

Abstract
Background
Recent experimental evidence suggests that the Rho/Rho-kinase (ROCK) system may play an important role in the pathogenesis of acute coronary syndrome (ACS) but there are little clinical data. This study examined if ROCK activity is increased in patients with acute coronary syndrome and if ROCK activity predicts long-term cardiovascular event.
Method
Blood samples were collected from 188 patients within 12 h after admission for ACS (53% men; aged 70±13) and from 61 control subject. The main outcome measures were all cause mortality, readmission with ACS or congestive heart failure (CHF) from presentation within around 2 years (mean:14.4±7.2 months; range: 0.5 to 26 months).
Results
ROCK activity increased in ST elevation myocardial infarction (STEMI, n=90) (3.33±0.93), non-STEMI (NSTEMI, n=68) (3.37±1.04) and unstable angina (UA, n=30) (2.53±0.59) groups when compared with disease controls (n=31) (2.06±0.38, all p<0.001) and healthy controls (n=30) (1.54±0.43, all p<0.001). There were 24 deaths, 34 readmissions with ACS and 15 admissions with CHF within 2 years. Patients with a high N-terminal pro-B-type natriuretic peptide (NT-proBNP) and high ROCK activity on admission had a five-fold risk of a cardiovascular event (RR: 5.156; 95% CI: 2.180–12.191) when compared to those with low NT-proBNP and low ROCK activity.
Conclusion
ROCK activity was increased in patients with ACS, particularly in those with myocardial infarction. The combined usage of both ROCK activity and NT-proBNP might identify a subset of ACS patients at particularly high risk.
Keywords: ROCK activity, Acute coronary syndrome, Rho kinase activity
1. Introduction
The risk of death or new ischemic events varies widely in patients presenting with acute coronary syndromes (ACS) [1,2], probably because the term covers a wide range of patients with varying disease severity including ST elevation myocardial infarction (STEMI), non-ST elevation myocardial infarction (NTSEMI) and unstable angina (UA). Several factors have been proposed to assess this heterogeneous risk profile among ACS patients including Global Registry of Acute Coronary Events (GRACE) risk score [3] or some cardiac biomarkers. The most widely studied biomarkers are C-reactive protein (CRP), an acute-phase reactant produced by hepatocytes in response to stimulation by inflammatory cytokines and N-terminal pro-B-type natriuretic peptide type (NT-proBNP) which is released in response to increased myocardial stretch secondary to ischemia induced left ventricular systolic and/or diastolic dysfunction [4].
Recently, evidence increasingly suggests that the Rho/Rho-kinase (ROCK) system may play an important role in the pathogenesis of ACS [5]. Preclinical studies showed that inhibition of Rho/ROCK pathway can limit early atherosclerotic plaque development [6], reduce the size of ischemic-reperfusion injury [7] and protect against vasopressin [8]. However, it is uncertain whether ROCK activity is elevated in patients during the acute phase of myocardial ischemia. Therefore, the objective of the present study was to investigate whether ROCK activity is elevated in ACS patients and determine the factors associated with increased ROCK activity in ACS. We also hypothesized that a multi-marker approach with the simultaneous assessment of CRP, NTpro-BNP and ROCK activity would provide complementary information to GRACE risk score in terms of prognosis in a population of patients with the diagnosis of ACS.
2. Methods
2.1. Patients
188 consecutive patients (53% men; aged 70±13 years) admitted to a university teaching hospital for ACS were enrolled between December 2007 and May 2009. ACS was diagnosed based on the ACC/AHA guideline [9]. Patients entered in the registry had to be ≥18 years old and alive at the time of hospital presentation, be admitted for ACS as a presumptive diagnosis, and had to have ≥1 of the following: electrocardiographic changes consistent with ACS, serial increases in serum biochemical markers of cardiac necrosis, and/or documentation of coronary artery disease. The qualifying ACS must not have been precipitated or accompanied by a significant comorbidity, trauma, or surgery. Patients were divided into ST elevation myocardial infarction (STEMI), non-STEMI (NSTEMI) and unstable angina (UA) groups. All the ACS subgroups matched for age, gender, and smoking status. Calculation of the GRACE score was based on clinical history, ECG and laboratory values upon first arrival to the CCU or the acute medical admissions unit [10]. All the patients were followed up for a mean of 14.4±7.2 months; range: 0.5 to 26 months, or until the occurrence of major events (death from any cause, readmission with ACS or admission with congestive heart failure).
2.2. Disease and normal controls
Sixty-one volunteers were subdivided into disease control group (n=31) (59% men; aged 69±8 years) and healthy control group (n=30) (67% men; aged 67±9 years) depending on the presence or absence of hypertension or smoking status which have been proved to influence ROCK activity [11,12]. The criteria for normal controls were: no history of cardiovascular or systemic illness, normal physical examination including blood pressure, as well as normal hemoglucostix and ECG, no echocardiographic evidence of structure or functional heart disease and no need for regular medications. All disease control subjects proceeded coronary angiography but showed normal epicardial coronary arteries. Written informed consents were obtained from all subjects.
2.3. Analysis of ROCK1, ROCK2 and ROCK activity
Blood samples were collected in the first 12 h after admission. Fasting glucose and lipid were taken in the first morning after admittance. Leukocytes were isolated from 10 mL peripheral blood at the admission following a validated and standardized protocol. NIH 3 T3 cell lysates were used as a positive control and to standardize the results of Western blot analyses from several membranes. Samples were stored at −80 °C and tested together. To avoid overphosphorylation, 1 mM of hydroxyfasudil is added to the fixative solution [13]. The resulting samples were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and bound proteins were detected by immunoblotting. The samples were analyzed by the rabbit anti-MBS polyclonal antibody (Covance, Princeton, New Jersey), anti-ROCK1 monoclonal antibody, anti-ROCK2 monoclonal antibody (BD Biosciences, San Jose, California), anti-actin monoclonal antibody (Sigma), anti-phospho-specific Ser854-myosin binding subunit (MBS) [13] polyclonal antibody (kindly provided by Prof. Liao JK (Boston, Massachusetts)). Rho kinase activity was expressed as the ratio of phosphorylation levels of myosin binding subunit (pMBS) in each sample per pMBS in each positive control divided by MBS in each sample per MBS in each positive control(ROCK activity=[sample pMBS/positive control pMBS]/[sample total MBS/positive control total MBS]) [13].
