

Abstract
The spatiotemporal dynamics of opioid signaling in the brain remain poorly defined. Photoactivatable opioid ligands provide a means to quantitatively measure these dynamics and their underlying mechanisms in brain tissue. Although activation kinetics can be assessed using caged agonists, deactivation kinetics are obscured by slow clearance of agonist in tissue. To reveal deactivation kinetics of opioid signaling we developed a caged competitive antagonist that can be quickly photoreleased in sufficient concentrations to render agonist dissociation effectively irreversible. Carboxynitroveratryl-naloxone (CNV-NLX), a caged analog of the competitive opioid antagonist NLX, was readily synthesized from commercially available NLX in good yield and found to be devoid of antagonist activity at heterologously expressed opioid receptors. Photolysis in slices of rat locus coeruleus produced a rapid inhibition of the ionic currents evoked by multiple agonists of the μ-opioid receptor (MOR), but not of α-adrenergic receptors, which activate the same pool of ion channels. Using the high-affinity peptide agonist dermorphin, we established conditions under which light-driven deactivation rates are independent of agonist concentration and thus intrinsic to the agonist-receptor complex. Under these conditions, some MOR agonists yielded deactivation rates that are limited by G protein signaling, whereas others appeared limited by agonist dissociation. Therefore, the choice of agonist determines which feature of receptor signaling is unmasked by CNV-NLX photolysis.
Introduction
Opioid receptors are a family of G protein–coupled receptors (GPCRs) that participate in analgesia, behavioral reinforcement, and drug abuse. G protein–mediated signaling is highly dynamic, exemplified by the susceptibility of opioid receptors and other GPCRs to undergo desensitization, trafficking, and long-term tolerance with prolonged exposure to agonist (Williams et al., 2001; von Zastrow et al., 2003; von Zastrow and Williams, 2012). We previously reported photoactivatable peptide agonists for opioid receptors (Banghart and Sabatini, 2012), which enabled fundamental signaling properties such as G protein–coupled K+ (GIRK) channel activation kinetics to be measured. These are limited by the kinetics of GPCR signaling and are largely ligand-independent (Ingram et al., 1997). In contrast, deactivation kinetics can be ligand-dependent, likely reflecting ligand-receptor dissociation rates (Ingram et al., 1997). However, neither ligand affinity nor GIRK/GPCR coupling kinetics are necessarily constant and may reflect the state of the receptor and the cell. Understanding the determinants of signaling deactivation kinetics is necessary to fully understand the molecular mechanisms that regulate G protein–mediated signaling; however, slow diffusional clearance of agonists in brain tissue, which results in rebinding of dissociated agonist during washout, prolongs receptor activation and limits the temporal resolution of such measurements. In contrast, rapid delivery of antagonist in a diffusion-independent manner should uncover intrinsic deactivation kinetics by preventing rebinding of dissociated agonist. To provide a means for measuring opioid signaling deactivation kinetics as well as a method to block opioid signaling in a spatiotemporally delimited manner, we developed a photoactivatable analog of the broad-spectrum opioid antagonist naloxone (NLX).
Materials and Methods
Chemical Synthesis and Characterization.
Commercial reagents were obtained from reputable suppliers and used as received. Naloxone hydrochloride dihydrate was obtained from Sigma-Aldrich (St. Louis, MO), and [2-(trimethylsilyl)ethoxy]methyl (SEM)-protected carboxynitroveratryl (CNV)-bromide (SEMO-CNV-Br) was prepared by Medicilon (Shanghai, China). All solvents were purchased in septum-sealed bottles stored under an inert atmosphere. All reactions were sealed with septa through which an argon atmosphere was introduced unless otherwise noted. Reactions were conducted in round-bottomed flasks or septum-capped amber screw-cap vials containing Teflon-coated magnetic stir bars. Reactions were monitored by thin layer chromatography (TLC) on precoated TLC glass plates (silica gel 60 F254, 250 μm thickness) or by liquid chromatography–mass spectrometry (4.6 mm × 150 mm, 5 μm C18 column; 5 μl injection; 30–95% CH3CN/H2O linear gradient with constant 0.1% v/v trifluoroacetic acid (TFA) additive; 10-minute run; 1 ml/min flow; electrospray ionization; positive ion mode; and UV detection at 210 nm, 254 nm, and 350 nm). TLC chromatograms were visualized by UV illumination. Flash chromatography was performed on an automated purification system using prepacked silica gel columns. High-resolution mass spectrometry was performed by the Small Molecule Mass Spectrometry Facility at the Harvard FAS Center for Systems Biology on an Agilent 6210 Time-of-Flight liquid chromatography/mass spectrometer (Agilent Technologies, Santa Clara, CA). NMR spectra were recorded on a Varian 400 MHz spectrometer (Agilent Technologies). 1H and 13C chemical shifts (δ) were referenced to residual solvent peaks. Data for 1H-NMR spectra are reported as follows: chemical shift (δ ppm), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, dd = doublet of doublets, m = multiplet), coupling constant (Hz), integration. Data for 13C-NMR spectra are reported by chemical shift. UV-visible spectra were recorded on the NanoDrop 2000 UV-VIS Spectrophotometer (Thermo Scientific, Waltham, MA). Room lights were covered with Roscolux Canary Yellow #312 film (Rosco Laboratories, Stamford, CT) to filter out wavelengths of light that would lead to unintentional photolysis during purification and handling.
