

Abstract
The only small-molecule compound demonstrated to substantially extend survival in prion-infected mice is a biaryl hydrazone termed “Compd B” (4-pyridinecarboxaldehyde,2-[4-(5-oxazolyl)phenyl]hydrazone). However, the hydrazone moiety of Compd B results in toxic metabolites, making it a poor candidate for further drug development. We developed a pharmacophore model based on diverse antiprion compounds identified by high-throughput screening; based on this model, we generated biaryl amide analogs of Compd B. Medicinal chemistry optimization led to multiple compounds with increased potency, increased brain concentrations, and greater metabolic stability, indicating that they could be promising candidates for antiprion therapy. Replacing the pyridyl ring of Compd B with a phenyl group containing an electron-donating substituent increased potency, while adding an aryl group to the oxazole moiety increased metabolic stability. To test the efficacy of Compd B, we applied bioluminescence imaging (BLI), which was previously shown to detect prion disease onset in live mice earlier than clinical signs. In our studies, Compd B showed good efficacy in two lines of transgenic mice infected with the mouse-adapted Rocky Mountain Laboratory (RML) strain of prions, but not in transgenic mice infected with human prions. The BLI system successfully predicted the efficacies in all cases long before extension in survival could be observed. Our studies suggest that this BLI system has good potential to be applied in future antiprion drug efficacy studies.
Introduction
Prion diseases are a class of progressive and uniformly fatal neurodegenerative disorders, which include Creutzfeldt-Jakob disease (CJD) in humans, bovine spongiform encephalopathy (BSE) in cattle, chronic wasting disease (CWD) in elk and deer, and scrapie in sheep (Aguzzi et al., 2008; Prusiner, 2013). The vast majority (∼85%) of CJD cases occur sporadically, but over 40 point mutations and octarepeat expansions have been identified in the prion protein (PrP) that cause familial prion diseases, which account for ∼15% of cases; additionally, a small subset (<1%) of human PrP prion diseases have occurred through infection. Conversely, BSE, chronic wasting disease, and scrapie are predominantly spread through an infectious etiology, although a genetic case of BSE has also been observed (Richt and Hall, 2008) and the periodic identification of new BSE cases likely represent spontaneous occurrences. All prion diseases result from refolding of the endogenous cellular prion protein (PrPC) into a self-propagating form, denoted PrPSc (Prusiner, 1998). Stochastic refolding likely accounts for sporadic and genetic cases, whereas the infectious etiology results from an exogenous PrPSc nidus initiating template-dependent refolding of PrPC. Currently, no treatments exist that halt or even slow any prion disease.
Numerous compounds have been reported to show antiprion activity in prion-infected cell-culture models, including pentosan polysulfate (PPS), dextran sulfate, heteropolyanion-23, Congo red, quinacrine, and 2-aminothiazoles (for a comprehensive review, see Trevitt and Collinge, 2006; Sim, 2012). However, the only small molecule demonstrated to substantially extend survival in prion-infected mice is “Compound B” (4-pyridinecarboxaldehyde,2-[4-(5-oxazolyl)phenyl]hydrazone) (Kawasaki et al., 2007), hereafter referred to as “Compd B.”
Despite its low effective concentration (EC50) in vitro, advancement of Compd B as a promising drug candidate is limited by the high-liability aryl hydrazone moiety present in its structure (Hwu et al., 2004). Therefore, we developed analogs of Compd B guided by a pharmacophore model, and generated more potent compounds with lower EC50 values. Pharmacokinetic studies conducted on these analogs indicated that several may prove to be suitable candidates as antiprion therapeutics.
While developing safe and more effective compounds for antiprion treatment, we were also seeking novel methods to monitor and predict drug efficacy in mouse models. Currently, the only method to determine efficacy of candidate compounds in vivo is to compare the survival times of treated and untreated prion-infected mice. Because wild-type mice generally have incubation periods of at least ∼120 to 150 days, such studies are time consuming and expensive. Previously, we showed that bioluminescence imaging (BLI) using transgenic (Tg) mice expressing a luciferase (luc) reporter driven by the glial fibrillary acidic protein (GFAP) promoter enabled prion disease progression to be monitored in vivo. Tg(Gfap-luc) mice showed upregulation of the BLI signal at ∼60 days postinoculation (dpi) with the Rocky Mountain Laboratory (RML) strain of prions, approximately half the time to onset of clinical signs (Tamgüney et al., 2009). In this study, we applied the BLI technique to monitor disease progression while a small molecule compound was administered as an antiprion therapeutic agent to determine whether drug efficacy could be assessed more rapidly.
We monitored the efficacy of Compd B in two lines of prion-infected mice—wild-type and PrP-overexpressing Tg mice—both expressing the luciferase reporter gene under the control of the Gfap promoter. We report that monitoring GFAP induction in vivo by measuring the luminescence signal enabled early detection of drug efficacy. To develop a more realistic in vivo model, we also performed intervention studies, administering compounds at different time points after prion infection (60–80 dpi). Disappointingly, we found that delayed treatment resulted in shorter extensions in survival.
Materials and Methods
Ethics Statement.
All animal studies were approved by the Institutional Animal Care and Use Committee at the University of California San Francisco.
Cellular PrPSc Reduction Assay.
Mouse N2a neuroblastoma cells (American Type Culture Collection) were transfected with full-length mouse PrP and infected with the RML strain of mouse-adapted scrapie prions, yielding ScN2a-cl3 cells (Ghaemmaghami et al., 2010). ScN2a-cl3 cells were maintained in tissue culture flasks (175 cm2) containing 32 ml of filter-sterilized (0.2 µm) minimum essential medium (MEM) with Earle’s salts and l-glutamine, supplemented with 10% fetal bovine serum, 250 µg/ml Geneticin (G418), 50 IU/ml penicillin, and 50 μg/ml streptomycin (supplemented MEM) in a humidified and CO2-enriched (5%) environment at 37°C. On day 1, the growth medium (supplemented MEM) was aspirated from the flasks, the cells were washed twice with 10 ml of calcium- and magnesium-free Dulbecco’s phosphate-buffered saline (PBS), and then they were detached by addition of 3 ml of cell dissociation buffer. After incubation at room temperature for 5 minutes, the dissociation buffer was aspirated, and the cells suspended in 10 ml of growth medium before counting using a Cellometer Auto T4 (Nexcelom Biosciences, Lawrence, MA). ScN2a-cl3 cells were seeded either into new, 175-cm2 tissue culture flasks for continued cell culture (9 × 106 cells into 32 ml of growth medium) or onto 96-well, tissue culture–treated, white polystyrene plates (Greiner Bio-One, Monroe, NC) for treatment with test compounds (40,000 cells/well in 100 μl of growth medium for ScN2a-cl3 cells were allowed to adhere for 4 hours at 37°C before compound addition.
Test compounds (100 µl) were added to each well to attain a final concentration of 10 µM. Two positive controls were used: quinacrine and PAMAM-G4. Quinacrine [2 mM in 100% dimethylsulfoxide (DMSO)] was added and then diluted to a final concentration of 20 µM in growth medium (0.2% DMSO, final concentration). PAMAM-G4 was diluted from a 1% stock solution (in MeOH) to achieve a final concentration of 10 µg/ml. As a negative control, 0.2% DMSO in growth medium was used. Media was aspirated on day 5, and cells were washed with PBS (250 µl/well) and aspirated dry. The cells were lysed by addition of 20 µl of lysis buffer (10 mM Tris HCl, 150 mM NaCl, 0.5% sodium deoxycholate, 0.5% Nonidet P-40) containing 7.5 U/ml benzonase; the plates were placed on a shaker at 37°C for 1 hour. Proteinase K [PK; 5 µl of 125 μg/ml in a Tris buffer (10 mM Tris HCl, 20 mM calcium chloride, 50% glycerol)] was added and incubated at 37°C for 1 hour, with shaking. PK digestion was stopped by addition of 5 µl of cold (4°C) 20 mM phenylmethylsulfonyl fluoride in ethanol. After 10 minutes at room temperature, 10 µl of 5 M guanidine isothiocyanate was added at 37°C for 1 hour (with shaking) to denature the protein. The lysate in each well was diluted with 120 μl of PBS, and 150 µl from each well was transferred to 96-well polystyrene enzyme-linked immunosorbent assay plates previously coated with D18 antibody [5 μg/ml/well in 300 μl of acidified PBS overnight at room temperature in a humidified chamber (Safar et al., 2002)]. The plates were sealed and incubated overnight at 5°C. The next day, the plates were washed three times with Tris-buffered saline/Tween 20 (TBST) buffer (20 mM Tris HCl, 137 mM NaCl, 0.05% Tween 20, pH 7.5), and the contents of each well were aspirated completely. Then 100 μl of a 1:1000 dilution of horseradish peroxidase–conjugated D13 antibody was added and incubated at 37°C for 1 hour. The plates were washed four times with TBST buffer, the contents were aspirated completely, and 100 μl of 2,2′-azinobis [3-ethylbenzothiazoline-6-sulfonic acid]-diammonium salt (ABTS) peroxidase substrate was added to each well. After 15 minutes of development at room temperature, the enzymatic reaction was stopped by addition of 100 μl of ABTS stop solution, and the plates immediately loaded onto a SpectraMax M5 plate reader (Molecular Devices, Sunnyvale, CA) for measurement of absorbance at 405 nm.
