

Abstract
Cytochrome P450 (P450) function requires the interaction of P450 and NADPH-cytochrome P450 reductase (CPR) in membranes, and is frequently studied using reconstituted systems composed solely of phosphatidylcholine. There is increasing evidence that other endoplasmic reticulum (ER) lipids can affect P450 structure, activity, and interactions with CPR. Some of these lipid effects have been attributed to the formation of organized liquid-ordered (lo) domains. The goal of this study was to determine if lo domains were formed in P450 reconstituted systems mimicking the ER membrane. CYP1A2, when incorporated in “ER-like” lipid vesicles, displayed detergent insolubility after treatment with Brij 98 and centrifugation in a sucrose gradient. Lipid probes were employed to identify domain formation in both ER-like vesicles and model membranes known to form lo domains. Changes in fluorescence resonance energy transfer (FRET) using an established donor/acceptor FRET pair in both ER-like and model lo-forming systems demonstrated the coexistence of lo- and liquid-disordered domains as a function of cholesterol and sphingomyelin content. Similarly, 6-dodecanoyl-2-dimethylaminonaphthalene (laurdan), a probe that reports on membrane organization, showed that cholesterol and sphingomyelin increased membrane order. Finally, brominated-phosphatidylcholine allowed for monitoring of the location of both CPR and CYP1A2 within the lo regions of ER-like systems. Taken together, the results demonstrate that ER-like vesicles generate microdomains, and both CYP1A2 and CPR predominantly localize into lo membrane regions. Probe fluorescent responses suggest that lipid microdomains form in these vesicles whether or not enzymes are included in the reconstituted systems. Thus, it does not appear that the proteins are critical for stabilizing lo domains.
Introduction
Cytochromes P450 (P450s) are membrane-bound proteins predominately found in the endoplasmic reticulum (ER) and are essential enzymes in the oxidation of endogenous and exogenous compounds (Nelson, 2003). P450s must interact with their redox partner, NADPH-cytochrome P450 reductase (CPR) to receive electrons that are required for substrate metabolism (Gigon et al., 1969). Another obligatory component of this monooxygenase system is phospholipid. Although phospholipid has been known to be an important element in P450 function for many years (Strobel et al., 1970), more recently there has been increasing evidence that specific phospholipids affect this system significantly. A number of different lipids have been demonstrated to directly affect catalytic activity, stability of the enzymes, and incorporation of P450 into the membrane (Blanck et al., 1984; Ingelman-Sundberg et al., 1996; Reed et al., 2006). Furthermore, a few studies have suggested that P450s can promote anionic lipid segregation and domain formation, which in turn affects P450 function (Kim et al., 2003; Kim et al., 2007; Jang et al., 2010).
Lipid microdomains in the plasma membrane also have been linked to protein function and a multitude of cellular events (Brown and London, 1998). There are two types of lipid domains that exist in the plasma membrane under physiologic conditions. Liquid-ordered (lo) phases are enriched in saturated fatty acid chains and cholesterol, which decreases the fluidity of the lipid environment and prevents them from being solubilized by nonionic detergents, leading to their informal name, detergent-resistant membranes (DRMs) (Brown and London, 1998). Conversely, liquid-disordered (ld) membrane regions are mainly composed of unsaturated fatty acid chains that allow for increased membrane fluidity (Quinn and Wolf, 2009). DRMs have been extensively characterized in the plasma membrane but only recently have been described in the ER (Pielsticker et al., 2005; Browman et al., 2006; Hayashi and Fujimoto, 2010; Brignac-Huber et al., 2011).
Our laboratory has previously characterized ER detergent-resistant microdomains (ER-DRMs) in rabbit liver microsomes that, similar to plasma membrane DRMs (Pike, 2004), are enriched in cholesterol and sphingomyelin (Brignac-Huber et al., 2011). CYP1A2 and CPR were found to reside in these ER-DRMs, which also were determined to be cholesterol-dependent. Moreover, upon using previously published protocols (Reed et al., 2008; Reed, 2010) to integrally incorporate CYP1A2 and CPR into purified vesicles that mimicked DRM, the apparent Km of CPR-CYP1A2 complex was decreased when compared with vesicles composed solely of bovine liver phosphatidylcholine. These findings suggested that the specific lipid composition of the ER-DRMs increased CYP1A2-dependent metabolism by altering the efficiency of CPR and CYP1A2 interaction. Although these studies showed that the function of P450 was influenced by its incorporation into vesicles composed of lipids with a composition similar to ER-DRMs, ordered lipid domain formation in these artificial membranes was not conclusively established.
Numerous studies have analyzed lipid microdomain formation in model lipid systems that mimic the plasma membrane (Dietrich et al., 2001; Loura et al., 2001; de Almeida et al., 2005; Baumgart et al., 2007). However, very few studies have done analogous experiments in lipid systems that mimic the ER. Therefore, in this study, we provide physical evidence that lipid domains are formed in vesicles made up of physiologic lipids at the relative concentrations observed in the ER. First, we demonstrate that purified CYP1A2 and CPR, when incorporated together, localize to DRMs in these vesicular reconstituted systems in a manner that is similar to their localization in rabbit liver microsomes (Brignac-Huber et al., 2011). In addition, the existence of lipid microdomains is demonstrated in the “ER-like” reconstituted systems by employing several fluorescent probes that have been shown to preferentially localize to either lo or ld regions in plasma membrane models. Alternatively, one of the probes used in this study, 6-dodecanoyl-2-dimethylaminonaphthalene (laurdan), displays different fluorescent characteristics when present in lo and ld domains. Studies using these types of probes typically have been performed in plasma membrane model systems containing three well defined lipid components and have led to the construction of phase diagrams showing the lipid proportions that define the boundaries for the lo, ld, and gel phases as well as those that delineate the boundaries of coexisting phases (de Almeida et al., 2003).
By using a ternary-lipid model system (de Almeida et al., 2003) as a reference and lipid vesicles containing only phosphatidylcholine (PC) as a “negative” control, our results using the fluorescent probes provide evidence for the existence of lipid microdomains in the ER-like reconstituted systems in the absence of proteins. When proteins were integrally incorporated into the vesicular reconstituted systems, the results indicate that CYP1A2 and CPR both localize to DRMs. Furthermore, the addition of proteins to the vesicular reconstituted systems does not seem to affect lipid microdomain distribution, suggesting that lipid domain formation occurs independently of protein and in turn influences the behavior of the enzymes of the P450 system.
Materials and Methods
Lipids were purchased from Avanti Polar Lipids (Alabaster, AL). The lipids used for the reconstituted system mimicking the ER (natural membrane systems) were natural extracts from a variety of sources: phosphatidylcholine (liver, bovine), phosphatidylethanolamine (liver, bovine), phosphatidylinositol (liver, bovine), phosphatidylserine (brain, porcine), phosphatidic acid (egg, chicken), and sphingomyelin (brain, porcine). The lipids used for reconstituted systems of model membranes were the synthetic lipids, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) and palmitoyl-sphingomyelin (PSM). Cholesterol was added to reconstituted systems using water-soluble cholesterol from Sigma-Aldrich (St. Louis, MO). Brij 98 was also obtained from Sigma-Aldrich. N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)-1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine, triethylammonium salt (NBD-PE), Lissamine rhodamine B 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine, triethylammonium salt (Rh-PE), and laurdan were purchased from Invitrogen/Life Technologies (Grand Island, NY). Protease cocktail inhibitor was purchased from Roche (Indianapolis, IN). The 5-μm syringe filters were purchased from Osmonics (Greenville, SC). YM-30 filters were purchased from Millipore (Billerica, MA), and 96-well, half-area flat bottom plates were purchased from Corning (Lowell, MA). Bio-Beads SM-2 were purchased from Bio-Rad (Hercules, CA).
Enzyme Source.
CYP1A2 was isolated and purified from β-naphthoflavone–treated rabbit liver microsomes as described previously (Coon et al., 1978). P450 levels were determined by measuring the absorbance of the carbon monoxy-ferrous complex at 450 nm (Omura and Sato, 1964).
Lipid Vesicle Preparation.
