

Abstract
Impaired transmission through glutamatergic circuits has been postulated to play a role in the underlying pathophysiology of schizophrenia. Furthermore, inhibition of the N-methyl-d-aspartate (NMDA) subtype of ionotropic glutamate receptors (NMDAR) induces a syndrome that recapitulates many of the symptoms observed in patients with schizophrenia. Selective activation of metabotropic glutamate receptor subtype 5 (mGlu5) may provide a novel therapeutic approach for treatment of symptoms associated with schizophrenia through facilitation of transmission through central glutamatergic circuits. Here, we describe the characterization of two novel N-aryl piperazine mGlu5 positive allosteric modulators (PAMs): 2-(4-(2-(benzyloxy)acetyl)piperazin-1-yl)benzonitrile (VU0364289) and 1-(4-(2,4-difluorophenyl)piperazin-1-yl)-2-((4-fluorobenzyl)oxy)ethanone (DPFE). VU0364289 and DPFE induced robust leftward shifts in the glutamate concentration-response curves for Ca2+ mobilization and extracellular signal-regulated kinases 1 and 2 phosphorylation. Both PAMs displayed micromolar affinity for the common mGlu5 allosteric binding site and high selectivity for mGlu5. VU0364289 and DPFE possessed suitable pharmacokinetic properties for dosing in vivo and produced robust dose-related effects in reversing amphetamine-induced hyperlocomotion, a preclinical model predictive of antipsychotic-like activity. In addition, DPFE enhanced acquisition of contextual fear conditioning in rats and reversed behavioral deficits in a mouse model of NMDAR hypofunction. In contrast, DPFE had no effect on reversing apomorphine-induced disruptions of prepulse inhibition of the acoustic startle reflex. These mGlu5 PAMs also increased monoamine levels in the prefrontal cortex, enhanced performance in a hippocampal-mediated memory task, and elicited changes in electroencephalogram dynamics commensurate with procognitive effects. Collectively, these data support and extend the role for the development of novel mGlu5 PAMs for the treatment of psychosis and cognitive deficits observed in individuals with schizophrenia.
Introduction
Schizophrenia affects approximately 1% of the population worldwide and is characterized by three core symptom clusters: positive symptoms, such as delusions and thought disorders; negative symptoms, including anhedonia and social withdrawal; and cognitive impairments (American Psychiatric Association, 2000). Poor functional and social outcomes are directly correlated with the cognitive deficits observed in patients with schizophrenia (Wu et al., 2005; Green, 2006). Currently available typical and atypical antipsychotic therapies are effective in decreasing the psychotic symptoms, but provide little or no relief for the negative symptoms or cognitive impairments and are associated with multiple adverse side effects (Swartz et al., 2008; Adrianzen et al., 2010; Pramyothin and Khaodhiar, 2010). Accumulating evidence suggests that impaired transmission through glutamatergic circuits may contribute to all three major symptom clusters observed in patients with schizophrenia (Krystal et al., 2002; Lewis and Moghaddam, 2006). For example, selective antagonists of the N-methyl-d-aspartate (NMDA) subtype of glutamate receptor (NMDAR), such as phencyclidine (PCP), induce psychosis and cognitive impairments in humans comparable to the symptoms observed in schizophrenia (Luby et al., 1959; Javitt and Zukin, 1991; Javitt, 2007). Moreover, NMDAR antagonists or genetic knockdown of the NR1 subunit of the NMDAR results in a mouse model of NMDAR hypofunction, which displays locomotor hyperactivity, sensorimotor gating deficits, aberrant social interactions, and spatial learning deficits (Ramsey, 2009). Numerous cellular, behavioral, and anatomic studies suggest that the metabotropic glutamate receptor subtype 5 (mGlu5) is a closely associated signaling partner with the NMDAR and may provide critical regulation of transmission through forebrain circuits that are thought to mediate the psychotomimetic effects of NMDAR antagonists (Herman et al., 2012). Based on this finding, it has been postulated that highly selective activators of mGlu5 may provide a novel approach to treatment of schizophrenia that has potential for treatment of all major symptom clusters (positive, negative, and cognitive) of this disorder. While attempts to develop selective mGlu5 agonists have not been fruitful, there has been tremendous progress in discovery of highly selective positive allosteric modulators (PAMs) of mGlu5. These mGlu5 PAMs can influence the binding and/or function of the receptor when simultaneously occupied by an orthosteric ligand and offer a number of potential advantages over orthosteric agonists. PAMs that enhance receptor activity, but have no intrinsic efficacy, have the capacity to maintain the spatial and temporal aspects of synaptic transmission and enhance mGlu5-mediated synaptic plasticity without altering normal activity-dependence of specific forms of synaptic plasticity (Ayala et al., 2009; Noetzel et al., 2013). Furthermore, recent studies reveal that pure mGlu5 PAMs have clear advantages over mGlu5 agonists in terms of their safety profiles (Rook et al., 2013). Several different mGlu5 PAM chemotypes have been identified, and previous studies have shown that mGlu5 PAMs from different scaffolds exhibit efficacy in animal models predictive of antipsychotic-like and cognition-enhancing activity (Kinney et al., 2005; Liu et al., 2008b; Spear et al., 2011; Gastambide et al., 2012; Gastambide et al., 2013; Gilmour et al., 2013). Herein, we describe the in vitro and behavioral characterization of two N-aryl piperazine mGlu5 PAMs: 2-(4-(2-(benzyloxy)acetyl)piperazin-1-yl)benzonitrile (VU0364289), which was discovered in-house from an evolved high-throughput screening negative allosteric modulator lead (Zhou et al., 2010), and 1-(4-(2,4-difluorophenyl)piperazin-1-yl)-2-((4-fluorobenzyl)oxy)ethanone (DPFE) (disclosed in a patent application by AstraZeneca and NPS [WO 2007/087135; Slassi et al., 2007]), both structural analogs of the recently published mGlu5 PAM, CPPZ (Spear et al., 2011; Xiong et al., 2010). We found that these N-aryl piperazines were potent cooperativity-driven mGlu5 PAMs with favorable pharmacokinetic and solubility properties for dosing in vivo and efficacious in preclinical models of NMDAR hypofunction, antipsychotic-like activity, and cognition enhancement.
Materials and Methods
Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS), and antibiotics were purchased from Invitrogen (Carlsbad, CA). [3H]MethoxyPEPy (76.3 Ci/mmol) was custom synthesized by PerkinElmer Life and Analytical Sciences (Waltham, MA). Unless otherwise stated, all other reagents were purchased from Sigma-Aldrich (St. Louis, MO) and were of an analytical grade.
Cell Culture and Receptor Mutagenesis
Mutations were introduced into the wild-type rat mGlu5 in pCI:Neo using site-directed mutagenesis (QuikChange II; Agilent Technologies, Santa Clara, CA) and verified by sequencing. Wild-type and mutant rat mGlu5 receptor constructs were transfected into human embryonic kidney (HEK) 293A cells, using FuGENE6 (Promega, Madison, WI) as the transfection reagent. Polyclonal stable cell lines were derived for rat mGlu5 mutant constructs by maintaining the cells at subconfluence for a minimum of four passages in the presence of 1 mg/ml G418. Stably transfected cell lines were subsequently maintained in complete DMEM supplemented with 10% FBS, 2 mM l-glutamine, 20 mM HEPES, 0.1 mM nonessential amino acids, 1 mM sodium pyruvate, antibiotic-antimycotic, and G418 (500 ug/ml; Mediatech, Manassas, VA) at 37°C in a humidified incubator containing 5% CO2, 95% O2. HEK293 cells stably expressing rat mGlu1 were maintained in the same media as HEK293A-rat mGlu5 cells. HEK293 cells stably expressing G protein–coupled inwardly-rectifying potassium channels (HEK293-GIRK) and each of the group II and group III mGlu subtypes; cell lines were maintained in growth media containing 45% DMEM, 45% F-12, 10% FBS, 20 mM HEPES, 2 mM l-glutamine, antibiotic/antimycotic, nonessential amino acids, 700 μg/ml G418, and 0.6 μg/ml puromycin.
Ca2+ Mobilization Assays
The day prior to assay, HEK293A-rat mGlu5 cells were seeded at 50,000 cells/well in poly(d-lysine)-coated black-walled, clear-bottom 96-well plates in assay medium (DMEM supplemented with 10% dialyzed fetal bovine serum, 20 mM HEPES, and 1 mM sodium pyruvate). The cell-permeable Ca2+ indicator dye Fluo-4 was used to assay receptor-mediated Ca2+ mobilization as described previously (Hammond et al., 2010) using a FlexStation II (Molecular Devices, Sunnyvale, CA). A 5-point smoothing function was applied to the raw fluorescent Ca2+ traces and basal fluorescence of individual wells determined during the first 20 seconds. The increase in fluorescence over basal was determined for both peak and plateau phases of the response before being normalized to the maximal peak response elicited by glutamate.
Phosphorylation of Receptor-Mediated Extracellular Signal-Regulated Kinases 1 and 2
Receptor-mediated extracellular signal-regulated kinases 1 and 2 (ERK1/2) phosphorylation was determined using the AlphaScreen-based ERK SureFire kit (PerkinElmer Life and Analytical Sciences, Boston, MA; and TGR Biosciences, Thebarton, Australia) as described previously (Gregory et al., 2012). Briefly, HEK293A-rat-mGlu5 cells were plated at a density of 40,000 cells/well in clear 96-well poly(d-lysine)-coated plates in assay medium 16–24 hours before assay. Media were aspirated, and cells were washed once with serum-free media (DMEM supplemented with 16 mM HEPES) and then serum-starved for 6 hours before assay. Serum-free media were exchanged for fresh 20 minutes prior to exposure to modulators and/or glutamate at room temperature. For interaction experiments with allosteric modulators, cells were exposed to allosteric modulator or vehicle 1 minute prior to stimulation with glutamate for 7 minutes. The assay was terminated by aspiration of ligand-containing media and addition of 50 μl/well lysis buffer. Following agitation for 10 minutes, 4 μl of lysate was transferred to a white 384-well plate (Costar, Corning Life Sciences, Tewksbury, MA). Under light-diminished conditions, 7 μl/well reaction buffer mixture (containing 1 part activation buffer to 6 parts reaction buffer and 1:250 [v/v] donor and acceptor beads) was added. After 90-minute incubation at 37°C, AlphaScreen signal was measured using a H4 synergy reader (Biotek, Winooski, VT) with standard AlphaScreen settings. Data are expressed as fold increase over basal levels of phosphorylated ERK1/2 and/or normalized to the maximal response elicited by a glutamate.
Selectivity Screening
Rat mGlu1.
The day prior to assay, HEK293 cells stably expressing rat mGlu1 were plated in black-walled, clear-bottomed, poly(d-lysine)-coated 384-well plates (Greiner Bio-One, Monroe, NC) in assay medium at a density of 20,000 cells/well. Assays were performed within Vanderbilt University’s High-Throughput Screening Center as described previously (Hammond et al., 2010; Rodriguez et al., 2010). Ca2+ flux was measured using the Functional Drug Screening System 6000 (FDSS6000, Hamamatsu, Japan); vehicle or test compound (10 µM) was added followed by a concentration response curve to glutamate. The change in relative fluorescence over basal was calculated before normalization to the maximal response to glutamate.
Group II and Group III mGlus.
Compound activity at the group II and III mGlu receptors was assessed using thallium flux through GIRK channels as previously described (Niswender et al., 2008; Hammond et al., 2010). Briefly, the day prior to assay HEK293-GIRK cells expressing mGlu subtype 2, 3, 4, 6, 7, or 8 were plated into 384-well, black-walled, clear-bottom poly(d-lysine)-coated plates at a density of 15,000 cells/well in assay medium. The following day, the medium was aspirated and replaced with assay buffer (Hanks’ Balanced Salt Solution, 20 mM HEPES, pH 7.4) supplemented with 1 µM Fluozin2-AM (Invitrogen, Carlsbad, CA). For these assays, vehicle or test compound (10 µM) was added followed by a concentration response curve to glutamate (or l(+)-2-amino-4-phosphonobutanoic acid (L-AP4) in the case of mGlu7) diluted in thallium buffer (125 mM NaHCO3, 1 mM MgSO4, 1.8 mM CaSO4, 5 mM glucose, 12 mM thallium sulfate, and 10 mM HEPES) and fluorescence was measured using an FDSS 6000. Data were analyzed as described previously (Niswender et al., 2008).
Radioligand Binding Assays: mGlu5.
Membranes were prepared from HEK293A cells expressing rat mGlu5 and mutants thereof as follows. Cells were harvested by trypsinization and pelleted by centrifugation for 3 minutes at 300g. Cell pellets were resuspended in ice-cold homogenization buffer (50 mM Tris-HCl, 10 mM EDTA, 0.9% NaCl, pH 7.4) and homogenized by three 10-second bursts with a Tekmar TP-18/10S1 homogenizer (Tekmar, Cincinnati, OH) separated by 30 seconds on ice. Cell fractions were separated by centrifugation at 1,000g for 10 minutes. Supernatant was then centrifuged for 1 hour at 30,000g and the resulting pellet resuspended in ice-cold Ca2+ assay buffer. For saturation binding experiments, membranes (20–50 µg/well) were incubated with a range of [3H]methoxyPEPy concentrations (0.5–60 nM) for 1 hour at room temperature with shaking in binding buffer (50 mM Tris-HCl, 0.9% NaCl, pH 7.4). Nonspecific binding was determined with 10 µM 2-methyl-6-(phenylethynyl)-pyridine (MPEP). For inhibition binding experiments, membranes were incubated with ∼2 nM [3H]methoxyPEPy and a range of concentrations of VU0364289 and DPFE (300 pM–100 µM) for 1 hour at room temperature with shaking in Ca2+ assay with 1% dimethylsulfoxide final. Binding assays were terminated by rapid filtration through GF/B Unifilter plates (PerkinElmer Life and Analytical Sciences) using a 96-well plate harvester (Brandel, Inc., Gaithersburg, MD) and three washes with ice-cold binding buffer, separating bound from free radioligand. Plates were allowed to dry overnight prior to addition of MicroScint 20 (40 µl/well; PerkinElmer Life and Analytical Sciences). Radioactivity was counted after at least 2-hour incubation using a TopCount Scintillation Counter (PerkinElmer Life and Analytical Sciences).
Plasma Protein Binding.
The protein binding of each compound was determined in human and rat plasma via equilibrium dialysis employing single-use rapid equilibrium dialysis plates with inserts (Thermo Fisher Scientific, Rochester, NY). Plasma (220 µl) was added to the 96-well plate containing test compound (5 µl) and mixed thoroughly. Subsequently, 200 µl of the plasma-compound mixture was transferred to the cis-chamber (red) of the rapid equilibrium dialysis plate, with an accompanying 350 µl of phosphate buffer (25 mM, pH 7.4) in the trans-chamber. The rapid equilibrium dialysis plate was sealed and incubated for 4 hours at 37°C with shaking. At completion, 50-µl aliquots from each chamber were diluted 1:1 (50 µl) with either plasma (cis) or buffer (trans) and transferred to a new 96-well plate, at which time ice-cold acetonitrile (2 volumes) was added to extract the matrices. The plate was centrifuged (3000g for 10 minutes), and supernatants were transferred and diluted 1:1 (supernatant:water) into a new 96-well plate, which was then sealed in preparation for liquid chromatography with tandem mass spectrometry analysis. Each compound was assayed in triplicate within the same 96-well plate.
In Vivo Studies
Subjects.
Adult male Sprague-Dawley rats, weighing 250–275 g, (Harlan Laboratories, Indianapolis, IN) were used for pharmacokinetic, locomotor, prepulse inhibition, contextual fear conditioning, and Irwin neurologic studies. The studies were approved by the Vanderbilt University Institutional Animal Care and Use Committee and conducted in accordance with the National Institutes of Health regulations of animal care covered in Principles of Laboratory Animal Care. In addition, adult male Sprague-Dawley rats (Harlan Laboratories, Horst, The Netherlands), weighing 250–300 g were used for the in vivo microdialysis and electroencephalography studies, which were carried out in strict accordance with the European Communities Council Directive of 24th November 1986 (86/609/EEC) and were approved by the Animal Care and Use Committee of Janssen Research and Development, a division of Janssen Pharmaceutica NV, and by the local ethical committee. Food and water were available ad libitum for all animals. All rats were group-housed in a large colony room under a 12-hour light-dark cycle with food and water available ad libitum unless otherwise indicated under the specific behavioral test. For the majority of behavioral studies in rats and mice, we used a dose range between 10 and 100 mg/kg i.p. for both DPFE and VU0364289, which provided suitable pharmacokinetics for evaluation of these compounds in vivo; in the case of the translational approaches, such as the electroencephalogram (EEG) studies, we evaluated the effects of these compounds after an oral route as would be used in a clinical setting. Finally, we choose to use substantially lower doses of DPFE to evaluate potential cognitive enhancing effects in the contextual fear conditioning study, since higher doses decreased performance in this task.
In Vivo Pharmacokinetic Study.
Rats were treated with either VU0364289 or DPFE (10 mg/kg i.p.), and then and trunk blood were collected at 0.25, 0.5, 1, 3, and 6 hours after being killed. Removed brains were thoroughly washed in saline and immediately frozen on dry ice and stored at −80°C. Plasma and brain homogenate samples were then processed and subjected to liquid chromatography with tandem mass spectrometry analysis as described in detail previously (Noetzel et al., 2012).
Drugs.
d-Amphetamine hemisulfate was obtained from Sigma-Aldrich and dissolved in double deionized water; risperidone was dissolved in acidified double deionized water; and VU0364289 and DPFE were dissolved 20% hydroxyl-propyl betacyclodextrin (HPBCD) in sterile water at 1 mg/ml. All solutions were adjusted to a pH of approximately 6 to 7 using 1 N sodium hydroxide and administered in a volume of 1 ml/kg i.p. or 10 ml/kg p.o. for studies in rats and 10 ml/kg i.p. for studies in mice.
Amphetamine-Induced Hyperlocomotion.
Amphetamine-induced hyperlocomotor activity studies were conducted using a SmartFrame Open Field System (Kinder Scientific, San Diego, CA) equipped with 32 horizontal (x- and y-axes) infrared photobeams located 1 cm above the floor of the chamber as previously described (Jones et al., 2008). Changes in ambulation or locomotor activity were measured as the number of total photobeam breaks, expressed in 5-minute intervals, and were recorded with a Pentium I computer equipped with the Motor Monitor System software (Kinder Scientific). Briefly, rats were habituated to the locomotor activity chambers for 30 minutes and then pretreated with either vehicle or dose of either VU0364289 or DPFE (10–100 mg/kg i.p.). Thirty minutes later, rats were administered with either vehicle or amphetamine (1 mg/kg s.c.) and monitored for another 60 minutes.
Apomorphine-Induced Disruption of Prepulse Inhibition of the Acoustic Startle Reflex.