A vial was charged with naloxone hydrochloride dihydrate (110 mg, 276 nmol) and 4 Å, 2.5 μm activated molecular sieve powder (50 mg) and then flushed with argon. Dimethylformamide (400 μl) was added, and the resulting suspension was sonicated and then allowed to stir for 15 minutes before the addition of K2CO3 (84 mg, 607 nmol). SEMO-CNV-Br (128 mg, 304 nmol) dissolved in dimethylformamide (500 μl) was added dropwise, and the reaction was allowed to stir for 10 hours at room temperature while covered with aluminum foil. It was subsequently diluted with ethyl acetate (20 ml) and extracted with saturated NaHCO3 (20 ml, 2×), washed with brine (20 ml, 3×) dried (MgSO4), and filtered. Silica gel (2.5 g) was added, and the mixture was concentrated to dryness. Flash chromatography (dry load with silica gel, 0–3% CH3OH/CH2Cl2, linear gradient) provided 178 mg of a pale pink solid, 175 mg of which was immediately dissolved in CH2Cl2 (15 ml) and covered with aluminum foil. TFA (2.5 ml) was added dropwise, which caused a precipitate to form, and the reaction was allowed to stir for 4 hours at room temperature. The reaction mixture was concentrated to dryness to provide CNV-NLX as the TFA salt (119 mg, 175 nmol, 66% overall yield). To remove traces of naloxone, this material was further purified in 10–20 mg batches by reverse-phase high-performance liquid chromatography (21.2 mm × 150 mm 5 μm Zorbax SB-C8 column (Agilent Technologies); 400 μl injections of ∼20 mM solution in 5% CH3CN/H2O with 0.1% v/v TFA; 5–60% CH3CN/H2O linear gradient with constant 0.1% v/v TFA additive; 45-minute run; 5 ml/min flow; with the UV-visible detectors off to avoid photolysis of the purified product). The material eluting between 32 and 36 minutes was collected, combined, removed of solvent in vacuo, and lyophilized to dryness. Typically, excellent recoveries were obtained (>95% by weight). Diastereomers were not resolved under these conditions. 1H-NMR (methanol-d4, 400 MHz): 7.45–7.61 (m, 1H), 7.19–7.33 (m, 1H), 6.73–6.87 (m, 1H), 6.6–6.71 (m, 1H), 6.47–6.60 (m, 1H), 5.76–5.92 (m, 1H), 5.48–5.61 (m, 2H), 4.78–4.85 (m, 1H), 3.76–3.87 (m, 7H), 3.66–3.74 (m, 1H), 3.45–3.64 (m, 1H), 3.23–3.40 (m, 1H), 2.98–3.15 (m, 1H), 2.78–2.98 (m, 2H), 2.43–2.74 (m, 2H), 2.07–2.17 (m, 1H), 1.79–1.92 (m, 1H), 1.41–1.71 (m, 2H); 13C-NMR (methanol-d4, 100 MHz): 206.61, 206.5, 153.48, 153.19, 148.63, 148.59, 145.58, 145.55, 141.05, 140.99, 140.80, 140.51, 128.30, 128.00, 127.05, 126.58, 126.52, 125.11, 124.35, 123.73, 121.25, 120.42, 120.26, 119.26, 111.66, 110.25, 108.00, 107.97, 107.67, 107.64, 89.75, 89.69, 89.48, 89.42, 69.87, 69.65, 63.03, 62.55, 62.50, 55.52, 55.43, 49.08, 46.07, 34.22, 30.80, 30.72, 27.36, 22.65; high-resolution mass spectrometry (electrospray ionization) calculated for C29H31N2O10 [M+H]+ 567.1973, found 567.1986.
Photolysis in Solution.
Several 40 μl samples of 0.5 mM CNV-NLX in phosphate-buffered saline, pH 7.2, contained in a quartz cuvette, were irradiated for various periods of time with ∼150 mW of light from a 0.22 NA fiber optic cable (ThorLabs, Newton, NJ) coupled to a 405-nm laser (Vortran Stradus 405–250; Vortran Laser Technology, Sacramento, CA) with periodic mixing (Supplemental Fig. 1). Each sample was then analyzed by reverse-phase high-performance liquid chromatography (Agilent Technologies) on a 4.6 mm × 150 mm Zorbax SB-C8 column (5 μm particle size) in H2O, CH3CN, 0.1% TFA at 1 ml/min. NLX and CNV-NLX were resolved by a gradient from 20% to 100% CH3CN over 10 minutes. Samples were monitored at 210 nm, 254 nm, and 350 nm. Mass spectrometry (Agilent 6420 Triple Quadrupole liquid chromotography–mass spectrometry) confirmed photolytic conversion of CNV-NLX to NLX (Supplemental Fig. 1).
Secreted Alkaline Phosphatase Assay.
Human embryonic kidney 293 cells stably transfected with a chimeric Gs-Gi protein that causes Gi-coupled receptors to activate rather than inhibit adenylate cyclase (Liberles and Buck, 2006; S. D. Liberles and L. B. Buck, personal communication) were grown in Dulbecco’s modified Eagle’s medium (Invitrogen, Carlsbad, CA) containing 5% fetal bovine serum (Invitrogen) and 500 μg/ml G-418 (Invitrogen) and maintained at 37°C in an atmosphere of 5% CO2. Cells were plated in 96-well plates at 50,000 cells/well and cotransfected with the GPCR- and cAMP-dependent reporter plasmid using Lipofectamine (Invitrogen) and PLUS reagent (Invitrogen). The transfection medium was replaced with ligand-containing Dulbecco’s modified Eagle’s medium (200 μl/well), and cells were incubated for 36–48 hours at 37°C and 5% CO2. Care was taken to avoid exposure to bright, nonfiltered light. Each plate was then wrapped in plastic wrap and incubated at 68°C for 2 hours to heat-inactivate native phosphatases. After transferring 100-μl aliquots from each well to a fresh 96-well plate, 100 μl of an aqueous buffer containing 2 M diethanolmine bicarbonate and 1.2 mM methylumbelliferone phosphate, pH 10.0, was added to each well. Plates were imaged using a PerkinElmer Envision plate reader (PerkinElmer, Waltham, MA) using optical settings for methylumbelliferone fluorescence, taking care to image all conditions to be compared on the same day for a single receptor at the same time-point after methylumbelliferone phosphate addition. In every individual experiment, NLX was evaluated alongside CNV-NLX at the same receptor, and within each plate an agonist-only and no-ligand control were included for normalization between plates. cDNAs encoding human δ- (OPRD1) and human κ- opioid receptors (OPRK1) contained an N-terminal 3xHA epitope tag (Missouri S&T cDNA Resource Center, Rolla, MO) and the murine μ-opioid receptor (MOR) (OPRM1) contained an N-terminal FLAG epitope tag. Data shown reflect the average of six replicates run in parallel for each condition.
Brain Slice Preparation.