Except for the details described below, vesicular lipid reconstituted systems containing 5 μM CYP1A2 were prepared at a 500:1 total lipid/P450 ratio as previously described (Reed et al., 2008). Various lipid compositions were prepared in our reconstituted systems containing either naturally occurring lipids or synthetic lipid to study model membranes. Phosphatidylcholine vesicles (V-PC), mixed phospholipid vesicles (V-Mix PL), vesicles with lipid composition similar to that of the ER membrane (V-ER lipids), and vesicles with the lipid composition similar to that of the DRM fractions (V-DRM) were prepared as previously described (Brignac-Huber et al., 2011). Model membranes were prepared with the lipid compositions (mol%) listed in Table 1. All lipids except for cholesterol (added as described below) were dissolved in chloroform and slowly dried under a stream of N2 at room temperature until chloroform was completely removed (∼1 hour). Lipid was rehydrated (at 1/5th the final volume of the vesicle preparation) with 250 mM HEPES (pH 7.5) containing 5% sodium glycocholate and bath-sonicated until it clarified. When appropriate, the solubilized lipid solution was then added to the purified enzymes (to achieve final protein concentrations of 5 μM P450 and 1 μM CPR) in four equal aliquots of the rehydrated lipid suspension. This step limited the destruction of P450 by detergent. N2 was layered over the P450/lipid solution between each addition before mixing the sample by inversion. When proteins were not present, the lipid solution was added to deionized water. The mixture was then incubated at 4°C for 1 hour before adding 125 mg of Biobeads SM-2 to remove the detergent. The samples were rocked for 2 hours at 4°C and the vesicles were subsequently separated from the Biobeads by drawing the sample into a syringe using a 26.5-gauge needle. The beads were then washed twice with 100 μl of reaction buffer (50 mM HEPES, 15mM MgCl2, 5mM EDTA, pH 7.5) to recover any residual vesicle volume. These washes were added to the original volume extracted from the beads and the entire sample was filtered through a 5-μm syringe filter. Vesicles that required the addition of cholesterol (indicated in Table 1) were treated as previously described using water-soluble cholesterol (Niu and Litman, 2002; Brignac-Huber et al., 2011). The water-soluble cholesterol was dissolved in H2O for a final cholesterol concentration of 4 μg/μl. Water-soluble cholesterol was added to the appropriate reconstituted systems and samples were rocked at room temperature for 2 hours, upon which they were filtered using an YM-30 filter at 1300g for 15 minutes. This centrifugation allows for the methyl-β-cyclodextrin (MβC) molecule to flow through the filter, leaving the vesicle system above the filter. The vesicle preparation was removed and the filter was washed twice with 75 μl of reaction buffer. Measurement of the ferrous-CO P450 complex (Omura and Sato, 1964) was used to determine P450 recovery after vesicle preparation. All lipid probes, in a chloroform stock solution, were added to lipid mixtures before drying. For fluorescence resonance energy transfer (FRET) interactions between NBD-PE and Rh-PE, the fluorescent probes were added at concentrations of 0.1 mol% and 0.5 mol%, respectively. Laurdan was added to the lipid mixture in 1:1000 dye/lipid ratio. Brominated PC was added at 15 mol%.
TABLE 1.
Lipid composition (mol%) of synthetic reconstituted systems
Purified lipid vesicles were prepared with the lipid composition listed to analyze the potential of lipid domain formation.
| Vesicle | PC | PE | PI | PS | PA | SM | Chol |
|---|---|---|---|---|---|---|---|
| PC | 100a | — | — | — | — | — | — |
| Mix PL | 68a | 20 | 10 | 1 | 1 | — | — |
| ER | 60a | 20 | 10 | 1 | 1 | 4c | 5 |
| DRM | 42a | 18 | 1.5 | 2 | 1.5 | 12c | 23 |
| Model 1 | 100b | — | — | — | — | — | — |
| Model 2 | 80b | — | — | — | — | 5d | 15 |
| Model 3 | 50b | — | — | — | — | 20d | 30 |
| Model 4 | 30b | — | — | — | — | 30d | 40 |
—, not present; Chol, cholesterol; PI, phosphatidylinositol; PS, phosphatidylserine.
Phosphatidylcholine in the “natural membrane” systems was a bovine liver extract.
The phosphatidylcholine in the model systems was 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC).
Sphingomyelin in the “natural membrane” systems was porcine brain sphingomyelin.
In the model systems, palmitoyl-sphingomyelin (PSM) was used.
Isolation of Detergent-Resistant Membranes by Brij 98 Solubilization and Discontinuous Sucrose Gradient Centrifugation.
A portion (0.2 ml) of the vesicular reconstituted systems containing CYP1A2 and/or CPR (described above) was taken to 1 ml with 0.75 ml of reaction buffer and 0.05 ml of either 1% or 10% Brij 98 stock solution in water (as indicated in the results). The vesicles were solubilized, and DRMs were isolated by centrifugation (19 hours at 210,000g at 4°C) on a discontinuous sucrose density gradient as described in a previous study (Brignac-Huber et al., 2011). Eleven, 1-ml fractions were removed from the top of the gradients, and the pellets were rehomogenized in reaction buffer, and protein localization was determined by Western blotting of the individual fractions. DRMs were isolated in fractions 2–5 of the discontinuous gradient.
Fluorescence Spectroscopy.
All fluorescence spectroscopy measurements, unless otherwise stated, were carried out at room temperature. The excitation wavelength for NBD-PE is 450 nm with an emission at 535 nm, whereas Rh-PE is excited at 535 nm and emits at 605 nm. Therefore, in FRET studies between Rh-PE and NBD-PE, the excitation wavelength was 450 nm and emission was monitored at 605 nm. Values of the relative FRET efficiencies (%) were determined by the following (eq. 1):
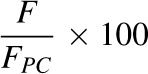 |
(1) |
where F is the fluorescence intensity at 605 nm of each mixed lipid system and FPC is the fluorescence intensity at 605 nm of either 100% bovine liver PC for the “physiologic” membrane systems or 100% POPC for the model membrane systems (de Almeida et al., 2003).
The laurdan emission spectrum provides an assessment of the degree of membrane order. Laurdan is a lipophilic dye that distributes equally into lo and ld membrane regions, but exhibits spectral properties that are dependent on lipid packing (Parasassi et al., 1986; Dinic et al., 2011). The laurdan emission spectrum exhibits a blue shift in the presence of lo domains, which is attributed to a dipolar relaxation phenomenon (Parasassi et al., 1986). The laurdan emission spectra were measured from 370 to 620 nm with excitation at 350 nm. General polarization (GP) of laurdan is a spectroscopic property, which can be used to quantify the relative proportion of coexisting lipid phases, with an increase in GP being reflective of an increase in membrane order. The GP values are measured as follows (eq. 2):
| (2) |
where IB and IR are the peaks of emission intensity at the blue and red edges of the emission spectra.
The fluorescence of tryptophan residues in CYP1A2 and CPR was measured at 341 nm after excitation at 295 nm. The effect of brominated PC on protein tryptophan fluorescence was expressed as the percent fluorescence relative to that observed when proteins were incorporated in lipid vesicles that did not contain brominated PC (eq. 3):
| (3) |
where F is the fluorescence intensity at 341 nm with Br-PC present and Fo is the fluorescence intensity at 341 nm in the absence of Br-PC.
All of the lipid components were included at identical relative concentrations in the two sets of vesicles, as bovine liver PC was substituted for brominated PC in the control vesicles. When the tryptophan fluorescence of CPR was studied, the reconstituted system was centrifuged through an Illustra Microspin S-400 HR size-exclusion spin column (GE Healthcare, Pittsburgh, PA) according to the manufacturer’s specifications. This step was performed to remove any CPR that was not physically incorporated in the lipid bilayer of the reconstituted system (Reed et al., 2006, 2008). It was determined that brominated lipids did not affect the incorporation of CPR into the lipid vesicles (data not shown).
Results
Isolation of DRMs from Purified, Reconstituted P450 Systems Containing Physiologic Lipids.