For cell viability assays, mouse N2a-cl3 cells were seeded into 96-well, black polystyrene plates (Greiner Bio-One) and treated with compound as described earlier for the enzyme-linked immunosorbent assay plates. After 5 days, the growth medium was aspirated, the plates washed once with PBS (250 μl/well), and the plates aspirated dry. Calcein acetoxymethyl ester (calcein-AM 100 μl/well, 5 μg/ml solution in calcium- and magnesium-free PBS) was added, and the plates were incubated at 37°C for 45 minutes. Fluorescent emission intensity was quantified using a Spectramax M5 plate reader (Molecular Devices), excitation/emission spectra of 485/530 nm.
Computational Methods.
The Compd B structure was prepared and minimized in the Ligprep module of Schrödinger Software Suite 2010, after which it was subjected to geometry optimization in Jaguar (version 7.8; Schrödinger, LLC, New York, NY) using density functional theory (DFT) (Hohenberg and Kohn, 1964; Kohn and Sham, 1965) with B3LYP functional and 6-31G** basis set. The direct inversion of the iterative subspace (DIIS) convergence scheme and default convergence criteria were used: maximum iteration number of 48 and energy change of 5 × 10−5 hartrees.
Pharmacophore modeling was performed using Phase (version 3.2; Schrödinger). The ligands were prepared in Ligprep, where all stereoisomers and ionized species at pH 7 were generated and minimized. Next, conformers for each molecule were generated using a mixed Monte Carlo multiple minima/low mode conformation (MCMM/LMOD) method (Chang et al., 1989; Kolossváry and Guida, 1999) and OPLS 2005 force-field (Jorgensen and Tirado-Rives, 1988; Kaminski et al., 2001). Parameters used in this step included a maximum of 1000 conformers per molecule, rapid sampling, and 100 minimization steps. The root-mean-square deviation (RMSD) cutoff of 1.0 Å was used to eliminate redundant conformers.
Chemistry of Compd B and Analogs.
One batch of Compd B was a generous gift from Professor Katsumi Doh-ura. The second batch was synthesized by ChemPartner (Shanghai, People's Republic of China). Amide Compd B isosteres that were not commercially available were synthesized using 2-(1H-7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HATU)–assisted amide coupling reactions between an aryl carboxylic acid and 4-(2-alkyl/cyclopropyloxazol-5-yl)anilines. The synthesis and characterization of these compounds are detailed in the Supplemental Data.
Mice.
FVB and CD-1 mice were purchased from Charles River (Hollister, CA). All Tg mice were on the FVB genetic background and bred at the Hunter’s Point animal facility at the University of California–San Francisco. Tg(MHu2M,M111V,M165V,E167Q)1014/Prnp0/0 mice have been described previously elsewhere (Giles et al., 2010). Tg(Gfap-luc) mice expressing the firefly luciferase reporter driven by a 12-kb fragment of the murine Gfap promoter (Zhu et al., 2004) were a gift from Caliper Life Sciences (Hopkinton, MA), from which a homozygous line was generated (Stöhr et al., 2012). Tg(MoPrP:Gfap-luc)4053 mice were obtained by crossing Tg mice overexpressing full-length MoPrP [Tg(MoPrP)4053] mice (Carlson et al., 1994; Telling et al., 1996) with homozygous Tg(Gfap-luc) animals. The mice were housed with free access to food and water and were maintained on alternating 12-hour light/dark cycles.
Pharmacokinetic Studies.
Single-dose pharmacokinetic studies were performed by oral gavage, with a compound concentration of 10 mg/kg in 200 µl of a formulation containing 20% propylene glycol, 5% labrosol, 5% ethanol, and 70% polyethylene glycol 400 (PEG400). At specified time points after dosing (0.5, 2, 4, and 6 hours), the mice were euthanized by CO2, and brain and plasma samples were collected and stored at −80°C until analysis.
For quantification of Compd B and analogs, samples were injected onto a BDS Hypersil C8 column maintained at room temperature. The amount of acetonitrile (ACN) in the gradient was increased from 25% ACN to 95% ACN over 2.0 minutes, held for 1.0 minute, and then reequilibrated to 25% ACN over 1.4 minutes. Data acquisition used multiple reaction monitoring (MRM) in the positive-ion mode, and the transitions monitored were m/z 265 → 160 for Compd B and m/z 321 → 253 for internal standard.
Microsomal incubations were performed with pooled female CD-1 mouse and human microsomes, obtained from BD Biosciences (San Jose, CA). Stock solutions (0.5 mM) of each compound prepared in DMSO were diluted 500-fold into 1 ml of microsomal incubation mixture (100 mM phosphate buffer, pH 7.4) and NADPH-regenerating system (BD Biosciences) to yield a final concentration of 1 µM. This was preincubated at 37°C for 5 minutes in an Eppendorf Thermo mixer, and the reaction was initiated by addition of 0.5 mg (25 μl of a 20 mg/ml solution) of liver microsomes. Aliquots (50 μl) were withdrawn at 0, 5, 15, 30, and 60 minutes and added to 100 μl of ACN containing internal standard. These were then centrifuged at ∼12,000g for 10 minutes. The supernatants were analyzed by liquid chromatography–tandem mass spectrometry (LC-MS/MS). Duplicate incubations were run for each time point. The percentage of solute remaining at the end of the incubation was used to calculate the in vitro half-life (t1/2), using t1/2 = −0.693/k, where (−k) is the slope of the linear regression line from the plot of log percent remaining versus incubation time.
Mouse Inoculation.
Weaned mice (∼8 weeks old) were inoculated intracerebrally with 30 μl of 1% brain homogenate, prepared either from mice infected with the RML prion strain or from a patient with sCJD (type MM1) then diluted in filtered PBS containing 5% (wt/vol) bovine albumin fraction V, by use of a 1-ml syringe and 27-gauge needle. The inoculated mice were examined every day, and prion disease was diagnosed based on standard diagnostic criteria every 2 to 3 days (Carlson et al., 1988; Scott et al., 1993).
Long-Term Dosing of Compounds.
All calculations were based on typical wild-type FVB mouse weight of 25 g and liquid diet consumption of 20 ml per day. In practice, both mouse weight and food consumption increased over the duration of the study.
The rodent liquid diet was prepared by mixing 900 g of powdered diet (product no. F1256SP; Bio-Serv, Frenchtown, NJ), 50 g of chocolate powder (Bio-Serv), and 4 liters of water in a large stainless steel blender. For studies with Compd B, 2200 mg of compound was added to 200 ml of 100% PEG400. To ensure dissolution, vortexing and sonicating were applied. The solution was stored at −4°C until use and then diluted 1:80 into the previously prepared rodent liquid diet to make a final dosing concentration of 110 mg/kg per day with 1.25% PEG400. For untreated mice, pure PEG400 was diluted 1:80 into the previously prepared rodent liquid diet. The amount of food was served based on the number of animals in each cage and was replaced by new batches every 2 to 3 days.
Bioluminescence Imaging.
Mice were imaged weekly using an in vivo imaging system (Caliper Life Sciences). For each image, mice were intraperitoneally injected with 50 μl of 30 mg/ml d-luciferin potassium salt solution (Gold Biotechnology, St. Louis, MO) in calcium- and magnesium-free PBS (Invitrogen, Carlsbad, CA). They were then anesthetized by an isoflurane-oxygen gas mix, and after 10 minutes were imaged for 60 seconds. Black construction-paper cutouts were used as ear covers to minimize noise signals from the ears. Bioluminescence values were quantified from Living Image 3.0 software (Caliper Life Sciences). BLI was initiated at 4 to 6 weeks after inoculation and was continued weekly until the animals were killed. For BLI curves, mean values were calculated from the same group of mice (n ≥ 3) at each time point.
PrPSc Detection by Immunoblot.
Twenty percent (wt/vol) brain homogenates were prepared from individual animals in calcium- and magnesium-free PBS by three cycles of 60 seconds bead beating at 6500 rpm, placing the tubes on ice between cycles with a tissue homogenizer (Precellys; Bertin Technologies, Montigny-le-Bretonneux, France). Protein concentrations were normalized using a bicinchoninic acid assay, and 750-μg aliquots were diluted to 1 ml with lysis buffer (10 mM Tris, pH 8.0, 150 mM NaCl, 0.5% deoxycholic acid, 0.5% Nonidet P-40). Digestion was performed by addition of 20 μg of PK and then incubated at 37°C with shaking for 1 hour. To stop the digestion, phenylmethylsulfonyl fluoride was added to a final concentration of 1 mM. After ultracentrifugation at 100,000g at 4°C, the supernatant was discarded, and the pellets were dissolved in 80 μl of NuPAGE sample buffer (Invitrogen), boiled, and then run on a 4–12% Tris-glycine SDS gel (Invitrogen). The gel was transferred to a polyvinylidene difluoride (PVDF) membrane using an iBlot (Invitrogen), and the membrane was blocked with 5% milk for 1 hour at room temperature. The membranes were subsequently incubated overnight with human-mouse P Fab conjugated to horseradish peroxidase and washed three times with TBST for 15 minutes before developing with the enhanced chemiluminescent reagent. Immunoblotting of CJD samples was performed as previously described (Giles et al., 2010).
