.3, with and without 50 mM glucose 6-phosphate (Na+ salt; Sigma-Aldrich) and incubated for 30 minutes at 37°C. Inorganic phosphate standards were prepared from 0.1–2 mM and added to the plate (20 μl) at the end of the incubation. The reaction was terminated by the addition of 200 μl detection reagent [3:1 1 mg/ml malachite green (oxalate salt; Sigma-Aldrich) in 1 N HCl/4.2% (w/v) ammonium molybdate in 5 N HCl] to samples and standards, followed immediately by the addition of 10 μl 0.05% (v/v) Tween-20. The reaction mixture was shaken briefly and allowed to equilibrate for 10–15 minutes, and absorbance was determined at 660 nm on a plate-based spectrophotometer. Phosphate formation was calculated as the difference between samples incubated with and without glucose 6-phosphate substrate to correct for endogenous phosphate content in the samples. Recovery of each fraction was calculated as the percentage of the total organelle-specific enzyme activity detected in each subfraction compared with the whole lysate.
Binding.
Binding was determined in whole-tissue lysates and cytosolic fractions by equilibrium dialysis. Initial studies were performed to determine the time to equilibrium and test for protein leakage or potential volume shifts with whole-liver tissue (data not shown). Aliquots were loaded into a 96-well equilibrium dialysis apparatus (HTDialysis, LLC, Gales Ferry, CT) and dialyzed against phosphate buffer for 6 hours with shaking at 37°C. Binding replicates (n = 3) consisted of 3-fold dilutions of each sample (1-, 3-, and 9-fold original sample). The unbound fraction (fu) was back-extrapolated to account for dilution during the homogenization/fractionation process, as well as subsequent dilutions, as described previously (Kalvass et al., 2007):
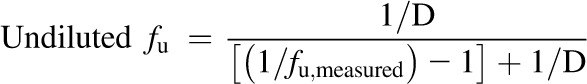 |
(1) |
This approach provides the best precision in the linear range, and that precision is lost when the measured unbound fraction becomes high (fu,measured > 80%). In cases where fu,measured was >80% at the lowest dilution, the value was reported as “greater than” the undiluted fu, calculated according to the equation above.
Sample Analysis.
Ritonavir, rosuvastatin, and furamidine were quantified by liquid chromatography–tandem mass spectrometry as described previously; rosuvastatin was quantified by liquid scintillation counting for SCH studies (Abe et al., 2008; Rezk et al., 2009; Lee and Brouwer, 2010; Yan et al., 2011). Total cellular concentrations in SCH were calculated by dividing the quantified substrate mass in the whole lysate by 7.4 μl/mg protein in SCH, the estimated hepatocellular volume determined by [3H]3-O-methyl-d-glucose (Lee and Brouwer, 2010).
Data Analysis.
The observed partition coefficient (Kpobserved) was calculated as the total tissue concentration (liver or SCH) divided by the outflow perfusate (IPL) or buffer (SCH) total concentration measured at the end of the perfusion or incubation, respectively. The predicted partition coefficient (Kppredicted) was calculated as the reciprocal of the tissue unbound fraction (fu,tissue), assuming 1) no binding in the extracellular matrix (buffer; fu,extracell = 1) and 2) equilibration between intracellular (tissue) and extracellular Cunbound (Cu,tissue = Cu,extracell):
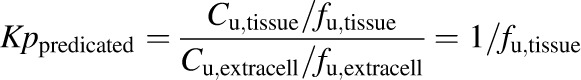 |
(2) |
where the tissue unbound fraction was determined using whole lysate or cytosol [fu,tissue = fu,lysate = (fcytosol • fu,cytosol)], and fcytosol represents the fraction of the total drug mass residing in the tissue (whole liver or SCH) that was recovered in the cytosol (final supernatant following fractionation): (Ccytosol • Vcytosol)/(Clysate • Vlysate); C and V represent concentration and volume, respectively; subscripts represent the cytosolic fraction (cytosol; final supernatant at the end of the fractionation procedure) and the whole lysate prior to fractionation (lysate). The unbound tissue concentration was calculated as the product of the tissue unbound fraction determined in whole lysate or cytosol and the total tissue concentration (Cu,tissue = fu,tissue • Ctissue),where Ctissue = Clysate • Vlysate/Vtissue; tissue volume (Vtissue) for whole liver (IPLs) was determined gravimetrically prior to homogenization, or calculated as 7.4 μl/mg protein in SCH, as indicated above. The ratio of intracellular and extracellular Cunbound (Kpu,u) was calculated as the unbound tissue concentration divided by the unbound concentration in perfusate or buffer (fu,buffer was assumed to be 1 in the absence of protein in both systems).