2.4. Statistical analysis
All statistical analyses were conducted with the SPSS statistical package for Vista version 15.0 (SPSS Inc., Chicago, Illinois). One-way analysis of variance (ANOVA) was used for comparing of mean values of continuous variables among groups, and post-hoc analysis was performed by Scheffe’s test to examine for inter-group differences. Dummy variable was used to adjusted age effect between different groups as healthy control subjects were inevitably younger than other diseased controls. Univariate linear regression models were used to assess the relation between parametric clinical variables and ROCK activity. Multivariate Cox regression analysis using the enter method was used to look at the independent predictors of clinical endpoint. Event rates for clinical outcomes were also determined using the Kaplan–Meier method and compared using the log rank test. Data were expressed as mean±SD and two-sided p value of p<0.05 was considered statistically significant.
3. Results
3.1. Clinical characteristics
The baseline clinical characteristics and laboratory parameters of the subjects in different groups were summarized in Table 1. There was no significant difference among the ACS groups, disease control and normal control groups for their body mass index, diastolic blood pressure and heart rate. All the ACS groups, the disease control and normal control groups were matched for age, gender, and smoking status. No significant difference of the medications was observed among ACS groups.
Table 1.
Baseline Characteristics of acute coronary syndrome and control subjects.
| Baseline characteristic | STEMI (n=90) | NSTEMI (n=68) | UA (n=30) | Disease control (n=31) | Normal control (n=30) | ANOVA p |
|---|---|---|---|---|---|---|
| Age(yrs) | 69±13 | 72±10 | 72±10 | 69±8 | 67±9 | NA |
| Gender (male) | 46(51%) | 38 (56%) | 15 (50%) | 16 (52%) | 20 (67%) | NA |
| Current smoker | 18 (20%) | 9 (11%) | 5 (17%) | 4 (13%) | 0 | NA |
| Medications | ||||||
| Hypertension | 36 (40%) | 39 (48%) | 16 (53%) | 17 (81%) | 0 | NA |
| DM | 19 (21%) | 20 (25%) | 9 (30%) | 0 | 0 | NA |
| Hyperlipidemia | 12 (13%) | 10 (12%) | 6 (22%) | 0 | 0 | NA |
| CVA | 6 (7%) | 3 (4%) | 3 (10%) | 0 | 0 | NA |
| Chronic renal failure | 7 (8%) | 3 (4%) | 2 (7%) | 0 | 0 | NA |
| LVEF (%) | 46.5±9.6§ | 50.2±12.0 | 58.0±10.0 | 64.2±4.1 | 68.5±3.5 | <0.001 |
| SBP (mmHg) | 135±29 | 147±29 | 151±30 | 143±20 | 129±22|| | 0.007 |
| DBP (mmHg) | 76±18 | 77±18 | 77±15 | 82±10 | 77±13 | 0.857 |
| HR (/minute) | 80±21 | 87±29 | 78±19 | 69±13 | 87±11 | 0.080 |
| BMI (kg/m2) | 23.9±3.4 | 24.2±3.6 | 25.8±2.9 | 25.3±8.5 | 23.6±3.2 | 0.629 |
| Heart failure symptom on presentation | 11 (14%) | 21 (46%) | 4 (15%) | NA | NA | NA |
| Laboratory test | ||||||
| Fasting glucose | 7.6±1.9§ | 6.6±1.7 | 5.8±0.9 | 5.2±0.8 | 5.2±0.3 | <0.001 |
| TC (mmol/l) | 4.9±1.1 | 4.7±1.2 | 4.2±0.9 | 4.8±0.8 | 5.1±0.5 | 0.050 |
| LDL-C (mmol/l) | 2.8±1.0 | 2.9±1.0 | 2.4±0.8 | 2.9±1.0 | 3.0±0.3 | 0.119 |
| TG (mmol/l) | 1.7±1.3 | 1.6±1.0 | 1.6±0.8 | 1.7±1.9 | 1.3±0.2 | 0.317 |
| HDL-C (mmol/l) | 1.3±0.5 | 1.2±0.4 | 1.2±0.3 | 1.4±0.4 | 1.5±0.6 | 0.051 |
| WBC (109/l) | 13.1±3.8# | 11.5±4.4§ | 8.6±3.3 | 8.4±3.7 | 5.5±1.8 | <0.001 |
| Creatinine (μmol/l) | 154±187 | 155±97 | 183±184 | 91±13** | 71±19** | 0.031 |
| eGFR | 67.2±31.3 | 57.4±26.5 | 53.4±25.3 | 79.8±2.5** | 82.8±4.1** | 0.023 |
| Peak cTnT (μg/l) | 3.80±5.77† | 0.76±1.12 | 0.023±0.03 | NA | NA | <0.001 |
| Peak CPK (U/l) | 2392.7±2026.0* | 658.0±1397.0 | 110.7±80.1 | 127.3±62.9 | NA | <0.001 |
p<0.05 vs NSTEMI, UA & Disease Control.
p<0.05 vs NSTEMI & UA.
p<0.05 vs ETEMI & Disease Control.
p<0.05 vs UA, Disease Control & Normal Control.
p<0.05 vs NSTEMI.
p<0.05 vs NSTEMI, UA, Disease control & Normal Control.
p<0.05 vs STEMI, NSTEMI & UA.
All results are presented as mean±SD.