Conformational Stability Assay.
Twenty microliters of 10% (wt/vol) brain homogenates were incubated with GdnHCl in increments of 0.5 M between 0 and 4 M at 22°C for 2 hours. The samples were subsequently diluted with lysis buffer to a final concentration of 0.4 M of GdnHCl. Digestion and immunoblotting were performed as described earlier. The immunoblot gels were scanned, and the densities of PrPSc bands were measured by ImageJ software (National Institutes of Health, Bethesda, MD). The denaturation curves were generated based on band densities. The concentration at which 50% of PrPSc in the sample was denatured (GdnHCl1/2) was then determined in Prism 6 (GraphPad Software, La Jolla, CA).
Statistical Analysis.
Statistical significance of differences observed between Compd B–treated and control samples from the conformational-stability assay was determined by using the Student’s t test. P < 0.05 was considered statistically significant.
Pathology.
After the mice were killed, their right half-brains were fixed in 10% formalin for a minimum of 3 days. The fixed brains were processed and embedded in paraffin, and 8-µm sections were cut from four representative brain regions as follows: cortex, cerebellum, hippocampus, and thalamus. The slides were deparaffinized, and the endogenous peroxidases were blocked with 3% H2O2 in methanol for 30 minutes. The slides were washed three times for 5 minutes using PBS with 0.2% Tween 20 (PBST) with three buffer changes. Immunohistochemistry for PrP required antigen retrieval by hydrolytic autoclaving (1 mM citrate buffer, pH 6.0 at 121°C for 10 minutes). Nonspecific antibody binding was blocked with 10% normal goat serum in PBST for 30 minutes. Slides were then incubated with the primary antibody: rabbit polyclonal anti-GFAP antibody (Dako North America, Carpinteria, CA) at 1:500 dilution or the R2 monoclonal anti-PrP antibody (Williamson et al., 1998) at 5 µg/ml in PBST at room temperature overnight. After washing, the sections were incubated with the appropriate biotinylated goat secondary antibody (Vector Laboratories, Burlingame, CA) at 1:100 in PBST with 10% normal goat serum for 30 minutes at room temperature. After additional washing, the ABC kit (Vector Laboratories) was used per the manufacturer’s instructions.
Sections were washed and developed with the DAB kit (Vector Laboratories) for 1 minute and washed again. Tween 20 was not present in the third wash. The sections were counterstained for 10 seconds in hematoxylin (Thermo Fisher Scientific, Waltham, MA), taken through graded alcohols to xylene, and coverslipped using Permount (Thermo Fisher Scientific). Images were taken at 20× and 40× magnifications using the SPOT Flex camera (SPOT Imaging Solutions, Sterling Heights, MI) and program on a Leica DM-IRB microscope (Leica, Wetzlar, Germany).
Results
Cellular PrPSc Reduction by Compd B.
The concentration-effect relationship of two independent batches of Compd B were determined in a 5-day PrPSc reduction assay using ScN2a-cl3 cells, which overexpress PrP (Fig. 1). Compd B, provided by Dr. Doh-ura, almost entirely eliminated protease-resistant PrPSc with an EC50 of 0.31 ± 0.14 µM (n = 4; Fig. 1A). This value is different from the original potency of 60 pM reported in ScN2a cells, but similar to results using two clones of RML-infected N2a-58 cells, which also overexpress PrP (EC50 = 0.32 µM in N002, and EC50 = 0.3 µM in Ch2) (Kawasaki et al., 2007). Differences in potency might be caused by the heterogeneity of N2a subclones (Mahal et al., 2007), or by the overexpression of PrP in the ScN2a-cl3 cells and N2a-58 cells relative to the wild-type expression of PrP in ScN2a cells. A second batch of Compd B, synthesized commercially at 100-g scale, had an EC50 of 0.76 ± 0.01 µM (n = 3; Fig. 1B) and was used for all subsequent studies.
Fig. 1.

Potency of two independent batches of Compd B in reducing PrPSc levels in ScN2a-cl3 cells. (A) Batch provided by Dr. Doh-ura. (B) Batch synthesized commercially and used in subsequent studies. EC50 values are mean ± S.E.M., determined from n = 4 (Doh-ura) or n = 3 (commercial) independent experiments.
Pharmacokinetic Liabilities of Compd B.
Despite the low EC50 values of Compd B in cell culture and the promising efficacy indicated by the large extension in survival of treated mice (Kawasaki et al., 2007), further development of this compound into a drug candidate is unlikely, largely because of the potential liability associated with the hydrazone functionality. This is a known toxicophore that undergoes Schiff base hydrolysis under physiologic conditions and releases aryl hydrazine, a toxic metabolite and carcinogen (Powell and Gannett, 2002). Aryl hydrazine is known to form DNA adducts, act as a DNA-cleaving agent (Hwu et al., 2004), and cause hemolytic anemia (Shalev et al., 1981). Thus, we sought new Compd B analogs that lack the liability associated with the undesirable hydrazone moiety yet maintain in vivo antiprion properties.
Pharmacophore Model.
New Compd B analogs were developed using a pharmacophore model built based on novel and diverse antiprion lead compounds discovered by high-throughput screening of RML-infected ScN2a-cl3 cells. Preliminary structure-activity relationship analysis of these leads suggested that a coplanar conjugated aromatic core structure is required for antiprion activity (Silber et al., in press). Lead compounds with a coplanar core structure were found to possess good antiprion potency (EC50 < 1 µM), whereas compounds with a nonplanar scaffold structure showed only moderate activity (EC50 > 1 µM). To build the pharmacophore model of PrPSc leads, we started with 440 compounds for which EC50 values of PrPSc reduction were determined in ScN2a-cl3 cells. We defined actives as those with EC50 < 10 μM (n = 274) and inactives as EC50 ≥ 10 μM (n = 166). The pharmacophore modeling was performed using Phase (version 3.2; Schrödinger, Inc.) (Dixon et al., 2006). Pharmacophore sites were assigned using a set of features defined in Phase as hydrogen-bond acceptor (HBA), hydrogen-bond donor (HBD), hydrophobic group, negatively charged group, positively charged group, and aromatic ring. Common pharmacophore hypotheses were identified using tree-based partitioning techniques and were consequently scored and ranked. Final pharmacophore analysis indicated that more than half of the active compounds, n = 150, fell within a four-site pharmacophore (Fig. 2A), with one HBD (blue), one HBA (red), and two aromatic sites (rings). The HBD and HDA are arranged into a linker that connects the two aromatic sites.
Fig. 2.

(A) A four-site pharmacophore model was generated containing one hydrogen-bond donor (blue), one-hydrogen bond acceptor (red), and two aromatic rings (orange rings). From 274 active compounds screened previously, 150 fell within this four-site model. (B) Overlay of the pharmacophore model on Compd B.
Computational analysis of the Compd B structure suggests that the molecule adopts a near coplanar conformation similar to the pharmacophore model (Fig. 2B). However, the HBA atom of Compd B, unlike in the model, links directly to the HBD atom and is displaced from the optimal location described by the model. Thus, for new Compd B analogs, we decided to replace the three-atom linker with a two-atom linker, such as an amide bond that contains a HBA (C=O) and HBD (NH), fitting well with the pharmacophore model.
Analogs of Compd B.
To test the hypothesis we developed from the pharmacophore model above, we first synthesized a Compd B analog, replacing the hydrazone linker by an amide (1). However, this compound was completely inactive in ScN2a-cl3 cells (Table 1). Replacing the pyridyl with a phenyl group restored the antiprion activity. The unsubstituted benzamide 2 had an EC50 of 0.64 ± 0.07 µM. Addition of an electron-donating group resulted in greatly improved potency: the 4-methoxy analog 3 had an EC50 of 0.12 ± 0.01 µM. Several other benzamide analogs (4–6) were tested; 4-Cl (4) and 3-MeO,4-Me (5) had similar potency to 3, whereas the 2,4-diF analog 6 was approximately 3-fold less potent than 3.
TABLE 1.
EC50 values of biaryl amide analogs of Compd B in ScN2a-cl3 cells
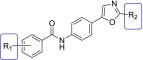
| Compound No. | R1 | R2 | EC50* |
|---|---|---|---|
| µM | |||
| 1 | 4-pyridyl (in place of R1-phenyl) | H | Inactive |
| 2 | H | H | 0.64 ± 0.07 |
| 3 | 4-MeO | H | 0.12 ± 0.01 |
| 4 | 4-Cl | H | 0.12 ± 0.07 |
| 5 | 3-MeO,4-Me | H | 0.16 ± 0.03 |
| 6 | 2,4-diF | H | 0.34 ± 0.06 |
| 7 | 4-CN | Me | 0.51 ± 0.11 |
| 8 | 4-OCHF2 | Me | 0.19 ± 0.05 |
| 9 | 2-NCCH2S | Me | Inactive |
| 10 | 2-F | 0.30 ± 0.01 | |
| 11 | 3-F | 0.54 ± 0.19 | |
| 12 | 4-F | 0.08 ± 0.02 | |
| 13 | 2,4-diF | 0.23 ± 0.05 | |
| 14 | 4-Cl | 0.10 ± 0.03 | |
| 15 | 4-MeOCH2CH2O | 0.06 ± 0.02 | |
| 16 | 3-CN | 0.36 ± 0.15 | |
| 17 | 4-CN | 0.54 ± 0.13 | |
| 18 | 3-Me | 0.39 ± 0.08 |
Average ± S.D. of three independent runs.