Results
To characterize the subcellular distribution of the probe compounds, differential centrifugation was performed on lysates from whole-liver tissue and SCH following drug treatment. Marker enzyme activities specific to mitochondria, lysosomes, endoplasmic reticulum (microsomes), and cytosol were examined in tissue lysate and subfractions to assess the purity and recovery of the various fractions. The distribution of these organelles in subfractions of whole-liver tissue and SCH is shown in Fig. 2. Using whole-liver tissue, separation of subfractions was demonstrated, with organelle-specific enzyme activity detected largely in the expected fractions. The lysosomal marker, acid phosphatase, was recovered primarily in the final supernatant instead of the 35,000g pellet, as would have been expected. Following fractionation of SCH lysates, the majority of membrane-bound organelles (including mitochondria and microsomes) were recovered in the initial low-speed spin (600g). Cytosolic separation and recovery was efficient and successful, as shown in Fig. 2, with ∼90% of the total LDH activity in the crude lysate recovered in the cytosolic fraction of SCH lysates.
Fig. 2.

Distribution and recovery of organelle-specific marker enzyme activity following differential centrifugation of whole-liver tissue (A) and SCH (B). Data are expressed as % total activity in the initial lysate recovered in each pellet; mean ± S.D. of n = 3 (whole liver) or n = 9 (SCH). (See Materials and Methods for more details.) super, supernatant.
Recovery of the probe drugs in all fractions was compared with the total mass in the whole lysate prior to fractionation. Recovery of probe drugs after fractionation of whole-liver tissue and SCH was approximately 100% (±10%). The average subcellular distribution of each drug in whole tissue and SCH is shown in Fig. 3. In all cases, the coefficient of variation was less than 20% when at least 10% of the drug was recovered in a given fraction. In whole-liver tissue, approximately 36% of the ritonavir was found in the cytosolic fraction, 31% in the microsomal fraction, and 18% in the mitochondrial fraction, with the remainder distributed fairly evenly among the remaining fractions. In SCH lysates, cytosolic recovery of ritonavir was similar to that in whole-liver tissue (43%). The remaining ritonavir was recovered primarily in the initial low-speed spin, consistent with binding/sequestration to organelles (including microsomes), which were difficult to separate in SCH lysates. Rosuvastatin distribution was confined primarily to the cytosol, with recovery of 72% in the cytosolic fraction of whole-liver tissue and 88% in SCH lysates. Rosuvastatin distribution to specific organelles was minimal (<11%). Furamidine was localized in the mitochondrial (43%), nuclear (18%), and lysosomal (15%) fractions of tissue lysate from the rat IPL; the remaining material was recovered in the cytosolic fraction, with minimal localization in the microsomal fraction. Furamidine was recovered predominantly (85%) in the initial low-speed spin (600g) following fractionation of SCH lysates, with minor recovery in the remaining fractions, including cytosol (3%). This is consistent with extensive binding and/or sequestration within membrane-bound organelles (including mitochondria), which were recovered in the 600g pellet following initial centrifugation of SCH lysates.
Fig. 3.

Subcellular distribution and recovery of ritonavir, rosuvastatin, and furamidine following differential centrifugation of whole-liver tissue (IPL) and SCH. Data are presented as % recovery of total drug in lysate. Open bars, 600g pellet; light gray, 10,000g pellet; cross-hatch, 35,000g pellet; diagonal hash, 100,000g pellet; black, 100,000g supernatant. (See Materials and Methods for more details.)
Concentrations of probe drugs in tissue, along with binding data and calculated concentration ratios (Kp and Kpu,u), are listed in Table 1. Total tissue accumulation (Kp) ranged over 3 orders of magnitude in the compound set selected for investigation. Ritonavir binding was extensive in whole-liver tissue and SCH (fu,lysate of 1.0% ± 0.1% and 3.0% ± 1.0%, respectively). Predicted accumulation (Kppredicted) of ritonavir based on binding and subcellular distribution (91–100 in IPL, 22–35 in SCH) was in good agreement with observed accumulation (Kpobserved; 110 and 33 in IPL and SCH, respectively). The estimated intracellular Cunbound approximated the extracellular concentration, resulting in a Kpu,u of ∼1, suggesting that binding to cellular components can explain the observed tissue accumulation. Rosuvastatin displayed moderate accumulation in liver tissue and SCH (Kpobserved of 33 and 17, respectively) in the absence of extensive binding and/or sequestration. Kppredicted (∼3–4 in IPL and SCH) underestimated the observed accumulation, as might be expected for a compound that is a substrate for hepatic uptake transporters. The estimated intracellular Cunbound and resulting Kpu,u (∼8–11) suggested accumulation of unbound rosuvastatin within the liver. Calculation of a precise Kpu,u in SCH was complicated by the low degree of binding in the diluted SCH lysate and cytosolic fraction, but accumulation of unbound drug was evident based on a Kpu,u value >5–6. Furamidine accumulation was extensive in whole-liver tissue and SCH (Kp of 8400 and 6900, respectively). Although furamidine also was extensively bound or sequestered, Kppredicted underestimated the observed accumulation by more than 10-fold. The estimated intracellular Cunbound and resulting Kpu,u (>16) confirmed accumulation of unbound drug within hepatocytes.
TABLE 1.