Studies were conducted using startle chambers with a Plexiglas cylinder mounted on a piezoelectric accelerometer for detecting motion as previously described (Jones et al., 2005). After a 20-minute pretreatment with vehicle, risperidone (3 mg/kg p.o.), or a dose of DPFE (30–100 mg/kg p.o.), rats were injected with apomorphine (1 mg/kg s.c.), and after an additional 10 minutes, they were placed into individual startle chambers for the following paradigm. After a 5-minute acclimation period, the rats were presented with five presentations of the startle stimulus alone, followed by seven randomized presentations of the following trial types: no stimulus, startle pulse alone (120 dayB, 40-millisecond broadband noise burst), prepulse noise alone (82dB, 40-millisecond broadband noise burst), and three prepulse (72, 77, or 82 dayB; 20 millisecond) plus startle pulse combinations. The intertrial interval varied pseudorandomly between 15 and 45 seconds, and the interstimulus interval was 100 milliseconds. Background noise of 67 dayB was presented continuously. Percent prepulse inhibition was calculated as 100 × (mean acoustic startle reflex [ASR] − mean ASR in prepulse plus pulse trials) / mean ASR in startle pulse trials.
Enhancement of Contextual Fear Conditioning.
Contextual fear conditioning studies were conducted in conditioning chambers housed in a sound-attenuating cubicle (Med Associates, St. Albans, VT) as previously described (Digby et al., 2012). All rats were handled and injected with saline for 2 days prior to conditioning. Rats were pretreated with DPFE (0–0.56 mg/kg i.p.) or vehicle 30 minutes before conditioning. The rats were placed in individual conditioning chambers and following a 2-minute habituation period, one footshock (1 second, 0.5 mA) was delivered through the stainless steel grid floor, and after 45 second rats were returned to their home cage. After ∼24 hours, the animals were exposed to the identical context for assessment of freezing behavior. All trials were video recorded (VID-CAM-MONO-2A; Med Associates) and scored blinded. The potential effect of DPFE on nociceptive response to the footshook stimulus was also separately measured. Rats were pretreated with vehicle or DPFE (1 mg/kg i.p.) for 30 minutes and then placed in the conditioning chambers, and behavioral changes in footshock threshold were assessed using increasing current from 0 to 0.5 mA in increments of 0.05 mA.
Nr1neo−/− Animals (NR1 Knockdown).
Since NMDAR antagonists are use-dependent channel blockers, we found that pretreatment or cotreatment of the mGlu5 PAMs used in the present studies in combination with phencyclidine resulted in enhanced disruptive effects of PCP on the behavioral endpoints measured (i.e., increased PCP-induced hyperlocomotion). For this reason, we choose to evaluate the effects of our mGlu5 PAMs in a genetic model of NMDR hypofunction to avoid confounds of using use-dependent NMDAR channel blockers after acute challenge. NR1 knockdown (NR1KD) animals were generated as previously described (Mohn et al., 1999) to harbor a hypomorphic mutation of the Grin1 gene that encodes the NR1 subunit of NMDAR. This mutation has been previously shown to cause a 90% reduction in NR1 subunit levels in the homozygous state (Mohn et al., 1999). Heterozygous mutant mice were intercrossed to generate wild-type and homozygous mutant (NR1KD) littermates on a genetically defined F1 background (C57Bl/6J:129X1Sv/J) as previously described (Ramsey et al., 2008).
Locomotor Activity.
Mice were individually tested for motor activity at 10–14 weeks of age under standardized environmental conditions using an automated Omnitech Digiscan apparatus (AccuScan Instruments, Columbus, OH), in a 42 cm2 Plexiglas chamber. Changes in ambulation or locomotor activity were measured as the number of total photobeam breaks, expressed in 5-minute intervals. Mice were pretreated for 30 minutes with vehicle or dose of DPFE (30 or 56.6 mg/kg i.p.), then placed into locomotor activity chambers and monitored for 120 minutes.
Y-Maze Test.
Mice were pretreated for 30 minutes with vehicle or dose of DPFE (10.0 mg/kg i.p.) and then placed in one arm of a standard Y-maze (San Diego Instruments, San Diego, CA) and allowed to navigate the maze for 8 minutes with 20 lux indirect lighting and monitored by video-based tracking software (Viewer2, Biobserve). Spontaneous alternations were recorded each time the mouse entered the three arms of the maze consecutively before re-entering an arm. The number of triplet entries (spontaneous alternation) was divided by the total number of arm entries to calculate the percentage of spontaneous alternation.
Sociability Test.
Sociability was measured using a modified method as previously described test (Moy et al., 2004). Mice were pretreated for 30 minutes with vehicle or DPFE (10.0 mg/kg i.p.) and then placed into a rectangular arena measuring 60 × 40.5 × 22 cm with 20 lux indirect lighting. Within the arena, a novel sex- and age-matched mouse was contained within an inverted wire cup so that the novel mouse was visible to the test mouse and could be accessed for sniffing and social investigation. Over a 10-minute testing period, the time that the test mouse spent in social investigation within a 5-cm circular zone around the novel mouse was measured using video-based tracking software (Viewer2; Biobserve, St. Augustin, Germany).
In Vivo Microdialysis.
Rats were singly housed, then surgically implanted with two guide cannula (CMA Microdialysis AB, Kista, Sweden); one into the left nucleus accumbens (NAC) (AP +1.7 mm bregma, ML 1.4 mm, DV 6.0 mm) and the other into the right prefrontal cortex (PFC) (AP +2.75 mm, ML 0.75 mm, DV 0.75 mm) (Paxinos and Watson, 1998), and allowed to recover for 7 days. One day prior to testing, rats were transferred to the experimental room and inserted with microdialysis probes (PFC with 4 mm and NAC with 2-mm membrane length) containing a 20-Kd cut-off polyarylthersulfone membrane. Starting at 7:00 AM on the next experimental day, dialysates were collected in four consecutive 30-minute samples, followed by eight consecutive 30-minute samples postadministration of vehicle or dose of DPFE (10, 30, and 100 mg/kg p.o.) in 20% HPBCD + Tween 20; n = 9–13 each treatment). Norepinehrine (NE), dopamine (DA), and serotonin (5-HT) levels were determined from brain dialysates by high-performance liquid chromatography with a Waters Alliance HT 2795 system equipped with a Waters electrochemical detector W 2465 (Waters Corporation, Milford, MA). Chromatographic separation was achieved on an Atlantis (Waters, Milford, MA) reversed-phase column (C18, 2.1 mm ID × 50 mm, 3 mm). The mobile phase was a mixture of acetonitrile (10%) and an aqueous solution (90%) of 60 mM sodium acetate, 0.1 mM EDTA, and 2 mM DSA, pH-adjusted to 5.8 by 100% acetic acid. The flow rate was 1.0 ml/min. The capillary electrochemical cell was set at a potential 0.70 V and sensitivity of 1nA with filter at 0.1s. Data were analyzed as the area under curve after vehicle or DPFE treatment.
Sleep/Wake EEG Study.
Rats were singly housed and maintained under controlled environmental conditions throughout: 22 ± 2°C ambient temperature, relative humidity 60%, 12:12 light cycle regimen (lights on 12 a.m, illumination intensity ∼100 lux). Rats were implanted with electroencephalogram recording apparatus using the following method: under anesthesia, rats were placed in a stereotaxic apparatus and three small cavities drilled into the cranial bone without perforating the dura to receive three stainless steel fixing screw-electrodes (diameter 1 mm) for the polygraphic recording of frontal and parietal EEG; two electrodes were placed on each side of the sagittal suture (AP +2 mm, ML –2 mm; and AP –6 mm, ML +3 mm from the bregma) (Paxinos and Watson, 1998), while the third (reference) electrode was positioned over the cerebellum. For recording the electrooculogram (EOG) and electromyogram (EMG), stainless steel wires (51/46 Teflon Bilaney, Germany) were placed in the periorbital and the nuchal muscle, respectively. All electrodes were fitted into an eight-hole connector and fixed with dental cement to the cranium. After a 10-day recovery period postsurgery, rats were given an additional 7 days habituation under home cage recording conditions before EEG data collection. Next, rats were connected with a rotating swivel to a bipolar recorder amplifier (Embla MedCare, Reykjavik, Iceland) with an input range of ±500 mV. The frontal, parietal EEG, EOG, and EMG signals and body movement activities were monitored for 20 h, directly digitized at 2 kHz, and subsequently managed by a software package (Somnologica Science, MedCare, Iceland).
Rats were pretreated with vehicle or dose of DPFE (10, 30, 100 mg/kg p.o.). Two EEG recording sessions were performed in 32 animals, randomly assigned to four treatment conditions (n = 8, each treatment group). The first recording session started at 1:30 PM and lasted 20 continuous hours after the administration of vehicle to all 32 rats. At 1:30 PM the next day, the second recording session was performed for the same duration following administration of vehicle or dose of DPFE as outlined above. Off-line, sleep-wake staging was automatically executed for each 2 second epoch and averaged for 30-minute periods, based on five EEG frequency bandings (δ: 0.4-4 Hz, θ: 4.2-8 Hz, α: 8.2-12 Hz, σ: 12.2-14 Hz, β: 14.2-30 Hz), integrated EMG, EOG, and body activity level. Each EEG epoch was assigned one of six sleep stages, classified as being indicative of active wakefulness, passive wakefulness, light non–rapid eye movement (REM)/slow wave sleep (lSWS), deep non-REM/slow wave sleep (dSWS), intermediate stage, or paradoxical/REM sleep. The amount of time spent in each vigilance state, the number and duration of episodes in each state, latencies for lSWS, dSWS, and REM, and the number of shifts from one state to another one were calculated. Latency was defined as the time between the beginning of the recording and the appearance of the first sleep period lasting at least 30 seconds. Time spent in each vigilance state (active wakefulness, passive wakefulness, lSWS, dSWS, intermediate stage, and REM) was expressed as a percentage of the recording period.
PharmacoEEG Study.
PharmacoEEG (pEEG) changes were determined in another group of rats under identical surgical and testing conditions as outlined under the sleep/wake EEG methods with the exception of the montage; pEEG animals were equipped with EEG and EMG electrodes as depicted in Supplemental Fig. 1. PharmacoEEG recording was performed in the dark phase of the circadian time following pretreatment with vehicle or dose of DPFE (10, 30, 100 mg/kg p.o.) (n = 8 for each treatment group). A stable baseline EEG recording was collected for 30 minutes (animals were picked up and a marker was introduced to tag the start of baseline period), followed by the administration of vehicle or dose of DPFE with a second marker indicating the start of the pharmacologic response and the subsequent 2h sampling period following the treatment. Continuous EEG and EMG field potentials were acquired at 2 kHz sample rate with an input range of ±500 mV through a Biosemi ActiveTwo system (Biosemi, Amsterdam, Netherlands) referenced to the CMS-DRL ground (common mode reference for online data acquisition and impedance measures, which is a feedback loop driving the average potential across the montage close to the amplifier zero). The signals were amplified and analog bandpass filtered between 1 and 100 Hz and digitized with 24-bit resolution.
Offline, the EEG power spectra were computed for consecutive 4s epochs in the frequency range of 0.5–100 Hz (Fast-Fourier transform routine, Hanning window, 0.25 Hz resolution). Epochs with artifacts were discarded on the basis of automatic detection. Adjacent 0.25 Hz bins were summed into 1 Hz bins up to 100 Hz for the full power spectrum. The data between 48 and 52 Hz were removed due to 50 Hz artifacts. Absolute power spectra of EEG signals were also computed by integrating the power spectrum in the following frequency windows: δ band (1–4 Hz), θ band (θ1: 4–6.5 Hz, θ2: 6.5–8 Hz), α band (α1: 8–11, α2: 11–14 Hz), β band (14–32), and γ band (γ1: 32–48; γ2: 52–100 Hz). Next, the spectral power values of consecutive 4-second epochs were averaged, and drug-induced changes in EEG oscillations were calculated in blocks of 15 minutes for 2 hours as the ratio of mean spectral power obtained following the administration of test drug versus the mean spectral power obtained during the 30-minute baseline period. This procedure allows for assessment of compound-induced changes in EEG power expressed at each frequency band as a percentage of the original power, compared with the vehicle condition.
Data Analysis.
All computerized nonlinear regression was performed using Prism 5.01 (GraphPad Software, San Diego, CA). Agonist and PAM concentration-response curves were analyzed using either a three- or four-parameter logistic equation as appropriate to determine estimates of potency (pEC50) and maximal agonist effect (Emax).
Shifts of glutamate concentration-response curves by allosteric modulators were globally fitted to an operational model of allosterism (Leach et al., 2007):
 |
(1) |
where A is the molar concentration of orthosteric agonist glutamate; B is the molar concentration of the allosteric modulator; KA is the equilibrium dissociation constant of the orthosteric agonist, glutamate; and KB is the allosteric modulator equilibrium dissociation constant. Affinity modulation is governed by the cooperativity factor α, and efficacy modulation is governed by β, where values >1 describe positive cooperativity, values <1 (but greater than 0) denote negative cooperativity, and values equal to 1 denote neutral cooperativity. The parameters τA and τB relate to the ability of the orthosteric and allosteric ligands, respectively, to engender receptor activation. Em and n denote the maximal possible system response and the transducer function that links occupancy to response, respectively. Unless otherwise stated all parameters were derived from global fitting glutamate concentration-response curves in the absence and presence of allosteric modulators.
An alternative, simplified, version of this operational model was applied to estimate a composite cooperativity parameter (αβ) for PAMs (Leach et al., 2010):
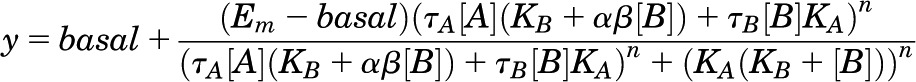 |
(2) |
where basal denotes the baseline level (ligand-independent) of the system response and all other parameters are as described above for eq. 1.
To derive estimates of neutral ligand affinity from translocation of PAM concentration-response curves, a competitive model (1) was applied as the Schild slope (s) was not significantly different to unity over the neutral allosteric ligand concentration range used
 |
(3) |
where bottom and top are the lower and upper plateaus, respectively, of the PAM concentration response curve; A is the molar concentration of PAM; B is the molar concentration of neutral modulator; and EC50 is the molar concentration of PAM required to generate a response halfway between the top and bottom. pA2 is the negative logarithm of the molar concentration of antagonist that requires a 2-fold higher concentration of PAM to achieve the same response. The Schild slope factor is represented by s (Arunlakshana and Schild, 1959). If s was found to be not significantly different from 1, then it was constrained to equal 1 and the resulting pA2 value is equal to the pKB (the negative logarithm of the KB).
Inhibition-binding data sets were fitted to a one-site inhibition binding model. Estimates of inhibitor dissociation constants (KI) were derived using the Cheng-Prusoff equation (Cheng and Prusoff, 1973).
All affinity, cooperativity, and potency parameters were estimated as logarithms and are expressed as the mean ± S.E.M. (Christopoulos, 1998). For all behavioral studies, statistical analyses were performed using one-way analysis of variance (ANOVA) or two-way ANOVA with a Dunnett’s post-test, and statistical significance was taken as P < 0.05. Sleep/wake EEG data were analyzed by a nonparametric analysis of variance of each 30-minute period, followed by a Wilcoxon-Mann–Whitney rank sum test of comparisons with the control group. Time course EEG spectral changes were submitted to two-way multivariate ANOVA for repeated measures with two main factors (treatment and period of recordings), followed by a one-way ANOVA with main factor treatment of each of the 15-minute periods. Plots for behavior studies were executed using SigmaPlot 11 (Systat Software, Inc. Chicago, IL) with statistical analyses performed using the JMP version 8 (SAS Institute Inc., Cary, NC).
Results
Characterization of Molecular Pharmacology of N-aryl Piperazine PAMs.
We recently reported the discovery of VU0364289, an N-aryl piperazine mGlu5 PAM, which evolved from derivatization of a high throughput screening mGlu5 negative allosteric modulator lead (Zhou et al., 2010). First, we sought to definitively characterize the pharmacology of VU0364289 along with DPFE, a structural analog disclosed in a patent application by AstraZeneca (Slassi et al., 2007) (Fig. 1A). Exposure of HEK293A cells stably expressing the rat mGlu5 receptor to glutamate results in mobilization of intracellular Ca2+, characterized by an initial transient peak followed by a sustained phase. Both DPFE and VU0364289 dose-dependently potentiated the peak response (Fig. 1B) and plateau phase (Supplemental Fig. 2) of Ca2+ mobilization in response to glutamate. PAM concentration-response curves performed in the presence of a low concentration of glutamate (∼EC25) yielded pEC50 values of 6.92 ± 0.15 (0.12 µM) and 6.35 ± 0.12 (0.45 µM) for DPFE and VU0364289, respectively. Both compounds maximally potentiated the EC25 glutamate peak response and, in the absence of glutamate, partial agonism was observed at 30 µM and 100 µM only (Fig. 1C). DPFE was assessed for its ability to enhance glutamate potency in both low and high mGlu5-expressing cell lines (Fig. 1, D and E). In line with the data reported for VU0364289 (Gregory et al., 2012), in the low-expressing cell line, this interaction was characterized by a progressive leftward shift that approached a limit, with no change in the maximal response (Fig. 1D). DPFE possessed a higher degree of cooperativity with glutamate than VU0364289, inducing 17.2 ± 5.1–fold compared with 7.3 ± 0.1–fold leftward shifts of the glutamate concentration response curve, respectively, at 30 µM. In the high-expressing HEK-mGlu5 cell line, DPFE also increased the Emax of glutamate as well as inducing a similar degree of leftward shift of the glutamate concentration-response curve (Fig. 1E). Analysis of these progressive fold-shift data utilizing an operational model of allosterism (Leach et al., 2007; Gregory et al., 2012) confirmed that DPFE has higher positive cooperativity (logβ) than VU0364289 (Table 1) and, similarly, low affinity for mGlu5 (5 µM). Furthermore, in agreement with previous findings for VU0364289 among other mGlu5 PAMs (Gregory et al., 2012), the assumption that affinity cooperativity was neutral (α = 1) had no impact on modulator affinity or cooperativity estimation, suggesting that DPFE, like VU0364289, elicits its potentiation primarily via efficacy modulation (Table 1).
Fig. 1.