3.2. Expression of ROCK activity, ROCK1 and ROCK2 in ACS and control subjects
In all the 3 ACS groups, the ROCK activity (STEMI=3.33±0.93, NSTEMI=3.37±1.04 and UA=2.53±0.59) was significantly higher than the disease control and normal control groups (disease control=2.06±0.38 and normal control=1.54±0.43) (all p<0.001). Interestingly, there was no significant difference of ROCK activity between the STEMI and NSTEMI groups, though they were significantly higher than the UA group (both p<0.001) (Fig. 1). Similarly, protein levels of ROCK1 and ROCK2 of ACS subgroups were significantly higher than that of disease control and normal control (all p<0.001), while no difference between disease and normal controls (Fig. 2).
Fig. 1.

Comparison of ROCK activity in different ACS groups, disease and normal control groups.
Fig. 2.
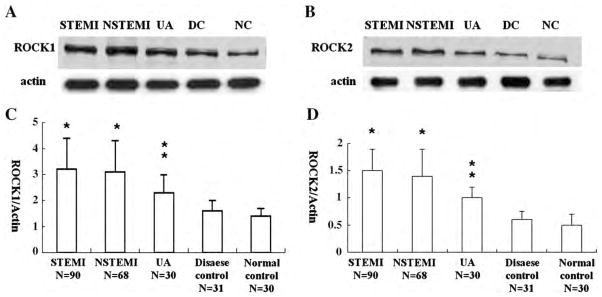
Expression of ROCK1 and ROCK2 protein levels in different groups. ROCK1/actin and ROCK2/actin in STEMI and NSTEMI were significantly higher than that in UA, disease and normal controls (all p<0.001). ROCK1/actin and ROCK2/actin in UA were significantly higher than that in disease and normal controls (all p<0.001). However, no significant difference of ROCK1 and ROCK2 between STEMI and NSTEMI, similarly between disease and normal controls.
3.3. Independent predictors of ROCK activity in ACS subjects
On multivariate analysis, heart failure symptom on presentation (p=0.002), LDL-C level (p=0.001), and number of diseased coronary vessels (p=0.048) were independent predictors of the ROCK activity in ACS patients (Table 2).
Table 2.
Prediction of ROCK activity in univariate and multivariate regression models.
| Variables | Univariate
|
Multivariate (stepwise)
|
||
|---|---|---|---|---|
| Coefficient | p | â | p | |
| Age (yrs) | 0.285 | <0.001 | – | – |
| Gender (male) | 0.056 | 0.410 | – | – |
| Current smoker | 0.143 | 0.050 | – | – |
| Heart failure symptom on presentation | 0.211 | 0.010 | 0.287 | 0.002 |
| LVEF (%) | −0.387 | <0.001 | – | – |
| cTnT (μg/l) (peak) | 0.403 | <0.001 | – | – |
| CPK (U/l) (peak) | 0.301 | <0.001 | – | – |
| Creatinine (μmol/l) | 0.260 | <0.001 | – | – |
| eGFR (60 ml/min/1.73 m2) | −0.209 | 0.010 | – | – |
| Fasting Glucose | 0.164 | 0.040 | – | – |
| WBC (×109/l) | 0.375 | <0.001 | – | – |
| LDL-C (mmol/L) | 0.270 | 0.001 | 0.315 | 0.001 |
| TC (mmol/l) | 0.082 | 0.250 | – | – |
| CRP | 0.277 | <0.001 | – | – |
| NT-proBNP | 0.296 | <0.001 | – | – |
| Number of diseased coronary vessels | 0.383 | <0.001 | 0.227 | 0.048 |
| Current usage of Statin | −0.118 | 0.137 | – | – |
3.4. Univariate and multivariate predictors of clinical endpoint
Totally, there were 24 deaths, 34 readmissions with ACS and 15 admissions with CHF within 2 years. Table 3 shows the univariate correlation between different clinical variables and long-term cardiovascular endpoint including the medications on ACS patients at discharge. Variables with the correlation of clinical endpoint less than 0.05 were chosen into the multivariate model. PCI treatment in admission (HR: 0.312; 95% CI: 0.121–0.554), ROCK activity (HR: 1.402; 95%CI: 1.010–1.908), NT-proBNP (per 200 pg/ml increased) (HR: 1.003; 95%CI: 1.001–1.005) and statins usage after discharge (HR: 0.483; 95%CI: 0.211–0.846) were independent predictors for long-term clinical endpoint (Table 4).
Table 3.
Baseline clinical characteristics (including medications at discharge) between no event and event groups.
| Characteristic | No event
|
Event
|
ANOVA |
|---|---|---|---|
| (n=104) | (n=69) | p | |
| Age(yrs) | 65±12 | 73±13 | <0.001 |
| Gender(male) | 73 (78%) | 37 (54%) | 0.020 |
| Current Smoker | 24 (23%) | 12 (17%) | 0.406 |
| Heart failure on admission | 8 (8%) | 27 (39%) | <0.001 |
| PCI done during hospitalization | 75 (72%) | 20 (29%) | <0.001 |
| Medical history | |||
| Hypertension | 59 (57%) | 38 (55%) | 0.578 |
| DM | 20 (19%) | 29 (42%) | 0.001 |
| Hyperlipidemia | 17 (16%) | 13 (19%) | 0.393 |
| CIHD | 14 (13%) | 20 (29%) | 0.011 |
| Clinic Test | |||
| LVEF (%) | 51±10 | 47±13 | 0.028 |
| SBP (mmHg) | 145±27 | 142±32 | 0.868 |
| DBP (mmHg) | 80±18 | 75±17 | 0.045 |
| HR (/minute) | 83±24 | 82±24 | 0.864 |
| BMI | 24±3 | 24±4 | 0.737 |
| Laboratory test | |||
| Fasting glucose (mmol/l) | 6.8±1.7 | 6.8±2.0 | 0.893 |
| TC (mmol/l) | 4.8±1.2 | 4.5±1.1 | 0.018 |
| LDL-C (mmol/l) | 3.0±1.0 | 2.5±0.9 | 0.004 |
| TG (mmol/l) | 1.7±1.2 | 1.7±1.0 | 0.998 |
| HDL-C (mmol/l) | 1.3±0.4 | 1.2±0.4 | 0.493 |
| WBC (109/l) | 11.8±4.3 | 11.6±4.2 | 0.768 |
| Creatinine (umol/l) | 111±46 | 218±202 | <0.001 |
| eGFR (ml/min/1.73 m2) | 61±21 | 46±32 | <0.001 |
| TnT (peak) (μg/l) | 1.7±2.9 | 2.5±5.6 | 0.440 |
| Creatine phoshokinase (peak) (U/l) | 1721±2049 | 830±1485 | 0.002 |
| ROCK activity | 3.07±0.83 | 3.49±1.17 | 0.011 |
| NT-proBNP | 3169±7479 | 15749±31329 | <0.001 |
| CRP | 26.2±36.7 | 37.9±49.2 | 0.074 |
| GRACE score 6 month predictor | 0.29±0.15 | 0.34±0.16 | 0.026 |
| Medications at discharge | |||
| Aspirin | 100 (96.7%) | 62 (90.4%) | 0.110 |
| Clopidogrel | 70 (67.4%) | 15 (21.2%) | <0.001 |
| Statins | 88 (84.6%) | 40 (57.7%) | <0.001 |
| Beta blockers | 68 (65.2%) | 36 (51.9%) | 0.117 |
| Calcium channel blockers | 7 (6.5%) | 19 (26.9%) | 0.001 |
| ACEI | 56 (54.3%) | 29 (42.3%) | 0.165 |
| Diuretics | 11 (10.4) | 24 (34.7%) | 0.001 |
| Regular Nitrates | 16 (15.2%) | 27 (38.5%) | 0.002 |
Table 4.
