

Abstract
Statins are 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors, which are widely used to lower serum cholesterol levels in the primary and secondary prevention of cardiovascular disease. Recent experimental and clinical evidence suggest that the beneficial effects of statins may extend beyond their cholesterol lowering effects, to include so-called pleiotropic effects. These cholesterol-independent effects include improving endothelial function, attenuating vascular and myocardial remodeling, inhibiting vascular inflammation and oxidation, and stabilizing atherosclerotic plaques. The mechanism underlying some of these pleiotropic effects is the inhibition of isoprenoid synthesis by statins, which leads to the inhibition of intracellular signaling molecules Rho, Rac and Cdc42. In particular, inhibition of Rho and one of its downstream targets, Rho kinase (ROCK), may be a predominant mechanism contributing to the pleiotropic effects of statins. In this review, we provide an update on the non-cholesterol-dependent statin effects in the cardiovascular system and highlight some of the recent findings from bench to bedside to support the concept of statin pleiotropy.
Keywords: Statin, cholesterol, pleiotropic effects, rho kinase, vascular
Statins or 3-hydroxy-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors are potent inhibitors of cholesterol biosynthesis and are established therapies for the primary and secondary prevention of coronary artery disease. Because serum cholesterol level is strongly associated with coronary heart disease, it has been generally assumed that the beneficial effects underlying statin therapy are entirely due to cholesterol reduction. However, clinical studies suggest that the overall benefits observed with statins may not be mediated solely by their lipid-lowering properties, but possibly through cholesterol-independent or pleiotropic effects 1–3.
PHARMACOKINETIC PROPERTIES OF STATINS
Statins were initially isolated and identified as secondary metabolites of fungi 4, 5. They inhibit the rate-limiting step of cholesterol biosynthesis, the conversion of HMG-CoA to L-mevalonic acid, through binding to HMG-CoA reductase’s active site and blocking the substrate product transition state of the enzyme 6. This leads to decreased hepatic cholesterol synthesis, upregulation of low-density lipoprotein (LDL) receptor, and increased clearance of plasma LDL-cholesterol. In addition, by inhibiting HMG-CoA reductase, statins could also inhibit the synthesis of important isoprenoid intermediates, such as farnesylpyrophosphate (FPP) and geranylgeranylpyrophosphate (GGPP) that lie downstream from L-mevalonic acid 7. These intermediates serve as important lipid attachments for the post-translational modification of intracellular proteins such as nuclear lamins, Ras, Rho, Rac and Rap 8. Thus, it is possible that, in addition to cholesterol lowering, the inhibition of these intracellular isoprenoid-dependent proteins may contribute to some of the biological effects of statins (Figure 1).
Figure 1. Biological actions of isoprenoids.

Statins inhibit HMG-COA reductase acitivity and decreases the isoprenylation of intracellular signaling molecules, such as RhoA, Rac1 and Cdc42.
Because statins differ in their lipophilicity, half-life, and potency 9, they possess different potencies for extra-hepatic HMG-CoA reductase inhibition. These differences in tissue permeability and metabolism may account for some of the observed differences in their peripheral side effects 10, but at the same time, enabling them to have more pleiotropic effects. Lipophilic statins such as simvastatin and fluvastatin are considered more likely to enter vascular cells by passive diffusion than hydrophilic statins such as pravastatin and rosuvastatin, which are primarily targeted to the liver. However, the observation that hydrophilic statins have similar pleiotropic effects as lipophilic statins puts into question whether there are really any cholesterol-independent effects of statins. Indeed, recent evidence suggests that some of the cholesterol-independent effects of these agents may be mediated by inhibition of hepatic HMG-CoA reductase leading subsequent reduction in circulating isoprenoid levels 11. This hypothesis may help explain why hydrophilic statins such as pravastatin and rosuvastatin are still able to exert cholesterol-independent benefits on the vascular wall without directly entering vascular wall cells. In this respect, the word “pleiotropic” may not reflect the hepatic versus non-hepatic effects of these agents.
DIVERSE TARGET POINTS FOR STATIN ACTIONS
Statin and Rho/ROCK
Rho kinases (ROCKs) are protein serine/threonine kinases of ~160 kDa that are downstream effectors of the small GTPase Rho. A critical step for intracellular trafficking and function of these proteins is their post-translational modification through isoprenylation 12, which can be inhibited by statins 13. By inhibiting mevalonate synthesis, statins prevent membrane targeting of Rho and its subsequent activation of ROCKs (Figure 2). Indeed, statins, at clinically relevant concentrations that are used to reduce LDL-cholesterol, have been shown to inhibit Rho isoprenylation 14 and ROCK activity in humans 15. It is therefore interesting to speculate whether some of the clinical benefits of statin therapy could be mediated by inhibition of the Rho/ROCK pathways.