Horizontal brain slices containing locus coeruleus (LC) neurons were prepared as described previously (Williams and North, 1984). Briefly, rats were killed and the brain was removed, blocked, and mounted in a VT 1200S vibratome chamber (Leica Biosystems, Buffalo Grove, IL). Horizontal slices (230–240 μm) were prepared in an ice-cold cutting solution containing (in mM): 126 NaCl, 2.5 KCl, 1.2 MgCl2, 2.6 CaCl2, 1.2 NaH2PO4, 11 d-glucose, 21.4 NaHCO3, and 0.01 (5S,10R)-(+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine (MK-801; Abcam, Cambridge, MA), equilibrated with 95% O2 and 5% CO2. Slices were stored at 34°C in glass vials with oxygenated (95% O2 and 5% CO2) artificial cerebrospinal fluid containing (in mM): 126 NaCl, 2.5 KCl, 1.2 MgCl2, 2.6 CaCl2, 1.2 NaH2PO4, 11 d-glucose, and 21.4 NaHCO3.
Electrophysiologic Recordings.
After an incubation period of at least 30 minutes, slices were hemisected and transferred to the recording chamber, which was superfused with 34°C artificial cerebrospinal fluid at a rate of 1.5–2 ml/min. Whole-cell recordings were made from LC neurons with an Axopatch 1D amplifier (Molecular Devices, Sunnyvale, CA) in voltage-clamp mode (Vhold = −60 mV). Recording pipettes (1.7–2.1 MΩ) were filled with internal solution containing (in mM): 115 potassium methanesulfonate or potassium methyl sulfate, 20 KCl, 1.5 MgCl2, 5 HEPES (K), 10 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA) or 0.1 EGTA, 2 Mg-ATP, 0.2 Na-GTP, pH 7.4, 275–280 mOsM. Series resistance was monitored without compensation and remained <15 MΩ for inclusion. Current was continuously recorded at 200 Hz with PowerLab (Chart version 5.4.2; AD Instruments, Colorado Springs, CO). Episodic currents were recorded at 10 kHz for 1 minute using AxoGraphX (1.4.3; AxoGraph Scientific, Sydney, NSW, Australia). Drugs were applied by bath superfusion.
Uncaging was carried out with full-field illumination using a 405-nm light-emitting diode (ThorLabs) coupled through a 60× objective (Olympus, Shinjuku, Tokyo, Japan) of either 0.9 or 1.0 numerical aperture. Light power was 10 mW at the back aperture of the objective and 1 mW at the focal point. A solution containing CNV-NLX (5 µM, in 6 ml) was recirculated for at least 5 minutes before an uncaging experiment. Opioid agonists were added to the solution containing CNV-NLX prior to photolysis. In most experiments slices were exposed to only a single light flash. In all experiments a saturating concentration of the α2-adrenoceptor agonist 5-bromo-N-(2-imidazolin-2-yl)-6-quinoxalinamine (UK14304; 3 µM) was applied and the resulting current was used as a postsynaptic control. In many experiments the amplitude of opioid-induced current was presented as a fraction of the current induced by UK14304. The small-molecule opioids oxycodone, codeine, morphine, etorphine, and methadone were obtained from the National Institute on Drug Abuse. [Met5]-enkephalin (ME), [D-Ala2, N-MePhe4, Gly-ol]-enkephalin (DAMGO), and endomorphin-1 were obtained from Sigma-Aldrich. β-Endorphin was obtained from Peptides International (Louisville, KY). Dermorphin (DERM) was obtained from Phoenix Peptides (Burlingame, CA). [D-Ser2, Leu5, Thr6]-enkephalin (DSLET), UK14304 and idazoxan were obtained from Tocris (Bristol, UK).
Data Analysis.
Summary data are presented as mean ± S.E.M. The time constant of decay was fit by a single exponential function using AxoGraph X. The time courses covered the area from at least 5 to 95% of the total time course of the decline in agonist-induced current. Summary data were analyzed using the indicated statistical tests, and differences were considered significant when P < 0.05.
Results
Design and Synthesis of Caged Naloxone.
We previously found that appending an α-carboxy-2-nitrobenzyl (CNB) caging group to the phenol on the conserved N-terminal tyrosine of the opioid peptides dynorphin A (1–8) and [Leu5]-enkephalin (LE) sufficiently reduced agonist potency to achieve strong photoactivation (i.e., carboxynitrobenzyl-tyrosine-[Leu5]-enkephalin (1); Fig. 1) (Banghart and Sabatini, 2012). The significance of this phenol in opioid ligand-receptor interactions is underscored in the morphinan family of opiate alkaloids, which contain a small-molecule surrogate of the N-terminal tyrosine in the form of a tyramine moiety that includes the oxygenated A-ring (Kane et al., 2006). For example, the potent MOR agonist morphine (2) is converted to the low-affinity agonist codeine (3), simply by addition of a methyl group to the 3-hydroxy moiety (Toll et al., 1998). By extension, we reasoned that the commercially available opioid antagonist NLX (4; Fig. 1), which contains a similar morphinan core structure, could be readily caged at the same position.
Fig. 1.

Design and synthesis of CNV-NLX, a caged opioid antagonist. (A) Chemical structures of carboxynitrobenzyl-tyrosine-[Leu5]-enkephalin (CYLE) (1), morphine (2), and codeine (3). (B) Two-step synthesis of CNV-NLX (6) from NLX (4).
Although NLX contains several reactive groups, the nucleophilic 3-phenol can be chemoselectively alkylated due to its low pKa and steric accessibility (Krassnig et al., 1998). Thus, commercially available NLX was treated under mildly basic conditions with SEMO-CNV-Br (5; Fig. 1) to install the α-carboxy-6-nitroveratryl (CNV) caging group (Russell et al., 2010, 2012). After protecting group removal with trifluoroacetic acid, CNV-NLX (6) was isolated in 66% overall yield as a mixture of diastereomers.