Univariate and multivariate analyses of clinical endpoints.
| Variables
|
Multivariate (enter)
|
||||
|---|---|---|---|---|---|
| Coefficient | p | HR | 95%CI | p value | |
| Age(yrs) | 0.385 | <0.001 | – | – | – |
| Gender (male) | −0.169 | 0.020 | – | – | – |
| Current Smoker | 0.437 | 0.406 | – | – | – |
| Heart failure on admission | 0.478 | <0.001 | – | – | – |
| Medications | |||||
| Hypertension | 0.381 | 0.578 | – | – | – |
| DM | 0.248 | 0.001 | – | – | – |
| Hyperlipidemia | 0.318 | 0.393 | – | – | – |
| CVA | 0.192 | 0.154 | – | – | – |
| Chronic renal failure | 0.512 | 0.010 | – | – | – |
| CIHD past medical history | 0.191 | 0.011 | – | – | – |
| LVEF (%) | −0.177 | 0.028 | – | – | – |
| SBP (mmHg) | 0.142 | 0.868 | – | – | – |
| DBP (mmHg) | 0.213 | 0.045 | – | – | – |
| HR (/minute) | 0.261 | 0.864 | – | – | – |
| BMI (kg/m2) | 0.314 | 0.737 | – | – | – |
| Laboratory test | |||||
| Fasting glucose | 0.131 | 0.893 | – | – | – |
| TC (mmol/l) | 0.251 | 0.018 | – | – | – |
| LDL-C (mmol/l) | 0.352 | 0.004 | – | – | – |
| TG (mmol/l) | 0.106 | 0.998 | – | – | – |
| HDL-C (mmol/l) | −0.267 | 0.493 | – | – | – |
| WBC (109/l) | 0.412 | 0.768 | – | – | – |
| Creatinine (μmol/l) | 0.576 | <0.001 | – | – | – |
| eGFR (ml/min/1.73 m2) | 0.561 | <0.001 | – | – | – |
| Peak cTnT (μg/l) | 0.302 | 0.44 | – | – | – |
| Peak CPK (U/l) | 0.471 | 0.002 | – | – | – |
| CRP | 0.361 | 0.074 | – | – | – |
| Baseline ROCK activity | 0.208 | 0.011 | 1.402 | 1.010–1.908 | 0.039 |
| NT-proBNP (per 200 pg/ml increased) | 0.312 | <0.001 | 1.003 | 1.001–1.005 | 0.032 |
| PCI treatment in admission | −0.42 | <0.001 | 0.312 | 0.121–0.554 | 0.001 |
| GRACE score | 0.19 | 0.026 | – | – | – |
| Medications at discharge | |||||
| Aspirin | −0.114 | 0.110 | – | – | – |
| Clopidogrel | 0.215 | <0.001 | – | – | – |
| Statins | −0.321 | <0.001 | 0.483 | 0.211–0.846 | 0.017 |
| Beta blockers | −0.387 | 0.117 | – | – | – |
| Calcium channel blockers | −0.219 | 0.001 | – | – | – |
| ACEI | 0.107 | 0.165 | – | – | – |
| Diuretics | −0.351 | 0.001 | – | – | – |
| Regular Nitrates | 0.241 | 0.002 | – | – | – |
3.5. ROCK activity and NT-proBNP as a composite measure
1,986 pg/ml and 3.03 (best cut-off value for long-term event) was used as a cut-off value of NT-proBNP and ROCK activity in our study population. As demonstrated by the Kaplan–Meier survival curves (Fig. 3), patients with a high NT-proBNP-high ROCK activity on admission were approximately five times more likely to experience a cardio-vascular event at around two years (RR: 5.156; 95% CI: 2.180–12.191) compared to those with low NT-proBNP and low ROCK activity. In addition, patients with high NT-proBNP-high ROCK activity were also more likely to die or experience a cardiovascular event at two years compared to those with high NT-proBNP-low ROCK activity (RR: 2.624; 95% CI: 1.035–6.651) (Fig. 3). From the incremental statistics results, combination of ROCK activity and NT-proBNP (Log rank X2=40.3) significantly predicted long-term outcome more accurately when comparing with NT-proBNP alone (Log rank X2=36.1) (p =0.022).
Fig. 3.