Total tissue concentrations, unbound fraction, and calculated tissue-to-medium or -perfusate partition coefficients (Kp and Kpu,u)
| Outcome | Ritonavir | Rosuvastatin | Furamidine | |||
|---|---|---|---|---|---|---|
| IPL | SCH | IPL | SCH | IPL | SCH | |
| Ctissue (μM) | 56 | 15 ± 2 | 18 | 17 ± 3 | 24c | 1500 ± 500 |
| Cmedium (μM) | 0.49 | 0.47 ± 0.12 | 0.54 | 1 | 0.0032c,d | 0.27 ± 0.19 |
| Kpobserved | 110 | 33 | 33 | 17 | 8400c,d | 6900 |
| Whole lysatea | ||||||
| fu,lysate (%) | 1.0 ± 0.1 | 3.0 ± 1.0 | 23 ± 1 | >36 ± 3b | 0.3 ± 0.1c | 0.9 ± 0.2 |
| Kppredicted | 100 | 35 | 4.3 | <2.8 | 330 | 110 |
| Cu,tissue (μM) | 0.56 | 0.46 ± 0.16 | 4.1 | >6.1 | 0.073 | 14 ± 6 |
| Kpu,u | 1.1 | 1.1 | 7.9 | >6.1 | 23 | 53 |
| Cytosola | ||||||
| fcytosol (%) | 36 | 43 ± 8 | 72 | 88 ± 1 | 20 | 3 ± 1 |
| fu,cytosol (%) | 3.3 ± 0.2 | 11 ± 1 | 46 ± 8 | >34 ± 3b | 6.2 ± 1.2c | 11 ± 4 |
| Kppredicted | 91 | 22 | 3.3 | <3.4 | 81 | 330 |
| Cu,tissue (μM) | 0.62 | 0.76 ± 0.31 | 5.9 | >5.0 | 0.30 | 4.3 ± 1.6 |
| Kpu,u | 1.3 | 1.7 | 11 | >5.0 | 93 | 16 |
Kppredicted and Cu,tissue calculated for lysate and cytosol as described in Materials and Methods.
Undiluted fu and subsequent calculations represented by inequalities when fu,measured > 80%, as described in Materials and Methods.
From Yan et al., 2011.
Taking into account fu of 44% in perfusate, containing 20% whole blood, as determined by Yan et al., 2011.
Discussion
The present study evaluated an organ-specific in vitro model system to estimate Cunbound and predict cellular partitioning and accumulation of drugs. The SCH system exhibits properly localized and functional transport proteins as well as metabolic and regulatory machinery. This system may be able to recapitulate the relevant cellular disposition of drugs whose distribution is influenced by the interplay of these processes in hepatocytes in vivo.
Total accumulation (Kpobserved) in whole tissue was well predicted by the SCH model (within 2- to 3-fold) for the limited compound set selected for investigation. A system that enables accurate estimates of hepatic Kp values would be a valuable addition to physiologically based pharmacokinetic modeling efforts to predict hepatic exposure in humans. Although tissue partitioning can be predicted using physicochemical properties, such as the method of Rodgers and Rowland (Rodgers et al., 2005; Rodgers and Rowland, 2006), exceptions and inaccuracies inevitably exist, particularly for compounds that may rely on active processes, such as transport and/or metabolism.
Although the Kp value is useful for estimating total tissue concentration, it does not provide information on the mechanism(s) of tissue accumulation. For mechanistic information, one must assess tissue binding and subcellular distribution. Kpu,u is an informative parameter for describing the potential role of active uptake; however, a Kpu,u ≈1 does not necessarily imply that passive processes (e.g., diffusion) are solely responsible for hepatocellular distribution. Although it is tempting to dismiss the potential contribution of active transport for such compounds, it is possible that a net balance exists between uptake and efflux processes. In such a case, impaired transport function (e.g., DDIs, genetic variation, and/or disease) may result in altered hepatic and/or systemic exposure, with corresponding implications for changes in efficacy, toxicity, and DDI potential. For example, while the Kpu,u of ritonavir in the current study was ∼1, ritonavir is excreted into bile in rats, dogs, and humans (Denissen et al., 1997; Pfeifer et al., 2013) and inhibits P-glycoprotein- and multidrug resistance–associated protein (MRP)-mediated efflux in vitro (Gutmann et al., 1999; Vourvahis and Kashuba, 2007). Although the role of hepatic uptake in ritonavir disposition has not been reported conclusively, uptake into suspended hepatocytes is sensitive to temperature (Parker and Houston, 2008), and ritonavir is a competitive inhibitor of OATP-mediated uptake (Annaert et al., 2010). Ritonavir accumulation (Kp) and Kpu,u (∼600 and ∼10, respectively) in suspended hepatocytes (Parker and Houston, 2008; Yabe et al., 2011) was greater than total accumulation reported in whole liver [10- to 100-fold in vivo (Denissen et al., 1997) and ∼50 in the present study] and the Kpu,u of ∼1 in SCH and IPLs in the present study. This discrepancy is likely due, at least in part, to internalization of the canalicular membrane in suspended hepatocytes, rendering biliary excretion nonfunctional (Bow et al., 2008), and underscores the importance of using a model that recapitulates the totality of liver function as closely as possible. Interestingly, the Kp of ∼100 and Kpu,u of 1.7 reported recently for ritonavir in human embryonic kidney (HEK)293 cells were in excellent agreement with the present studies in whole rat liver and SCH, despite the fact that HEK293 cells largely lack the assortment of transporters and metabolizing enzymes expected to influence the hepatocellular disposition of drugs (Mateus et al., 2013). We demonstrated recently that ritonavir did not inhibit MRP2-mediated biliary excretion of the imaging agent 99mTc-mebrofenin in humans (Pfeifer et al., 2013). This apparent in vitro–in vivo disconnect was reconciled by accounting for the Cunbound of ritonavir in hepatocytes relative to its inhibitory potency against MRP2 (Pfeifer et al., 2013), and highlights the importance of estimating accurately the relevant hepatocellular Cunbound to predict potential DDIs.