Similar to the CNB caging group used in carboxynitrobenzyl-tyrosine-[Leu5]-enkephalin, the CNV moiety contains a carboxylic acid to reduce the net hydrophobicity of the caging group, which improves solubility, and may advantageously interact electrostatically with the receptor to impair binding. In contrast to CNB, CNV contains two methoxy moieties at the 4 and 5 positions on the nitroaromatic ring, which extend the absorbance spectrum into the visible wavelength range (Supplemental Fig. 1A) (Russell et al., 2010, 2012). This offers advantages over caging groups that only respond to UV light in terms of the cost of effective light sources for photolysis, reduced scattering in tissue, and lower phototoxicity (Trigo et al., 2009). Liquid chromatography–mass spectrometry analysis confirmed that CNV-NLX cleanly converts to NLX upon illumination at 405 nm (Supplemental Fig. 1, B–F).
Pharmacological Evaluation in Heterologously-Expressed Opioid Receptors.
The functional inactivity of CNV-NLX as an antagonist of opioid receptors was assayed in a heterologous expression system. Human embryonic kidney 293 cells were transfected with MOR along with a chimeric Gs-Gi α subunit that causes Gi-coupled receptors to activate, rather than inhibit, adenylate cyclase, and a cAMP response element-binding protein–dependent reporter plasmid that leads to the production of secreted alkaline phosphatase as a function of receptor activation through the cAMP pathway (Liberles and Buck, 2006; Banghart and Sabatini, 2012; S. D. Liberles and L. B. Buck, personal communication). Antagonist dose-response curves were obtained by treating cells with 1 µM LE, a concentration that produces near-maximal activation, along with various concentrations of NLX or CNV-NLX. Although NLX produced dose-dependent inhibition, CNV-NLX exhibited no antagonist activity (Fig. 2A). Separately, agonist dose-response curves were obtained for LE in the presence of 5 μM NLX and 5 μM CNV-NLX. Although NLX completely suppressed the LE response, CNV-NLX was without effect (Fig. 2B). Similar results were obtained at δ- and κ-receptors (Supplemental Fig. 2).
Fig. 2.

CNV-NLX is inactive at the μ-opioid receptor expressed in human embryonic kidney 293 cells. (A) Dose-response curves of NLX (black circles) and CNV-NLX (blue squares) in the presence of 1 μM LE (n = 12 wells). (B) Dose-response curves of LE alone (black squares), and in the presence of 5 μM NLX (upward red triangles) and CNV-NLX (downward green triangles) (n = 6 wells).
Light-Triggered Antagonism in Brain Tissue.
We next asked whether photolysis of CNV-NLX in brain tissue leads to rapid antagonism of opioid receptors and whether this antagonism is specific for opioid receptors over α-adrenergic receptors, which activate the same signaling pathway. To this end, whole-cell voltage-clamp recordings were obtained from acute slices of rat locus coeruleus, which exhibit large outward GIRK currents when exposed to MOR agonists, as well as α-adrenergic receptor agonists (Williams and North, 1984; Fiorillo and Williams, 1996). High concentrations of various agonists were applied to activate GIRK channels in the presence of CNV-NLX (5 μM), followed by full-field delivery of a 5-second flash of 405-nm light (Fig. 3). The currents evoked by the MOR agonists DAMGO, dermorphin, morphine, methionine enkephalin, and methadone were rapidly inhibited immediately following the light flash, followed by a gradual recovery, consistent with photorelease of NLX, the dilution of NLX, and agonist rebinding over time (Fig. 3A). In contrast, the current evoked by the α-adrenergic receptor agonist UK14303 was not affected by light (Fig. 3, A and B; Supplemental Fig. 3). These data confirm that photolysis of CNV-NLX can strongly suppress opioid signaling evoked by high concentrations of many common MOR agonists, and that this suppression is not due to a nonspecific action on GPCRs.
Fig. 3.
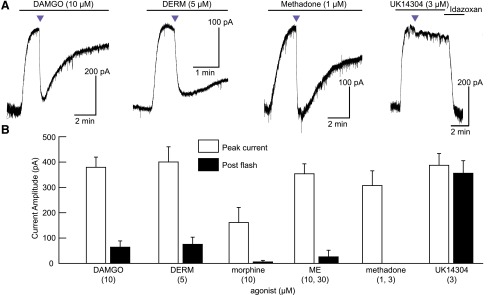
Light-driven antagonism of opioid receptors by CNV-NLX. (A) Photolysis inhibits currents evoked by high concentrations of the MOR agonists DAMGO, DERM, and methadone, but causes only an artifactual response at α-adrenergic receptors in the presence of UK14304. (B) Summary plot comparing currents evoked by MOR agonists and the α-adrenergic agonist UK14304, before and after CNV-NLX photolysis (n = 4–8 cells per agonist). Two concentrations were pooled for both ME and methadone. Purple triangles indicate 5-second flashes of 405-nm light.
To examine the ability to control the amount of released antagonist with light, subsaturating concentrations of DAMGO (100 nM) and CNV-NLX (5 μM) were applied, and the duration of the light flash was varied. Subsaturating concentrations of MOR agonists activate sustained outward currents that do not desensitize substantially over tens of minutes, which facilitates quantification of the light-driven antagonism. Under these conditions, a 100-millisecond flash inhibited the DAMGO-evoked current to 39.2 ± 7.6% (n = 13) of the peak, whereas a subsequent 2-second flash decreased the current to 0.7 ± 0.1% of the peak (n = 10; Fig. 4A). To ensure that DAMGO did not saturate MORs, which would confound these interpretations, a saturating concentration of UK14304 (3 μM) was applied, followed by the selective α-adrenergic antagonist idazoxan (1 μM) at the end of each recording. α-Adrenergic receptors and MORs activate the same pool of GIRK channels in LC neurons (Williams and North, 1984; Fiorillo and Williams, 1996). Whereas a saturating concentration of DAMGO activates a current that is about 120% that of the peak current induced by UK14304 (Williams and North, 1984; Fiorillo and Williams, 1996), 100 nM DAMGO resulted in a current that was about 70% as large, confirming that the 100 nM DAMGO-evoked current was not maximal (Fig. 4B).
Fig. 4.

Analog photocontrol of NLX release. (A) Varying the duration of the light flash produces graded inhibition of the outward current evoked by bath perfusion of a subsaturating concentration of DAMGO. (B) Summary plot comparing the current amplitudes produced by DAMGO (100 nM) before and after each uncaging stimulus to that produced by a saturating concentration of UK14304 (3 µM, n = 4 cells). Because DAMGO (100 nM) produced smaller peak currents than UK14304 (3 µM), the opioid stimulus is confirmed to be subsaturating.