Potentiation of glutamate-mediated Ca2+ mobilization by N-aryl piperazines. (A) Chemical structures of VU0364289 and DPFE. Changes in intracellular Ca2+ mobilization following exposure to modulators in the absence and presence of glutamate were investigated in HEK293A cells stably expressing rat mGlu5 using a Fluo-4 fluorescence-based Ca2+ assay. (B) Concentration-response curves for VU0364289 and DPFE to a low dose of glutamate (sub-EC30) are shown. (C) Response to VU0364289 and DPFE in absence of glutamate; concentration response curve to glutamate is shown (closed squares) for reference. HEK293A cells stably expressing rat mGlu5, at relatively low (D) and high (E) levels were exposed to different concentrations of DPFE (as indicated below figure) for 60 seconds prior to generation of a glutamate concentration-response curve for intracellular Ca2+ mobilization. Data represent the mean ± S.E.M. of four to eight independent experiments performed in duplicate. Error bars not shown lie within the dimensions of the symbol.
TABLE 1.
Summary of in vitro potency, affinity, and cooperativity parameters for N-aryl piperazine PAMs
| mGlu5-low (Ca2+) |
mGlu5-high (Ca2+) |
mGlu5-low (pERK1/2) |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| pEC50a | pKBb,c | Logαβb,c | pKBb,d | logβd | pKBb d | logβd | pKBb,d | logβd | |
| DPFE | 6.92 ± 0.15 (0.12 μM) | 5.32 ± 0.08 (4.8 μM) | 1.12 ± 0.11 | 5.33 ± 0.07 (4.7 μM) | 1.14 ± 0.05 | 5.99 ± 0.12 (1.0 μM) | 0.94 ± 0.09 | 6.87 ± 0.09 (135 nM) | 0.12 ± 0.02 |
| VU0364289 | 6.35 ± 0.12e (0.45 μM) | 5.18f (6.6 μM) | 0.97f | 5.22f (6.0 μM) | 0.90# | 5.83f (1.5 μM) | 0.78f | 6.29f (513 nM) | 0.16f |
Negative logarithm of the EC50.
Modulator affinity and cooperativity estimates derived using eq. 2, where a composite cooperativity parameter (logαβ) is derived.
Negative logarithm of the allosteric modulator equilibrium dissociation constant.
Modulator affinity and efficacy cooperativity (β) estimate derived using eq. 1, with the assumption that α = 1.
VU0364289 potency previously reported in Gregory et al., 2012.
Data from Gregory et al, 2012; provided here for benefit of comparison.
These N-aryl piperazines were also assessed in a second assay of mGlu5 activation, namely, phosphorylation of ERK1/2 (pERK1/2). In the HEK293-mGlu5 low-expressing cell line, pERK1/2 levels transiently increase in response to glutamate, peaking at 7 minutes and returning to baseline levels by 15 minutes (Fig. 2A). In contrast to the Ca2+ mobilization assay, at 3 µM, both VU0364289 and DPFE acted as agonists in inducing pERK1/2, displaying a similar time course to glutamate (Fig. 2A). As agonists, both PAMs showed greater efficacy than glutamate with respect to pERK1/2 (Fig. 2B). Commensurate with previous reports for VU0364289 (Gregory et al., 2012), DPFE also potentiates glutamate-induced pERK1/2, shifting the concentration-response curve to the left and increasing the Emax (Fig. 2C). Application of the operational model of allosterism (Eq. 1) to these data reveals that, compared with the Ca2+ assay data, both VU0364289 and DPFE have higher affinity estimates and lower cooperativity values for pERK1/2 (Table 1).
Fig. 2.