To elucidate the role of lipid microdomains in the function of the P450 enzyme system, it would be useful to replicate the domains that occur within a natural membrane in a reconstituted system. The advantage of this approach lies in the ability to examine the interactions of these proteins under defined and controlled conditions. In our previous study (Brignac-Huber et al., 2011), we showed that the interaction of CYP1A2 and CPR was facilitated and in turn the catalytic activity was stimulated when the enzymes were integrally reconstituted in vesicles that had the same lipid composition as DRMs. However, the previous study did not demonstrate that lipid microdomains were formed in the vesicular reconstituted systems or that the P450 and CPR interacted with putative domains in these systems. As a result, these possibilities are addressed in this study by using our previously published method to prepare vesicular, reconstituted P450 systems that have the enzymes integrally incorporated in the lipid bilayer (Reed et al., 2008; Reed, 2010).
The most direct way to demonstrate that DRMs were present in the reconstituted systems and were associated with the P450 enzyme system was to solubilize the reconstituted vesicles with Brij 98 and isolate the DRMs using discontinuous sucrose density centrifugation (Brignac-Huber et al., 2011). Due to their high lipid-to-protein ratio, detergent-resistant membranes coalesce around the interface between the 5 and 38% sucrose layers (i.e., fractions 2–5) (Pielsticker et al., 2005). Figure 1 shows the results of this type of experiment and compares P450 reconstituted systems made up of only bovine liver PC (V-PC) and those made up of a mixture of natural lipids at the same relative proportions occurring in the ER (V-ER).
Fig. 1.

Discontinuous sucrose density gradient profiles of enzyme-lipid reconstituted systems after detergent solubilization with Brij 98. Reconstituted systems containing CYP1A2 and/or CPR (as indicated in the center of the figure) were prepared and solubilized with Brij 98 (at the concentrations indicated) as described in Materials and Methods. The resulting suspensions were layered on a discontinuous sucrose gradient, and 1-ml fractions from the top to bottom of the gradient (indicated by the lane numbering) were analyzed by Western blotting for either CPR or CYP1A2. The asterisk indicates the lane location of the 50-and 70-kDa molecular weight marker for CYP1A2 and CPR, respectively. (A) Profiles generated from V-PC reconstituted systems, and (B) profiles generated from V-ER systems. When both enzymes were included in the reconstituted system, the word “Both” is used, and the enzyme detected in the Western blots is indicated in parentheses. The results are representative of at least three separate experiments.
Figure 1 (top two profiles) shows the gradient fractions obtained after detergent treatment and centrifugation of a lipid reconstituted system containing only CYP1A2. To demonstrate the progressive solubilization of the vesicles, two concentrations of Brij 98 (0.05 and 0.5%) were used in the experiments. The left panel shows the results when the vesicles were composed of only PC, and Fig. 1B shows the corresponding results with V-ER reconstituted systems. It may be noted that lower Brij 98 concentrations were used to solubilize the model membranes than those used to solubilize rabbit liver microsomes (Brignac-Huber et al., 2011). This is consistent with previous studies of model membrane vesicles showing that lower detergent concentrations are necessary for solubilization relative to those needed for natural membrane solubilization (Dietrich et al., 2001; Sengupta et al., 2008; Pathak and London, 2011). The results showed complete solubilization of the CYP1A2 in PC vesicles (none of the protein was isolated in fractions 2–5). Thus, when the reconstituted system was composed of a single phospholipid, PC, and not capable of forming segregated lipid domains, all of the CYP1A2 was solubilized by treatment with 0.5% Brij 98. Conversely, a significant proportion of the enzyme (approximately 25%) was still located in the uppermost fractions of the gradient in the V-ER system treated with 0.5% Brij 98. Thus, similar to what was observed in rabbit liver microsomes, CYP1A2 was resistant to solubilization with Brij 98 when it was incorporated in a reconstituted system that had the lipid composition of the endoplasmic reticulum.
Figure 1 also shows the corresponding sucrose density gradient profiles using reconstituted systems containing only CPR (third and fourth profiles from the top). Unlike the results with CYP1A2 reconstituted systems, CPR was solubilized from both V-PC (A) and V-ER (B) vesicles at 0.5% Brij 98. Although a small amount of CPR still remained in fractions 4 and 5, the great majority of the protein was no longer found in the fractions representing the DRM. This finding suggests that CPR does not have the same inherent affinity for DRMs as CYP1A2 when reconstituted alone into the V-ER vesicles.
In addition, Fig. 1 shows the sucrose gradient profiles when both CPR and CYP1A2 were reconstituted into the V-PC and V-ER systems (fifth and sixth profiles from the top). As observed in the reconstituted systems containing CYP1A2 alone, the P450 also isolated to domains when reconstituted with CPR. Interestingly, CPR also was localized to domains (approximately 28%) in the presence of CYP1A2, suggesting that the presence of CYP1A2 “draws” CPR into the domain. Because CPR was also observed to be in DRMs when examined in microsomes, it seems the presence of P450s in the domains either changes the localization of CPR or increases the tightness by which CPR binds to the DRM regions.
Assessment of the Degree of Membrane Order Using Fluorescent Lipid Probes.
We examined the potential of reconstituted vesicles to form lipid domains by utilizing several lipid probes in the absence and presence of either CYP1A2 or CPR. The general structures of the fluorescent lipid probes and brominated phospholipids used in this study are illustrated in Fig. 2. There were two types of comparisons made with regard to different reconstituted systems. One set of comparisons used model systems composed of different concentrations of a chemically defined phospholipid, POPC, a chemically defined sphingomyelin, PSM, and cholesterol. The second set of comparisons used reconstituted systems composed of different proportions of naturally occurring phospholipids and sphingomyelin (with or without cholesterol).
Fig. 2.

Lipid probes. Chemical structure of the fluorescent lipid analogs employed and preference of lipid domain location.
Domain formation has been well characterized using the model system lipids, and the processes involved in microdomain formation using vesicles derived from the model lipids are believed to be representative of those that occur in the plasma membranes (de Almeida et al., 2003, 2005). The ternary diagram shown in Fig. 3 indicates the lipid compositions that correspond to vesicles composed entirely of the ld phase; a mixture of ld and lo phases; entirely of the lo phase; a mixture of ld, lo, and gel (so) phases; and a mixture of lo and so phases. As a basis of comparison for the behavior of fluorescent lipid probes in membranes composed of natural lipids, we examined lipid probe fluorescence in four types of vesicles made from the model lipids. The compositions (shown as black numbered dots in Fig. 3) were representative of membranes containing varying proportions of lipid phases that would be expected to form in physiologic membranes. More specifically, we wanted to study probe fluorescence with the following types of membrane: 1) one composed entirely of liquid-disordered lipid (POPC); 2) one composed of a mixture of lipids that forms coexisting liquid-ordered and liquid-disordered regions but represents a composition near the phase boundary for liquid-disordered membranes (and thus, only a small proportion of the membrane would consist of the lo phase); 3) same as 2) but the lipid mixture represents a composition in the middle of the domain for coexisting lipid phases; and 4) same as 2) but the mixture represents a composition near the liquid-ordered phase region. The proportion of lipid in the lo phase would be expected to increase with the proportion of cholesterol (de Almeida et al., 2003). The lipid composition of the vesicles tested is reported in Table 1.
Fig. 3.

Phase diagram of model lipid systems. Lipid phase diagram of POPC/PSM/Chol at 23°C. Black numbered circles are experimental points used for vesicles in the model lipid systems and represent the following conditions: 1) one composed entirely of liquid-disordered lipids; 2) one composed of a mixture of lipids that forms a membrane with coexisting liquid-ordered and liquid-disordered regions but represents a composition near the phase boundary for liquid-disordered membranes; 3) same as 2) but the lipid mixture represents a composition in the middle of the domain for coexisting lipid phases; 4) same as 2) but the mixture represents a composition near the liquid-ordered phase region. Lipid compositions are described in Table 1. [Adapted from (de Almeida et al., 2003) with permission from Elsevier.]
Probe fluorescence was also examined in the following four types of lipid vesicles composed of natural lipids (i.e., those extracted from animal tissues): 1) PC alone (V-PC), 2) a mixture of phospholipids in proportions comparable to those of the ER (V-Mix PL), 3) a mixture of phospholipids, cholesterol, and sphingomyelin in proportions equal to those of the ER (V-ER), and 4) a mixture of phospholipids, cholesterol, and sphingomyelin in proportions equal to those of DRM isolated from the ER (V-DRM) as described previously (Brignac-Huber et al., 2011). By observing the behavior of lipid probes in model systems with different extents of phase separation, we could compare the probe responses in “physiologic” vesicles to assess whether similar processes were occurring.