Rosuvastatin accumulation in whole liver (Kpobserved = 33) and SCH (Kpobserved = 17) was in good agreement with previous reports of 18- to 45-fold hepatic accumulation after oral administration in rats (Nezasa et al., 2002b) and 15-fold accumulation in isolated hepatocytes (Nezasa et al., 2003). Although rosuvastatin binding did account for a portion of tissue accumulation (Kppredicted ∼3–4), the majority of accumulation was due to unbound drug (Kpu,u of 8–11), consistent with efficient hepatic uptake and rate-limited efflux, and previous Kpu,u values ranging from ∼10–60 in vitro (Nezasa et al., 2003; Yabe et al., 2011; Shitara et al., 2013). Rosuvastatin binding data in SCH demonstrated a limitation associated with the required dilution of small in vitro tissue samples when performing these studies. For compounds such as rosuvastatin that are not extensively bound (fu,measured > 80%), dilution of SCH samples upon collection resulted in observed binding data that were difficult to extrapolate accurately. Therefore, the resulting Kpu,u value in SCH is a conservative minimum and potentially underestimates the true value. However, the observed SCH fu,lysate of >36% ± 3% is similar to the 48% reported previously in suspended hepatocytes (Yabe et al., 2011), so the reported Kpu,u of >5–6 from SCH (Table 1) is likely less than a 2-fold underestimate, and still indicates the contribution of active uptake.
Extensive hepatic accumulation of the active metabolite, furamidine, was observed in both rat IPLs and SCH, as reported previously (Yan et al., 2011). Previous studies using differential centrifugation revealed that furamidine was localized primarily in the mitochondrial fraction of the liver and kidney (Midgley et al., 2007). Similarly, furamidine was localized primarily in the mitochondrial fraction (43%) following subcellular fractionation of tissue lysate from the rat IPL. Despite extensive binding and sequestration, these processes underpredict the observed accumulation in liver tissue. Accumulation of unbound furamidine (Kpu,u) was predicted to be ≥16-fold, suggesting permeability-limited efflux of this charged metabolite formed in the hepatocyte. The extensive hepatic binding/sequestration may limit the systemic exposure to furamidine; in contrast, a structural analog had a 5-fold greater hepatic fu, which could contribute, in part, to the enhanced systemic exposure of this agent compared with furamidine (Yan et al., 2011).
Subcellular fractionation has not been reported in SCH. An established method to determine subcellular localization and sequestration of drugs and/or metabolites in a relevant in vitro system would be useful. This is particularly true of the liver, where drug-induced liver injury via idiosyncratic hepatocellular injury is the most frequent cause of regulatory action on drugs, including failure to approve, labeling changes, and withdrawal from the market (Watkins et al., 2008). Hepatotoxic drugs are known to cause hepatocellular injury through diverse pathways, including organelle-specific sequestration and toxicity, such as phopholipidosis (lysosomes) (Reasor and Kacew, 2001) and mitochondrial disruption/dysfunction (Labbe et al., 2008), among others (Gunawan and Kaplowitz, 2007). Although isolation of subcellular fractions consisting of membrane-bound organelles proved challenging in SCH, cell lysis, with release and isolation of soluble cytosolic proteins and contents, was successful as determined by LDH activity and rosuvastatin recovery. Enzyme markers of subcellular components were recovered primarily in the initial, low-speed centrifugation step from SCH lysates, indicating that these organelles were retained in large, dense conglomerates. It is possible that the collagen matrix, indispensible to the sandwich configuration, prevented the plasma membrane from separating enough to release larger, membrane-bound bodies. This limitation may provide an advantage for rapid isolation of the cytosolic fraction in SCH. The present data suggest that a single, low-speed (600g) spin, which can be performed on any benchtop microcentrifuge, will reliably exclude cellular organelles from the resulting supernatant of SCH lysate. This single-step method has the additional advantage of minimizing potential redistribution of drugs during multiple centrifugation steps and associated dilutions. Although the specific site(s) of subcellular sequestration would remain unknown, extensive distribution/recovery of drug in the pellet could indicate the need to consider intracellular pharmacokinetics. Meanwhile, the cytosolic fraction would contain the unbound drug content in the tissue (fcytosol), which could be evaluated further by determining the extent of binding to cytosolic protein (fu,cytosol), as reported in the present studies.