Kinetic Analysis of Opioid Signaling Deactivation.
By pre-equilibrating tissue with the caged antagonist, photorelease results in nearly immediate delivery directly to receptor binding sites without long-range diffusion through complex and dense neuropil. During experiments in which agonists were applied at several concentrations, we observed that the rate of light-driven current decay varied with agonist identity as well as concentration for a given agonist. Time constants (tau) of current decay ranged from up to ∼10 seconds at high concentrations, down to ∼1 second with low concentrations of some agonists. Across experiments utilizing identical photolysis conditions, the kinetics of current decay should reflect four parameters: 1) agonist dissociation rate (koff), which is determined by its affinity (Kd) and association rate (kon), as Kd = koff/kon; 2) naloxone association rate, which is constant across conditions and much faster than the observed decay rates (Supplemental Fig. 4); 3) the rate of G protein–mediated biochemical signaling deactivation (kdeact); and 4) the relative concentrations of agonist and photoreleased competitive antagonist, the latter of which is fixed by the experimental conditions (Fig. 5). Studies employing rapid application and washout of MOR agonists in acutely isolated LC neurons indicate that, with the exception of the high-affinity agonist etorphine, GIRK deactivation had a time constant ranging from 0.7 to 1.4 seconds following washout (Ingram et al., 1997). Therefore the deactivation time constants observed on the scale of several seconds likely reflect competitive binding between agonist and antagonist. At high agonist concentrations, rebinding of agonist after NLX photorelease presumably slows the rate of current decay. However, with photorelease of sufficient NLX to prevent rebinding, the observed kinetics of deactivation should remain constant over a range of low agonist concentrations.
Fig. 5.
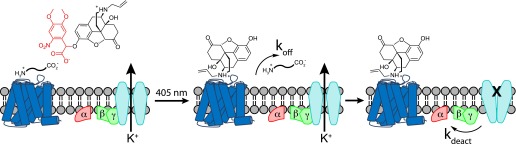
Schematic illustrating the molecular events that underlie opioid signaling deactivation in response to antagonist uncaging in the presence of agonist. The photo-released competitive antagonist can bind only after agonist dissociation, which occurs with a rate constant of koff. In the agonist-free state, the G protein subunits that support GIRK channel opening dissociate from the activated channels, allowing channel closure with a rate constant of kdeact.
To determine if photolysis of CNV-NLX can resolve such ligand-receptor interaction kinetics in brain slices, we obtained a kinetic dose-response curve of the effects of NLX photorelease on currents evoked by the high-affinity peptide agonist DERM (Fig. 6). The concentration of DERM was varied from 10 nM to 10 μM and applied in the presence of 5 μM CNV-NLX, which was photolyzed by a 5-second flash at the peak of the agonist-evoked outward current. Saturating MOR-mediated currents are, on average, 1.2-fold larger than α-adrenergic receptor–mediated currents in the same cells (Williams and North, 1984; Fiorillo and Williams, 1996). By measuring the current evoked by each concentration of DERM in comparison with that evoked by UK14304 (3 μM) in the same cells, DERM was found to activate MORs with an EC50 of ∼10 nM, and saturating responses were reached at ∼500 nM. As predicted, the currents evoked by low concentrations of DERM decayed faster after photolysis of CNV-NLX than those evoked by large concentrations (Fig. 6A), and complete block was achieved at all but the highest concentrations. Furthermore, the time constant of deactivation did not vary significantly at the three lowest concentrations tested (10, 30, and 100 nM; one-way analysis of variance, P = 0.26) and only slowed upon reaching near-saturating concentrations (500 nM–10 µM; Fig. 6B). This likely reflects rebinding of agonist due to reversible, competitive receptor occupancy by the photoreleased NLX at high agonist concentrations, whereas at EC50 agonist concentrations and lower, the photoreleased NLX outcompetes agonist at the receptor, thus eliminating rebinding. These results suggest that the rate of deactivation measured at low, subsaturating concentrations of agonists can reflect the rate of agonist dissociation (koff). Furthermore, the postphotolysis current amplitude dose-response curve closely matches that obtained for the same agonist in the presence of 100 nM NLX (normalized to UK14304), suggesting that at least 100 nM NLX is released under these photolysis conditions (Supplemental Fig. 5).
Fig. 6.

Low agonist concentrations reveal DERM ligand dissociation kinetics. (A) Superimposition of the currents evoked by 10 nM and 10 μM DERM (left); amplitude-normalized currents reveal slower deactivation at saturating agonist concentration (right). (B) Summary plot of the DERM current, normalized to the current induced by UK14304 (3 µM, black squares), and tau decay (open circles) as a function of DERM concentration (n = 4–8 cells per condition). The dashed line indicates the average of the time constants measured at 10, 30, and 100 nM, which did not vary significantly.
DERM is known to have a high affinity for MOR and thus is expected to have a relatively slow koff in comparison with other opioid agonists. To compare the kinetics of agonist unbinding between agonists, deactivation kinetics of a panel of 11 agonists was examined at subsaturating, near EC50 concentrations that should avoid contributions from agonist rebinding. The opioid-evoked currents were determined to be below saturation by comparing them to the currents evoked by UK14304 (3 μM) in the same cells (Fig. 7, A and B). Varying deactivation rates were observed with no obvious correlation to current amplitude, suggesting that agonist rebinding was not occurring. To determine if deactivation kinetics were predictive of receptor affinity, the corresponding values for each agonist were plotted on a logarithmic scale (Fig. 7C). Affinities are presented as pKi determined at rat MORs in radioligand competition experiments (Supplemental Table 1 and associated discussion) (Childers and Snyder, 1980; Raynor et al., 1994, 1995; McPherson et al., 2010).
Fig. 7.

Light-induced deactivation of currents produced by various MOR agonists at subsaturating concentrations. (A) Superimposition of the normalized currents evoked by subsaturating concentrations of various small-molecule (left) and peptide (right) agonists. (B) Summary plot of the normalized currents (white bars) and corresponding light-induced deactivation rates (black bars) at the indicated subsaturating concentrations. Opioid agonist–induced peak currents are normalized to the peak currents produced by UK14303 (3 µM) in the same cells (n = 4–8 cells per condition). (C) Summary plot of the deactivation time constant versus agonist affinity according to compound class (n = 4–8 cells per condition).