Encouraged by the potency of 3–5, we dosed these compounds in mice to evaluate their pharmacokinetic profiles and determined in vitro metabolic stability in human and mouse hepatic microsomes (Table 2). Compound 3 displayed poor pharmacokinetic characteristics, relative to Compd B, with low plasma and brain exposures likely caused by its low metabolic stability in mouse hepatic microsomes (t1/2 = 7 minutes). Compounds 4 and 6 showed slightly improved metabolic stability as well as brain and plasma exposures compared with 3. Compound 5 displayed better metabolic stability (t1/2 = 20 minutes) and thus better brain and plasma exposures.
TABLE 2.
Pharmacokinetic parameters of Compd B and analogs
Brain and plasma exposure were measured in vivo after a single dose of 10 mg/kg administered by oral gavage. Microsomal stability was measured in vitro in mouse and human microsomal preparations.
| Compound | Brain Exposure |
Plasma Exposure |
Microsomal Stability* |
|||
|---|---|---|---|---|---|---|
| Cmax | AUClast | Cmax | AUClast | Mouse | Human | |
| μM | μM*h | μM | μM*h | |||
| Compd B | 0.46 ± 0.16 | 1.23 ± 0.15 | 0.83 ± 0.38 | 2.36 ± 0.25 | 15 (6) | 57 (49) |
| 1 | 0.31 ± 0.01 | 0.08 ± 0.01 | 0.34 ± 0.01 | 0.47 ± 0.02 | 15 (6) | >60 (80) |
| 2 | 0.36 ± 0.08 | 0.94 ± 0.30 | 0.55 ± 0.05 | 1.54 ± 0.44 | 12 (3) | >60 (60) |
| 3 | 0.02 ± 0.00 | 0.03 ± 0.04 | 0.04 ± 0.04 | 0.15 ± 0.11 | 7 (0.1) | >60 (49) |
| 4 | 0.08 ± 0.08 | 0.14 ± 0.11 | 0.05 ± 0.06 | 0.08 ± 0.08 | 11 (2) | >60 (70) |
| 5 | 0.89 ± 0.47 | 1.97 ± 0.43 | 1.36 ± 0.76 | 3.13 ± 0.87 | 20 (12) | >60 (51) |
| 6 | 0.13 ± 0.03 | 0.26 ± 0.07 | 0.11 ± 0.04 | 0.26 ± 0.07 | 16 (7) | >60 (70) |
| 7 | 0.05 ± 0.04 | 0.18 ± 0.12 | 0.05 ± 0.04 | 0.18 ± 0.10 | 16 (7) | 9 (0.9) |
| 8 | 0.03 ± 0.00 | 0.01 ± 0.00 | 0.04 ± 0.01 | 0.06 ± 0.01 | 5 (0.2) | 9 (1) |
| 9 | 0.97 ± 0.03 | 2.35 ± 0.28 | 3.77 ± 0.87 | 7.86 ± 1.46 | 29 (24) | >60 (54) |
| 10 | 6.12 ± 1.75 | 16.8 ± 0.52 | 2.53 ± 0.85 | 7.59 ± 0.76 | 21 (14) | >60 (80) |
| 11 | 1.28 ± 0.11 | 5.97 ± 0.83 | 0.87 ± 0.12 | 3.74 ± 0.04 | 21 (15) | >60 (81) |
| 12 | 1.14 ± 0.03 | 4.83 ± 0.95 | 0.71 ± 0.06 | 3.13 ± 0.67 | 17 (9) | >60 (81) |
| 13 | 2.74 ± 0.65 | 13.5 ± 0.23 | 1.20 ± 0.78 | 6.10 ± 0.26 | 18 (11) | >60 (87) |
| 14 | 0.46 ± 0.38 | 0.88 ± 0.32 | 0.16 ± 0.13 | 0.33 ± 0.10 | 7 (0.4) | >60 (74) |
| 15 | 0.28 ± 0.17 | 0.48 ± 0.14 | 0.10 ± 0.06 | 0.30 ± 0.06 | 11 (3) | 36 (33) |
| 16 | 5.16 ± 2.93 | 18.7 ± 5.69 | 6.39 ± 3.10 | 23.9 ± 6.23 | >60 (70) | >60 (94) |
| 17 | 2.86 ± 0.35 | 6.61 ± 0.01 | 1.54 ± 0.56 | 4.09 ± 0.61 | 24 (18) | >60 (79) |
| 18 | 1.09 ± 0.11 | 4.22 ± 0.71 | 1.05 ± 0.11 | 3.90 ± 0.89 | 25 (20) | >60 (63) |
Half-life in minutes and percentage remaining after 60-minute incubation.
Cmax, maximum peak drug concentration; AUClast, area under the time concentration curve from time zero to time of last measurable concentration.
To improve the metabolic stability and pharmacokinetic profiles of these analogs, we sought to introduce a methyl or cyclopropyl group at the 2-position of the oxazole ring to block any oxidation at this site. A total of 12 analogs were purchased or synthesized (7–18). Substitution at the para-position of the phenyl ring is favored for antiprion potency, with analogs 8, 12, 14, and 15 displaying the best potency (EC50 = 0.06–0.19 µM; Table 1). In the case of fluoro-substituted benzamides, 12 (4-F) was up to 7-fold more potent than its positional isomers 10 (2-F), 11 (3-F), and 13 (2,4-diF). Cyano analog 16 and 3-Me 18 had EC50 values of ∼0.35 µM. However, two para-substituted cyano analogs (7 and 17) displayed only moderate antiprion potency (EC50 of ∼0.5 µM). Analog 9, with 2-NCCH2S, was inactive in our assays.
When tested in vitro for metabolic stability and in vivo for brain and plasma exposure, many of these methyl- or cyclopropyl-substituted oxazole analogs showed improved pharmacokinetic profiles (Table 2). Fluoro analogs 10 and 11, cyano analogs 16 and 17, and 3-Me 18 had t1/2 longer than 20 minutes in mouse hepatic microsomes as well as improved brain and plasma exposures compared with Compd B. All four fluoro analogs (10–13) showed higher brain exposures (Cmax and area under the time concentration curve) compared with Compd B. Electron-rich analog 15 (4-MeOEtO) had poor stability in mouse hepatic microsomal preparations (t1/2 = 11 minutes). Interestingly, compound 14 (4-Cl) with an electron-withdrawn group also showed poor microsomal stability (t1/2 = 7 minutes). Not surprisingly, both 14 (4-Cl) and 15 (4-MeOEtO) displayed poor plasma and brain exposures in mouse pharmacokinetic studies. More importantly, the brain Cmax values for several analogs (10, 12, 13, and 16) were at least 10-fold their corresponding EC50 values.
Overall, six analogs (10, 12, 13, and 16–18) showed improved antiprion potency (Table 1) and better pharmacokinetic results (Table 2) than Compd B, suggesting that they may be more suitable candidates as antiprion therapeutics.
Long-Term Dosing of Compd B for Efficacy Studies.
While designing more potent and less toxic compounds as therapeutics for prion disease, we pursued finding a more efficient technique to measure the efficacy of candidate molecules in mouse models, with the hope that a well-validated animal system will be in place when new candidate drugs are identified. Because Compd B is the only small molecule demonstrated to substantially extend survival in prion-infected mice, it was used as a positive indicator for the purpose of setting up and evaluating our animal testing procedure.
From previous studies, dosing at 100 mg/kg per day of Compd B to FVB mice for 3 days in a liquid diet resulted in brain concentrations of ∼10 μM, 25 times higher than the EC50 value of 0.4 μM in ScN2a-cl3 cells. However, doses of 150 mg/kg per day for 8 days resulted in two of four animals dying (Silber et al., 2013). Therefore, we selected a dose between the two (of 110 mg/kg per day) to be used in future long-term in vivo dosing studies. To apply continuous dosing, Compd B was mixed into a liquid diet along with 1.25% of PEG400 as an excipient to enable the dissolution of the compound. In preliminary studies, we observed male mice had a high mortality rate after 2–3 months when PEG400 was included in their food. From whole-body pathologic examination of two individual cases, both animals showed a distended urinary bladder that contained globular substance lining the lumen. It is likely that PEG400 was excreted into the urine and precipitated in the bladder, resulting in diminished bladder function and distension. In comparison, female mice dosed for up to 280 days showed no signs of intolerance to PEG400. As a result, all further experiments were performed in female mice only.
To determine concentrations of Compd B in the brain upon longer-term dosing, we collected brains from mice dosed at 110 mg/kg per day for 7, 14, 21, and 28 days (Supplemental Table 1). Unexpectedly, there was no correlation between duration of dosing and the concentration of Compd B in the brain. Occasionally, values above 10 µM were observed, but the majority of mice had very low levels in the brain. This lack of correlation likely reflects the rapid metabolic half-life of Compd B (15 minutes) and the variability between when mice last ate (and thus were dosed) and when they were killed for analysis. These observations imply that brain concentrations likely vary widely over the course of each day.