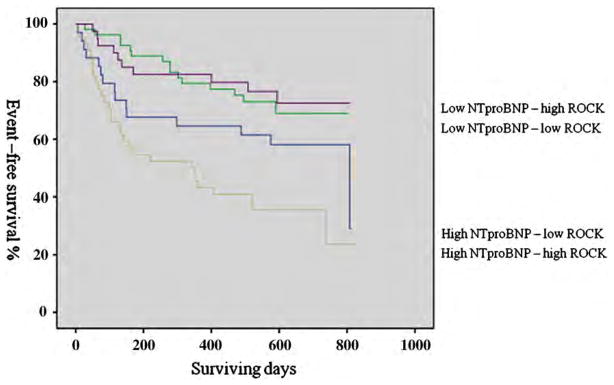
Kaplan–Meier survival curves for cardiovascular events. High NT-proBNP defined as 1986 pg/ml (sensitivity is 63.8% and specificity is 67.3%) and high ROCK activity defined as 3.03 (sensitivity is 56.5% and specificity is 56.7%). High NT-proBNP-high ROCK activity group has higher risk (RR: 5.156; 95% CI: 2.180 to 12.191) to low NT-proBNP-low ROCK activity group. Similarly, high NT-proBNP-high ROCK activity group has higher risk (RR: 2.624; 95% CI: 1.035 to 6.651) to high NT-proBNP-low ROCK activity group. Log rank=20, p<0.001.
4. Discussion
Our study demonstrated that peripheral leukocyte ROCK activity was increased in ACS compared to normal or at-risk subjects, especially in those with elevated cTnT ≥0.1 μg/l. In addition, heart failure symptom on admission, LDL-C level and number of diseased coronary vessels were independent predictors for increased ROCK activity in ACS. We also defined the utility of the combined measurement of baseline ROCK activity and NT-proBNP as biomarkers to predict adverse events.
4.1. ROCK activity across spectrum of coronary artery disease
Elevated ROCK activity has been shown to be involved in many cardiovascular diseases [5]. In addition, Feska demonstrated that ROCK activity was also increased in acute stroke patients, suggesting its pathogenesis role in acute ischemia [14]. Our findings corroborate and extend the above by demonstrating that there is a gradation of ROCK activity from normal in the healthy control, mildly elevated in at-risk group without ACS to those with unstable angina and myocardial infarction. Interestingly, there was no significant difference in ROCK activity between STEMI and NSTEMI according to differing peak TnT levels which represents the degree of myocardial injury. A previous study has found that ROCK-2 phosphororylated the Tn complex, most likely at cTnT, which means that the Rho/ROCK pathway is involved during the whole process of myocardial injury [15]. We could not demonstrate a difference of ROCK activity between STEMI and NSTEMI, probably because ROCK activation is nonspecific, and occurs in many thrombotic, inflammatory, and malignant neoplastic disorders, including acute stroke [14]. ROCK activity increase might be due to the atherosclerosis burden and degrees of inflammation.
One of our novel findings was that LDL-cholesterol and severity of coronary artery disease were two independent predictors for ROCK activity in the ACS patients. Atherosclerosis is a progressive disease characterized by the accumulation of lipid, such as the increase of LDL-cholesterol. Inflammation plays a key role during this whole pathological process. In this study, triple-vessel disease may represent more severe underlying atherosclerosis and inflammation. Evidence indicates that ROCK-mediated pathway is involved at all stages of the inflammatory process. Activated ROCK down-regulates eNOS [16], whereas ROCK inhibition by hydroxylfasudil rapidly increases endothelial eNOS activity [17]. Nitric oxide itself antagonizes the vasoconstrictor effect of ROCK through activation of myosin phosphatase [18]. ROCK activation leads to endothelial hyperpermeability and hence enhances atherosclerosis [19]. Furthermore, ROCK1 plays a key role in macrophage chemotaxis, cholesterol uptake and foam cell formation, all of which are hallmark events in the pathogenesis of atherosclerosis [20]. ROCK1 also mediates neointimal proliferation via recruitment of circulating leukocytes and infiltration of inflammatory cells into the vessel wall [21]. In a low-density lipoprotein (LCL) receptor knockout mice model, activation of the transcription factor NF-κB via Rho/ROCK pathway was enhanced after a high-fat cholate-free diet while inhibition of ROCK significantly was associated with suppression of early atherosclerotic plaque development [6]. In this study, triple-vessel disease may represent more severe underlying atherosclerosis and inflammation. Furthermore, long-term inhibition of ROCK has been shown to cause marked regression of coronary arteriosclerosis in a pig model [22]. Rho/ROCK signaling inhibition by HMG-CoA reductase inhibitors (statins) offers a potential mechanism for some of the pleotropic effects of these agents [23]. Importantly, Nohira et al. (2008) were first to demonstrate that statins inhibit ROCKs activity and improve endothelial function in patients with stable atherosclerosis [24].
In the study, protein levels of ROCK1 and ROCK2 were significantly higher in ACS groups than that of disease and normal controls. Similar observations were found in ischemia/reperfusion injury animal model [25]. However, no obvious increase of ROCK1 and ROCK2 was found between disease and normal control, which was similar in previous studies [26,27]. Thus the increase of ROCK activity might be due to the elevated ROCK itself as well as the rise of protein level. Severe inflammation during atherosclerosis will cause many leukocytes recruitment and endothelial cell injury. But it is relatively mild and stable in subjects with only smoking habit or hypertension. The increase of ROCK protein level and ROCK activity are probably more from the results although current study could not clarify it.