Figure 2. Regulation of the Rho GTPase cycle.

Rho protein cycle between an inactive GDP-bound and an active GTP-bound state. Inhibition of the mevalonate synthesis by statins prevents memrbane targeting of Rho and its subsequent activation of ROCK. The inhibition of the Rho/ROCK pathway may mediate some of the pleiotropic effects of statins on the vascular wall.
Statins and Rac
Rac is a 20–39 kDa monomeric G-protein and also a member of the small GTPase subfamily. The major Rac signaling pathway includes remodeling of the actin cytoskeleton and generation of reactive oxygen species (ROS). Since the development of myocardial hypertrophy is exhibited by ventricular remodeling and increased oxidative stress, Rac may be an important mediator of cardiac hypertrophy 16. Furthermore, results from animal and human studies suggest that some of the pleiotropic effects of statins may be mediated through inhibition of Rac1. For example, in a mouse model, simvastatin prevented angiotensin II (Ang II) or pressure overload induced hypertrophy through inhibition of Rac1-mediated NADPH oxidase activity in vascular smooth muscle and heart 17, 18. This finding is further supported by analysis of failing human heart tissues, where increased ROS generation is associated with increased Rac1 activity, both of which are attenuated by statin treatment 19. Indeed, a recent study indicates that statins at the clinical dosage used among Asians, mainly exhibits it pleiotropic effect through the inhibition of Rac120.
Statins and the Peroxisome Proliferator-Activated Receptor
Statins may also exert pleiotropic effects through activation of peroxisome proliferator-activated receptors (PPARs). In macrophages, statins induce PPAR-gamma transcription activity, inhibit LPS-induced TNF-alpha and MCP-1 expression, and repress the transcriptional activity of nuclear transcription factor and activator protein (AP1) through PPAR-alpha and PPAR-gamma 21. These findings are supported by data demonstrating that statins stabilize atherosclerotic plaques through the activation of PPAR-gamma and that combined administration of simvastatin with PPAR-gamma agonists elicit additive effects on atherosclerotic plaque regression 22–24.
CELLULAR EFFECTS OF STATINS
Statins and the endothelium
Several clinical studies have shown that statins can improve endothelial function in patients with hypercholesterolemia and atherosclerosis through cholesterol-dependent and -independent pathways 25–27. Because endothelium-derived nitric oxide (NO) is an important mediator of endothelial function, there are several different mechanisms by which statins could upregulate endothelial NO synthase (eNOS) (Figure 3).
Figure 3. Upregulation of eNOS by statins.
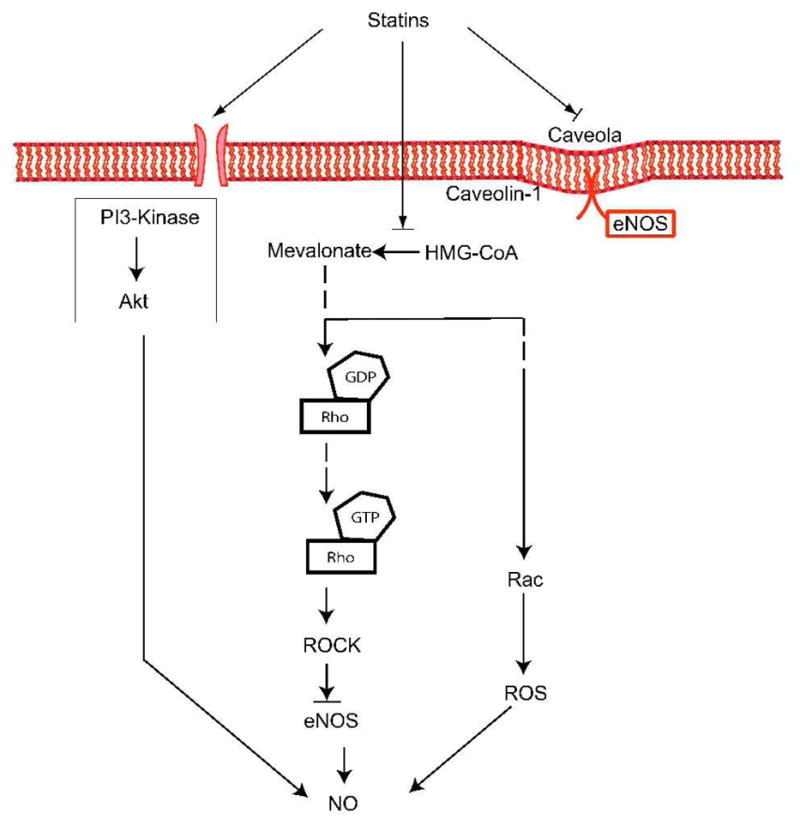
Statins modulate eNOS expression through three major mechanisms: 1) Increased eNOS mRNA stability through inhibition of Rho isoprenylation. 2) Increased eNOS phosphorylation through PI3K-dependent signaling. 3) Restoration of eNOS activity through reduction of caveolin-1 abundance.