A number of potential limitations are associated with the subcellular fractionation approach, including the following. 1) There is some degree of cross-contamination among isolated cellular fractions. This can be assessed primarily by recovery of marker enzyme activity specific for each organelle, but adds significant time- and labor-intensive efforts to sample analysis. The recovery of acid phosphatase activity in multiple fractions (Fig. 2A) is an example of potential cross-contamination. Lysosomes are quite fragile, a characteristic that is noted and/or exploited in traditional fractionation methods (Burnside and Schneider, 1982; Ohsumi et al., 1983); therefore, it is likely that a portion of the lysosomes were disrupted during the homogenization process in both SCH and whole-tissue matrices. 2) Potential disruption of binding equilibrium between cellular compartments occurs during the fractionation process. Tissue homogenization and fractionation inherently involve dilution, which can shift binding equilibrium. Despite the potential limitations of the reported method, assessment of intracellular Cunbound and mechanisms of hepatic accumulation is challenging, and in vitro methods are needed for general application to compounds that are not amenable to visualization by imaging techniques such as fluorescence. Other methods are not without limitations, such as assessment of cellular accumulation at 4°C versus 37°C (Shitara et al., 2013). The assumption of the latter method is that all active processes are inert at 4°C and that cellular accumulation represents passive equilibration of unbound drug. However, the effect of temperature and potential artifacts on nonspecific binding (Igari et al., 1981; Kodama et al., 1999; Zeitlinger et al., 2011) and membrane fluidity and partitioning (Herbette et al., 1983; Palmeira and Oliveira, 1992; Liu et al., 2001) are well established. The methods applied to rat SCH in the present investigation to determine intracellular Cunbound and Kpu,u were reported recently in immortalized (HEK293) cells (Mateus et al., 2013), which lack the full complement of mechanisms present in hepatocytes that influence cellular accumulation (e.g., active uptake, metabolism, and biliary excretion).
Hepatic intracellular Cunbound, Kpu,u, and subcellular localization can be used to improve prediction of clinical efficacy, toxicity, and DDIs. A straightforward in vitro method was developed to determine hepatocellular accumulation of total and unbound drug in the SCH model. This method was used successfully to differentiate the contribution of active transport versus binding/sequestration as mechanisms of hepatocellular accumulation for a set of probe drugs with distinct mechanisms of hepatocellular uptake and accumulation. Further validation of this approach using SCH to determine the hepatocellular Cunbound and Kpu,u of additional compounds is ongoing.
Abbreviations
- Clysate
whole lysate concentration prior to fractionation
- Ctissue
total tissue concentration
- Cunbound
unbound concentration
- Cu,extracell
extracellular unbound concentration
- Cu,tissue
unbound tissue concentration
- DDI
drug-drug interaction
- fcytosol
fraction of total drug mass recovered in the cytosol
- fu
unbound fraction
- fu,buffer
unbound fraction in perfusate or buffer
- fu,cytosol
unbound fraction in cytosol
- fu,extracell
unbound fraction in extracellular matrix
- fu,lysate
unbound fraction in whole lysate
- fu,measured
measured unbound fraction
- fu,tissue
unbound fraction in tissue
- HBSS
Hanks’ balanced salt solution
- HEK
human embryonic kidney
- IPL
isolated perfused liver
- Kp
ratio of intracellular and extracellular total concentration
- Kpobserved
observed partition coefficient
- Kppredicted
predicted partition coefficient
- Kpu,u
ratio of intracellular and extracellular unbound concentration
- LDH
lactate dehydrogenase; MRP, multidrug resistance–associated protein
- OATP
organic anion–transporting polypeptide
- SCH
sandwich-cultured hepatocytes
- Vcytosol
volume of cytosolic fraction (supernatant volume following fractionation)
- Vlysate
lysate volume prior to fractionation
- Vtissue
tissue volume prior to homogenization
Authorship Contributions
Participated in research design: Pfeifer, Yan, Brouwer.
Conducted experiments: Pfeifer, Harris, Yan.
Performed data analysis: Pfeifer, Harris, Yan.
Wrote or contributed to the writing of the manuscript: Pfeifer, Yan, Brouwer.
Footnotes
This work was supported by the National Institutes of Health National Institute of General Medical Sciences [Grant R01-GM41935]. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. N.P. was supported, in part, by the University of North Carolina Royster Society of Fellows.