Although two agonists (β-endorphin and etorphine) exhibited dramatically slower deactivation kinetics, most agonists exhibited deactivation time constants ranging from 1–2 seconds, and none were faster than 1 second. Agonists with the highest affinities generally exhibited the slowest decay rates (β-endorphin, DERM, and etorphine), and the lowest affinity agonist, codeine, produced the fastest decay rate. Although DERM is reported as the highest affinity peptide ligand in the series, β-endorphin currents decayed >3-fold more slowly. At the other extreme, although DAMGO, ME, and endomorphin-1 are reported to exhibit very similar affinities, the DAMGO- and ME-induced currents decayed ∼3-fold more rapidly and were among the fastest to deactivate in the entire series. When considered according to compound class, the small molecules, which are most likely to exhibit a similar kon, trend according to affinity, but with a slope of 0.3 rather than 1. For example, the decay rates measured for morphine and codeine differ by a factor of 1.6 rather than a factor of 10, as would be predicted for a constant kon. The peptides showed even greater variability, consistent with their structural diversity and complexity, which could give rise to various forms of multistep ligand-receptor interactions that determine kon and koff.
Discussion
In this study we describe the development of a caged analog of the opioid antagonist NLX and demonstrate that it provides a means to study opioid receptor deactivation dynamics in brain tissue. This reagent lacks residual antagonist activity at concentrations up to at least 100 μM, and photolysis in brain slices affords rapid and nearly complete inhibition of MOR-mediated outward currents at both subsaturating and near-saturating concentrations of agonist. Using full-field uncaging with a 405-nm light-emitting diode as a light source and 5 μM CNV-NLX in the bath, light flashes ranging from 100 milliseconds to 5 seconds in duration produced graded inhibition of the agonist-evoked currents, with the largest stimulus affording at least 100 nM NLX at the recorded neurons, corresponding to about 2% photolysis. In principle, the use of more powerful light sources would allow for even greater concentration jumps and would enable shorter light flashes to produce comparable levels of inhibition. At subsaturating agonist concentrations, the inhibition produced by a single light flash was stable for several tens of seconds, presumably reflecting slow diffusion of NLX away from the illuminated area. At saturating agonist concentrations, the light-induced antagonism reversed more quickly, which is likely facilitated by competitive interactions between the ligands.
In LC neurons, MORs and α-adrenergic receptors activate the same population of GIRK channels (Williams and North, 1984; Fiorillo and Williams, 1996). The convergence of Gi/o-coupled receptor signaling onto GIRK channels in LC neurons allowed us to examine the selectivity of light-driven antagonism with CNV-NLX for opioid receptors. Although photolysis of CNV-NLX during the application of the α-adrenoreceptor agonist UK14304 appeared to produce a small and transient inhibition in the form of an inward current, this effect persisted in the absence of CNV-NLX, indicating that it is an artifact of the intense light flash and is not due to nonspecific actions of the photolysis products. The precise origin of this artifact is not clear from these data.
NLX is well established to function as a competitive antagonist at MORs and has also been characterized as an inverse-agonist under certain circumstances (Connor and Traynor, 2010). In our experiments, NLX was photoreleased in the presence of agonist, so the photoreleased NLX requires agonist dissociation before it can bind to receptors, according to the simplest models of ligand-receptor interactions that contain a single agonist binding site per functional receptor. In the absence of antagonist, the unbound agonist would simply rebind to sustain a constant level of receptor activation. However, if a sufficiently high concentration of antagonist is released to outcompete the agonist, agonist rebinding is overcome such that MORs cease to activate GIRK channels. Our race experiments (Supplemental Fig. 4) involving simultaneous uncaging of agonist and antagonist indicate that NLX binding occurs much faster than GIRK activation, which occurs with a time constant of ∼300 milliseconds under these conditions. In acutely isolated LC neurons, GIRK channels deactivated in the absence of bound agonist with a time constant of ∼1 second (Ingram et al., 1997), so deactivation rates slower than 1 second were interpreted to reflect, at least in part, agonist dissociation from receptors. A similar time course of GIRK current deactivation following GABAB receptor activation was also found in acutely isolated hippocampal pyramidal neurons (Sodickson and Bean, 1996). The deactivation of GIRK current is dependent on multiple mechanisms that can include agonist/receptor interactions, downstream mechanisms such as the activity of regulator of G protein–signaling proteins, and the experimental conditions. The similarity in the rate of GIRK inactivation between the MOR and GABAB receptors may be of interest given that the GABAB receptor is an obligate dimer (Jones et al., 1998; Kaupmann et al., 1998; White et al., 1998; Pin et al., 2003). There is structural evidence that MOR can form dimers (Manglik et al., 2012), raising the possibility that NLX may initiate deactivation prior to agonist dissociation by binding to unoccupied allosterically coupled orthosteric sites in multimeric receptors (Albizu et al., 2006; Springael et al., 2007); however, the functional consequences of these associations remain to be determined. What is known is that single MORs isolated in nano discs have the ability to activate G proteins, such that dimers of MOR are not strictly necessary at a functional level (Kuszak et al., 2009). Thus our interpretations of the measured deactivation rates are currently restricted to the simplest scenario of monomeric receptors and competitive interactions at a single orthosteric binding site.
Using the high-affinity peptide agonist DERM and a fixed light-driven dose of CNV-NLX (5 µM, 5-second flash), we determined an agonist concentration window in which the measured current deactivation rates appear to reflect agonist unbinding from the receptor (10–100 nM), occurring with a time constant of ∼4 seconds. Although the amplitude of the DERM-evoked currents varied according to agonist concentration, the current deactivation rates did not vary significantly. Apparently, the amount of NLX released by this light stimulus and dose (∼100 nM, 100 × Kd) is sufficient to fully outcompete DERM and prevent agonist rebinding within this concentration range. At higher DERM concentrations, the rate of deactivation slowed significantly. The agonist concentration range over which photolysis of CNV-NLX will provide an accurate measure of dissociation kinetics without a contribution from agonist rebinding will vary with agonist affinity, concentration of CNV-NLX, and the quantity of light applied. In principle, kinetic studies can be conducted on large concentrations of high-affinity agonists using large concentrations of CNV-NLX and/or high-intensity light, but it may be more practical to use subsaturating doses of agonist to conserve material and avoid phototoxicity.