Compd B Prolonged Survival in Tg(Gfap-luc) Mice.
Despite the lack of sustained high levels of Compd B in the brain, Compd B doses of 300 mg/kg per day were reported to double survival of RML-infected Tga20 mice that overexpress MoPrP (Kawasaki et al., 2007). Compd B increased the incubation time from 69 ± 6 to 154 ± 20 days. By use of mice that did not overexpress wild-type MoPrP, we administered Compd B at 110 mg/kg per day PO; the drug was diluted in 1.25% PEG400 in a chocolate liquid diet.
To determine whether extended survival could be predicted by BLI, FVB mice expressing a transgene encoding luciferase under control of the Gfap promoter were inoculated intracerebrally with RML prions. Compd B treatment was initiated 1 day after inoculation with RML prions and continued until mice showed signs of neurologic dysfunction, at which time they were euthanized. Untreated prion-infected Tg(Gfap-luc) mice were fed 1.25% PEG400 dissolved in the chocolate liquid diet. The bioluminescence signal measured from untreated, RML-infected Tg(Gfap-luc) mice showed a sustained increase beginning at ∼55 dpi, which is consistent with previous findings (Tamgüney et al., 2009); these mice showed signs of neurologic dysfunction at 108 ± 1 dpi (n = 8; Fig. 3A and C). In contrast, the BLI signal in Tg(Gfap-luc) mice treated with Compd B remained unchanged for ∼200 dpi, providing an early, noninvasive indication that treatment was likely to be efficacious (Fig. 3C). The average BLI signal increased about 6-fold beginning at ∼200 dpi and plateaued for 30 to 40 days before the appearance of signs of neurologic dysfunction. The mean incubation time for the Compd B–treated group was 219 ± 21 days (n = 10) (Table 3), indicating that Compd B prolonged survival of RML-infected mice by more than 100 days and that its efficacy could be monitored by BLI.
Fig. 3.

Kaplan-Meier survival (A and B) and BLI (C and D) curves of Tg(Gfap-luc) mice (A and C) and Tg(MoPrP:Gfap-luc)4053 mice (B and D) infected with RML prions and treated with a 110 mg/kg per day concentration of Compd B beginning at 1 dpi. Compd B treatment (gray) extended survival by 90–100 days compared with untreated mice (black). Treatment with Compd B suppressed the BLI signal at 60 days (C), when infected untreated mice showed upregulation, which predicted efficacy of the compound. Mean brain bioluminescence signal expressed as a percentage of the maximum; error bars represent SEM. Bars under each graph indicate the initiation and duration of treatment.
TABLE 3.
Incubation periods for prion-infected mice treated with Compd B
| Mouse Line |
Inoculum |
Dose |
Treatment Started |
Mean Incubation Period ± S.E.M. |
n/n0* |
|---|---|---|---|---|---|
| mg/kg per day | dpi | days | |||
| Tg(Gfap-luc) | RML | 0 | n/a | 108 ± 1 | 8/8 |
| Tg(Gfap-luc) | RML | 110 | 1 | 219 ± 21 | 10/10 |
| Tg(Gfap-luc) | RML | 110 | 60 | 182 ± 9 | 9/9 |
| Tg(Gfap-luc) | RML | 110 | 78 | 163 ± 14 | 9/9 |
| Tg(MoPrP:Gfap-luc)4053 | RML | 0 | n/a | 52 ± 2 | 15/15 |
| Tg(MoPrP:Gfap-luc)4053 | RML | 110 | 1 | 143 ± 7 | 12/12 |
| Tg(MHu2M,M111V,M165V,E167Q)1014 | sCJD | 0 | n/a | 80 ± 1 | 13/13 |
| Tg(MHu2M,M111V,M165V,E167Q)1014 | sCJD | 110 | 1 | 78 ± 1 | 5/5 |
n0, number of inoculated mice; n/a, not applicable.
n, number of ill mice.
Compd B Prolonged Survival in Tg Mice Overexpressing MoPrP.
To determine whether BLI could also be predictive of Compd B efficacy in mice overexpressing MoPrP, we used mice expressing two transgenes: MoPrP and Gfap-luc. These bigenic mice, designated Tg(MoPrP:Gfap-luc)4053 mice, express full-length MoPrP at a level ∼4-fold higher than that of endogenous PrP. The Tg(MoPrP:Gfap-luc)4053 mice have an incubation time of ∼50 days after intracerebral inoculation with RML prions and showed a sustained increase in bioluminescence beginning at ∼30 dpi (Fig. 3B and D). In these bigenic mice, Compd B treatment extended the time interval from inoculation to an increase in the BLI signal from ∼30 to ∼120 dpi and prolonged the time at which signs of neurologic dysfunction were detected from 52 ± 2 days (n = 15) to 143 ± 7 days (n = 12). Notably, Compd B extended both the time from inoculation to an increase in the BLI signal and to the manifestation of neurologic dysfunction by ∼90 days (Fig. 3, B and D; Table 3).
Biochemical and Neuropathologic Analysis of Brains from Compd B–Treated Mice.
Immunoblotting of brain samples from Tg(Gfap-luc) mice treated with Compd B showed protease-resistant PrPSc bands (Fig. 4A). Overall, mice developing clinical signs at earlier time points showed less PrPSc in their brains compared with mice at later time points, but some interanimal variabilities were observed.
Fig. 4.

Biochemical and neuropathologic analysis of Compd B–treated Tg(Gfap-luc) mouse brains. (A) Immunoblot of brain homogenates prepared from RML-infected Tg(Gfap-luc) mice without Compd B treatment (lane 1) and treated with 110 mg/kg/d of Compd B beginning at 1 dpi (lanes 2–9). Each lane shows brain homogenate from a single animal, whose incubation time is indicated. Samples were probed for protease-resistant PrPSc after PK digestion (upper panel); actin is shown as a control (lower panel). Apparent molecular masses based on migrated protein standards are shown in kilodaltons. (B) Conformational-stability curves of PrPSc in untreated (black) and Compd B–treated (gray), RML-infected Tg(Gfap-luc) mice. GdnHCl1/2 values were 1.4 ± 0.2 M (n = 6) for the untreated group and 1.8 ± 0.3 M (n = 5) for the treated group, which were significantly different (P = 0.014). (C–H) Immunohistochemical analysis of brain sections from untreated (C–E, 112 dpi) and treated (F–H, 284 dpi) Tg(Gfap-luc) mice. H&E staining showed a small amount of vacuolation in the hippocampus in both untreated (C) and treated (F) mice. Immunostaining for PrPSc showed uniformly distributed and mildly intense PrPSc in virtually all regions of the brain of untreated mice (D). In contrast, PrPSc accumulated focally in treated mice (G), particularly in the thalamus, but also in the brainstem (not shown); no substantial PrPSc was found in the hippocampus or the neocortex. GFAP staining showed moderately intense astrocytic gliosis in virtually all regions of the brain of untreated mice (E) but minimal astrocytic gliosis was seen in the treated mice (H). Nc, neocortex; Hp, hippocampus; Th, thalamus. Bar in H represents 200 μm and applies to C–G.
To determine whether Compd B altered the prion strain characteristics, we measured the conformational stability of RML prions in the brains of Compd B–treated Tg(Gfap-luc) mice. GdnHCl denaturation curves for PrPSc were determined using homogenates prepared from untreated and Compd B–treated Tg(Gfap-luc) mice (Fig. 4B). PrPSc in untreated mice exhibited a GdnHCl1/2 value of 1.4 ± 0.2 M (n = 6), whereas the treated mice showed a GdnHCl1/2 value of 1.8 ± 0.3 M (n = 5). This analysis demonstrated that the PrPSc in treated mice was significantly more stable than that in the untreated mouse brains (P = 0.014). These findings argue that a conformational change occurred in PrPSc as a result of exposure to Compd B.
Consistent with a difference in conformational stability, several neuropathologic differences were also observed between untreated and Compd B–treated Tg(Gfap-luc) mice (Fig. 4, C–H). Untreated mice showed uniform deposition of PrPSc in all regions of the brain and brainstem (Fig. 4D). In contrast, the treated mice showed a patchy distribution of PrPSc, which was most evident in the thalamus and brainstem but largely absent from the hippocampus and cerebral cortex, which was consistent even for mice that had high levels of PK-resistant PrP (Fig. 4G). GFAP staining showed moderately intense astrocytic gliosis in all brain areas of untreated mice (Fig. 4E), but minimal astrocytic gliosis was found in 4 of 6 Compd B–treated mice (Fig. 4H). H&E staining revealed only a few vacuoles in the hippocampus and no nerve cell loss in both untreated and treated mice (Fig. 4, C and F).
Immunoblotting of brain samples from Tg(MoPrP:Gfap-luc)4053 mice treated with Compd B showed protease-resistant PrPSc bands (Fig. 5A). As with Tg(Gfap-luc) mice, animals developing signs of neurologic dysfunction at earlier time points harbored less protease-resistant PrPSc in their brains compared with ill mice at later time points.
Fig. 5.