The first pathway involves the Rho/ROCK signaling, through which statins increase the stability of eNOS mRNA, leading to increased eNOS expression 28, 29. In-vitro studies show that direct inhibition of Rho by Clostridium botulinum C3 transferase or overexpression of a dominant-negative mutant of RhoA increases eNOS expression, confirming that RhoA negatively regulates eNOS expression and activity 29. Furthermore, inhibition of ROCK by the ROCK inhibitors, fasudil or Y27632, also leads to increased eNOS expression and activity 15, 30, 31.
A second important mechanism by which statins activate eNOS is mediated through the serine-threonine protein kinase Akt. Statins rapidly promote the activation of Akt in endothelial cells leading to eNOS phosphorylation and increased angiogenesis 32. Because this process is inhibited by the phosphatidylinositol-3 kinase (PI3K) inhibitors, wortmannin and LY294002, these findings indicate that statins activate Akt by upregulating PI3K signaling 32. Interestingly, inhibition of the Rho/ROCK pathway also leads to the rapid activation of PI3/Akt pathway and cardioprotection, suggesting a similar mechanism by which statins upregulate eNOS expression 31.
A third mechanism, through which statins regulate eNOS activity, is through their effects on caveolin-1. Caveolin-1 is an integral membrane protein that binds to eNOS in caveolae and thereby inhibit NO production directly 33. Its allosteric competitor calmodulin (CaM) promotes the calcium-dependent activation of eNOS through binding to the CaM-binding motif, and therefore can displace an adjacent auto-inhibitory loop on eNOS 34, 35. In-vitro data with atorvastatin show that caveolin-1 abundance is reduced after statin treatment, leading to restoration of eNOS activity in endothelial cells. This effect is completely reversed by the addition of mevalonate 36.
Finally, statins may exert beneficial non-cholesterol effects through enhancing the mobilization of EPCs 37, 38. Indeed, statins, by stimulating the PI3K/Akt pathway, induce angiogenesis by promoting the mobilization, proliferation, migration, and survival of circulating EPCs 39, 40. Interestingly, this effect is observed at lower concentrations of statins only, while higher concentrations of statins elicit anti-angiogenic effects 41.
Statins and vascular smooth muscle
Vascular smooth muscle cells (VSMC) contribute to vascular proliferative diseases and recent studies have shown that statins can attenuate cytokine-mediated VSMC proliferation in coronary artery smooth muscle cells and also inhibit pathological proliferation such as that observed in transplant-associated arteriopathy 42, 43. The ability of statins to inhibit cell proliferation through an isoprenoid-dependent way is demonstrated in fibroblasts where G1 cell cycle arrest induced by lovastatin is reversed by the addition of mevalonate or GGPP 44. Furthermore, DNA synthesis in VSMC induced by platelet-derived growth factor (PDGF) is reversed by isoprenoid, but not cholesterol 45. It appears that inhibition of Rho may be the predominant effect of statins on VSMC proliferation as the inhibition is reversed by GGPP, but not by farnesyl pyrophosphate or LDL cholesterol 46. Indeed, direct inhibition of Rho by Clostridium botulinum C3 transferase or by a dominant-negative Rho mutant increases p27Kip1 and inhibits SMC proliferation after PDGF stimulation 46. Similarly, another study showed that atorvastatin inhibited serotonin-induced mitogenesis and migration through inhibition of GTP-RhoA formation in pulmonary artery smooth muscle cells 47. This effect is also reversed by GGPP, but not FPP. Taken together, these findings suggest that Rho/ROCK pathway mediates SMC proliferation and that inhibition of Rho isoprenylation by statins may be the predominant mechanism by which statins inhibit vascular SMC proliferation.