It should be noted that the lipid vesicles prepared with naturally occurring lipids contain a mixture of fatty acid chains. Studies have shown that this aspect of these systems make lipid phase transitions more subtle than those occurring in the otherwise comparable model systems. For instance, Jin et al. (2006) used a probe (di-4-ANEPPDHQ) that responds to alterations in membrane organization through shifts in its emission scan. This study found that, although the reporter probe was useful for vesicles, the emission spectrum shift was far less dramatic for vesicles composed of natural lipids when compared with vesicles made of synthetic lipids. This effect was attributed to the more heterogeneous mixture of acyl chains found with natural lipid systems and would likely lessen the order of lo domains when compared with lo domains formed from synthetic lipids (Jin et al., 2006). Similarly, a calorimetric study by Barenholz et al. (1976) examined the thermotropic behavior of lipid vesicles composed of natural and synthetic sphingomyelin lipid species. It was reported that membrane organization of sphingomyelin is a complex function of its specific composition and that the transition temperature of natural sphingomyelin was much broader than the synthetic forms. Because it is less likely to obtain data with fluorescent probes that implicate lipid phase transition in vesicles made from naturally occurring lipids, more confidence can be placed in the data that do support the occurrence of these processes in these systems.
Segregation of Fluorescent Probes into Lipid Microdomains Using FRET.
There are a number of useful probes to evaluate phase separation in pure lipid systems. Two such probes which have been extensively used and characterized are NBD-PE and Rh-PE (Fig. 2). These probes are fluorescently labeled at the head group of the lipid and have different acyl chain properties. NBD-PE has two saturated acyl chains that allow this lipid to reside in more ordered domains (lo) of the membranes. Conversely, Rh-PE has two unsaturated acyl chains that are kinked and therefore prefers to reside in more fluid regions (ld) of the membrane (Dietrich et al., 2001; de Almeida et al., 2003; Crane and Tamm, 2004). FRET between the two probes is dependent on the distance between them and can be measured by exciting NBD-PE at 450 nm (emission 535 nm) and monitoring Rh-PE emission at 605 nm. Rh-PE is excited directly through irradiation at 535 nm. Because the probes selectively partition into the ordered and disordered membrane regions, respectively, the extent of FRET varies in the presence of cholesterol and sphingomyelin when these lipid constituents lead to microdomain formation. Incorporation of the probes into a homogeneous fluid membrane would produce a baseline FRET signal between the Rh and NBD probes. As the proportions of cholesterol and sphingomyelin are increased across the ld/lo phase boundary (Fig. 3), the FRET interaction would be expected to decrease. The maximal decrease in FRET fluorescence would be observed where the size of the lipid domains leads to the greatest segregation of the probes; however, as the size the domains vary on either side of this point of optimal segregation, the FRET signal would be expected to increase. Thus, by comparing the FRET signal in vesicles consisting entirely of an ld environment with mixed lipid systems that possess lo and ld domains, the potential for lipid microdomain formation can be assessed.
First, potential FRET interactions in the model system were monitored as described above. In the pure POPC vesicles (Model 1), there would be no phase separation and thus the two probes should be free to interact. The Model 2 lipid vesicles (Table 1) are located on the phase diagram (Fig. 3) at a point at which phase separation changes from ld to ld + lo. The last two systems, Model 3 and Model 4 contain higher proportions of cholesterol and sphingomyelin and correspond to compositions at which the lo regions comprise larger proportions of the entire lipid system.
FRET values were calculated relative to the FRET signal in Model 1 (100% POPC). As shown in Fig. 4, the relative FRET signal at 23°C (Fig. 4A) and 37°C (Fig. 4B) between NBD-PE and Rh-PE was significantly decreased in the Model 2 vesicles suggesting that phase segregation of the two probes was greatest in these vesicles. As the sphingomyelin and cholesterol concentrations were increased (Model 3), the degree of FRET between the two probes appeared to increase and was even greater in the Model 4 vesicles. This effect was likely due to relocalization of the NBD-PE probe into the ld regions as suggested by Crane and Tamm (2004), who performed fluorescence recovery after photobleaching studies in model membranes to monitor the localization and mobility of specific probes in lo and ld domains of model membranes. Consistent with the predicted outcome described above, the NBD-PE probe partitioned less selectively into lo regions as the cholesterol composition was increased above 15% in the model membranes. The Crane and Tamm study used higher sphingomyelin (SM) concentrations than in the present study, therefore there cannot be a direct comparison of the two results, but it is envisaged that the same effect could occur in these lipid systems with high cholesterol content. Ideally, to confirm a FRET interaction, an increase in the fluorescence signal of the acceptor molecule (in this case, Rh-PE) of the FRET pair should be matched by a decrease in the fluorescence signal of the donor molecule (NBD-PE). However, this relationship is complicated with this FRET pair by the well-known ability of NBD-PE to self-quench its fluorescence (Rodgers and Glaser, 1991; Brown et al., 1994). The selective partitioning of NBD-PE into relatively small lo domains formed at lipid compositions near the ld/lo + ld boundary (Model 2 lipids) would be expected to result in a greater degree of crowding of the probe in the domains and consequently a greater degree of self-quenching of its fluorescence. Thus, in this study, the fluorescence emission of the acceptor is used as an indicator of the physical interaction of this FRET pair and not the fluorescence emission of the donor.
Fig. 4.

FRET interaction of NBD-PE and Rh-PE in model lipid vesicles. Degree of FRET interaction was calculated as stated in Materials and Methods for vesicles composed of model lipids. Measurements were done at 23°C (A) and 37°C (B). These data represent the mean ± S.E.M. for three determinations, where significant differences from 100% POPC (Model 1) vesicles are represented by *P ≤ 0.05; **P ≤ 0.01.
The interaction between NBD-PE and Rh-PE also was monitored in lipid systems that were composed of various natural lipids at physiologically relevant compositions (Brignac-Huber et al., 2011) (Table 1). Relative to vesicles made up of 100% PC (V-PC), the addition of other phospholipids (V-Mix PL) did not significantly influence the FRET response at either 23°C (Fig. 5A) or 37° (Fig. 5B). However, the addition of 5 mol% cholesterol and 4 mol% sphingomyelin to the mixed phospholipids (as shown in the V-ER lipid system) produced a 15 and 20% decrease in the FRET signal at 23°C and 37°C, respectively. These results suggest effective segregation of the NBD-PE and Rh-PE probes into lo and ld domains, respectively. The FRET quenching in the V-ER system was most comparable to the FRET results associated with Model 2 and was consistent with a lipid composition in the lo + ld region of the phase diagram near the boundary for the ld region (Fig. 3), favoring the formation of relatively small ordered domains. Similar to the model system, as the cholesterol and sphingomyelin content was increased in the DRM vesicles, the FRET signal was increased. Again, this suggests that the selective partitioning of NBD-PE into the lo domains was diminished as the size of the domains increased with increasing cholesterol and sphingomyelin content.
Fig. 5.

FRET interaction of NBD-PE and Rh-PE in lipid vesicles mimicking the ER membrane. Degree of FRET interaction was calculated as stated in Materials and Methods for vesicles composed of natural lipids mimicking the ER membrane. Measurements were done at 23°C (A) and 37°C (B). These data represent the mean ± S.E.M. for three determinations, where significant differences from V-PC vesicles are represented by ***P ≤ 0.001.
Detection of Lipid Microenvironment Using the Laurdan Fluorescent Probe.