Biochemical and neuropathologic analysis of Compd B–treated Tg(MoPrP:Gfap-luc)4053 mouse brains. (A) Immunoblots of brain homogenates prepared from prion-infected Tg(MoPrP:Gfap-luc)4053 mice without Compd B treatment (lane 1) and treated with 110 mg/kg/d of Compd B beginning at 1 dpi (lanes 2–13). Each lane shows brain homogenate from a single animal, whose incubation time is indicated. Samples were probed for PK-resistant PrPSc (upper panel); actin is shown as a control (lower panel). Apparent molecular masses based on migrated protein standards are shown in kilodaltons. (B) Conformational-stability curves of PrPSc in untreated (black) and Compd B–treated (gray) Tg(MoPrP:Gfap-luc)4053 mice. GdnHCl1/2 concentrations were 1.4 ± 0.3 M (n = 6) for the untreated group and 1.7 ± 0.2 M (n = 6) for the treated group. (C–H) Immunohistochemical analysis of brain sections from untreated (C–E) and treated (F–H) Tg(MoPrP:Gfap-luc)4053 mice. H&E staining showed mild vacuolation in untreated (C) and treated mice (F). Immunostaining for PrPSc showed intense, uniformly distributed PrPSc throughout the brain of untreated mice (D) and multifocal patches of PrPSc accumulation in treated mice (G). GFAP staining showed intense astrocytic gliosis, particularly in the hippocampus, of untreated mice (E), but multifocal, moderately intense astrocytic gliosis resembling the patchy distribution of PrPSc in treated mice (H). Nc, neocortex; Hp, hippocampus; Th, thalamus. Bar in H represents 200 μm and applies to C–G.
To determine whether Compd B altered the prion strain characteristics in Tg(MoPrP:Gfap-luc)4053 mice, we measured the conformational stability of RML prions in the brains of these bigenic mice. GdnHCl denaturation curves for PrPSc were determined using homogenates prepared from untreated and Compd B–treated mice (Fig. 5B). PrPSc in untreated Tg(MoPrP:Gfap-luc)4053 mice exhibited a GdnHCl1/2 value of 1.4 ± 0.3 M (n = 6) while that in the treated mice possessed a GdnHCl1/2 value of 1.7 ± 0.2 M (n = 6). In contrast to the increase in conformational stability of PrPSc attributed to Compd B in Tg(Gfap-luc) mice (Fig. 4B), the increased stability of PrPSc in Tg(MoPrP:Gfap-luc)4053 mice was not statistically significant (P = 0.079).
Similar to the differences in neuropathology with untreated and Compd B–treated Tg(Gfap-luc) mice (Fig. 4, C–H), substantial differences were found in the neuropathology of untreated and Compd B–treated Tg(MoPrP:Gfap-luc)4053 mice (Fig. 5, C–H). Untreated Tg(MoPrP:Gfap-luc)4053 mice showed substantially more intense deposition of PrPSc (Fig. 5D) compared with treated mice, which had broader distribution of patchy PrPSc clusters, including a greater area of accumulation in the thalamus and patchy accumulation in the hippocampus, cerebral cortex, and brainstem (Fig. 5G). GFAP staining revealed moderate astrocytic gliosis in all regions and intense astrocytic gliosis in the hippocampus of untreated mice (Fig. 5E). In contrast, the treated group showed a moderate degree of astrocytic gliosis in areas where patchy deposits of PrPSc appeared (Fig. 5H). Similar to Tg(Gfap-luc) mice, H&E staining of Tg(MoPrP:Gfap-luc)4053 brains did not show any pathology, with only a few vacuoles seen in the hippocampus and no nerve cell loss (Fig. 5, C and F).
Compd B Was Ineffective against CJD Prions.
Although the foregoing results with Tg mice inoculated with RML prions were encouraging, we asked whether Compd B could extend the lives of Tg mice inoculated with CJD prions. To investigate this possibility, we chose Tg(MHu2M,M111V,M165V,E167Q)1014/Prnp0/0 mice, which have abbreviated incubation times for human CJD prions (Giles et al., 2010). The Tg1014 mice express a chimeric mouse/human PrP transgene and develop signs of neurologic dysfunction in ∼80 days after inoculation with the MM1 subtype of CJD prions. After we administered a concentration of 110 mg/kg per day of Compd B to Tg1014 mice infected with CJD(MM1) prions beginning at 1 dpi, we observed no extension in survival of the Tg1014 mice. Those Tg1014 mice receiving Compd B displayed signs of neurologic dysfunction at 78 ± 1 days compared with 80 ± 1 days for the untreated controls (Fig. 6A). Analysis of brains, both biochemically and neuropathologically, also failed to show any differences. By immunoblotting, levels of PrPSc in the brains of treated mice were indistinguishable from the untreated controls (Fig. 6B). Neuropathologic analysis showed that both untreated (Fig. 6, C–E) and treated (Fig. 6, F–H) Tg1014 mice infected with CJD prions had widespread vacuolation, punctate PrPSc deposition, and strong astrocytic gliosis.
Fig. 6.

Survival, biochemical, and pathologic analyses of Tg(MHu2M,M111V,M165V,E167Q)1014 mice infected with sCJD prions. (A) Kaplan-Meier survival curve of Tg1014 mice infected with sCJD prions and treated with 110 mg/kg/d of Compd B (gray) beginning at 1 dpi. Untreated mice are shown as controls (black). No extension in survival was observed. (B) Immunoblot of brain homogenates prepared from the sCJD (MM1) inoculum (lane 1), prion-infected Tg1014 mice without Compd B treatment (lanes 2 and 3), and prion-infected Tg1014 mice treated with 110 mg/kg/d of Compd B beginning at 1 dpi (lanes 4 and 5). Samples were probed for PK-resistant PrPSc. Apparent molecular masses based on migrated protein standards are shown in kilodaltons. (C–H) Immunohistochemical analysis of brain sections from untreated (C–E) and treated (F–H) Tg1014 mice. H&E staining showed robust vacuolation in untreated (C) and treated mice (F). Immunostaining for PrPSc showed intense, uniformly distributed PrPSc throughout the brains of both untreated (D) and treated (G) mice. GFAP staining showed severely intense astrocytic gliosis, particularly in the hippocampus, of untreated mice (E) and treated mice (H). Nc, neocortex; Hp, hippocampus; Th, thalamus. Bar in H represents 200 μm and applies to C–G.
Delayed Administration of Compd B.
Tg(Gfap-luc) mice were treated with Compd B beginning 60 or 78 days after intracerebral inoculation with RML prions (Fig. 7; Table 3). Compared with mice treated with Compd B beginning 1 dpi that had incubation periods of 219 dpi, those receiving Compd B beginning 60 dpi showed a reduced survival time of 182 dpi. A further reduction in survival time to 163 dpi was found when the Tg(Gfap-luc) mice were treated with Compd B beginning 78 dpi. These reductions in the effectiveness of Compd B are consistent with results from an earlier study (Kawasaki et al., 2007).
Fig. 7.

Kaplan-Meier (A and B) and BLI (C and D) curves of Tg(Gfap-luc) mice infected with RML prions and treated with 110 mg/kg per day of Compd B (gray) beginning at 60 dpi (A and C) or 78 dpi (B and D). Untreated mice are shown as controls (black). Delayed treatment resulted in shorter extensions in survival time, compared with Tg(Gfap-luc) mice treated with Compd B beginning at 1 dpi (see Fig. 3A). Mean brain bioluminescence signal expressed as a percentage of the maximum; error bars represent SEM. Bars under each graph indicate the initiation and duration of treatment.
Immediately after initiation of Compd B therapy at either 60 or 78 dpi, we found that the BLI signal increased rapidly to very high levels (Fig. 7, C and D). After 30 to 50 days, the bioluminescence level fell to ∼2 × 106 photons/s and remained there until approximately 150 dpi. Over the next 50 days, the BLI signal remained above ∼8 × 106 photons/s as the mice developed signs of neurologic dysfunction. Of note, three mice in the 60-dpi group were found dead in the cage, and one mouse in the 78-dpi group showed clinical signs of neurologic dysfunction, including ataxia, circling, weight loss, and dull hair coat shortly after initiating dosing with Compd B. Those mice were excluded in the calculations of mean survival.
Discussion
Growing evidence argues that many neurodegenerative disorders, including Alzheimer’s and Parkinson’s diseases as well as the tauopathies and amyotrophic lateral sclerosis, result from a particular protein becoming a prion (Meyer-Luehmann et al., 2006; Clavaguera et al., 2009; Frost et al., 2009; Grad et al., 2011; Münch et al., 2011; Luk et al., 2012; Mougenot et al., 2012; Prusiner, 2012). The single major risk factor for all these diseases is age, and changing demographics imply that neurodegenerative diseases will become the major burden to healthcare systems in the coming decades. Developing effective therapeutics against neurodegenerative disease therefore represents an urgent unmet need.