Interestingly in this study, ACS patients with heart failure symptom had higher ROCK activity than those without heart failure. Recently, many cellular and molecular biology studies have proved an involvement of RhoA/ROCK signaling pathway in many aspects of cardiovascular functions such as cardiac hypertrophy and ventricular remodeling after myocardial infarction [28]. Sauzeau et al. reported that human urotensin II-induced VSMC proliferation was inhibited by a ROCK inhibitor, suggesting that RhoA and ROCK mediate the stimulation of VSMC growth [29]. In the adult rat myocardium, pressure overload induces a rapid activation of ROCK, suggesting that it could play a critical role in the coordination of initial mechanisms and adaptive changes triggered by mechanical stress in cardiac myocytes [30]. Recent genetic studies by Wei L’s laboratory and others support the concept that ROCK1 and ROCK2 have distinct non-redundant functions in cardiac hypertrophy and remodeling [31]. ROCK1 deletion did not impair compensatory hypertrophic response, but significantly reduced cardiomyocyte apoptosis and fibrosis in response to pressure overload induced by transverse aortic constriction [32]. A latest study provided the long-term beneficial effects of ROCK1 deficiency in hypertrophic decompensation and suggested that ROCK1 may be an attractive therapeutic target to limit heart failure progression [33].
4.2. Combination of NT-proBNP and ROCK activity can identify a subset of ACS patients at particularly high risk
In this study, the GRACE score and CRP levels were also examined. The GRACE score significantly correlated with long-term events but it was not an independent predictor. This might be because our sample size was small and the follow-up period was more than 2 years. In our cohort, in-hospital PCI was also independent predictors and strongly correlated with the future outcome. It appears to be better than the GRACE score in predicting long-term outcomes. In this study, clopidogrel was routinely used for with PCI. On the other hand, among these ACS patients, the usage of statins after discharge had a greater protection against death or major cardiovascular events, which is consistent with previous studies [34]. The more severely ill ACS subjects often get better treatment in hospital. This might be why in this study, PCI treatment and statins usage were independent predictors. However, the predictive value of GRACE score may be increased when combined with other biomarkers, such as NT-proBNP [35]. It is probable that biomarkers might be more useful than the GRACE score alone for predicting long-term outcome. Similarly, no correlation between CRP and long-term outcome was found. This might also due to the small sample size as previously this kind of acute response marker has been shown to be a strong independent predictor of future cardiovascular events [36]. CRP also correlates with the number of angiographically complex coronary artery stenoses. Similar results were found in this study between ROCK activity and the severity of coronary arterial disease. Probably, this indicates that ROCK activity might be a stronger predictor than CRP for future cardiovascular events.
Our study showed that both ROCK activity and NT-proBNP are independent predictors for future cardiovascular events but not elevated cardiac biomarkers (cTnT and CPK). Congestive heart failure (CHF) is a common complication in patients with an acute coronary syndrome [37]. Patients with unstable angina who present with CHF have 4-fold higher hospital death rates than those without CHF at admission [38]. NT-proBNP level could be used to detect the presence of heart failure on admission. Clinical studies have clearly demonstrated that NT-proBNP levels are increased after episodes of ischemia: elevated NT-proBNP level has been observed in patients with unstable angina [39] and during and after percutaneous coronary intervention. Thus, NT-proBNP reflects ischemia as well as haemodynamics and the magnitude and duration of the increase in plasma concentrations of NT-proBNP after ACS are proportional to myocardial infarct size and the degree of left ventricular dysfunction (although of course ischemia impairs LV function). Additionally, elevated ROCK activation is involved in the pathogenesis of not only atherosclerosis but also inflammation and the contraction of the smooth muscle cells, which plays an important role in the pathogenesis of ACS. Thus, ROCK activity could represent the severity of ACS, as well as number of actively diseased coronary arteries. Knowing more about how ROCK is involved in the disease processes of ACS may help to understand the underlying mechanism and guide the treatment and improve the prognosis of ACS.
Finally, in the current study, we demonstrated that ROCK activity increased in ACS patients and proved that ROCK activity might be a good biomarker to predict long-term event. ROCK activity plus NT-proBNP can identify risk better than NT-proBNP alone in the majority of ACS patients. This is especially true for the ACS patients who have high NT-proBNP but low ROCK activity.
5. Limitations
The main limitation of this study is the relatively small number of patients and a moderately short duration of follow-up. Usually for hazard ratios of 2, a much sample size of patients and events should be needed. Comparison with other biomarkers such as myeloperoxidase and lipocalin would also be interesting.
6. Conclusions
ACS is associated with increased ROCK activity. In addition, both ROCK activity and NT-proBNP can be used synergistically to identify a subset of ACS patients who are at a particularly high risk of future cardiovascular events.
Acknowledgments
I especially appreciate the guidance of Prof. James K. Liao during this project and thank Prof. John E Sanderson for his modifying the manuscript.
The authors of this manuscript have certified that they comply with the Principles of Ethical Publishing in the International Journal of Cardiology [40].
Funding: None.
Abbreviations
- ACS
Acute coronary syndrome
- cTnT
Cardiac troponin T
- ANOVA
One-way analysis of variance
- BH
Body height
- BMI
Body mass index
- BW
Body weight
- CHD
Coronary heart disease
- CHF
Congestive heart failure
- CIHD
Coronary ischemic heart disease
- CPK
Creatine phosphokinase
- CRP
C-reactive protein
- CVA
Cardio vascular accidents
- DBP
Diastolic blood pressure
- DM
Diabetes mellitus
- eGFR
Estimated Glomerular Filtration Rate
- GRACE
Global Registry of Acute Coronary Events
- HDL-C
High-density lipoprotein cholesterol
- HR
Heart rate
- LDL-C
Low-density lipoprotein cholesterol
- LVEF
Left ventricular ejection fraction
- NSTEMI
Non-ST elevation myocardial infarction
- NT-proBNP
N-terminal pro-B-type natriuretic peptide
- PCI
Percutaneous coronary intervention
- ROCK
Rho kinase
- SBP
Systolic blood pressure
- STEMI
ST elevation myocardial infarction
- TC
Total cholesterol
- TG
Triglycerides
- WBC
White blood cells
- UA
Unstable angina
Footnotes
Competing interests: None.
Patient consent: Obtained.