Statins and the myocardium
Cardiac hypertrophy is mediated, in part by myocardial oxidative stress. Because Rac1 is required for NADPH oxidase activity, it is likely that statins inhibit cardiac hypertrophy through an antioxidant mechanism involving inhibition of Rac1 geranylgeranylation. Indeed, statins inhibit AngII-induced oxidative stress and cardiac hypertrophy in rodents 17, 48. Data from in-vivo studies also demonstrate a protective function for statins against ischemic myocardial injury 49, 50. Several follow-up studies confirmed these findings in normocholesterolemic as well as hypercholesterolemic animal models 51, 52. One of the main contributors to this protective effect of statins is the increase in NO bioavailability, resulting in increased vasodilation and facilitating regional myocardial blood flow under hypoxic conditions53, 54. Furthermore, statin-induced NO production inhibits the upregulation of adhesion molecules involved in leukocyte-endothelial cell interactions 55. Finally, statins could also preserve mitochondrial membrane potential in response to oxidative stress in cardiac myocytes in a NO-dependent manner 56.
Statins and platelets
A critical role in the manifestation of acute coronary syndromes is mediated by platelets. Circulating platelets are associated with mural thrombus formation at the site of plaque rupture and vascular injury 57. Hypercholesterolemia has been associated with increases in platelet reactivity 58. Although statins could decrease platelet reactivity through the reduction of serum cholesterol levels, recent studies suggest that some of the effects may be non-cholesterol-dependent. For example, in the recent JUPITER trial, treatment with rosuvastatin was associated with decreased venous thromboembolism 59, an effect, which is probably unrelated to cholesterol reduction since hypercholesterolemia is not a risk factor for venous thromboembolism.
One of the non-cholesterol-dependent mechanism includes the statin-mediated upregulation of eNOS in platelets leading to decreased platelet activation 60. This is demonstrated for atorvastatin in a mouse model 60, as well as for fluvastatin in a clinical study with hypercholesterolemic patients 61. Furthermore, statins inhibit tissue factor expression by macrophages and thereby reduce the thrombotic potential of the vascular wall 62. Interestingly, a recent study suggests that PPARs may also mediate some of the antiplatelet actions of statins and fibrates 63.
DIRECT NON-LDL EFFECTS OF STATINS IN CARDIOVASCULAR DISEASE
Statins and atherosclerosis
Atherosclerosis is a complex inflammatory process that is characterized by the cross-talk between excessive inflammation and lipid accumulation 64. Statins have been found to modulate immune activation and to exert anti-inflammatory effects on the vascular wall by decreasing the number of inflammatory cells in atherosclerotic plaques 65. The mechanism is due, in part, to the immunomodulatory ability of statins to decrease the expression of endothelial adhesion molecules, such as ICAM-1, VCAM-1 and E-selectin directly 66, 67. Furthermore, statins attenuate P-selectin expression and leukocyte adhesion by increasing endothelial NO production 68. Interestingly, this cholesterol-independent effect of statins was absent in eNOS-deficient mice, suggesting that eNOS mediates some of the vascular protective effects of statins 69.
T-lymphocytes also play an important role in the pathogenesis of atherosclerosis. The activation of T-lymphocytes and the control of the immune response are mediated by the major histocompatibility complex class II (MHC-II) and CD40/CD40L. Statins inhibit MHC-II expression on endothelial cells and monocyte-macrophages via inhibition of the promoter IV of the transactivator CIITA and thereby repress MHC-II mediated T-cell activation 70. In addition, statins decrease INF-gamma induced CD40 expression and CD40-related activation of vascular cells 71, 72. Interestingly, statins could also modulate LFA, a major counter receptor for ICAM-1 on leukocytes, through a non-Rho/ROCK pathway 73. By binding directly to its regulatory site in the beta-2 integrin, statin inhibit T-cell activation and suppress the inflammatory response independent of HMG-CoA reductase inhibition and small GTPases 73, 74.
Statins and stroke
Although the correlation between elevated cholesterol level and cerebrovascular disease remains controversial 75, 76, statins have become one of the most important therapies for stroke prevention and treatment. In humans, statins may prevent ischemic strokes through their vascular effects. Because studies with other lipid-modifying drugs have not been successful in reducing the incidence of stroke, these findings suggest that statins may protect against stroke through non-cholesterol-dependent mechanisms 77, 78.