mGlu5 PAMs potentiate Glu-mediated pERK1/2 and show robust agonist activity. (A) In HEK293A cells expressing low levels of mGlu5, 1 mM glutamate, 3 µM DPFE or 3 µM VU0364289 transiently increase levels of phosphorylated ERK1/2, peaking at 7 to 8 minutes and returning to baseline by 15 minutes, while vehicle has no effect. (B) Concentration response-curves reveal that DPFE and VU0364289 are more efficacious agonists than glutamate. (C) Increasing concentrations of DPFE raise baseline levels of pERK1/2 and shift the glutamate concentration-response curve to the left. Data represent the mean ± S.E.M. of three to six independent experiments performed in triplicate. Error bars not shown lie within the dimensions of the symbol.
N-Aryl Piperazine PAMs Are Competitive with the Neutral Allosteric Ligand 5MPEP.
Given that these PAMs are based on a structurally distinct chemical scaffold to those previously identified, we were interested in whether they interacted with the common allosteric site on mGlu5 to which prototypical allosteric ligands, such as MPEP and the 3-cyano-N-(1,3-diphenyl-1H-pyrazol-5-yl)benzamide (CDPPB) series of PAMs, are known to bind. We first assessed the ability of the neutral allosteric ligand 5MPEP to shift the PAM concentration-response curve in the presence of a low concentration of glutamate (Fig. 3). 5MPEP alone had no effect on the response to glutamate; however, induced parallel rightward shifts of both VU0364289 (Fig. 3A) and DPFE (Fig. 3B) concentration response curves with no change in the Emax. This is consistent with a competitive mode of interaction between 5MPEP and these N-aryl piperazine PAMs. Furthermore, Schild regression of PAM EC50 values in the absence and presence of 5MPEP yielded Schild slopes not significantly different from unity (Fig. 3C). Direct application of a competitive model to these data gave similar affinity estimates for 5MPEP, pKB = 6.17 ± 0.11 (versus DPFE; 679 nM) and 6.10 ± 0.11 (versus VU0364289; 802 nM), consistent with those previously reported (Rodriguez et al., 2005).
Fig. 3.

5MPEP inhibition of N-aryl piperazine potentiation of glutamate. HEK293A cells stably expressing rat mGlu5 were exposed to different concentrations of 5MPEP for 60 seconds prior to addition of a concentration range of PAM followed by addition of a low dose of glutamate (sub-EC30). (A and B) Rightward shifts in potentiator concentration-response curves caused by co-addition of 5MPEP. (C) Schild regression of changes in VU0364289 (closed circles) and DPFE (open circles) concentration-response curve EC50 values in presence of 5MPEP. Data represent the mean ± S.E.M. of four to six independent experiments performed in duplicate. Error bars not shown lie within the dimensions of the symbol.
N-Aryl Piperazine PAMs Interact with the Common Allosteric (MPEP-Binding) Site.
As confirmation of a competitive mode of interaction with the common allosteric site of mGlu5, the effect of two point mutations was examined on the ability of these PAMs to potentiate the response to glutamate (Fig. 4, A, C, and E). In agreement with previous studies, substitution of Ala809 for Val (A809V) at the top of TM7 significantly reduces glutamate potentiation by VU29 (a CDPPB series PAM) and inhibition by MPEP (Fig. 4B; Table 2). This mutation also abolished [3H]methoxyPEPy-specific binding up to 30 nM (data not shown). At A809V, potentiation of glutamate by 3 µM DPFE and VU0364289 was decreased 3-fold compared with wild-type (Table 2). In TM1, F585I decreases potentiation by N-[4-chloro-2-[(1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)methyl]phenyl]-2-hydroxybenzamide but has no effect on PAMs interacting with the common allosteric “MPEP” site such as VU29 and the nicotinamide and picolinamide acetylenes (Chen et al., 2007; Rodriguez et al., 2012; Gregory et al., 2012, 2013) (Fig. 4E; Table 2). Consistent with the hypothesis that these N-aryl piperazines interact with the common allosteric site, F585I had no effect on the ability of DPFE or VU0364289 to potentiate glutamate responses (Fig. 4C).
Fig. 4.

Effect of point mutations on mGlu5 allosteric modulators. Polyclonal HEK293A cells stably expressing rat mGlu5 wild-type and mutant constructs were exposed to a single concentration of allosteric modulator prior to generation of concentration-response curves for glutamate-mediated Ca2+ mobilization. (A and B) Glutamate concentration-response curves at the wild-type receptor in the absence and presence of modulators. (C and D) Glutamate concentration-response curves at mGlu5-A809V in the absence and presence of modulators. (E and F) Glutamate concentration-response curves at mGlu5-F585I in the absence and presence of modulators. In all panels the concentration-response curve for glutamate in the absence of modulator is shown in closed circles, in the presence of 3 µM VU0364289 (closed squares), 3 µM DPFE (open squares), 3 µM N-[4-chloro-2-[(1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)methyl]phenyl]-2-hydroxybenzamide (closed triangles), 3 µM VU29 (open triangles), and 10 nM MPEP (open circles). Data represent the mean ± S.E.M. of four to eight independent experiments performed in duplicate. Error bars not shown lie within the dimensions of the symbol.
TABLE 2.
Effect of mutations on potentiation of glutamate-mediated Ca2+ mobilization by allosteric modulators
Data represent mean ± S.E.M. of at least four independent experiments performed in duplicate.
| Fold Increase in Glutamate pEC50 in Presence of 3 µM PAM |
||||
|---|---|---|---|---|
| CPPHA | VU29 | DPFE | VU0364289 | |
| mGlu5-wt | 3.2 ± 0.1 | 6.1 ± 1.4 | 7.7 ± 0.8 | 4.2 ± 0.7 |
| F585I | 1.2 ± 0.1* | 11.2 ± 4.8 | 8.8 ± 2.1 | 5.6 ± 1.3 |
| A809V | 2.8 ± 0.4 | 2.0 ± 0.3* | 2.6 ± 0.2* | 1.3 ± 0.1* |
Significantly different from mGlu5-wt value, P < 0.05, one-way ANOVA, Dunnett’s post-test.
These N-aryl piperazine PAMs were also investigated for their ability to inhibit [3H]methoxyPEPy binding. In preliminary studies, VU0364289 did not fully displace [3H]methoxyPEPy, with ∼49% specific binding remaining at 30 µM (Zhou et al., 2010); however, when conducted under different experimental conditions (in Ca2+ assay buffer with 1% DMSO final), 25.4 ± 2.5% specific binding remained in the presence of 100 µM VU0364289 (Gregory et al., 2012). Under these same conditions, 100 µM DPFE almost fully displaced the radioligand with only 9.7 ± 1.2% specific binding remaining (Fig. 5). Assuming a competitive interaction with [3H]methoxyPEPy, the apparent affinity of DPFE, pKi = 5.10 ± 0.08 (Ki = 8 µM), agreed well with functional estimates from progressive fold-shift assays (Table 1).
Fig. 5.

Inhibition of [3H]methoxyPEPy binding by N-aryl piperazines in comparison with MPEP. Membranes prepared from HEK293A cells stably expressing rat mGlu5 were incubated with 1–3 nM [3H]methoxyPEPy in the absence and presence of a range of concentrations of MPEP, VU0364289, or DPFE for 1 hour at room temperature. VU0364289 and MPEP data previously reported (Gregory et al., 2012) are provided for reference. Data represent the mean ± S.E.M. of three to four independent experiments performed in triplicate. Error bars not shown lie within the dimensions of the symbol.
DPFE and VU0364289 Selectively Potentiate Activation of mGlu5.
Both of these N-aryl piperazine PAMs were also assessed for their ability to modulate glutamate activity of the other seven mGlu subtypes (preliminary data reported for VU0364289 in Zhou et al., 2010). For all subtypes, the potential for modulation of the glutamate concentration response curve was assessed at 10 µM of either PAM, with the exception of mGlu7, where L-AP4 was used as the agonist. Neither VU0364289 nor DPFE significantly modulated agonist activity at the other seven mGlu subtypes (Fig. 6). To investigate their propensity to interact with other receptors and ion channels, these PAMs were also examined for their ability to inhibit radioligand binding at 68 other targets. At 10 µM, DPFE only displayed significant inhibition (∼70%) at the σ1 receptor. VU0364289, at 10 µM, only caused significant inhibition (∼75%) at the 5-HT2B receptor (Supplemental Fig. 3).
Fig. 6.