While the FRET results demonstrate segregation of the lo and ld probes into different membrane regions, these experiments alone do not directly establish the phase state of the membrane (Bagatolli, 2006). In an effort to confirm microdomain formation in these membrane systems, laurdan was used to probe the effect of alterations in lipid composition on membrane characteristics. Laurdan is a lipophilic dye that distributes equally into lo and ld membrane regions but exhibits spectral properties that are specific to different types of lipid packing (Parasassi et al., 1986; Dinic et al., 2011). The laurdan emission spectrum exhibits a blue shift in the presence of lo domains (Parasassi et al., 1986). The relative proportions of lo and ld phases can be assessed by generalized polarization, which is a normalized intensity ratio of the two phases (Parasassi et al., 1991). Laurdan emission scans were first monitored with the model systems composed of different SM and cholesterol compositions. As illustrated in Fig. 3, 100% POPC vesicles (Model 1) form a fluid bilayer (ld) (de Almeida et al., 2003). The laurdan emission spectra for these vesicles (Fig. 6) produced a peak of fluorescence intensity around 461 nm. Increasing the concentration of PSM and cholesterol would be expected to produce a higher degree of membrane organization (Fig. 3), which would affect the relaxation properties of the laurdan probe. The incorporation of small amounts of PSM and cholesterol (to 5 and 15%, respectively) into POPC vesicles (Model 2) led to a depression of the fluorescence peak at 461 nm with an increase in the shoulder at 434 nm (Fig. 6). Further increases in PSM and cholesterol increased this response, causing a significant shift of the laurdan emission spectra, producing peak intensity at 434 nm with decreased fluorescence intensity at 461 nm. These data indicate that the presence of cholesterol and PSM increased the order of the membrane microenvironment surrounding the laurdan probe.
Fig. 6.

Laurdan emission spectra in model lipid vesicles. Laurdan emission spectra (400–620 nm) were monitored using 350-nm excitation wavelength in lipid vesicles prepared with model lipids. Scans are representative of four independent experiments.
Based on the results obtained from the model systems, the goal of the next experiments was to determine if membranes with lipid structure and composition similar to those found in the ER exhibit similar changes in laurdan emission. As expected, the laurdan emission spectra of 100% V-PC (Fig. 7) produced a similar spectrum as the 100% POPC system (Fig. 6), suggestive of a homogeneous, fluid membrane. Interestingly, vesicles composed of V-Mix PL produced a significant decrease in fluorescence intensity at 461 nm, and an alteration in the 434 nm/461 nm ratio. These changes are similar to those associated with the presence of cholesterol- and sphingomyelin-containing lo domains and may reflect the putative ability of anionic phospholipids to form ordered domains in mixed PL vesicles (Ahn and Yun, 1998; Kim et al., 2003, 2007). Vesicles having a lipid composition similar to the total ER membrane (V-ER) produced a laurdan emission spectrum with depressed fluorescence at 461 nm and a relative increase in the intensity at 434 nm, again suggesting transition to more ordered membrane regions. Lastly, V-DRM produced a further shift in the spectra to 434 nm with a concomitant decrease of fluorescence intensity at 461 nm. The GP values were calculated from each scan and are shown in Table 2. In both the model systems and the physiologic lipid systems, the maximum GP values were seen in the vesicles containing the highest amount of cholesterol and sphingomyelin. Based on the magnitudes of the GP values, domain formation in the physiologic lipid systems did not appear to be as dramatic as in the model systems; however, it should be recognized that the alterations in cholesterol concentrations were more modest than those made with the defined model systems. Nonetheless, these results are consistent with lo domain formation with the more complex lipid membranes.
Fig. 7.

Laurdan emission spectra in lipid vesicles mimicking the ER membrane. Laurdan emission spectra (400–620 nm) were monitored using 350-nm excitation wavelength in lipid vesicles prepared with lipids mimicking the ER membrane in the absence of CYP1A2 (solid lines) and presence of 5 μM CYP1A2 (dashed lines) as described in Materials and Methods. Scans are representative of four independent experiments.
TABLE 2.
Generalized polarization emission values
GP values from Figs. 5 and 6 were determined at 23°C for each lipid system as described in Materials and Methods. These data represent the mean ± S.E.M. for three determinations.
| Lipid | Generalized Polarization Values | |
|---|---|---|
| Model 1 | −0.057 ± 0.005 | |
| Model 2 | −0.014 ± 0.009 | |
| Model 3 | 0.087 ± 0.01 | |
| Model 4 | 0.16 ± 0.01 | |
| – CYP1A2 | + CYP1A2 | |
| V-PC | −0.067 ± 0.006 | −0.071 ± 0.006 |
| V-Mix PL | −0.009 ± 0.002 | −0.01 ± 0.005 |
| V-ER | −0.0048 ± 0.006 | −0.005 ± 0.007 |
| V-DRM | 0.064 ± 0.01 | 0.063 ± 0.006 |
The changes described above examined the potential for lipid domain formation in the absence of protein. Additionally, we examined the effect of the presence of the P450 protein CYP1A2 on the phase properties of these lipid vesicles (Fig. 7). A direct comparison of the laurdan emission scans in the absence (solid lines) and presence of CYP1A2 (dashed lines) showed no significant effect on the GP values for laurdan emission in the presence of CYP1A2. These results suggest that the addition of CYP1A2 to the vesicle systems did not significantly affect the lipid-packing properties of the vesicles. GP values in all lipid systems with and without CYP1A2 are shown in Table 2.
Generalized polarization of laurdan is strongly affected by membrane organization (Table 2) and also by temperature (Parasassi et al., 1991). Higher temperatures elicit a transition of the lo and lo + ld boundary in the phase diagram, causing membranes to become more fluid (ld). Laurdan emission scans were performed with all lipid systems as a function of temperature. This change in membrane fluidity is illustrated from the laurdan emission scan of POPC vesicles containing 20% PSM and 30% cholesterol (Model 3) at temperatures ranging from 23–60°C in Fig. 8A. The same scans were repeated for each model lipid system, generating similar results (Supplemental Fig. 1). The results demonstrate that as the temperature was increased, the maximum fluorescence intensity shifted from 434 nm to 461 nm, demonstrating a progressive decrease in membrane order (Parasassi et al., 1991). GP values were then calculated for each of the scans and plotted against temperature (Fig. 8B) and showed a break in the curve between 40° and 45°C, which is representative of the lipid transition from lo + ld to ld phases (Parasassi et al., 1991). The same experiment was done in the vesicles composed of natural lipids and results from the V-ER lipids are shown in Fig. 9A. The data for all the other vesicle systems can be found in Supplemental Fig. 2. Again, GP values were calculated for each scan and plotted against temperature (Fig. 9B). The data illustrate that vesicles composed of physiologically relevant lipid compositions exhibited similar changes in membrane organization and fluidity as a function of temperature as was observed with defined model systems. Therefore lo and ld phases coexist not only in the synthetic lipid systems but also in membranes with lipid compositions that more closely mimic the ER bilayer.
Fig. 8.

Laurdan emission spectra and GP profiles as a function of temperature in model systems. Laurdan emission scans (400–620 nm) were monitored using 350-nm excitation as a function of temperature. The effect of temperature increase on the laurdan emission spectra is illustrated from the Model 3 vesicle system (A). Generalized polarization values were determined for each lipid vesicle at each temperature (B). Emission scans are representative of four independent experiments and GP profiles are representative of the mean ± S.E.M. for four determinations. RT, room temperature.
Fig. 9.

Laurdan emission spectra and GP profiles as a function of temperature in “natural membrane” vesicles. Laurdan emission scans (400–620 nm) were monitored using 350-nm excitation as a function of temperature. The effect of temperature increase on the laurdan emission spectra is illustrated from the V-ER system (A). Generalized polarization values were determined for each lipid vesicle at each temperature (B). Emission scans are representative of four independent experiments and GP profiles are representative of the mean ± S.E.M for four determinations. RT, room temperature.
Brominated Phospholipid.