References
- Abe K, Bridges AS, Yue W, Brouwer KLR. (2008) In vitro biliary clearance of angiotensin II receptor blockers and 3-hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors in sandwich-cultured rat hepatocytes: comparison with in vivo biliary clearance. J Pharmacol Exp Ther 326:983–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annaert P, Ye ZW, Stieger B, Augustijns P. (2010) Interaction of HIV protease inhibitors with OATP1B1, 1B3, and 2B1. Xenobiotica 40:163–176 [DOI] [PubMed] [Google Scholar]
- Bow DA, Perry JL, Miller DS, Pritchard JB, Brouwer KLR. (2008) Localization of P-gp (Abcb1) and Mrp2 (Abcc2) in freshly isolated rat hepatocytes. Drug Metab Dispos 36:198–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouwer KLR, Thurman RG. (1996) Isolated perfused liver. Pharm Biotechnol 8:161–192 [DOI] [PubMed] [Google Scholar]
- Brown HS, Wilby AJ, Alder J, Houston JB. (2010) Comparative use of isolated hepatocytes and hepatic microsomes for cytochrome P450 inhibition studies: transporter-enzyme interplay. Drug Metab Dispos 38:2139–2146 [DOI] [PubMed] [Google Scholar]
- Burnside J, Schneider DL. (1982) Characterization of the membrane proteins of rat liver lysosomes. Composition, enzyme activities and turnover. Biochem J 204:525–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YJ, Huang SM, Liu CY, Yeh PH, Tsai TH. (2008) Hepatobiliary excretion and enterohepatic circulation of colchicine in rats. Int J Pharm 350:230–239 [DOI] [PubMed] [Google Scholar]
- Chu X, Korzekwa K, Elsby R, Fenner K, Galetin A, Lai Y, Matsson P, Moss A, Nagar S, Rosania GR, et al. International Transporter Consortium (2013) Intracellular drug concentrations and transporters: measurement, modeling, and implications for the liver. Clin Pharmacol Ther 94:126–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denissen JF, Grabowski BA, Johnson MK, Buko AM, Kempf DJ, Thomas SB, Surber BW. (1997) Metabolism and disposition of the HIV-1 protease inhibitor ritonavir (ABT-538) in rats, dogs, and humans. Drug Metab Dispos 25:489–501 [PubMed] [Google Scholar]
- Deshmukh SV, Harsch A. (2011) Direct determination of the ratio of unbound fraction in plasma to unbound fraction in microsomal system (fu p/fu mic) for refined prediction of phase I mediated metabolic hepatic clearance. J Pharmacol Toxicol Methods 63:35–39 [DOI] [PubMed] [Google Scholar]
- Dollery CT. (2013) Intracellular drug concentrations. Clin Pharmacol Ther 93:263–266 [DOI] [PubMed] [Google Scholar]
- Giacomini KM, Huang SM, Tweedie DJ, Benet LZ, Brouwer KLR, Chu X, Dahlin A, Evers R, Fischer V, Hillgren KM, et al. International Transporter Consortium (2010) Membrane transporters in drug development. Nat Rev Drug Discov 9:215–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Y, Zhao Z, McConn DJ, Beaudet B, Tallman M, Speake JD, Ignar DM, Krise JP. (2007) Lysosomes contribute to anomalous pharmacokinetic behavior of melanocortin-4 receptor agonists. Pharm Res 24:1138–1144 [DOI] [PubMed] [Google Scholar]
- Griffin L, Annaert P, Brouwer KLR. (2011) Influence of drug transport proteins on the pharmacokinetics and drug interactions of HIV protease inhibitors. J Pharm Sci 100:3636–3654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunawan BK, Kaplowitz N. (2007) Mechanisms of drug-induced liver disease. Clin Liver Dis 11:459–475 [DOI] [PubMed] [Google Scholar]
- Gupta A, Chatelain P, Massingham R, Jonsson EN, Hammarlund-Udenaes M. (2006) Brain distribution of cetirizine enantiomers: comparison of three different tissue-to-plasma partition coefficients: K(p), K(p,u), and K(p,uu). Drug Metab Dispos 34:318–323 [DOI] [PubMed] [Google Scholar]
- Gutmann H, Fricker G, Drewe J, Toeroek M, Miller DS. (1999) Interactions of HIV protease inhibitors with ATP-dependent drug export proteins. Mol Pharmacol 56:383–389 [DOI] [PubMed] [Google Scholar]
- Hammarlund-Udenaes M, Fridén M, Syvänen S, Gupta A. (2008) On the rate and extent of drug delivery to the brain. Pharm Res 25:1737–1750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbette L, Katz AM, Sturtevant JM. (1983) Comparisons of the interaction of propranolol and timolol with model and biological membrane systems. Mol Pharmacol 24:259–269 [PubMed] [Google Scholar]
- Igari Y, Sugiyama Y, Awazu S, Hanano M. (1981) Interspecies difference in drug protein binding-temperature and protein concentration dependency: effect on calculation of effective protein fraction. J Pharm Sci 70:1049–1053 [DOI] [PubMed] [Google Scholar]
- Jones HM, Barton HA, Lai Y, Bi YA, Kimoto E, Kempshall S, Tate SC, El-Kattan A, Houston JB, Galetin A, et al. (2012) Mechanistic pharmacokinetic modeling for the prediction of transporter-mediated disposition in humans from sandwich culture human hepatocyte data. Drug Metab Dispos 40:1007–1017 [DOI] [PubMed] [Google Scholar]