Selectivity of N-aryl piperazines across other mGlu subtypes. A single concentration (10 µM) of VU0364289 (closed squares) and DPFE (open squares) was assessed for modulation of orthosteric agonist activity (closed circles) (glutamate at all except mGlu7 where L-AP4 was used) across the other seven mGlu subtypes. Activity at mGlu1 was investigated using fluo-4 fluorescence-based measurement of Ca2+ mobilization. Activity at all other subtypes was assessed by a fluorescence-based assay of thallium-flux through GIRK channels (Niswender et al., 2008). Data represent the mean ± S.E.M. of three independent experiments performed in triplicate, with the exception of mGlu1 where the mean of two independent experiments are shown. Error bars not shown lie within the dimensions of the symbol.
Pharmacokinetic and Behavioral Toxicity Profiles of VU0364289 and DPFE.
Table 3 summarizes the in vitro and in vivo pharmacokinetic parameters of VU0364289 and DPFE. Free fraction of a compound in the presence of plasma proteins and the stability of a compound in liver microsomes are important qualities in a desirable drug metabolism and pharmacokinetic profile. DPFE and VU0364289 were tested for plasma protein binding in the presence of rat plasma and were found to exhibit favorable free fraction with 96% and 90% bound, respectively. In tests for stability in rat and human liver microsomes, there was 26% and 58%, respectively, of parent compound remaining after a 15-minute incubation period with DPFE; similar experiments with VU0364289 using a 60-minute incubation protocol (Zhou et al., 2010) revealed less than 5% of parent remaining in both rat and human liver microsomes. In vivo pharmacokinetic parameters of DPFE and VU0364289 were studied in rats following administration of 10 mg/kg i.p. in 20% HPBCD solution. At different time points, including 0.25, 0.5, 1, 3, and 6 hours after dosing, the concentrations of DPFE and VU0364289 were measured in systemic plasma (trunk blood) and whole-brain tissues. Both compounds were rapidly and significantly absorbed in rats as evident from systemic plasma concentrations (Fig. 7, A and B). The Cmax of both compounds was achieved in systemic plasma within 0.25 hour of dosing with values of 1093 ng/ml (∼3.0 µM) and 1,280 ng/ml (∼3.8 µM) for DPFE and VU0364289, respectively. Both compounds were characterized by excellent central nervous system penetration with area under the curve brain/area under the curve systemic plasma ratios of 1.26 for DPFE and 1.09 for VU0364289. Modified Irwin neurologic battery revealed little evidence of acute toxicity at 30 mg/kg and 56.6 mg/kg DPFE or 56.6 mg/kg VU0364289 (Supplemental Table 1).
TABLE 3.
Summary of in vitro and in vivo pharmacokinetic parameters
| VU0364289 | DPFE | |
|---|---|---|
| Cmax (ng/ml) | 1280 | 1093 |
| Tmax (hr) | 0.25 | 0.25 |
| Plasma protein binding | 94% (h); 90% (r) | 97% (h); 96% (r) |
| Rat Fu (free fraction) | 0.10 | 0.04 |
Fig. 7.

In vivo pharmacokinetic profiles of VU0364289 (panel A) and DPFE (panel B). Data represent mean determinations from two animals.
DPFE and VU0364289 Reverse Amphetamine-Induced Hyperlocomotion.
The ability of a compound to reverse amphetamine-induced hyperlocomotion in rodents, accomplished by both atypical and typical antipsychotics, is considered a predictive value of the antipsychotic efficacy of a compound (Kinney et al., 2003). Previous studies have established efficacy in this behavioral model for mGlu5 PAMs (Kinney et al., 2005; Liu et al., 2008a; Liu et al., 2008b). Administration of 56.6 and 100 mg/kg i.p. DPFE significantly reversed amphetamine-induced hyperlocomotion, with ∼60% fewer ambulations observed, over the entire time course of the study (Fig. 8A). This reversal was dose-dependent, with animals receiving 30 mg/kg i.p. DPFE in conjunction with amphetamine (1 mg/kg s.c.) showing significantly fewer ambulations than controls between the 80- and 95-minute time points only. Administration of VU0364289 also dose-dependently reversed amphetamine-induced hyperlocomotor activity (Fig. 8B). Animals receiving both the 56.6 and 100 mg/kg doses of VU0364289 in conjunction with amphetamine showed significantly fewer ambulations, achieving a similar level of reversal as DPFE.
Fig. 8.

VU0364289 and DPFE dose-dependently reversed amphetamine-induced hyperlocomotion. Rats were placed in the open-field chambers for 30-minute habituation intervals, prior to pretreatment with vehicle or indicated dose of test compound in 20% HPBCD vehicle intraperitoneally for an additional 30 minutes. All rats then received an injection of 1 mg/kg s.c. amphetamine, and locomotor activity was measured for an additional 60 minutes. (A) VU0364289 produced a significant dose-dependent reversal of amphetamine-induced hyperlocomotion when administered intraperitoneally and had no effect when administered alone. (B) DPFE produced a significant reversal of amphetamine-induced hyperlocomotion and had no effect when administered alone. Data are expressed as mean ± S.E.M. of the number of beam breaks/5 minutes; error bars not shown lie within the dimensions of the symbol (n = 8–14 per dose). *P < 0.05 versus vehicle + amphetamine, Dunnett’s test.
DPFE Does Not Reverse Apomorphine-Induced Disruption of PrePulse Inhibition.
The potential antipsychotic profile of DPFE was further profiled using prepulse inhibition disrupted with apomorphine (Fig. 9). As shown in Fig. 9A, the atypical antipsychotic, risperidone (3 mg/kg) significantly reversed apomorphine-induced disruption of prepulse inhibition to the acoustic startle reflex at 10 decibels (db) and 15db. In comparison, DPFE (30–100 mg/kg p.o.) did not significantly reverse apomorphine-induced disruption of prepulse inhibition. Further, neither risperidone nor DPFE influenced the apomorphine-induced decrease in startle amplitude (Fig. 9B). These data provide a point of deviation from the preclinical profile reported for another mGlu5 PAM, ADX47273 (Schlumberger et al., 2009). It is important to note that the oral route of administration may have reduced in vivo exposure to DPFE contributing to its lack of efficacy in this model.
Fig. 9.

DPFE does not reverse apomorphine-induced disruption of prepulse inhibition. (A) The effect of DPFE (30 , 56.6 , 100 mg/kg) on apomorphine-induced disruption of prepulse inhibition of the acoustic startle reflex was assessed in rats in parallel with the positive control risperidone (3 mg/kg). Data are expressed as changes in the % inhibition. (B) The startle amplitude was not significantly different for any of the treatment groups. Bars represent the mean ± S.E.M. value for six to seven rats per dose group. *P ≤ 0.05 versus vehicle/apomorphine group, Dunnett’s test.
DPFE Enhances the Acquisition of Contextual Fear Conditioning.
Contextual fear conditioning is a form of Pavlovian classic conditioning that is mediated by the hippocampus (Phillips and LeDoux, 1992). As shown in Fig. 10, pretreatment with DPFE (0.1–0.56 mg/kg i.p.) produced dose-related enhancement in the acquisition of contextual fear conditioning, significant at the 0.56 mg/kg dose (F3,17 = 4.95, P < 0.01; *P < 0.01). In a separate experiment, pretreatment with DPFE (1 mg/kg i.p.) had no effect on the threshold of sensitivity to the footshock stimulus as compared with vehicle treatment group (data not shown).
Fig. 10.

DPFE enhances acquisition of contextual fear conditioning. Pretreatment with DPFE (0.1–0.56 mg/kg i.p.) produced dose-related enhancement in the percentage of freezing behavior during the recording period, significant at the 0.56 mg/kg dose (F3,17 = 4.95 P < 0.01; *P < 0.01). Bars represent the mean ± S.E.M value for five to six rats per group.
Investigation of DPFE Efficacy in a Rodent Model of NMDA Receptor Hypofunction.
Knockdown of the NR1 subunit of NMDAR in mice (NR1KD) provides a genetic model of NMDAR hypofunction. These mice have 10% of normal NMDA receptor function and display hyperlocomotor activity in comparison with wild-type mice (Mohn et al., 1999). Administration of 30 mg/kg DPFE significantly reduced ambulatory counts in transgenic mice compared with vehicle-treated NR1KD animals (Fig. 11A). In addition, the number of ambulations of NR1KD mice receiving 56.6 mg/kg DPFE was significantly lower at all time points from 20 to 115 minutes, excluding the 45-minute point. Further, wild-type animals treated with vehicle or DPFE were not significantly different at any time point. NR1KD mice also show impairments in working memory (Y-maze; Fig. 11B) and social interactions (Fig. 11C). Compared with littermate controls, NR1KD mice traveled greater distances (12,209 ± 436 cm versus 8532 ± 324 cm) and had increased arm entries (51.8 ± 4.5 versus 28.7 ± 0.9) during the course of the Y-maze task. Administration of 10 mg/kg DPFE slightly increased arm entries and distance traveled by both groups (wild-type: 40.9 ± 3.2 entries, 10,587 ± 523 cm; NR1KD: 65.3 ± 5.1 entries, 13,286 ± 407 cm); however, this only reached significance for the wild-type animals. Cognitive performance in this task is assessed by the number of spontaneous alternations, treatment with 10 mg/kg DPFE had no significant effect on Y-maze performance. Social investigation by NR1KD mice and littermate controls was not influenced by administration of 10 mg/kg DPFE.
Fig. 11.

DPFE produced dose-dependent reversal of hyperlocomotion in NR1 knockdown mice. (A) Mice were administered vehicle or a 30 or 56.6 mg/kg i.p. dose of test compound and placed in the open-field chambers where locomotor activity was monitored for 120 minutes. DPFE at both doses produced a significant reversal of hyperlocomotion in −/− mice. Wild-type treatment groups did not differ significantly from wild-type vehicle groups. Data are expressed as mean ± S.E.M. of the number of beam breaks/ 5 minutes; error bars not shown lie within the dimensions of the symbol (n = 6–12 per dose). *P < 0.05 versus vehicle, Dunnett’s test. (B) NR1 knockdown mice have significantly impaired performance in the Y-maze task compared with littermates. Administration of DPFE (10 mg/kg) did not significantly restore performance of these animals. Data are expressed as mean ± S.E.M. percentage of spontaneous alternation (%SAP) of n = 10–13 mice. *P < 0.05 versus vehicle, unpaired t test. (C) NR1 knockdown mice have significantly impaired social interactions compared with littermates. Administration of DPFE (10 mg/kg) did not significantly restore performance of these animals. Data are expressed as mean ± S.E.M. of time spent investigating of n = 10–13 mice.
Effect of DPFE on Neurotransmitter Levels in Key Brain Regions.
Monoamine neurotransmitter levels were assessed in the nucleus accumbens and prefrontal cortex of male rats following dosing with DPFE using microdialysis (Fig. 12). Dose-dependent increases in PFC monoamine levels were observed. Administration of 10 mg/kg p.o. DPFE significantly increased PFC norepinephrine levels to 200% of control, while both 30 and 100 mg/kg p.o. doses elevated norepinephrine, dopamine and serotonin levels to ∼300–500% above baseline. These increases occurred in the first hour and were sustained up to 4 hours post dosing with DPFE. In the NAC, norepinephrine and serotonin, but not dopamine, levels were also significantly increased (∼300% over baseline levels), although only at the highest doses.
Fig. 12.