Although the FRET experiments demonstrated that lo domains existed in the presence of enzyme, we also wanted to determine whether CYP1A2 and CPR partitioned into ld or lo regions of these vesicles. To address this question, we used 1,2-di-(9,10-dibromo)stearoyl-sn-glycero-3-phosphocholine (BrPC), which contains brominated tags on the acyl chains (Fig. 2). The bulky bromine tags produce kinks in the tails causing this lipid to have properties similar to unsaturated PC chains that segregate into ld regions of lipid vesicles (East and Lee, 1982; Carney et al., 2007). Additionally, the bromine tags quench tryptophan fluorescence; therefore, we used this probe to assess its interaction with CYP1A2, given that this P450 contains eight tryptophan residues throughout its sequence (Ozols, 1986), with six being located on the enzyme surface (Sansen et al., 2007). Since BrPC prefers to reside in ld membrane regions, the location of CYP1A2 can be determined based on tryptophan quenching in the different lipid systems. We performed these experiments in V-PC and V-ER lipid systems because we achieved the greatest partitioning of phase-selective probes in the V-ER systems according to FRET measurements using Rh-PE and NBD-PE. This would increase the likelihood of being able to detect significant changes in the CYP1A2 Trp fluorescence. First, various amounts of BrPC were titrated into V-PC with 5 μM CYP1A2 to identify concentrations where efficient quenching of CYP1A2 tryptophan fluorescence was observed (Supplemental Fig. 3). Based on these results, 15 mol% BrPC was selected and substituted for PC in each of the vesicle systems. CYP1A2 tryptophan fluorescence at 341 nm was then monitored in each lipid system with and without the brominated lipid present. In V-PC, the fluorescence of tryptophan residues in CYP1A2 was quenched approximately 20% by the BrPC at 23°C (Fig. 10A). The degree of fluorescence quenching was significantly less in V-ER lipids, suggesting separation of the BrPC and CYP1A2 into different domains. Results were similar at 23°C and 37°C. Since it is known that the BrPC resides in the ld regions of the membrane (East and Lee, 1982; Carney et al., 2007), these data support the localization of CYP1A2 into the lo regions of the membrane and confirm our previous findings regarding the membrane localization of CYP1A2 in rabbit liver microsomes (Brignac-Huber et al., 2011).
Fig. 10.

CYP1A2 and CPR tryptophan quenching by brominated-PC in V-PC and V-ER vesicles. The tryptophan fluorescence of enzymes (A) CYP1A2 and (B) CPR were measured (excitation 295/emission 341) in the presence (F) and absence (Fo) of 15 mol% BrPC in V-PC and V-ER systems. Fluorescence quenching (F/Fo) was measured as stated in Materials and Methods at 23°C and 37°C. These data represent the mean ± S.E.M. for four determinations, where significant differences are represented by *P ≤ 0.05; **P ≤ 0.01.
The similar experiment was also performed with lipid reconstituted systems containing only CPR (Fig. 10B). Surprisingly, there was significantly less quenching of CPR tryptophan fluorescence by brominated lipids when the enzyme was reconstituted in the V-ER system (relative to that in V-PC). These findings suggest that CPR also localizes to a separate lipid domain from that containing the brominated lipids in the V-ER. These results are in contrast to those obtained when CPR-containing vesicles were solubilized and centrifuged on a discontinuous sucrose density gradient as the latter indicated that CPR was not localized to DRMs (Fig. 1).
Discussion
The ER lipid membrane contains low levels of sphingomyelin and cholesterol and has been described as an organelle of loosely packed lipids, allowing for insertion and export of lipids and proteins (van Meer et al., 2008). But many groups have shown that there is potential for organized microdomain formation within the ER membrane (Sevlever et al., 1999; Browman et al., 2006; Jang et al., 2010; Brignac-Huber et al., 2011). Our laboratory recently isolated ER-DRMs from rabbit liver microsomes and found that CYP1A2 and CPR reside primarily in these resistant domains (Brignac-Huber et al., 2011). Enzymatic studies both with microsomes and with reconstituted systems demonstrated that CYP1A2 activities were increased in the more complex systems containing SM and cholesterol—due to an increase in the apparent binding affinity of the CPR∙CYP1A2 complex. The goals of the present study were to determine whether ordered lipid microdomains could be detected in reconstituted lipid vesicles having compositions similar to those found in the ER and to demonstrate whether CYP1A2 and CPR, when incorporated in the vesicles, predominantly resided within the ordered domains.
The formation of lipid domains in model membranes has been investigated for many years with the use of fluorescent probes that preferentially reside in lo or ld lipid regions (de Almeida et al., 2003; Crane and Tamm, 2004; Baumgart et al., 2007). These investigations have led to the development of phase diagrams that identify the lipid compositions corresponding to different possible physical states of lipid organization within a membrane (de Almeida et al., 2003). Although microdomain formation was established with model systems, the concentrations and specific types of lipid constituents are commonly outside the physiologic range for biologic membranes. Therefore, we compared the behaviors of lipid probes in the cholesterol/POPC/sphingomyelin model system and in reconstituted systems with compositions similar to ER membranes to assess the potential for lipid microdomain formation in the systems containing physiologic lipids.
FRET measured between NBD-PE and Rh-PE probes in the vesicles composed of physiologic lipids was consistent with the existence of lipid microdomains and corresponded very well to the FRET measured in analogous model systems. The lowest degree of FRET was observed in the Model 2 system and in V-ER reconstituted systems which contained the lowest amount of cholesterol and sphingomyelin (Figs. 4 and 5). At this lipid composition, there is a predicted phase transition within the Model 2 membrane that allows the coexistence of ld and lo regions (Fig. 3). Because the lipid composition lies near the phase boundary, there is selective segregation and maximum separation of the two probes, leading to decreased FRET between the probes. It would be expected for FRET between NBD-PE and Rh-PE to be similar in the V-ER and Model 2 lipid reconstituted systems because it has been shown that biologic membranes have lipid compositions that are very close to the lo/ld phase transition (Lingwood et al., 2008). In addition, these results illustrate that the ER membrane composition is capable of phase separation and membrane heterogeneity at physiologic temperatures (Fig. 5B). The subsequent increase in FRET with increasing cholesterol and sphingomyelin compositions in V-DRM and Model 3 vesicles is a phenomenon that has been described elsewhere and is attributed to decreased selectivity of the NBD probe as cholesterol levels are increased (Crane and Tamm, 2004). This effect is further demonstrated by the fact that as the cholesterol concentration is additionally increased in the Model 4 system, there is a continued increase in the FRET signal.
While the selectivity of the probes discussed above can be dependent on the lipid composition, laurdan is a useful tool that indicates the proportions of lo and ld domains in a manner that is independent of the domain size. This particular probe reflects changes in lipid organization through variations in its fluorescence emission. Moreover, since laurdan distributes equally within lo and ld phases, a GP value can be calculated from the emission scan of laurdan, which quantifies the relative proportion of ld and lo phases (Parasassi et al., 1991). A higher degree of membrane organization will produce a higher GP value than disordered domains. Again, spectral shifts of laurdan were similar in the model systems and the “natural membrane” reconstituted systems. As the cholesterol and sphingomyelin compositions increased in either system, the excitation spectra blue-shifted, providing support for the existence of organized domains within the vesicles (Figs. 6 and 7). Furthermore, the GP values corroborated these results as the highest values were observed in V-DRM lipids.
The V-Mix PL preparation was tested because of research showing that different types of phospholipids, especially anionic phospholipids, stimulate P450 catalytic activity and promote the interaction of P450 and CPR (Ingelman-Sundberg et al., 1981, 1996; Blanck et al., 1984; Ahn et al., 1998, 2005). In addition, several studies have described domain formation by anionic phospholipids in the presence of P450s. For example, Kim et al. (2003) used various lipid probes to illustrate that the presence of phosphatidylethanolamine (PE) promoted anionic phospholipid-enriched domains in ternary systems composed of phosphatidylcholine (PC), PE, and phosphatidylserine or phosphatidic acid (PA). Moreover, this group found that the presence and clustering (domain formation) of anionic phospholipids caused CYP3A4 to be more catalytically active and more deeply inserted into the lipid vesicles. Furthermore, it was postulated that this phenomenon may be important in CYP3A4 interactions with other proteins in membranes (Kim et al., 2003). Another study found that CYP2B1 induced the formation of anionic lipid domains in vesicles containing PC, PE, and anionic phospholipid (PA, phosphatidylserine, or phosphatidylinositol were all tested) with PA having the greatest clustering effect in the presence of CYP2B1 (Kim et al., 2007). In addition, the presence of PA caused a conformational change in CYP2B1 and increased CYP2B1 activity. It should be noted that these studies used much higher anionic phospholipid concentrations than those found in the ER membrane.