Ethics approval: This study was conducted with the approval of Hong Kong ethics committees.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Armstrong PW, Fu Y, Chang WC, et al. Acute coronary syndromes in the GUSTO-IIb trial: prognostic insights and impact of recurrent ischemia. Circulation. 1998;98:1860–8. doi: 10.1161/01.cir.98.18.1860. [DOI] [PubMed] [Google Scholar]
- 2.Zaacks SM, Liebson PR, Calvin JE, Parrillo JE, Klein LW. Unstable angina and non-Q wave myocardial infarction: does the clinical diagnosis have therapeutic implications? J Am Coll Cardiol. 1999;33:107–18. doi: 10.1016/s0735-1097(98)00553-1. [DOI] [PubMed] [Google Scholar]
- 3.Tang EW, Wong CK, Herbison P. Global Registry of Acute Coronary Events (GRACE) hospital discharge risk score accurately predicts long-term mortality post acute coronary syndrome. Am Heart J. 2007;153:29–35. doi: 10.1016/j.ahj.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 4.Escudier B, Pluzanska A, Koralewski P, et al. AVOREN Trial investigators. Bevacizumab plus interferon alfa-2a for treatment of metastatic renal cell carcinoma: a randomised, double-blind phase III trial. Lancet. 2007;370:2103–11. doi: 10.1016/S0140-6736(07)61904-7. [DOI] [PubMed] [Google Scholar]
- 5.Dong M, Yan BP, Liao JK, Lam YY, Yip GW, Yu CM. Rho-kinase inhibition: a novel therapeutic target for the treatment of cardiovascular diseases. Drug Discov Today. 2010;15:622–9. doi: 10.1016/j.drudis.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mallat Z, Gojova A, Sauzeau V, et al. Rho-associated protein kinase contributes to early atherosclerotic lesion formation in mice. Circ Res. 2003;93:884–8. doi: 10.1161/01.RES.0000099062.55042.9A. [DOI] [PubMed] [Google Scholar]
- 7.Hamid SA, Bower HS, Baxter GF. Rho kinase activation plays a major role as a mediator of irreversible injury in reperfused myocardium. Am J Physiol Heart Circ Physiol. 2007;292:H2598–606. doi: 10.1152/ajpheart.01393.2006. [DOI] [PubMed] [Google Scholar]
- 8.Sato S, Ikegaki I, Asano T, Shimokawa H. Antiischemic properties of fasudil in experimental models of vasospastic angina. Jpn J Pharmacol. 2001;87:34–40. doi: 10.1254/jjp.87.34. [DOI] [PubMed] [Google Scholar]
- 9.Krumholz HM, Anderson JL, Brooks NH, et al. American College of Cardiology/American Heart Association Task Force on Performance Measures; Writing Committee to Develop Performance Measures on ST-Elevation and Non-ST-Elevation Myocardial Infarction. ACC/AHA clinical performance measures for adults with ST-elevation and non-ST-elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Performance Measures (Writing Committee to Develop Performance Measures on ST-Elevation and Non-ST-Elevation Myocardial Infarction) Circulation. 2006;113:732–61. doi: 10.1161/CIRCULATIONAHA.106.172860. [DOI] [PubMed] [Google Scholar]
- 10.de Araujo GP, Ferreira J, Aguiar C, Seabra-Gomes R. TIMI, PURSUIT, and GRACE risk scores: sustained prognostic value and interaction with revascularization in NSTE-ACS. Eur Heart J. 2005;26:865–72. doi: 10.1093/eurheartj/ehi187. [DOI] [PubMed] [Google Scholar]
- 11.Noma K, Goto C, Nishioka K, et al. Smoking, endothelial function, and Rho-kinase in humans. Arterioscler Thromb Vasc Biol. 2005;25:2630–5. doi: 10.1161/01.ATV.0000189304.32725.bd. [DOI] [PubMed] [Google Scholar]
- 12.Kanda T, Wakino S, Homma K, et al. Rho-kinase as a molecular target for insulin resistance and hypertension. FASEB J. 2006;20:169–71. doi: 10.1096/fj.05-4197fje. [DOI] [PubMed] [Google Scholar]
- 13.Liu PY, Liao JK. A method for measuring Rho kinase activity in tissues and cells. Methods Enzymol. 2008;439:181–9. doi: 10.1016/S0076-6879(07)00414-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feske SK, Sorond FA, Henderson GV, et al. Increased leukocyte ROCK activity in patients after acute ischemic stroke. Brain Res. 2009;1257:89–93. doi: 10.1016/j.brainres.2008.12.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vahebi S, Kobayashi T, Warren CM, de Tombe PP, Solaro RJ. Functional effects of rho-kinase-dependent phosphorylation of specific sites on cardiac troponin. Circ Res. 2005;96:740–7. doi: 10.1161/01.RES.0000162457.56568.7d. [DOI] [PubMed] [Google Scholar]
- 16.Takemoto M, Sun J, Hiroki J, Shimokawa H, Liao JK. Rho-kinase mediates hypoxia-induced downregulation of endothelial nitric oxide synthase. Circulation. 2002;106:57–62. doi: 10.1161/01.cir.0000020682.73694.ab. [DOI] [PubMed] [Google Scholar]
- 17.Yamakawa T, Tanaka S, Numaguchi K, et al. Involvement of Rho-kinase in angiotensin II-induced hypertrophy of rat vascular smooth muscle cells. Hypertension. 2000;35:313–8. doi: 10.1161/01.hyp.35.1.313. [DOI] [PubMed] [Google Scholar]
- 18.Bolz SS, Vogel L, Sollinger D, et al. Nitric oxide-induced decrease in calcium sensitivity of resistance arteries is attributable to activation of the myosin light chain phosphatase and antagonized by the RhoA/Rho kinase pathway. Circulation. 2003;107:3081–7. doi: 10.1161/01.CIR.0000074202.19612.8C. [DOI] [PubMed] [Google Scholar]
- 19.van Nieuw Amerongen GP, Vermeer MA, van Hinsbergh VW. Role of RhoA and Rho kinase in lysophosphatidic acid-induced endothelial barrier dysfunction. Arterioscler Thromb Vasc Biol. 2000;20:E127–33. doi: 10.1161/01.atv.20.12.e127. [DOI] [PubMed] [Google Scholar]