In vivo microdialysis in the nucleus accumbens and prefrontal cortex. Sprague-Dawley rats were orally administered DPFE at dose 0 (vehicle: ●, n = 11), 10 (▼, n = 12), 30 (◆, n = 13), and 100 (▲, n = 9) mg/kg in 20% HPBCD + Tween. Norepinehrine (top row), dopamine (middle row), and serotonin (bottom row) levels were determined from brain dialysates collected in 30-minute samples sampled from nucleus accumbens (left column) and prefrontal cortex (right column). *P < 0.05 versus vehicle, ANOVA of area under the curve (AUC). Data are expressed as median ± S.E.M. of percentage change of mean individual baseline.
DPFE Elicits Changes in Sleep/Wake Architecture and pEEG.
Acute treatment with DPFE exerted several effects on the sleep-wake architecture of rats (Fig. 13, line graphs). Most notably, administration of 30 mg/kg and 100 mg/kg DPFE significantly and dose-dependently increased time spent in passive wakefulness during the first 4-hours post administration (Fig. 13, inset bar graphs). This increase in passive waking persisted throughout the light phase and for the highest dose protruded into the dark phase more than 10 hours after administration at the expense of active waking; a significant increase in passive waking over the total recording time (20 hours) was consequently also found after 30 and 100 mg/kg compared with vehicle treatment.
Fig. 13.

Effects of DPFE on sleep/wake architecture of rats. The impact of oral administration of vehicle 20% HPBCD + Tween 20 (black) or DPFE: 10 mg/kg (green); 30 mg/kg (blue); or 100 mg/kg (red) with n = 8 for each treatment condition during the light phase on time spent in six different sleep-wake states (A) through (F). Line graphs indicate state amounts expressed as percentage of time spent in state (Y-axis) for consecutive 30-minute periods over 20-hour postadministration (x-axis); inset bar graphs indicate mean ± S.E.M. time (min) spent in state during the first 4 hours after administration. *Significantly different from vehicle (P < 0.05), Wilcoxon-Mann–Whitney rank sum test.
Furthermore, in the pEEG study dose-dependent effects DPFE were observed on θ1 and α1 oscillations as measured during the active (dark) phase (Fig. 14). Higher frequencies, especially in the γ bands, were largely unaffected. Effects were most pronounced in the parietal and occipital areas as compared with more frontal areas of the cortex (see Fig. 14; frontal versus occipital comparison), where transduction of underlying hippocampal neuronal activity is less pronounced.
Fig. 14.