The results from the present study differ from those of previous reports in that our data suggest that microdomain formation is governed by the lipids present and not created by the introduction of protein—at least for CYP1A2 at lipid compositions encountered in the ER. Actually, our study is consistent with that of Ahn and Yun (1998), which used the fluorescence of lipid probes to show that lipid microdomains were formed in the absence of any added protein. However, the Ahn and Yun study used anionic lipid concentrations that were much higher than would be encountered in physiologic membranes. Our study demonstrates the protein-independence of lipid microdomain formation by the comparison of the laurdan emission spectra in the absence and presence of CYP1A2 (Fig. 7). The presence of CYP1A2 did not affect the laurdan spectra or the resulting GP values (Table 2). Although there are numerous differences when comparing these results to literature reports, including the relative compositions of the lipids used and the different types of P450s added to the reconstituted system, our results are consistent with the localization of CYP1A2 into previously formed lo microdomains, rather than the microdomains being generated as a result of the presence of the P450 enzyme. Nonetheless, the laurdan experiments in the present study support the idea of anionic phospholipid domain formation. Interestingly, the laurdan emission scans were suggestive of an intermediate degree of membrane organization in the presence of physiologically relevant concentrations of anionic phospholipids (V-Mix PL) without the inclusion of cholesterol and sphingomyelin (Fig. 7). In accordance with the qualitative difference of the laurdan scan in the V-Mix PL vesicles, the GP value was 7-fold higher than that determined in the V-PC membranes (Table 2), suggesting a more ordered lipid bilayer in V-Mix PL vesicles.
Lastly, the results using brominated phospholipids established that both CYP1A2 and CPR preferentially resided in the lo regions of the V-ER systems (Fig. 10). These results are supportive of previous studies that showed CYP1A2 DRM residence in microsomal tissue (Bae et al., 2004; Brignac-Huber et al., 2011). However, the findings with CPR-containing systems contradicted the conclusions derived from detergent solubilization of the lipid vesicles as no CPR was observed in DRMs of the vesicles. This may indicate that CPR, like CYP1A2, preferentially associates with lo domains but may associate less tightly to the domains than the P450 binds, so that its localization is not maintained after solubilization of the V-ER with Brij 98.
The present study demonstrates that organized lipid domains do form in the presence of physiologically relevant ER lipids and that CYP1A2 preferentially resides in the ordered regions of the membrane. Furthermore, the presence of CYP1A2 does not appear to significantly influence microdomain formation. CPR appears to preferentially associate with domains loosely in the absence of CYP1A2, which allows it to be easily solubilized by detergent treatment; however, CPR may be drawn into the more ordered domains through its association with CYP1A2. Further work is needed to elucidate the nature of the interaction of CPR with DRMs. Taken together these results underscore the important roles that lipid microdomain formation play in the localization and function of the enzymes of the P450 monooxygenase system.
Supplementary Material
Abbreviations
- BrPC
1,2-di-(9,10-dibromo)stearoyl-sn-glycero-3-phosphocholine
- CPR
NADPH-cytochrome P450 reductase
- DRMs
detergent-resistant membranes
- ER
endoplasmic reticulum
- FRET
fluorescence resonance energy transfer
- GP
general polarization
- laurdan
6-dodecanoyl-2-dimethylaminonaphthalene
- ld
liquid-disordered domains
- lo
liquid-ordered domains
- MβC
methyl-beta-cyclodextrin
- NBD-PE
N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)-1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine, triethylammonium salt
- P450
cytochrome P450
- PA
phosphatidic acid
- PC
phosphatidylcholine
- PE
phosphatidylethanolamine
- POPC
1-palmitoyl-2-oleyl-sn-glycero-3-phosphocholine
- PSM
palmitoyl sphingomyelin
- Rh-PE
Lissamine rhodamine B 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine, triethylammonium salt
- SM
sphingomyelin
- V-DRM
vesicles with lipid composition similar to that of the detergent resistant membranes of the ER
- V-ER
vesicles with a lipid composition similar to the ER membrane
- V-Mix PL
mixed phospholipid vesicles
- V-PC
phosphatidylcholine vesicles
Authorship Contributions
Participated in research design: Brignac-Huber, Reed, Eyer, Backes.
Conducted experiments: Brignac-Huber, Eyer.
Performed data analysis: Brignac-Huber, Reed, Backes.
Wrote or contributed to the writing of the manuscript: Brignac-Huber, Reed, Backes.
Footnotes
This work was supported by a US Public Health Services research grant from the National Institutes of Health National Institute of Environmental Health Sciences [Grant R01 ES004344]; and a predoctoral research fellowship from the State of Louisiana Board of Regents [Grant LEQSF(2005-10) GF-08].
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.
References
- Ahn T, Guengerich FP, Yun CH. (1998) Membrane insertion of cytochrome P450 1A2 promoted by anionic phospholipids. Biochemistry 37:12860–12866 [DOI] [PubMed] [Google Scholar]
- Ahn T, Yun CH. (1998) Phase separation in phosphatidylcholine/anionic phospholipid membranes in the liquid-crystalline state revealed with fluorescent probes. J Biochem 124:622–627 [DOI] [PubMed] [Google Scholar]
- Ahn T, Yun CH, Oh DB. (2005) Involvement of nonlamellar-prone lipids in the stability increase of human cytochrome P450 1A2 in reconstituted membranes. Biochemistry 44:9188–9196 [DOI] [PubMed] [Google Scholar]
- Bae TJ, Kim MS, Kim JW, Kim BW, Choo HJ, Lee JW, Kim KB, Lee CS, Kim JH, Chang SY, et al. (2004) Lipid raft proteome reveals ATP synthase complex in the cell surface. Proteomics 4:3536–3548 [DOI] [PubMed] [Google Scholar]
- Bagatolli LA. (2006) To see or not to see: lateral organization of biological membranes and fluorescence microscopy. Biochim Biophys Acta 1758:1541–1556 [DOI] [PubMed] [Google Scholar]
- Barenholz Y, Suurkuusk J, Mountcastle D, Thompson TE, Biltonen RL. (1976) A calorimetric study of the thermotropic behavior of aqueous dispersions of natural and synthetic sphingomyelins. Biochemistry 15:2441–2447 [DOI] [PubMed] [Google Scholar]
- Baumgart T, Hunt G, Farkas ER, Webb WW, Feigenson GW. (2007) Fluorescence probe partitioning between Lo/Ld phases in lipid membranes. Biochim Biophys Acta 1768:2182–2194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanck J, Smettan G, Ristau O, Ingelman-Sundberg M, Ruckpaul K. (1984) Mechanism of rate control of the NADPH-dependent reduction of cytochrome P-450 by lipids in reconstituted phospholipid vesicles. Eur J Biochem 144:509–513 [DOI] [PubMed] [Google Scholar]
- Brignac-Huber LM, Reed JR, Backes WL. (2011) Organization of NADPH-cytochrome P450 reductase and CYP1A2 in the endoplasmic reticulum—microdomain localization affects monooxygenase function. Mol Pharmacol 79:549–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browman DT, Resek ME, Zajchowski LD, Robbins SM. (2006) Erlin-1 and erlin-2 are novel members of the prohibitin family of proteins that define lipid-raft-like domains of the ER. J Cell Sci 119:3149–3160 [DOI] [PubMed] [Google Scholar]
- Brown DA, London E. (1998) Functions of lipid rafts in biological membranes. Annu Rev Cell Dev Biol 14:111–136 [DOI] [PubMed] [Google Scholar]
- Brown RS, Brennan JD, Krull UJ. (1994) Self-quenching of nitrobenzoxadiazole labeled phospholipids in lipid membranes. J Chem Phys 100:6019–6027 [Google Scholar]
- Carney J, East JM, Lee AG. (2007) Penetration of lipid chains into transmembrane surfaces of membrane proteins: studies with MscL. Biophys J 92:3556–3563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coon MJ, van der Hoeven TA, Dahl SB, Haugen DA. (1978) Two forms of liver microsomal cytochrome P-450, P-450lm2 and P-450LM4 (rabbit liver). Methods Enzymol 52:109–117 [DOI] [PubMed] [Google Scholar]