Although many compounds have been reported that are effective against cell models of prion disease, the only small molecule to substantially extend survival in prion-infected mice is Compd B (Kawasaki et al., 2007). However, this molecule is rapidly metabolized, which requires frequent, high doses to sustain sufficient concentrations in the brain to achieve therapeutic efficacy. Furthermore, Compd B suffers from a toxicophore in the form of a hydrazone. On the basis of the analysis of diverse antiprion lead compounds, including Compd B, we developed a four-site pharmacophore model consisting of two aromatic rings separated by a two-atom linker containing a HBD and a HBA. Therefore, we developed Compd B analogs in which the hydrazone was replaced by an amide. The direct amide analog of Compd B (1) was inactive in ScN2a-cl3 cells; however, replacement of the pyridyl group with a phenyl group (2) restored antiprion activity. On the basis of this compound, we determined structure-activity relationships using selected modifications to the phenyl and oxazole rings (Table 1). Blocking the oxidation of the oxazole moiety by introduction of an alkyl group and introducing an electron-withdrawing group to the phenyl ring increased the potency and metabolic stability. Of the 18 novel compounds tested, four compounds (10, 12, 13, and 16) had maximal drug concentrations (Cmax) in the brain that were 10-fold greater than their respective EC50 values. The efficacy of these compounds in vivo remains to be determined.
Compd B administered to prion-infected Tg mice prolonged survival but with some variability (Figs. 3 and 7), which was also observed in immunoblots between individual animals treated with Compd B (Figs. 4 and 5). This variability might be explained by the dosing administration. Although dispensing Compd B in a liquid diet is convenient and enables continuous dosing in the long time scales required in mouse models of neurodegenerative disease, it might also result in wide fluctuations in drug concentrations in the brains of mice (Supporting Table 1). Mice feed at discrete intervals that are more frequent during the dark cycle; during the light cycle, gaps between feeding periods can be on the order of hours (Goulding et al., 2008). For a compound with t1/2 of ∼15 minutes, such as Compd B, tissue concentration can drop ∼4000-fold in 3 hours. Therefore, over the course of each day, treated mice are likely to have large variations in brain concentrations of Compd B, which may account for the disparities in survival extensions that we observed.
BLI has proved useful in monitoring neurodegeneration in various Tg mouse models (Keller et al., 2009; Tamgüney et al., 2009; Watts et al., 2011). Here we demonstrate that it can be used to predict drug efficacy before extension in survival is evident. When the BLI signal began to increase in untreated mice infected with the RML prion strain, the signal remained unchanged in RML-infected mice given Compd B beginning at 1 dpi (Fig. 3). BLI proved less useful when therapeutic intervention was initiated later (Fig. 7) due to the abrupt increase in bioluminescence. The BLI signal is an indirect measure of gene expression, relying on the amount of luciferin substrate crossing the blood-brain barrier (BBB) and the appropriate cofactor availability for luciferase. One possible scenario as prion disease progresses is that the BBB may be more susceptible to disruption upon introduction of Compd B; this might explain the rapid increases in bioluminescence observed when dosing was started at 60 or 78 dpi.
Brains from RML-infected mice treated with Compd B occasionally showed lower levels of PrPSc than untreated mice, but in all cases there was no difference in the sizes or proportions of PK-resistant PrP glycoforms. However, conformational stability of PrPSc, as determined by GdnHCl denaturation, suggested a change in strain (Peretz et al., 2001), with Compd B–treated mice exhibiting higher GdnHCl1/2 values (Figs. 4B and 5B). Moreover, neuropathologic analysis of brains from Compd B–treated mice differed substantially from untreated controls: even when treated and untreated mice showed similar levels of PrPSc by immunoblot, Compd B–treated mice showed sparse and patchy PrP by immunohistochemistry (Figs. 4G and 5G). Treatment with drugs can result in the transformation of prion strains (Ghaemmaghami et al., 2009; Li et al., 2010; Ghaemmaghami et al., 2011). In these studies, Compd B may directly or indirectly interact with PrPSc, resulting in an alternative PrPSc conformation.
Our studies with Compd B treatment showed an extension in survival, but all infected mice eventually succumbed to disease. If prion strains represent ensembles of conformations, then Compd B treatment might be effective against the major strain conformation but not against other conformations in the RML inocula. In an earlier study, Compd B showed a modest extension of the survival of mice inoculated with the mouse-passaged human prion strain Fukuoka-1 (Kawasaki et al., 2007). However, we observed no efficacy of Compd B against sCJD(MM1) prions inoculated into Tg1014 mice (Fig. 6); sCJD(MM1) prions are most common human strain. These findings emphasize the need to perform drug efficacy studies on human prion strains whenever possible.
Our findings with Compd B have important implications for the development of antiprion therapeutics. The new lead compounds developed here, and the application of BLI to drug efficacy studies, should advance these efforts. While this article was in press, two chemically unrelated compounds were shown to extend survival in prion-infected mice (Wagner et al., 2013; Berry et al., in press), further arguing that effective therapeutics can be developed.
Supplementary Material
Acknowledgments
The authors thank the staff of the Hunter’s Point animal facility. They also thank Dr. Katsumi Doh-ura for the generous gift of Compd B, Dr. Robert Wilhelm for helpful suggestions on the manuscript, Dr. Shenheng Guan, Sumita Bhadwaj, Smita Patel, and Shigenari Hayashi for technical assistance, and Hang Nguyen for expert editorial assistance.
Abbreviations
- ACN
acetonitrile
- BLI
bioluminescence imaging
- BSE
bovine spongiform encephalopathy
- CJD
Creutzfeldt-Jakob disease
- Compd B
4-pyridinecarboxaldehyde,2-[4-(5-oxazolyl)phenyl]hydrazone
- DMSO
dimethylsulfoxide
- dpi
days postinoculation
- GFAP
glial fibrillary acidic protein
- HBA
hydrogen-bond acceptor
- HBD
hydrogen-bond donor
- luc
luciferase
- MEM
minimum essential medium
- Mo
mouse
- PBS
phosphate-buffered saline
- PBST
PBS with 0.2% Tween 20
- PEG400
polyethylene glycol 400
- PK
proteinase K
- PrP
prion protein
- PrPC
cellular prion protein
- PrPSc
self-propagating prion protein
- RML
Rocky Mountain Laboratory
- sCJD
sporadic Creutzfeldt-Jakob disease
- TBST
Tris-buffered saline/Tween 20
- Tg
transgenic
Author Contributions
Participated in research design: Lu, Giles, Geva, Silber, Prusiner.
Conducted experiments: Lu, Li, Rao, Dolghih, Gever, Geva, Elepano, Oehler, DeArmond.
Contributed new reagents or analytic tools: Bryant, Renslo, Silber.
Performed data analysis: Lu, Giles, Li, Rao, Jacobson, DeArmond, Silber, Prusiner.
Wrote or contributed to the writing of the manuscript: Lu, Giles, DeArmond, Silber, Prusiner.
Footnotes
This work was supported by the National Institutes of Health National Institute on Aging [Grants AI064709, AG002132, AG010770, and AG021601]; by gifts from the Sherman Fairchild Foundation and Rainwater Charitable Foundation; and by a gift from the G. Harold and Leila Y. Mathers Charitable Foundation.
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.