Our kinetic data imply important considerations for the use of CNV-NLX to study GPCR deactivation kinetics using caged antagonists. None of the agonist-induced currents deactivated with time constants faster than 1 second, suggesting the presence of an instrumental limit. Thus, some ligands may dissociate with time constants faster than 1 second, but the rate-limiting step in signaling deactivation for such agonists is likely disassembly of the G protein–GIRK complex to enable channel closure. In contrast, several agonists, including DERM, yielded long time-constants (3–10 seconds), suggesting that slow ligand dissociation is rate-limiting in these cases, whereas those falling between the extremes likely reflect mixed contributions from both events on approximately the same time scale. Regardless of which mechanism constitutes the rate-limiting step, these deactivation time constants are intrinsic to each ligand-receptor complex and constitute a useful biophysical parameter for characterizing the receptor state and its associated signaling pathways. Thus in the context of studying mechanisms that regulate GPCR signaling, the choice of agonist determines which process is predominantly being measured: ligand dissociation or GIRK coupling (Fig. 5). Monitoring current deactivation with slowly dissociating agonists should reveal changes in receptor affinity that might occur during prolonged signaling (Birdsong et al., 2013) or during cross-talk between GPCRs. On the other hand, rapidly dissociating agonists should reveal changes in the intrinsic rate of GIRK deactivation.
Because NLX is not selective for opioid receptor subtypes, CNV-NLX can be used to probe δ- and κ-opioid receptors, in addition to MORs. Due to the necessity of the A-ring phenol for high-affinity binding of opiate alkaloids in general, this caging strategy should work equally well with other opiates that contain an analogous phenol, regardless of whether they function as agonists or antagonists. When considered with our success in caging the opioid peptide agonists LE and dynorphin A (1–8) at the N-terminal tyrosine phenol, these findings open the door to caged opioid ligands that exhibit receptor subtype selectivity, of which many candidates are available as either peptides or small molecules.
We anticipate that CNV-NLX will also be useful to study actions of endogenously released opioid peptides in brain slices and in vivo. Although little is known about the quantities of endogenous opioids released in vivo, there is no evidence that saturating concentrations of agonist are reached. Since CNV-NLX effectively antagonizes high concentrations of exogenous agonists, it should be effective in blocking endogenous opioid actions in vivo and in ex vivo tissue with high spatiotemporal precision. Since the dissociation rate of NLX from MORs and diffusional clearance is slow, photoreleased NLX remains active for several minutes after a light flash. Together these points suggest that photolysis of CNV-NLX in vivo may facilitate the identification of the functions of endogenous opioid signaling in behaving animals.
Supplementary Material
Acknowledgments
The authors thank Seksiri Arttamangkul, William Birdsong, and Nicolas Tritsch for insightful discussions; S. D. Liberles, L. B. Buck, and J. L. Whistler for plasmids; and the Institute of Chemistry and Cell Biology (ICCB)-Longwood screening facility for access to plate readers.
Abbreviations
- BAPTA
1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid
- CNB
carboxynitrobenzyl
- CNV
carboxynitroveratryl
- DAMGO
[D-Ala2, N-MePhe4, Gly-ol]-enkephalin
- DERM
dermorphin
- DSLET
[D-Ser2, Leu5, Thr6]-enkephalin
- GIRK
G protein–coupled K+ channel
- GPCR
G protein–coupled receptor
- LC
locus coeruleus
- LE
[Leu5]-enkephalin
- ME
[Met5]-enkephalin
- MK-801
(5S,10R)-(+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine
- MOR
μ-opioid receptor
- NLX
naloxone
- SEM
[2-(trimethylsilyl)ethoxy]methyl
- SEMO-CNV-Br
SEM-protected CNV-bromide
- tau
time constant
- TFA
trifluoroacetic acid
- TLC
thin-layer chromatography
- UK14303
5-bromo-N-(2-imidazolin-2-yl)-6-quinoxalinamine
Authorship Contributions
Participated in research design: Banghart, Williams, Sabatini.
Conducted experiments: Banghart, Williams, Shah.
Contributed new reagents or analytic tools: Banghart, Lavis.
Performed data analysis: Banghart, Williams.
Wrote or contributed to the writing of the manuscript: Banghart, Williams, Sabatini.
Footnotes
This work was supported by the Howard Hughes Medical Institute (to B.L.S. and L.D.L.); the National Institutes of Health National Institute of Mental Health [Grant MH085498] (to B.L.S.); and the National Institutes of Health National Institute on Drug Abuse [Grant DA08163] (to J.T.W.) and [Grant DA034648] (to M.R.B.); and postdoctoral fellowships from the Helen Hay Whitney Foundation and the Charles A. King Trust, N.A., Bank of America, Co-Trustee (to M.R.B.).
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.