Effect of DPFE on pharmaco-electroencephalographic oscillations. Effects of acute oral administration of vehicle: 20% HPBCD + Tween (row A), 10 mg/kg (row B), 30 mg/kg (row C), or 100 mg/kg (row D) of DPFE on pEEG relative spectral power in the frontal (left panels) and occipital (right panels) cortical regions in rats (n = 8 for each treatment group). Only effects on left hemispheric recordings are shown, as no clear differences were detected compared with the contralateral (right hemisphere) traces. Bar charts indicate average percentage changes from baseline for each EEG spectral band for eight consecutive 15-minute postadministration periods. Where δ:1.0–4.0 Hz; θ1: 4.0–6.5 Hz; θ2:6.5–8.0 Hz; α1:8.0–11.0 Hz; α2: 1.0–14.0 Hz; β1: 14.0–18.0 Hz; β2: 18.0–32.0 Hz; γ1: 32.0–48.0 Hz; γ2: 52.0–100 Hz. Values exceeding the 20% change line may be considered significantly different from vehicle (P < 0.05).
Discussion
The development of selective mGlu5 PAMs has advanced as an important potential therapeutic strategy for the treatment of schizophrenia through the facilitation of forebrain glutamatergic signaling thought to be disrupted in this disorder. Herein, we described the in vitro and behavioral characterization of two N-aryl piperazine mGlu5 PAMs, DPFE and VU0364289, both found to be selective, cooperativity-driven mGlu5 PAMs that interact with the common MPEP allosteric site on mGlu5. These N-aryl piperazines showed improved solubility and pharmacokinetic properties, efficacy in a preclinical model predictive of antipsychotic-like activity, and enhancement of dopamine release in the prefrontal cortex without producing behavioral toxicity. Moreover, DPFE enhanced acquisition of a hippocampal-mediated memory task and elicited changes in sleep/wake architecture consistent with a procognitive pharmacologic profile. Finally, we provide the first report of reversal of behavioral deficits in NR1 KD mice, a genetic model of NMDAR hypofunction, using the mGlu5 PAM DPFE. Collectively, these findings provide strong support for the hypothesis that selective mGlu5 activation may serve as a novel approach for the treatment of psychotic symptoms and cognitive deficits in patients with schizophrenia.
Rigorous pharmacologic approaches were applied to probe the mechanism of action of DPFE and VU0364289. While DPFE and VU0364289 have similar affinity for mGlu5, DPFE displays ∼2-fold higher cooperativity, which is greater than several reported PAM scaffolds (Kinney et al., 2005; Chen et al., 2008; Liu et al., 2008b; Vanejevs et al., 2008; Hammond et al., 2010; Rodriguez et al., 2010; Gregory et al., 2012; Gilmour et al., 2013). In agreement with our recent findings, potentiation by these N-aryl piperazine PAMs was attributable to a mechanism solely involving glutamate efficacy modulation. Furthermore, DPFE agonism for Ca2+ and pERK1/2 displayed a similarly biased agonist profile to that recently reported for other mGlu5 agonist-PAMs (Gregory et al., 2012). The implications of these differences between mGlu5 allosteric versus orthosteric agonism are yet to be determined. Given that DPFE and VU0364289 exhibit low affinity for mGlu5 (≥8 µM), we conclude their pharmacologic effect is largely driven via cooperativity.
Despite being from a distinct chemical scaffold, DPFE and VU0364289 interact with the common allosteric binding site on mGlu5. Specifically, interactions with the neutral allosteric ligand 5MPEP were consistent with competition and potentiation by both PAMs was decreased at the A809V mutant receptor, a mutation known to impact “MPEP-site” modulators (Gregory et al., 2013). Both compounds also displaced [3H]methoxyPEPy, although this was incomplete over the concentration range tested, likely attributable to low compound affinity. Indeed, another PAM from this same scaffold, the lead compound (2b) (also referred to as CPPZ) with an ∼3-fold higher affinity than DPFE, fully displaced [3H]MPEP (Xiong et al., 2010; Spear et al., 2011). Together these data illustrate that the common mGlu5 allosteric site can accommodate a diverse array of chemical scaffolds that show not only subtype selectivity but diverse pharmacology.
Both DPFE and VU0364289 produced robust dose-related reversals of amphetamine-induced hyperlocomotion, a preclinical model predictive of antipsychotic-like activity, consistent with a potential role for selective mGlu5 PAMs in the treatment of the positive symptoms in schizophrenia. Our findings with DPFE and VU0364289 confirm and extend previous studies with earlier generation mGlu5 PAMs, including CDPPB, ADX47273, and CPPZ (Kinney et al., 2005; Liu et al., 2008b; Schlumberger et al., 2009; Rosenbrock et al., 2010; Spear et al., 2011). In contrast, more recent studies using the mGlu5 PAMs LSN2463359 and LSN2814617 reported only marginal antipsychotic-like effects (Gilmour et al., 2013). Although the reasons for the apparent differences in antipsychotic-like activity across the different chemical classes of mGlu5 PAMs are not well understood, there are clear differences at the molecular level in the actions of the N-aryl piperazine mGlu5 PAMs relative to the LSN mGlu5 PAMs. While both DPFE and VU0364289 are less potent than either LSN PAM, they have higher cooperativity with glutamate as evidenced by a ≥ 10-fold leftward shift in the glutamate concentration response curve as compared with a 2–3-fold shift for LSN mGlu5 PAMs (Gilmour et al., 2013). This cooperativity difference is also reflected in the PAM potency curves Emax values; potentiation by DPFE and VU0364289 reached the glutamate Emax, while LSN mGlu5 PAMs achieved only 65–79%. Such differences suggest that the efficacy of DPFE and VU0364289 is cooperativity-driven in nature, whereas LSN mGlu5 PAMs may rely more on affinity, since previous studies reported a clear link between receptor occupancy and efficacy (Gilmour et al., 2013).
Activation of mGlu5 directly potentiates NMDAR function and also has independent actions that increase excitatory transmission and synaptic plasticity in a manner similar to that of NMDAR activation in forebrain circuits (Herman et al., 2012). To date, most preclinical studies evaluating mGlu5 PAMs efficacy on NMDAR hypofunction have focused primarily on pharmacological challenge models using different NMDAR antagonists. Unfortunately, these studies reported varying degrees of efficacy in reversing the behavioral disruptions induced by different NMDAR antagonists, likely dependent on the antagonist mode of action (Liu et al., 2008b; Rosenbrock et al., 2010; Schlumberger et al., 2010; Gastambide et al., 2013; Gilmour et al., 2013). Thus, we evaluated the ability of DPFE to reverse the behavioral deficits in NR1KD mice, a genetic model of NMDAR hypofunction (Ramsey, 2009), to avoid potential confounds observed with NMDAR antagonist challenge models. NR1KD mice display an approximately 90% decrease in functional NMDARs in limbic and forebrain regions and corresponding behavioral alterations, including hyperlocomotor activity and performance deficits in social interaction and cognition (Dzirasa et al., 2009; Halene et al., 2009; Ramsey, 2009; Moy et al., 2012), comparable to those induced with NMDAR antagonists. Treatment with DPFE reversed the hyperlocomotor activity observed in the NR1KD mice similar to the effects observed with clinically available antipsychotics (Mohn et al., 1999). Sociability and performance deficits in Y-maze memory tasks were not improved at the 10 mg/kg dose of DPFE, suggesting higher doses may be required to normalize these cognitive deficits. Collectively, these data in the NR1KD mice provide independent validation for a role for selective mGlu5 PAMs in reversing behavioral disruptions resulting from NMDAR hypofunction.
Interestingly, DPFE enhanced release of monoamines and serotonin in the PFC, consistent with previously reported effects of the atypical antipsychotic risperidone (Gessa et al., 2000; Ohoyama et al., 2011). Importantly, increased extracellular DA and NE in the PFC are associated with the cognition-enhancing doses of methylphenidate and atomoxetine (Bymaster et al., 2002; Berridge et al., 2006). In contrast to the PFC effects, only 5-HT and NE levels were elevated in nucleus accumbens; DA levels were unchanged. Prior in vivo microdialysis studies using mGlu5 PAMs reported mixed outcomes in nucleus accumbens (e.g., ADX47273, Liu et al., 2008b), but no findings on PFC neurotransmitter changes. The lack of effect on extracellular levels of DA in the nucleus accumbens suggests a low potential for abuse liability with DPFE. In addition, both DPFE and VU0364289 were well tolerated after acute administration with a lack of acute toxicity in the modified Irwin neurologic test battery. To further understand the future utility of mGlu5 PAMs in the clinic, it will also be important to evaluate the effects of DPFE and VU0364289 after repeated dosing. Here, we provide the first report of enhanced PFC monoamine levels induced by selective mGlu5 enhancement, comparable in magnitude to clinically efficacious antipsychotics.
Facilitation of mGlu5 plays a critical role in normal learning and memory functions, suggesting mGlu5 PAMs may improve cognitive impairments associated with schizophrenia (Herman et al., 2012). While previous studies focused on mGlu5 PAM reversal of psychostimulant-induced deficits in cognition (i.e., Reichel et al., 2011; Gastambide et al., 2013), we evaluated the ability of DPFE to facilitate the hippocampal-mediated acquisition of contextual fear conditioning, a classic Pavlovian conditioning task (Phillips and LeDoux, 1992). Interestingly, DPFE enhanced acquisition of this cognitive task within a dose range significantly lower than that required to reverse amphetamine-induced hyperlocomotion, consistent with the previous report of lower doses of the mGlu5 PAM ADX47273 needed to enhance performance in another cognition model (Liu et al., 2008b). Further, DPFE dose-dependently elicited changes in rat sleep-wake architecture, primarily increasing passive wakefulness, mirroring the previous report that CDPPB increased wakefulness and light sleep, although no distinction was made between active and passive wake (Parmentier-Batteur et al., 2012). Moreover, the novel mGlu5 PAMs, LSN2463359 and LSN2814617, also increased wakefulness (Gilmour et al., 2013). A compensatory rebound in REM without a non-REM rebound was reported, indicating a lack of rebound hypersomnolence of mGlu5 PAMs. This observation is in line with the present study design, which further differentiated between changes in light and deep non-REM sleep. Interestingly, the initial deep non-REM sleep reduction was accompanied by a light non-REM increase and slight deep non-REM compensation during the later hours of the 20-hour recording period. In the pharmacoEEG study, enhanced θ1 synchrony and decreased α1 synchrony was observed in response to acute dosing with DPFE. Increased θ rhythm in the PFC in response to several antipsychotics has been reported (Sebban et al., 1999). Clinically effective antipsychotics elicit decreases in α1 and γ2 EEG synchrony. It is noteworthy that DPFE, despite its antipsychotic profile, did not induce changes in γ2 EEG synchrony, suggesting different potential mechanisms underlying the efficacy of mGlu5 PAMs.
In summary, DPFE and VU0364289 represent an exciting structural class of selective and potent cooperativity-driven mGlu5 PAMs with favorable pharmacokinetic properties and robust efficacy in several behavioral models at doses that did not produce adverse side effects as observed with other mGlu5 activators. Our findings provide further support for mGlu5 PAM development for the potential treatment of all three symptom clusters observed in individuals with schizophrenia.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge the technical expertise of Kiran K. Gogi, Daryl F. Venable, and Rocio Zamorano for performing the mGlu selectivity assays; Wendy Horsfall for assistance with behavioral tests on NR1 KD mice; Christ Nolten and Gerd Van Den Kieboom for support with microdialysis studies; and Heidi Huysmans and Sofie Versweyveld for support with EEG studies.
Abbreviations
- L-AP4
l(+)-2-amino-4-phosphonobutanoic acid
- 5-HT
serotonin
- 5MPEP
5-methyl-2-(phenylethynyl)pyridine
- AP
anteroposterior
- ASR
acoustic startle reflex
- ANOVA
analysis of variance
- CDPPB
3-cyano-N-(1,3-diphenyl-1H-pyrazol-5-yl)benzamide
- DA
dopamine
- DMEM
Dulbecco’s modified Eagle’s medium
- DPFE
1-(4-(2,4-difluorophenyl)piperazin-1-yl)-2-((4-fluorobenzyl)oxy)ethanone
- dSWS
deep non-REM/slow wave sleep
- DV
dorsoventral
- EEG
electroencephalogram
- EMG
electromyogram
- EOG
electrooculogram
- ERK1/2
extracellular signal-regulated kinases 1 and 2
- FBS
fetal bovine serum
- GIRK
G protein–coupled inwardly-rectifying potassium channels
- HEK
human embryonic kidney
- HPBCD
hydroxyl-propyl betacyclodextrin
- lSWS
light non-REM/slow wave sleep
- mGlu
metabotropic glutamate receptor
- ML
mediolateral
- MPEP
2-methyl-6-(phenylethynyl)-pyridine
- NAC
nucleus accumbens
- NE
norepinehrine
- NMDA
N-methyl-d-aspartate
- NMDAR
NMDA receptor
- NR1KD
NR1 knockdown
- PAM
positive allosteric modulator
- PCP
phencyclidine
- pEEG
pharmacoEEG
- pERK1/2
phosphorylated ERK1/2
- PFC
prefrontal cortex
- REM
paradoxical/rapid eye movement sleep
- VU0364289
2-(4-(2-(benzyloxy)acetyl)piperazin-1-yl)benzonitrile
Authorship Contributions
Participated in research design: Gregory, Herman, Jones, Drinkenburg, Mackie, Daigle, Caron, Weaver, Steckler, Lavreysen, Niswender, Jones, Conn.
Conducted experiments: Gregory, Herman, Hammond, Ramsey, Byun, Hrupka, Daigle, Jones, Jadhav, Bridges.
Contributed new reagents or analytic tools: Lindsley, Stauffer, Bartolomé, Macdonald, Manka
Performed data analysis: Gregory, Herman, Ramsey, Ahnaou, Drinkenburg, Byun, Bridges, Daigle, Weaver, Jones, Lavreysen.
Wrote or contributed to the writing of the manuscript: Gregory, Herman, Jones, Drinkenburg, Ramsey.
Footnotes
This work was supported by the National Institutes of Health National Institute of Mental Health [Grants R01 MH062646 and R01 MH073853]; National Institutes of Health National Institute on Drug Abuse [Grant F32 DA 030026]; and National Institutes of Health National Institute of Neurologic Disorders and Stroke [Grant 5R01 NS031373-15]; an industry-sponsored contract from Johnson & Johnson; a National Health and Medical Research Council (Australia) Overseas Biomedical Postdoctoral Training fellowship (to K.J.G.); an American-Australian Association Merck Co. Foundation fellowship (to K.J.G.); a NARSAD-Maltz Investigator Award (to K.J.G.); and the Canadian Institutes of Health [Grant 258294] (to A.J.R.).
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.
References
- Adrianzén C, Arango-Dávila C, Araujo DM, Ruíz I, Walton RJ, Dossenbach M, Karagianis J. (2010) Relative association of treatment-emergent adverse events with quality of life of patients with schizophrenia: post hoc analysis from a 3-year observational study. Hum Psychopharmacol 25:439–447 [DOI] [PubMed] [Google Scholar]
- American Psychiatric Association (2000) Diagnostic and Statistical Manual of Mental Disorders, Ed. 4th American Psychiatric Publishing, Washington, D.C. [Google Scholar]
- Arunlakshana O, Schild HO. (1959) Some quantitative uses of drug antagonists. Br Pharmacol Chemother 14:48–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala JE, Chen Y, Banko JL, Sheffler DJ, Williams R, Telk AN, Watson NL, Xiang Z, Zhang Y, Jones PJ, et al. (2009) mGluR5 positive allosteric modulators facilitate both hippocampal LTP and LTD and enhance spatial learning. Neuropsychopharmacology 34:2057–2071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge CW, Devilbiss DM, Andrzejewski ME, Arnsten AF, Kelley AE, Schmeichel B, Hamilton C, Spencer RC. (2006) Methylphenidate preferentially increases catecholamine neurotransmission within the prefrontal cortex at low doses that enhance cognitive function. Biol Psychiatry 60:1111–1120 [DOI] [PubMed] [Google Scholar]