- Crane JM, Tamm LK. (2004) Role of cholesterol in the formation and nature of lipid rafts in planar and spherical model membranes. Biophys J 86:2965–2979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Almeida RF, Fedorov A, Prieto M. (2003) Sphingomyelin/phosphatidylcholine/cholesterol phase diagram: boundaries and composition of lipid rafts. Biophys J 85:2406–2416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Almeida RF, Loura LM, Fedorov A, Prieto M. (2005) Lipid rafts have different sizes depending on membrane composition: a time-resolved fluorescence resonance energy transfer study. J Mol Biol 346:1109–1120 [DOI] [PubMed] [Google Scholar]
- Dietrich C, Bagatolli LA, Volovyk ZN, Thompson NL, Levi M, Jacobson K, Gratton E. (2001) Lipid rafts reconstituted in model membranes. Biophys J 80:1417–1428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinic J, Biverståhl H, Mäler L, and Parmryd I (2011) Laurdan and di-4-ANEPPDHQ do not respond to membrane-inserted peptides and are good probes for lipid packing. Biochim Biophys Acta 1808:298–306. [DOI] [PubMed]
- East JM, Lee AG. (1982) Lipid selectivity of the calcium and magnesium ion dependent adenosinetriphosphatase, studied with fluorescence quenching by a brominated phospholipid. Biochemistry 21:4144–4151 [DOI] [PubMed] [Google Scholar]
- Gigon PL, Gram TE, Gillette JR. (1969) Studies on the rate of reduction of hepatic microsomal cytochrome P-450 by reduced nicotinamide adenine dinucleotide phosphate: effect of drug substrates. Mol Pharmacol 5:109–122 [PubMed] [Google Scholar]
- Hayashi T, Fujimoto M. (2010) Detergent-resistant microdomains determine the localization of sigma-1 receptors to the endoplasmic reticulum-mitochondria junction. Mol Pharmacol 77:517–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingelman-Sundberg M, Haaparanta T, Rydström J. (1981) Membrane charge as effector of cytochrome P-450LM2 catalyzed reactions in reconstituted liposomes. Biochemistry 20:4100–4106 [DOI] [PubMed] [Google Scholar]
- Ingelman-Sundberg M, Hagbjörk AL, Ueng YF, Yamazaki H, Guengerich FP. (1996) High rates of substrate hydroxylation by human cytochrome P450 3A4 in reconstituted membranous vesicles: influence of membrane charge. Biochem Biophys Res Commun 221:318–322 [DOI] [PubMed] [Google Scholar]
- Jang HH, Kim DH, Ahn T, Yun CH. (2010) Functional and conformational modulation of human cytochrome P450 1B1 by anionic phospholipids. Arch Biochem Biophys 493:143–150 [DOI] [PubMed] [Google Scholar]
- Jin L, Millard AC, Wuskell JP, Dong X, Wu D, Clark HA, Loew LM. (2006) Characterization and application of a new optical probe for membrane lipid domains. Biophys J 90:2563–2575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KH, Ahn T, Yun CH. (2003) Membrane properties induced by anionic phospholipids and phosphatidylethanolamine are critical for the membrane binding and catalytic activity of human cytochrome P450 3A4. Biochemistry 42:15377–15387 [DOI] [PubMed] [Google Scholar]
- Kim KH, Kim DH, Jang HH, Kim M, Kim DH, Kim JS, Kim JI, Chae HZ, Ahn T, Yun CH. (2007) Lateral segregation of anionic phospholipids in model membranes induced by cytochrome P450 2B1: bi-directional coupling between CYP2B1 and anionic phospholipid. Arch Biochem Biophys 468:226–233 [DOI] [PubMed] [Google Scholar]
- Lingwood D, Ries J, Schwille P, Simons K. (2008) Plasma membranes are poised for activation of raft phase coalescence at physiological temperature. Proc Natl Acad Sci USA 105:10005–10010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loura LM, Fedorov A, Prieto M. (2001) Fluid-fluid membrane microheterogeneity: a fluorescence resonance energy transfer study. Biophys J 80:776–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson DR. (2003) Comparison of P450s from human and fugu: 420 million years of vertebrate P450 evolution. Arch Biochem Biophys 409:18–24 [DOI] [PubMed] [Google Scholar]
- Niu SL, Litman BJ. (2002) Determination of membrane cholesterol partition coefficient using a lipid vesicle-cyclodextrin binary system: effect of phospholipid acyl chain unsaturation and headgroup composition. Biophys J 83:3408–3415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omura T, Sato R. (1964) The carbon monoxide-binding pigment of liver microsomes. I. Evidence for its hemoprotein nature. J Biol Chem 239:2370–2378 [PubMed] [Google Scholar]
- Ozols J. (1986) Complete amino acid sequence of a cytochrome P-450 isolated from beta-naphthoflavone-induced rabbit liver microsomes. Comparison with phenobarbital-induced and constitutive isozymes and identification of invariant residues. J Biol Chem 261:3965–3979 [PubMed] [Google Scholar]
- Parasassi T, Conti F, Gratton E. (1986) Time-resolved fluorescence emission spectra of Laurdan in phospholipid vesicles by multifrequency phase and modulation fluorometry. Cell Mol Biol 32:103–108 [PubMed] [Google Scholar]
- Parasassi T, De Stasio G, Ravagnan G, Rusch RM, Gratton E. (1991) Quantitation of lipid phases in phospholipid vesicles by the generalized polarization of Laurdan fluorescence. Biophys J 60:179–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathak P, London E. (2011) Measurement of lipid nanodomain (raft) formation and size in sphingomyelin/POPC/cholesterol vesicles shows TX-100 and transmembrane helices increase domain size by coalescing preexisting nanodomains but do not induce domain formation. Biophys J 101:2417–2425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pielsticker LK, Mann KJ, Lin WL, Sevlever D. (2005) Raft-like membrane domains contain enzymatic activities involved in the synthesis of mammalian glycosylphosphatidylinositol anchor intermediates. Biochem Biophys Res Commun 330:163–171 [DOI] [PubMed] [Google Scholar]
- Pike LJ. (2004) Lipid rafts: heterogeneity on the high seas. Biochem J 378:281–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn PJ, Wolf C. (2009) The liquid-ordered phase in membranes. Biochim Biophys Acta 1788:33–46 [DOI] [PubMed] [Google Scholar]
- Reed JR. (2010) The use of liposomes in the study of drug metabolism: a method to incorporate the enzymes of the cytochrome p450 monooxygenase system into phospholipid, bilayer vesicles. Methods Mol Biol 606:11–20 [DOI] [PubMed] [Google Scholar]
- Reed JR, Brignac-Huber LM, Backes WL. (2008) Physical incorporation of NADPH-cytochrome P450 reductase and cytochrome P450 into phospholipid vesicles using glycocholate and Bio-Beads. Drug Metab Dispos 36:582–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed JR, Kelley RW, Backes WL. (2006) An evaluation of methods for the reconstitution of cytochromes P450 and NADPH P450 reductase into lipid vesicles. Drug Metab Dispos 34:660–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers W, Glaser M. (1991) Characterization of lipid domains in erythrocyte membranes. Proc Natl Acad Sci USA 88:1364–1368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansen S, Yano JK, Reynald RL, Schoch GA, Griffin KJ, Stout CD, Johnson EF. (2007) Adaptations for the oxidation of polycyclic aromatic hydrocarbons exhibited by the structure of human P450 1A2. J Biol Chem 282:14348–14355 [DOI] [PubMed] [Google Scholar]
- Sengupta P, Hammond A, Holowka D, Baird B. (2008) Structural determinants for partitioning of lipids and proteins between coexisting fluid phases in giant plasma membrane vesicles. Biochim Biophys Acta 1778:20–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevlever D, Pickett S, Mann KJ, Sambamurti K, Medof ME, Rosenberry TL. (1999) Glycosylphosphatidylinositol-anchor intermediates associate with triton-insoluble membranes in subcellular compartments that include the endoplasmic reticulum. Biochem J 343:627–635 [PMC free article] [PubMed] [Google Scholar]
- Strobel HW, Lu AYH, Heidema J and Coon MJ (1970) Phosphatidylcholine requirement in the enzymatic reduction of hemoprotein P-450 and in fatty acid, hydrocarbon, and drug hydroxylation. J Biol Chem 245:4851–4854 [PubMed] [Google Scholar]
- van Meer G, Voelker DR, Feigenson GW. (2008) Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol 9:112–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.