References
- Aguzzi A, Sigurdson C, Heikenwaelder M. (2008) Molecular mechanisms of prion pathogenesis. Annu Rev Pathol 3:11–40 [DOI] [PubMed] [Google Scholar]
- Berry DB, Lu D, Geva M, Watts JC, Bhardwaj S, Oehler A, Renslo AR, DeArmond SJ, Prusiner SB, Giles K. (In press) Drug resistance confounding prion therapeutics. Proc Natl Acad Sci. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson GA, Ebeling C, Yang S-L, Telling G, Torchia M, Groth D, Westaway D, DeArmond SJ, Prusiner SB. (1994) Prion isolate specified allotypic interactions between the cellular and scrapie prion proteins in congenic and transgenic mice. Proc Natl Acad Sci USA 91:5690–5694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson GA, Goodman PA, Lovett M, Taylor BA, Marshall ST, Peterson-Torchia M, Westaway D, Prusiner SB. (1988) Genetics and polymorphism of the mouse prion gene complex: control of scrapie incubation time. Mol Cell Biol 8:5528–5540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang G, Guida WC, Still WC. (1989) An internal coordinate Monte Carlo method for searching conformational space. J Am Chem Soc 111:4379–4386 DOI: 10.1021/ja00194a035. [Google Scholar]
- Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A, Fraser G, Stalder AK, Beibel M, Staufenbiel M, et al. (2009) Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol 11:909–913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon SL, Smondyrev AM, Knoll EH, Rao SN, Shaw DE, Friesner RA. (2006) PHASE: a new engine for pharmacophore perception, 3D QSAR model development, and 3D database screening: 1. Methodology and preliminary results. J Comput Aided Mol Des 20:647–671 [DOI] [PubMed] [Google Scholar]
- Frost B, Jacks RL, Diamond MI. (2009) Propagation of tau misfolding from the outside to the inside of a cell. J Biol Chem 284:12845–12852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaemmaghami S, Ahn M, Lessard P, Giles K, Legname G, DeArmond SJ, Prusiner SB. (2009) Continuous quinacrine treatment results in the formation of drug-resistant prions. PLoS Pathog 5:e1000673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaemmaghami S, Ullman J, Ahn M, St. Martin S, Prusiner SB. (2010) Chemical induction of misfolded prion protein conformers in cell culture. J Biol Chem 285:10415–10423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaemmaghami S, Watts JC, Nguyen H-O, Hayashi S, DeArmond SJ, Prusiner SB. (2011) Conformational transformation and selection of synthetic prion strains. J Mol Biol 413:527–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giles K, Glidden DV, Beckwith R, Seoanes R, Peretz D, DeArmond SJ, Prusiner SB. (2008) Resistance of bovine spongiform encephalopathy (BSE) prions to inactivation. PLoS Pathog 4:e1000206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giles K, Glidden DV, Patel S, Korth C, Groth D, Lemus A, DeArmond SJ, Prusiner SB. (2010) Human prion strain selection in transgenic mice. Ann Neurol 68:151–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goulding EH, Schenk AK, Juneja P, MacKay AW, Wade JM, Tecott LH. (2008) A robust automated system elucidates mouse home cage behavioral structure. Proc Natl Acad Sci USA 105:20575–20582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grad LI, Guest WC, Yanai A, Pokrishevsky E, O’Neill MA, Gibbs E, Semenchenko V, Yousefi M, Wishart DS, Plotkin SS, et al. (2011) Intermolecular transmission of superoxide dismutase 1 misfolding in living cells. Proc Natl Acad Sci USA 108:16398–16403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohenberg P, Kohn W. (1964) Inhomogeneous electron gas. Phys Rev 136:B864–B871 DOI:10.1103/PhysRev.136.B864. [Google Scholar]
- Hwu JR, Lin CC, Chuang SH, King KY, Su TR, Tsay SC. (2004) Aminyl and iminyl radicals from arylhydrazones in the photo-induced DNA cleavage. Bioorg Med Chem 12:2509–2515 [DOI] [PubMed] [Google Scholar]
- Jorgensen WL, Tirado-Rives J. (1988) The OPLS potential functions for proteins. Energy minimizations for crystals of cyclic peptides and crambin. J Am Chem Soc 110:1657–1666 DOI:10.1021/ja00214a001. [DOI] [PubMed] [Google Scholar]
- Kaminski GA, Friesner RA, Tirado-Rives J, Jorgensen WL. (2001) Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J Phys Chem B 105:6474–6487 DOI:10.1021/jp003919d. [Google Scholar]
- Kawasaki Y, Kawagoe K, Chen CJ, Teruya K, Sakasegawa Y, Doh-ura K. (2007) Orally administered amyloidophilic compound is effective in prolonging the incubation periods of animals cerebrally infected with prion diseases in a prion strain-dependent manner. J Virol 81:12889–12898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller AF, Gravel M, Kriz J. (2009) Live imaging of amyotrophic lateral sclerosis pathogenesis: disease onset is characterized by marked induction of GFAP in Schwann cells. Glia 57:1130–1142 [DOI] [PubMed] [Google Scholar]
- Kohn W, Sham LJ. (1965) Self-consistent equations including exchange and correlation effects. Phys Rev 140:A1133–A1138 DOI:10.1103/PhysRev.140.A1133. [Google Scholar]
- Kolossváry I, Guida WC. (1999) Low-mode conformational search elucidated: Application to C39H80 and flexible docking of 9-deazaguanine inhibitors into PNP. J Comput Chem 20:1671–1684 DOI:10.1002/(SICI)1096-987X(19991130)20:15<1671::AID-JCC7>3.0.CO;2-Y. [Google Scholar]
- Li J, Browning S, Mahal SP, Oelschlegel AM, Weissmann C. (2010) Darwinian evolution of prions in cell culture. Science 327:869–872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk KC, Kehm VM, Zhang B, O’Brien P, Trojanowski JQ, Lee VMY. (2012) Intracerebral inoculation of pathological α-synuclein initiates a rapidly progressive neurodegenerative α-synucleinopathy in mice. J Exp Med 209:975–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahal SP, Baker CA, Demczyk CA, Smith EW, Julius C, Weissmann C. (2007) Prion strain discrimination in cell culture: the cell panel assay. Proc Natl Acad Sci USA 104:20908–20913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer-Luehmann M, Coomaraswamy J, Bolmont T, Kaeser S, Schaefer C, Kilger E, Neuenschwander A, Abramowski D, Frey P, Jaton AL, et al. (2006) Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science 313:1781–1784 [DOI] [PubMed] [Google Scholar]
- Mougenot A-L, Nicot S, Bencsik A, Morignat E, Verchère J, Lakhdar L, Legastelois S, Baron T. (2012) Prion-like acceleration of a synucleinopathy in a transgenic mouse model. Neurobiol Aging 33:2225–2228 [DOI] [PubMed] [Google Scholar]
- Münch C, O’Brien J, Bertolotti A. (2011) Prion-like propagation of mutant superoxide dismutase-1 misfolding in neuronal cells. Proc Natl Acad Sci USA 108:3548–3553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peretz D, Scott MR, Groth D, Williamson RA, Burton DR, Cohen FE, Prusiner SB. (2001) Strain-specified relative conformational stability of the scrapie prion protein. Protein Sci 10:854–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell JH, Gannett PM. (2002) Mechanisms of carcinogenicity of aryl hydrazines, aryl hydrazides, and arenediazonium ions. J Environ Pathol Toxicol Oncol 21:1–31 [PubMed] [Google Scholar]
- Prusiner SB. (1998) Prions. Proc Natl Acad Sci USA 95:13363–13383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusiner SB. (2013) Prions, in Fields Virology (Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B, eds) pp 2418–2456, Lippincott Williams & Wilkins, Philadelphia [Google Scholar]
- Prusiner SB. (2012) Cell biology. A unifying role for prions in neurodegenerative diseases. Science 336:1511–1513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richt JA, Hall SM. (2008) BSE case associated with prion protein gene mutation. PLoS Pathog 4:e1000156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safar JG, Scott M, Monaghan J, Deering C, Didorenko S, Vergara J, Ball H, Legname G, Leclerc E, Solforosi L, et al. (2002) Measuring prions causing bovine spongiform encephalopathy or chronic wasting disease by immunoassays and transgenic mice. Nat Biotechnol 20:1147–1150 [DOI] [PubMed] [Google Scholar]
- Scott M, Groth D, Foster D, Torchia M, Yang S-L, DeArmond SJ, Prusiner SB. (1993) Propagation of prions with artificial properties in transgenic mice expressing chimeric PrP genes. Cell 73:979–988 [DOI] [PubMed] [Google Scholar]
- Shalev O, Leida MN, Hebbel RP, Jacob HS, Eaton JW. (1981) Abnormal erythrocyte calcium homeostasis in oxidant-induced hemolytic disease. Blood 58:1232–1235 [PubMed] [Google Scholar]
- Silber BM, Rao S, Fife KL, Gallardo-Godoy A, Renslo AR, Dalvie DK, Giles K, Freyman Y, Elepano M, Gever JR, et al. (2013) Pharmacokinetics and metabolism of 2-aminothiazoles with antiprion activity in mice. Pharm Res 30:932–950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silber BM, Gever JR, Li Z, Gallardo-Godoy A, Renslo AR, Widjaja K, Irwin JJ, Rao S, Jacobson MP, Ghaemmaghami S, et al. (In press) Antiprion compounds that reduce PrPSc levels in dividing and stationary-phase cells. Bioorg Med Chem. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sim VL. (2012) Prion disease: chemotherapeutic strategies. Infect Disord Drug Targets 12:144–160 [DOI] [PubMed] [Google Scholar]
- Stöhr J, Watts JC, Mensinger ZL, Oehler A, Grillo SK, DeArmond SJ, Prusiner SB, Giles K. (2012) Purified and synthetic Alzheimer’s amyloid beta (Aβ) prions. Proc Natl Acad Sci USA 109:11025–11030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamgüney G, Francis KP, Giles K, Lemus A, DeArmond SJ, Prusiner SB. (2009) Measuring prions by bioluminescence imaging. Proc Natl Acad Sci USA 106:15002–15006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telling GC, Haga T, Torchia M, Tremblay P, DeArmond SJ, Prusiner SB. (1996) Interactions between wild-type and mutant prion proteins modulate neurodegeneration in transgenic mice. Genes Dev 10:1736–1750 [DOI] [PubMed] [Google Scholar]
- Trevitt CR, Collinge J. (2006) A systematic review of prion therapeutics in experimental models. Brain 129:2241–2265 [DOI] [PubMed] [Google Scholar]
- Wagner J, Ryazanov S, Leonov A, Levin J, Shi S, Schmidt F, Prix C, Pan-Montojo F, Bertsch U, Mitteregger-Kretzschmar G, et al. (2013) Anle138b: a novel oligomer modulator for disease-modifying therapy of neurodegenerative diseases such as prion and Parkinson's disease. Acta Neuropathol. 125:795–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts JC, Giles K, Grillo SK, Lemus A, DeArmond SJ, Prusiner SB. (2011) Bioluminescence imaging of Abeta deposition in bigenic mouse models of Alzheimer’s disease. Proc Natl Acad Sci USA 108:2528–2533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson RA, Peretz D, Pinilla C, Ball H, Bastidas RB, Rozenshteyn R, Houghten RA, Prusiner SB, Burton DR. (1998) Mapping the prion protein using recombinant antibodies. J Virol 72:9413–9418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L, Ramboz S, Hewitt D, Boring L, Grass DS, Purchio AF. (2004) Non-invasive imaging of GFAP expression after neuronal damage in mice. Neurosci Lett 367:210–212 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.