- Kalvass JC, Maurer TS, Pollack GM. (2007) Use of plasma and brain unbound fractions to assess the extent of brain distribution of 34 drugs: comparison of unbound concentration ratios to in vivo p-glycoprotein efflux ratios. Drug Metab Dispos 35:660–666 [DOI] [PubMed] [Google Scholar]
- Kodama H, Kodama Y, Shinozawa S, Kanemaru R, Todaka K, Mitsuyama Y. (1999) Temperature effect on serum protein binding kinetics of phenytoin in monotherapy patients with epilepsy. Eur J Pharm Biopharm 47:295–298 [DOI] [PubMed] [Google Scholar]
- Kudo N, Sakai A, Mitsumoto A, Hibino Y, Tsuda T, Kawashima Y. (2007) Tissue distribution and hepatic subcellular distribution of perfluorooctanoic acid at low dose are different from those at high dose in rats. Biol Pharm Bull 30:1535–1540 [DOI] [PubMed] [Google Scholar]
- Labbe G, Pessayre D, Fromenty B. (2008) Drug-induced liver injury through mitochondrial dysfunction: mechanisms and detection during preclinical safety studies. Fundam Clin Pharmacol 22:335–353 [DOI] [PubMed] [Google Scholar]
- Lee JK, Brouwer KR. (2010) Determination of intracellular volume of rat and human sandwich-cultured hepatocytes (Abstract ID 1595). Toxicol Sci 114:339 [Google Scholar]
- Li N, Bi YA, Duignan DB, Lai Y. (2009) Quantitative expression profile of hepatobiliary transporters in sandwich cultured rat and human hepatocytes. Mol Pharm 6:1180–1189 [DOI] [PubMed] [Google Scholar]
- Liu X, Chen C, Smith BJ. (2008) Progress in brain penetration evaluation in drug discovery and development. J Pharmacol Exp Ther 325:349–356 [DOI] [PubMed] [Google Scholar]
- Liu XY, Yang Q, Kamo N, Miyake J. (2001) Effect of liposome type and membrane fluidity on drug-membrane partitioning analyzed by immobilized liposome chromatography. J Chromatogr A 913:123–131 [DOI] [PubMed] [Google Scholar]
- Mateus A, Matsson P, Artursson P. (2013) Rapid measurement of intracellular unbound drug concentrations. Mol Pharm 10:2467–2478 [DOI] [PubMed] [Google Scholar]
- Midgley I, Fitzpatrick K, Taylor LM, Houchen TL, Henderson SJ, Wright SJ, Cybulski ZR, John BA, McBurney A, Boykin DW, et al. (2007) Pharmacokinetics and metabolism of the prodrug DB289 (2,5-bis[4-(N-methoxyamidino)phenyl]furan monomaleate) in rat and monkey and its conversion to the antiprotozoal/antifungal drug DB75 (2,5-bis(4-guanylphenyl)furan dihydrochloride). Drug Metab Dispos 35:955–967 [DOI] [PubMed] [Google Scholar]
- Nezasa K, Higaki K, Matsumura T, Inazawa K, Hasegawa H, Nakano M, Koike M. (2002a) Liver-specific distribution of rosuvastatin in rats: comparison with pravastatin and simvastatin. Drug Metab Dispos 30:1158–1163 [DOI] [PubMed] [Google Scholar]
- Nezasa K, Higaki K, Takeuchi M, Nakano M, Koike M. (2003) Uptake of rosuvastatin by isolated rat hepatocytes: comparison with pravastatin. Xenobiotica 33:379–388 [DOI] [PubMed] [Google Scholar]
- Nezasa K, Takao A, Kimura K, Takaichi M, Inazawa K, Koike M. (2002b) Pharmacokinetics and disposition of rosuvastatin, a new 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor, in rat. Xenobiotica 32:715–727 [DOI] [PubMed] [Google Scholar]
- Obach RS. (1996) The importance of nonspecific binding in in vitro matrices, its impact on enzyme kinetic studies of drug metabolism reactions, and implications for in vitro-in vivo correlations. Drug Metab Dispos 24:1047–1049 [PubMed] [Google Scholar]
- Obach RS. (1999) Prediction of human clearance of twenty-nine drugs from hepatic microsomal intrinsic clearance data: an examination of in vitro half-life approach and nonspecific binding to microsomes. Drug Metab Dispos 27:1350–1359 [PubMed] [Google Scholar]
- Ockerman PA. (1967) Glucose-6-phosphatase assay on microgram amounts of liver tissue. Clin Chim Acta 17:201–206 [DOI] [PubMed] [Google Scholar]
- Ohsumi Y, Ishikawa T, Kato K. (1983) A rapid and simplified method for the preparation of lysosomal membranes from rat liver. J Biochem 93:547–556 [PubMed] [Google Scholar]
- Paine MF, Wang MZ, Generaux CN, Boykin DW, Wilson WD, De Koning HP, Olson CA, Pohlig G, Burri C, Brun R, et al. (2010) Diamidines for human African trypanosomiasis. Curr Opin Investig Drugs 11:876–883 [PubMed] [Google Scholar]
- Palmeira CM, Oliveira CR. (1992) Partitioning and membrane disordering effects of dopamine antagonists: influence of lipid peroxidation, temperature, and drug concentration. Arch Biochem Biophys 295:161–171 [DOI] [PubMed] [Google Scholar]
- Parker AJ, Houston JB. (2008) Rate-limiting steps in hepatic drug clearance: comparison of hepatocellular uptake and metabolism with microsomal metabolism of saquinavir, nelfinavir, and ritonavir. Drug Metab Dispos 36:1375–1384 [DOI] [PubMed] [Google Scholar]