References
- Albizu L, Balestre MN, Breton C, Pin JP, Manning M, Mouillac B, Barberis C, Durroux T. (2006) Probing the existence of G protein-coupled receptor dimers by positive and negative ligand-dependent cooperative binding. Mol Pharmacol 70:1783–1791 [DOI] [PubMed] [Google Scholar]
- Banghart MR, Sabatini BL. (2012) Photoactivatable neuropeptides for spatiotemporally precise delivery of opioids in neural tissue. Neuron 73:249–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birdsong WT, Arttamangkul S, Clark MJ, Cheng K, Rice KC, Traynor JR, Williams JT. (2013) Increased agonist affinity at the μ-opioid receptor induced by prolonged agonist exposure. J Neurosci 33:4118–4127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childers SR, Snyder SH. (1980) Differential regulation by guanine nucleotides or opiate agonist and antagonist receptor interactions. J Neurochem 34:583–593 [DOI] [PubMed] [Google Scholar]
- Connor M, Traynor J. (2010) Constitutively active μ-opioid receptors. Methods Enzymol 484:445–469 [DOI] [PubMed] [Google Scholar]
- Fiorillo CD, Williams JT. (1996) Opioid desensitization: interactions with G-protein-coupled receptors in the locus coeruleus. J Neurosci 16:1479–1485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingram S, Wilding TJ, McCleskey EW, Williams JT. (1997) Efficacy and kinetics of opioid action on acutely dissociated neurons. Mol Pharmacol 52:136–143 [DOI] [PubMed] [Google Scholar]
- Jones KA, Borowsky B, Tamm JA, Craig DA, Durkin MM, Dai M, Yao WJ, Johnson M, Gunwaldsen C, Huang LY, et al. (1998) GABA(B) receptors function as a heteromeric assembly of the subunits GABA(B)R1 and GABA(B)R2. Nature 396:674–679 [DOI] [PubMed] [Google Scholar]
- Kane BE, Svensson B, Ferguson DM. (2006) Molecular recognition of opioid receptor ligands. AAPS J 8:E126–E137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaupmann K, Malitschek B, Schuler V, Heid J, Froestl W, Beck P, Mosbacher J, Bischoff S, Kulik A, Shigemoto R, et al. (1998) GABA(B)-receptor subtypes assemble into functional heteromeric complexes. Nature 396:683–687 [DOI] [PubMed] [Google Scholar]
- Krassnig R, Koch M, Jennewein HK, Greiner E, Schmidhammer H. (1998) A new and efficient synthesis of the μ opioid receptor antagonists 14-O-methyl- and 14-O-ethylnaloxone and -naltrexone. Heterocycles 47:1029–1032 [Google Scholar]
- Kuszak AJ, Pitchiaya S, Anand JP, Mosberg HI, Walter NG, Sunahara RK. (2009) Purification and functional reconstitution of monomeric mu-opioid receptors: allosteric modulation of agonist binding by Gi2. J Biol Chem 284:26732–26741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberles SD, Buck LB. (2006) A second class of chemosensory receptors in the olfactory epithelium. Nature 442:645–650 [DOI] [PubMed] [Google Scholar]
- Manglik A, Kruse AC, Kobilka TS, Thian FS, Mathiesen JM, Sunahara RK, Pardo L, Weis WI, Kobilka BK, Granier S. (2012) Crystal structure of the µ-opioid receptor bound to a morphinan antagonist. Nature 485:321–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPherson J, Rivero G, Baptist M, Llorente J, Al-Sabah S, Krasel C, Dewey WL, Bailey CP, Rosethorne EM, Charlton SJ, et al. (2010) μ-opioid receptors: correlation of agonist efficacy for signalling with ability to activate internalization. Mol Pharmacol 78:756–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pin JP, Galvez T, Prézeau L. (2003) Evolution, structure, and activation mechanism of family 3/C G-protein-coupled receptors. Pharmacol Ther 98:325–354 [DOI] [PubMed] [Google Scholar]
- Raynor K, Kong H, Chen Y, Yasuda K, Yu L, Bell GI, Reisine T. (1994) Pharmacological characterization of the cloned kappa-, delta-, and mu-opioid receptors. Mol Pharmacol 45:330–334 [PubMed] [Google Scholar]
- Raynor K, Kong H, Mestek A, Bye LS, Tian M, Liu J, Yu L, Reisine T. (1995) Characterization of the cloned human mu opioid receptor. J Pharmacol Exp Ther 272:423–428 [PubMed] [Google Scholar]
- Russell AG, Ragoussi ME, Ramalho R, Wharton CW, Carteau D, Bassani DM, Snaith JS. (2010) Alpha-carboxy-6-nitroveratryl: a photolabile protecting group for carboxylic acids. J Org Chem 75:4648–4651 [DOI] [PubMed] [Google Scholar]
- Russell AG, Sadler MJ, Laidlaw HJ, Gutiérrez-Loriente A, Wharton CW, Carteau D, Bassani DM, Snaith JS. (2012) Photorelease of tyrosine from α-carboxy-6-nitroveratryl (αCNV) derivatives. Photochem Photobiol Sci 11:556–563 [DOI] [PubMed] [Google Scholar]
- Sodickson DL, Bean BP. (1996) GABAB receptor-activated inwardly rectifying potassium current in dissociated hippocampal CA3 neurons. J Neurosci 16:6374–6385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springael JY, Urizar E, Costagliola S, Vassart G, Parmentier M. (2007) Allosteric properties of G protein-coupled receptor oligomers. Pharmacol Ther 115:410–418 [DOI] [PubMed] [Google Scholar]
- Toll L, Berzetei-Gurske IP, Polgar WE, Brandt SR, Adapa ID, Rodriguez L, Schwartz RW, Haggart D, O’Brien A, White A, et al. (1998) Standard binding and functional assays related to medications development division testing for potential cocaine and opiate narcotic treatment medications. NIDA Res Monogr 178:440–466 [PubMed] [Google Scholar]
- Trigo FF, Corrie JE, Ogden D. (2009) Laser photolysis of caged compounds at 405 nm: photochemical advantages, localisation, phototoxicity and methods for calibration. J Neurosci Methods 180:9–21 [DOI] [PubMed] [Google Scholar]
- von Zastrow M, Williams JT. (2012) Modulating neuromodulation by receptor membrane traffic in the endocytic pathway. Neuron 76:22–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Zastrow M, Svingos A, Haberstock-Debic H, Evans C. (2003) Regulated endocytosis of opioid receptors: cellular mechanisms and proposed roles in physiological adaptation to opiate drugs. Curr Opin Neurobiol 13:348–353 [DOI] [PubMed] [Google Scholar]
- White JH, Wise A, Main MJ, Green A, Fraser NJ, Disney GH, Barnes AA, Emson P, Foord SM, Marshall FH. (1998) Heterodimerization is required for the formation of a functional GABA(B) receptor. Nature 396:679–682 [DOI] [PubMed] [Google Scholar]
- Williams JT, North RA. (1984) Opiate-receptor interactions on single locus coeruleus neurones. Mol Pharmacol 26:489–497 [PubMed] [Google Scholar]
- Williams JT, Christie MJ, Manzoni O. (2001) Cellular and synaptic adaptations mediating opioid dependence. Physiol Rev 81:299–343 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.