- Bymaster FP, Felder C, Ahmed S, McKinzie D. (2002) Muscarinic receptors as a target for drugs treating schizophrenia. Curr Drug Targets CNS Neurol Disord 1:163–181 [DOI] [PubMed] [Google Scholar]
- Chen Y, Goudet C, Pin JP, Conn PJ. (2008) N-4-Chloro-2-[(1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)methyl]phenyl-2-hydroxybenzamide (CPPHA) acts through a novel site as a positive allosteric modulator of group 1 metabotropic glutamate receptors. Mol Pharmacol 73:909–918 [DOI] [PubMed] [Google Scholar]
- Chen Y, Nong Y, Goudet C, Hemstapat K, de Paulis T, Pin JP, Conn PJ. (2007) Interaction of novel positive allosteric modulators of metabotropic glutamate receptor 5 with the negative allosteric antagonist site is required for potentiation of receptor responses. Mol Pharmacol 71:1389–1398 [DOI] [PubMed] [Google Scholar]
- Cheng Y, Prusoff WH. (1973) Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol 22:3099–3108 [DOI] [PubMed] [Google Scholar]
- Christopoulos A. (1998) Assessing the distribution of parameters in models of ligand-receptor interaction: to log or not to log. Trends Pharmacol Sci 19:351–357 [DOI] [PubMed] [Google Scholar]
- Digby GJ, Noetzel MJ, Bubser M, Utley TJ, Walker AG, Byun NE, Lebois EP, Xiang Z, Sheffler DJ, Cho HP, et al. (2012) Novel allosteric agonists of M1 muscarinic acetylcholine receptors induce brain region-specific responses that correspond with behavioral effects in animal models. J Neurosci 32:8532–8544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzirasa K, Ramsey AJ, Takahashi DY, Stapleton J, Potes JM, Williams JK, Gainetdinov RR, Sameshima K, Caron MG, Nicolelis MA. (2009) Hyperdopaminergia and NMDA receptor hypofunction disrupt neural phase signaling. J Neurosci 29:8215–8224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gastambide F, Cotel MC, Gilmour G, O’Neill MJ, Robbins TW, Tricklebank MD. (2012) Selective remediation of reversal learning deficits in the neurodevelopmental MAM model of schizophrenia by a novel mGlu5 positive allosteric modulator. Neuropsychopharmacology 37:1057–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gastambide F, Gilmour G, Robbins TW, Tricklebank MD. (2013) The mGlu₅ positive allosteric modulator LSN2463359 differentially modulates motor, instrumental and cognitive effects of NMDA receptor antagonists in the rat. Neuropharmacology 64:240–247 [DOI] [PubMed] [Google Scholar]
- Gessa GL, Devoto P, Diana M, Flore G, Melis M, Pistis M. (2000) Dissociation of haloperidol, clozapine, and olanzapine effects on electrical activity of mesocortical dopamine neurons and dopamine release in the prefrontal cortex. Neuropsychopharmacology 22:642–649 [DOI] [PubMed] [Google Scholar]
- Gilmour G, Broad LM, Wafford KA, Britton T, Colvin EM, Fivush A, Gastambide F, Getman B, Heinz BA, McCarthy AP, et al. (2013) In vitro characterisation of the novel positive allosteric modulators of the mGlu₅ receptor, LSN2463359 and LSN2814617, and their effects on sleep architecture and operant responding in the rat. Neuropharmacology 64:224–239 [DOI] [PubMed] [Google Scholar]
- Green MF. (2006) Cognitive impairment and functional outcome in schizophrenia and bipolar disorder. J Clin Psychiatry 67:e12. [PubMed] [Google Scholar]
- Gregory KJ, Nguyen ED, Reiff SD, Squire EF, Stauffer SR, Lindsley CW, Meiler J, Conn PJ. (2013) Probing the metabotropic glutamate receptor 5 (mGlu5) positive allosteric modulator (PAM) binding pocket: Discovery of point mutations that engender a “molecular switch” in PAM pharmacology. Mol Pharmacol 83:991–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory KJ, Noetzel MJ, Rook JM, Vinson PN, Stauffer SR, Rodriguez AL, Emmitte KA, Zhou Y, Chun AC, Felts AS, et al. (2012) Investigating metabotropic glutamate receptor 5 allosteric modulator cooperativity, affinity, and agonism: enriching structure-function studies and structure-activity relationships. Mol Pharmacol 82:860–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halene TB, Ehrlichman RS, Liang Y, Christian EP, Jonak GJ, Gur TL, Blendy JA, Dow HC, Brodkin ES, Schneider F, et al. (2009) Assessment of NMDA receptor NR1 subunit hypofunction in mice as a model for schizophrenia. Genes Brain Behav 8:661–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond AS, Rodriguez AL, Townsend SD, Niswender CM, Gregory KJ, Lindsley CW, Conn PJ. (2010) Discovery of a novel chemical class of mGlu(5) allosteric ligands with distinct modes of pharmacology. ACS Chem Neurosci 1:702–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman EJ, Bubser M, Conn PJ, Jones CK. (2012) Metabotropic glutamate receptors for new treatments in schizophrenia. Handb Exp Pharmacol 213: 297–365 [DOI] [PubMed] [Google Scholar]
- Javitt DC. (2007) Glutamate and schizophrenia: phencyclidine, N-methyl-D-aspartate receptors, and dopamine-glutamate interactions. Int Rev Neurobiol 78:69–108 [DOI] [PubMed] [Google Scholar]
- Javitt DC, Zukin SR. (1991) Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry 148:1301–1308 [DOI] [PubMed] [Google Scholar]
- Jones CK, Brady AE, Davis AA, Xiang Z, Bubser M, Tantawy MN, Kane AS, Bridges TM, Kennedy JP, Bradley SR, et al. (2008) Novel selective allosteric activator of the M1 muscarinic acetylcholine receptor regulates amyloid processing and produces antipsychotic-like activity in rats. J Neurosci 28:10422–10433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones CK, Eberle EL, Shaw DB, McKinzie DL, Shannon HE. (2005) Pharmacologic interactions between the muscarinic cholinergic and dopaminergic systems in the modulation of prepulse inhibition in rats. J Pharmacol Exp Ther 312:1055–1063 [DOI] [PubMed] [Google Scholar]
- Kinney GG, Burno M, Campbell UC, Hernandez LM, Rodriguez D, Bristow LJ, Conn PJ. (2003) Metabotropic glutamate subtype 5 receptors modulate locomotor activity and sensorimotor gating in rodents. J Pharmacol Exp Ther 306:116–123 [DOI] [PubMed] [Google Scholar]
- Kinney GG, O’Brien JA, Lemaire W, Burno M, Bickel DJ, Clements MK, Chen TB, Wisnoski DD, Lindsley CW, Tiller PR, et al. (2005) A novel selective positive allosteric modulator of metabotropic glutamate receptor subtype 5 has in vivo activity and antipsychotic-like effects in rat behavioral models. J Pharmacol Exp Ther 313:199–206 [DOI] [PubMed] [Google Scholar]
- Krystal JH, Anand A, Moghaddam B. (2002) Effects of NMDA receptor antagonists: implications for the pathophysiology of schizophrenia. Arch Gen Psychiatry 59:663–664 [DOI] [PubMed] [Google Scholar]
- Leach K, Loiacono RE, Felder CC, McKinzie DL, Mogg A, Shaw DB, Sexton PM, Christopoulos A. (2010) Molecular mechanisms of action and in vivo validation of an M4 muscarinic acetylcholine receptor allosteric modulator with potential antipsychotic properties. Neuropsychopharmacology 35:855–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leach K, Sexton PM, Christopoulos A. (2007) Allosteric GPCR modulators: taking advantage of permissive receptor pharmacology. Trends Pharmacol Sci 28:382–389 [DOI] [PubMed] [Google Scholar]
- Lewis DA, Moghaddam B. (2006) Cognitive dysfunction in schizophrenia: convergence of gamma-aminobutyric acid and glutamate alterations. Arch Neurol 63:1372–1376 [DOI] [PubMed] [Google Scholar]
- Liu F, Grauer S, Kelley C, Navarra R, Graf R, Zhang G, Atkinson PJ, Popiolek M, Wantuch C, Khawaja X, et al. (2008a) ADX47273 [S-(4-fluoro-phenyl)-3-[3-(4-fluoro-phenyl)-[1,2,4]-oxadiazol-5-yl]-piperidin-1-yl-methanone]: a novel metabotropic glutamate receptor 5-selective positive allosteric modulator with preclinical antipsychotic-like and procognitive activities. J Pharmacol Exp Ther 327:827–839 [DOI] [PubMed] [Google Scholar]
- Liu F, Grauer S, Kelley C, Navarra R, Graf R, Zhang G, Atkinson PJ, Popiolek M, Wantuch C, Khawaja X, et al. (2008b) ADX47273 [S-(4-fluoro-phenyl)-3-[3-(4-fluoro-phenyl)-[1,2,4]-oxadiazol-5-yl]-piperidin-1-yl-methanone]: a novel metabotropic glutamate receptor 5-selective positive allosteric modulator with preclinical antipsychotic-like and procognitive activities. J Pharmacol Exp Ther 327:827–839 [DOI] [PubMed] [Google Scholar]
- Luby ED, Cohen BD, Rosenbaum G, Gottlieb JS, Kelley R. (1959) Study of a new schizophrenomimetic drug; sernyl. AMA Arch Neurol Psychiatry 81:363–369 [DOI] [PubMed] [Google Scholar]
- Mohn AR, Gainetdinov RR, Caron MG, Koller BH. (1999) Mice with reduced NMDA receptor expression display behaviors related to schizophrenia. Cell 98:427–436 [DOI] [PubMed] [Google Scholar]
- Moy SS, Nadler JJ, Perez A, Barbaro RP, Johns JM, Magnuson TR, Piven J, Crawley JN. (2004) Sociability and preference for social novelty in five inbred strains: an approach to assess autistic-like behavior in mice. Genes Brain Behav 3:287–302 [DOI] [PubMed] [Google Scholar]
- Moy SS, Nikolova VD, Riddick NV, Baker LK, Koller BH. (2012) Preweaning sensorimotor deficits and adolescent hypersociability in Grin1 knockdown mice. Dev Neurosci 34:159–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niswender CM, Johnson KA, Luo Q, Ayala JE, Kim C, Conn PJ, Weaver CD. (2008) A novel assay of Gi/o-linked G protein-coupled receptor coupling to potassium channels provides new insights into the pharmacology of the group III metabotropic glutamate receptors. Mol Pharmacol 73:1213–1224 [DOI] [PubMed] [Google Scholar]
- Noetzel MJ, Gregory KJ, Vinson PN, Manka JT, Stauffer SR, Lindsley CW, Niswender CM, Xiang Z, Conn PJ. (2013) A novel metabotropic glutamate receptor 5 positive allosteric modulator acts at a unique site and confers stimulus bias to mGlu5 signaling. Mol Pharmacol 83:835–847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noetzel MJ, Rook JM, Vinson PN, Cho HP, Days E, Zhou Y, Rodriguez AL, Lavreysen H, Stauffer SR, Niswender CM, et al. (2012) Functional impact of allosteric agonist activity of selective positive allosteric modulators of metabotropic glutamate receptor subtype 5 in regulating central nervous system function. Mol Pharmacol 81:120–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohoyama K, Yamamura S, Hamaguchi T, Nakagawa M, Motomura E, Shiroyama T, Tanii H, Okada M. (2011) Effect of novel atypical antipsychotic, blonanserin, on extracellular neurotransmitter level in rat prefrontal cortex. Eur J Pharmacol 653:47–57 [DOI] [PubMed] [Google Scholar]
- Parmentier-Batteur S, O’Brien JA, Doran S, Nguyen SJ, Flick RB, Uslaner JM, Chen H, Finger EN, Williams TM, Jacobson MA, et al. (2012) Differential effects of the mGluR5 positive allosteric modulator CDPPB in the cortex and striatum following repeated administration. Neuropharmacology 62:1453–1460 [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C.(1998) The Rat Brain in Stereotaxic Coordinates, 4th Ed Academic Press, San Diego, CA [Google Scholar]
- Phillips RG, LeDoux JE. (1992) Differential contribution of amygdala and hippocampus to cued and contextual fear conditioning. Behav Neurosci 106:274–285 [DOI] [PubMed] [Google Scholar]
- Pramyothin P, Khaodhiar L. (2010) Metabolic syndrome with the atypical antipsychotics. Curr Opin Endocrinol Diabetes Obes 17:460–466 [DOI] [PubMed] [Google Scholar]
- Ramsey AJ. (2009) NR1 knockdown mice as a representative model of the glutamate hypothesis of schizophrenia. Prog Brain Res 179:51–58 [DOI] [PubMed] [Google Scholar]
- Ramsey AJ, Laakso A, Cyr M, Sotnikova TD, Salahpour A, Medvedev IO, Dykstra LA, Gainetdinov RR, Caron MG. (2008) Genetic NMDA receptor deficiency disrupts acute and chronic effects of cocaine but not amphetamine. Neuropsychopharmacology 33:2701–2714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichel CM, Schwendt M, McGinty JF, Olive MF, See RE. (2011) Loss of object recognition memory produced by extended access to methamphetamine self-administration is reversed by positive allosteric modulation of metabotropic glutamate receptor 5. Neuropsychopharmacology 36:782–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez AL, Grier MD, Jones CK, Herman EJ, Kane AS, Smith RL, Williams R, Zhou Y, Marlo JE, Days EL, et al. (2010) Discovery of novel allosteric modulators of metabotropic glutamate receptor subtype 5 reveals chemical and functional diversity and in vivo activity in rat behavioral models of anxiolytic and antipsychotic activity. Mol Pharmacol 78:1105–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez AL, Nong Y, Sekaran NK, Alagille D, Tamagnan GD, Conn PJ. (2005) A close structural analog of 2-methyl-6-(phenylethynyl)-pyridine acts as a neutral allosteric site ligand on metabotropic glutamate receptor subtype 5 and blocks the effects of multiple allosteric modulators. Mol Pharmacol 68:1793–1802 [DOI] [PubMed] [Google Scholar]
- Rodriguez AL, Zhou Y, Williams R, David Weaver C, Vinson PN, Dawson ES, Steckler T, Lavreysen H, Mackie C, Bartolomé JM, et al. (2012) Discovery and SAR of a novel series of non-MPEP site mGlu₅ PAMs based on an aryl glycine sulfonamide scaffold. Bioorg Med Chem Lett 22:7388–7392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rook JM, Noetzel MJ, Pouliot WA, Bridges TM, Vinson PN, Cho HP, Zhou Y, Gogliotti RD, Manka JT, Gregory KJ, et al. (2013) Unique signaling profiles of positive allosteric modulators of metabotropic glutamate receptor subtype 5 determine differences in in vivo activity. Biol Psychiatry 73:501–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbrock H, Kramer G, Hobson S, Koros E, Grundl M, Grauert M, Reymann KG, Schröder UH. (2010) Functional interaction of metabotropic glutamate receptor 5 and NMDA-receptor by a metabotropic glutamate receptor 5 positive allosteric modulator. Eur J Pharmacol 639:40–46 [DOI] [PubMed] [Google Scholar]
- Slassi A, Isaac M, Arora J, Brown D. (2007) Piperazines and piperidines as mGluR5 potentiators. PCT International Application WO 2007087135. 2 Aug 2007. [Google Scholar]
- Schlumberger C, Pietraszek M, Gravius A, Danysz W. (2010) Effects of a positive allosteric modulator of mGluR5 ADX47273 on conditioned avoidance response and PCP-induced hyperlocomotion in the rat as models for schizophrenia. Pharmacol Biochem Behav 95:23–30 [DOI] [PubMed] [Google Scholar]
- Schlumberger C, Pietraszek M, Gravius A, Klein KU, Greco S, Morè L, Danysz W. (2009) Comparison of the mGlu(5) receptor positive allosteric modulator ADX47273 and the mGlu(2/3) receptor agonist LY354740 in tests for antipsychotic-like activity. Eur J Pharmacol 623:73–83 [DOI] [PubMed] [Google Scholar]
- Sebban C, Tesolin-Decros B, Millan MJ, Spedding M. (1999) Contrasting EEG profiles elicited by antipsychotic agents in the prefrontal cortex of the conscious rat: antagonism of the effects of clozapine by modafinil. Br J Pharmacol 128:1055–1063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spear N, Gadient RA, Wilkins DE, Do M, Smith JS, Zeller KL, Schroeder P, Zhang M, Arora J, Chhajlani V. (2011) Preclinical profile of a novel metabotropic glutamate receptor 5 positive allosteric modulator. Eur J Pharmacol 659:146–154 [DOI] [PubMed] [Google Scholar]
- Swartz MS, Stroup TS, McEvoy JP, Davis SM, Rosenheck RA, Keefe RS, Hsiao JK, Lieberman JA. (2008) What CATIE found: results from the schizophrenia trial. Psychiatr Serv 59:500–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanejevs M, Jatzke C, Renner S, Müller S, Hechenberger M, Bauer T, Klochkova A, Pyatkin I, Kazyulkin D, Aksenova E, et al. (2008) Positive and negative modulation of group I metabotropic glutamate receptors. J Med Chem 51:634–647 [DOI] [PubMed] [Google Scholar]
- Wu EQ, Birnbaum HG, Shi L, Ball DE, Kessler RC, Moulis M, Aggarwal J. (2005) The economic burden of schizophrenia in the United States in 2002. J Clin Psychiatry 66:1122–1129 [DOI] [PubMed] [Google Scholar]
- Xiong H, Brugel TA, Balestra M, Brown DG, Brush KA, Hightower C, Hinkley L, Hoesch V, Kang J, Koether GM, et al. (2010) 4-aryl piperazine and piperidine amides as novel mGluR5 positive allosteric modulators. Bioorg Med Chem Lett 20:7381–7384 [DOI] [PubMed] [Google Scholar]
- Zhou Y, Manka JT, Rodriguez AL, Weaver CD, Days EL, Vinson PN, Jadhav S, Hermann EJ, Jones CK, Conn PJ, et al. (2010) Discovery of N-Aryl Piperazines as Selective mGluR5 Potentiators with Improved In Vivo Utility. ACS Medicinal Chemistry Letters 1:433–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.