- 20.Wang HW, Liu PY, Oyama N, et al. Deficiency of ROCK1 in bone marrow-derived cells protects against atherosclerosis in LDLR−/− mice. FASEB J. 2008;22:3561–70. doi: 10.1096/fj.08-108829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Noma K, Rikitake Y, Oyama N, et al. ROCK1 mediates leukocyte recruitment and neointima formation following vascular injury. J Clin Invest. 2008;118:1632–44. doi: 10.1172/JCI29226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morishige K, Shimokawa H, Eto Y, et al. Adenovirus-mediated transfer of dominant-negative rho-kinase induces a regression of coronary arteriosclerosis in pigs in vivo. Arterioscler Thromb Vasc Biol. 2001;21:548–54. doi: 10.1161/01.atv.21.4.548. [DOI] [PubMed] [Google Scholar]
- 23.Laufs U, Liao JK. Targeting Rho in cardiovascular disease. Circ Res. 2000;87:526–8. doi: 10.1161/01.res.87.7.526. [DOI] [PubMed] [Google Scholar]
- 24.Nohria A, Prsic A, Liu PY, et al. Statins inhibit Rho kinase activity in patients with atherosclerosis. Atherosclerosis. 2009;205:517–21. doi: 10.1016/j.atherosclerosis.2008.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang J, Bian HJ, Li XX, et al. ERK-MAPK signaling opposes rho-kinase to reduce cardiomyocyte apoptosis in heart ischemic preconditioning. Mol Med. 2010;16:307–15. doi: 10.2119/molmed.2009.00121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hidaka T, Hata T, Soga J, et al. Increased leukocyte rho kinase (ROCK) activity and endothelial dysfunction in cigarette smokers. Hypertens Res. 2010;33:354–9. doi: 10.1038/hr.2010.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu PY, Chen JH, Lin JL, Liao JK. Increased Rho kinase activity in a Taiwannese population withmetabolic syndrome. J Am Coll Cardiol. 2007;49:1619–24. doi: 10.1016/j.jacc.2006.12.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hattori T, Shimokawa H, Higashi M, et al. Long-term inhibition of Rho-kinase suppresses left ventricular remodeling after myocardial infarction in mice. Circulation. 2004;109:2234–9. doi: 10.1161/01.CIR.0000127939.16111.58. [DOI] [PubMed] [Google Scholar]
- 29.Sauzeau V, Le ME, Bertoglio J, Scalbert E, Pacaud P, Loirand G. Human urotensin II-induced contraction and arterial smooth muscle cell proliferation are mediated by RhoA and Rho-kinase. Circ Res. 2001;88:1102–4. doi: 10.1161/hh1101.092034. [DOI] [PubMed] [Google Scholar]
- 30.Torsoni AS, Fonseca PM, Crosara-Alberto DP, Franchini KG. Early activation of p160ROCK by pressure overload in rat heart. Am J Physiol Cell Physiol. 2003;284:C1411–9. doi: 10.1152/ajpcell.00098.2002. [DOI] [PubMed] [Google Scholar]
- 31.Rikitake Y, Oyama N, Wang CY, et al. Decreased perivascular fibrosis but not cardiac hypertrophy in ROCK1+/− haploinsufficient mice. Circulation. 2005;112:2959–65. doi: 10.1161/CIRCULATIONAHA.105.584623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chang J, Xie M, Shah VR, et al. Activation of Rho-associated coiled-coil protein kinase 1 (ROCK-1) by caspase-3 cleavage plays an essential role in cardiac myocyte apoptosis. Proc Natl Acad Sci U S A. 2006;103:14495–500. doi: 10.1073/pnas.0601911103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shi J, Zhang YW, Yang Y, Zhang L, Wei L. ROCK1 plays an essential role in the transition from cardiac hypertrophy to failure in mice. J Mol Cell Cardiol. 2010;49:819–28. doi: 10.1016/j.yjmcc.2010.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cannon CP, Braunwald E, McCabe CH, et al. Pravastatin or Atorvastatin Evaluation and Infection Therapy-Thrombolysis in Myocardial Infarction 22 Investigators. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med. 2004;350:1495–504. doi: 10.1056/NEJMoa040583. [DOI] [PubMed] [Google Scholar]
- 35.Ang DS, Wei L, Kao MP, Lang CC, Struthers AD. A comparison between B-type natriuretic peptide, global registry of acute coronary events (GRACE) score and their combination in ACS risk stratification. Heart. 2009;95:1836–42. doi: 10.1136/hrt.2008.160234. [DOI] [PubMed] [Google Scholar]
- 36.Koc M, Karaarslan O, Abali G, Batur MK. Variation in high-sensitivity C-reactive protein levels over 24 hours in patients with stable coronary artery disease. Tex Heart Inst J. 37:42–48. [PMC free article] [PubMed] [Google Scholar]
- 37.The Long-term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group. Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. N Engl J Med. 1998;339:1349–57. doi: 10.1056/NEJM199811053391902. [DOI] [PubMed] [Google Scholar]
- 38.Steg PG, Dabbous OH, Feldman LJ, et al. Determinants and prognostic impact of heart failure complicating acute coronary syndromes: observations from the Global Registry of Acute Coronary Events (GRACE) Circulation. 2004;109:494–9. doi: 10.1161/01.CIR.0000109691.16944.DA. [DOI] [PubMed] [Google Scholar]
- 39.Talwar S, Squire IB, Downie PF, Davies JE, Ng LL. Plasma N terminal pro-brain natriuretic peptide and cardiotrophin 1 are raised in unstable angina. Heart. 2000;84:421–4. doi: 10.1136/heart.84.4.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Coats AJ, Shewan LG. Ethics in the authorship and publishing of scientific articles. Int J Cardiol. 2011;153:239–40. doi: 10.1016/j.ijcard.2011.10.119. [DOI] [PubMed] [Google Scholar]