- Pfeifer ND, Goss SL, Swift B, Ghibellini G, Ivanovic M, Heizer WD, Gangarosa LM, Brouwer KLR. (2013) Effect of ritonavir on (99m)Technetium-mebrofenin disposition in humans: a semi-PBPK modeling and in vitro approach to predict transporter-mediated DDIs. CPT Pharmacometrics Syst Pharmacol 2:e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reasor MJ, Kacew S. (2001) Drug-induced phospholipidosis: are there functional consequences? Exp Biol Med (Maywood) 226:825–830 [DOI] [PubMed] [Google Scholar]
- Rezk NL, White NR, Jennings SH, Kashuba AD. (2009) A novel LC-ESI-MS method for the simultaneous determination of etravirine, darunavir and ritonavir in human blood plasma. Talanta 79:1372–1378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers T, Leahy D, Rowland M. (2005) Physiologically based pharmacokinetic modeling 1: predicting the tissue distribution of moderate-to-strong bases. J Pharm Sci 94:1259–1276 [DOI] [PubMed] [Google Scholar]
- Rodgers T, Rowland M. (2006) Physiologically based pharmacokinetic modelling 2: predicting the tissue distribution of acids, very weak bases, neutrals and zwitterions. J Pharm Sci 95:1238–1257 [DOI] [PubMed] [Google Scholar]
- Sato K, Mizuki Y, Komuro S. (2010) Consideration of reliable concentrations for prediction of change in enzyme activity by mechanism-based inactivation using physiologically-based pharmacokinetic model simulations. Drug Metab Pharmacokinet 25:335–342 [DOI] [PubMed] [Google Scholar]
- Shitara Y, Maeda K, Ikejiri K, Yoshida K, Horie T, Sugiyama Y. (2013) Clinical significance of organic anion transporting polypeptides (OATPs) in drug disposition: their roles in hepatic clearance and intestinal absorption. Biopharm Drug Dispos 34:45–78 [DOI] [PubMed] [Google Scholar]
- Smith DA, Di L, Kerns EH. (2010) The effect of plasma protein binding on in vivo efficacy: misconceptions in drug discovery. Nat Rev Drug Discov 9:929–939 [DOI] [PubMed] [Google Scholar]
- Swift B, Pfeifer ND, Brouwer KLR. (2010) Sandwich-cultured hepatocytes: an in vitro model to evaluate hepatobiliary transporter-based drug interactions and hepatotoxicity. Drug Metab Rev 42:446–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchaparian EH, Houghton JS, Uyeda C, Grillo MP, Jin L. (2011) Effect of culture time on the basal expression levels of drug transporters in sandwich-cultured primary rat hepatocytes. Drug Metab Dispos 39:2387–2394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vourvahis M, Kashuba AD. (2007) Mechanisms of pharmacokinetic and pharmacodynamic drug interactions associated with ritonavir-enhanced tipranavir. Pharmacotherapy 27:888–909 [DOI] [PubMed] [Google Scholar]
- Ward ES, Pollack GM, Brouwer KLR. (2000) Probenecid-associated alterations in valproic acid pharmacokinetics in rats: can in vivo disposition of valproate glucuronide be predicted from in vitro formation data? Drug Metab Dispos 28:1433–1439 [PubMed] [Google Scholar]
- Watkins PB, Seligman PJ, Pears JS, Avigan MI, Senior JR. (2008) Using controlled clinical trials to learn more about acute drug-induced liver injury. Hepatology 48:1680–1689 [DOI] [PubMed] [Google Scholar]
- Yabe Y, Galetin A, Houston JB. (2011) Kinetic characterization of rat hepatic uptake of 16 actively transported drugs. Drug Metab Dispos 39:1808–1814 [DOI] [PubMed] [Google Scholar]
- Yamano K, Yamamoto K, Kotaki H, Sawada Y, Iga T. (1999) Quantitative prediction of metabolic inhibition of midazolam by itraconazole and ketoconazole in rats: implication of concentrative uptake of inhibitors into liver. Drug Metab Dispos 27:395–402 [PubMed] [Google Scholar]
- Yan GZ, Brouwer KLR, Pollack GM, Wang MZ, Tidwell RR, Hall JE, Paine MF. (2011) Mechanisms underlying differences in systemic exposure of structurally similar active metabolites: comparison of two preclinical hepatic models. J Pharmacol Exp Ther 337:503–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeitlinger MA, Derendorf H, Mouton JW, Cars O, Craig WA, Andes D, Theuretzbacher U. (2011) Protein binding: do we ever learn? Antimicrob Agents Chemother 55:3067–3074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Zhou F, Lu M, Ji W, Niu F, Zha W, Wu X, Hao H, Wang G. (2012) Pharmacokinetics-pharmacology disconnection of herbal medicines and its potential solutions with cellular pharmacokinetic-pharmacodynamic strategy. Curr Drug Metab 13:558–576 [DOI] [PubMed] [Google Scholar]
- Zhou F, Zhang J, Li P, Niu F, Wu X, Wang G, Roberts MS. (2011) Toward a new age of cellular pharmacokinetics in drug discovery. Drug Metab Rev 43:335–345 [DOI] [PubMed] [Google Scholar]