One possible target for statin in stroke prevention could be the effects of statin on eNOS. Following cerebrovascular occlusion, eNOS−/− mice develop greater proliferative, inflammatory response to vascular injury and larger cerebral infarcts 79, 80. However, mice pre-treated with statins, show higher cerebral blood flow and smaller cerebral infarct sizes following cerebrovascular occlusion 81. In contrast, no increase in cerebral blood flow or neuroprotection was observed in eNOS−/− mice treated with statins, indicating that the upregulation of eNOS accounts for most of the neuroprotective effects of these agents 80.
EVIDENCE OF STATIN PLEIOTROPY IN CLINICAL TRIALS
Traditionally, it has been assumed that the beneficial effects underlying statin therapy are predominantly due to cholesterol reduction. However, several clinical trials have suggested that their benefits may extend beyond cholesterol, supporting the notion that the clinical benefits of statin therapy may be independent of LDL-cholesterol reduction. Recently, the JUPITER (Justification for the Use of statin in Primary prevention: an Intervention Trial Evaluating Rosuvastatin) trial showed that statins could be beneficial in the primary prevention of cardiovascular disease in patients with elevated hsCRP, but relatively low cholesterol levels and other cardiovascular risks 82. This study evaluated the effect of rosuvastatin (20 mg/day) in reducing the rate of cardiovascular events among apparently healthy subjects with LDL-cholesterol levels of less than 130mg/dl (3.4 mmol/L), but hsCRP levels of greater than 2 mg/L 83. Subanalysis of this trial show that asymptomatic individuals randomly allocated to rosuvastatin benefited particularly if low concentrations of both LDL-cholesterol and C-reactive proteins were achieved 84. These finding supports the importance of inflammatory components in mediating cardiovascular diseases, but also suggests a non-cholesterol-dependent effect of statins since the reduction in hsCRP by rosuvastatin was not related to the reduction in LDL-cholesterol. Interestingly, recent findings from the COSMOS (Multicenter coronary atherosclerosis study measuring effects of Rosuvastatin using intravascular ultrasound in Japanese subjects) trial confirm these data. Using intravascular ultrasound (IVUS) to investigate the effect of rosuvastatin on coronary atherosclerosis in Japanese subjects with ischemic heart diseases, the authors found that rosuvastatin treatment significantly reduced intracoronary plaque volume and increased in lumen volume. However, there was no significant correlation between the reduction in plaque volume and the reduction in plasma LDL, supporting the idea of effects beyond cholesterol lowering 85.
In addition, two clinical trials with ezetimibe further support the notion of statin pleiotropy in humans. Ezetimibe which acts by decreasing cholesterol absorption in the intestine is used alone or in combination with statin therapy to enhance lipid lowering 86, 87. Interestingly, several clinical studies have demonstrated beneficial effects of statins over ezetimibe despite comparable reduction in LDL-cholesterol 27, 88. In the ENHANCE (Ezetimibe and Simvastatin in Hypercholesterolemia Enhances Atherosclerosis Regression) study – a double blind, randomized trial comparing ezetimibe/simvastatin (10 mg/80 mg) to the highest recommended dose of 80 mg simvastatin, ezetimibe/simvastatin combination did not reduce the intima-media thickness of the carotid artery wall in patients with familial hypercholesterolemia, despite significant incremental reductions in levels of both LDL-cholesterol and C-reactive protein 89. In addition, our group has recently compared high-dose statin monotherapy with equivalent cholesterol-lowering efficacy of the same statin at a lower dose plus ezetimibe. Two recent studies demonstrated that high dose statins alone improve flow-mediated dilation (FMD) more than dual therapy with low-dose statin and ezitimibe despite comparable reductions in LDL-cholesterol 25, 90.
FUTURE DIRECTIONS
Because of the potential for pleiotropic effects, statin therapy is currently being considered for medical conditions beyond cardiovascular disease. Statin pleiotropy, however, is still an evolving concept and clinical studies have yet to show how much of the benefits of statins are non-LDL-dependent. Part of the frustration is due to the lack of a definitive biomarker for statin pleiotropy. With the molecular insights obtained from basic studies regarding isoprenoid synthesis and inhibition of the Rho/ROCK and Rac pathways, it may be possible in the near future to actually define the clinical benefits of statin therapy attributed to non-LDL effects. For example, measurement of leukocyte ROCK activity is currently being utilized to identify patients at cardiovascular risks and to gauge statin efficacy on vascular function that is non-LDL-dependent 25, 91, 92. Whether ROCK activity could also independently predict cardiovascular outcomes of statin therapy beyond cholesterol reduction remains to be determined.
Acknowledgments
This work is supported by grants from the National Institutes of Health HL052233 to JK. Liao and the Deutsche Forschungsgesellschaft (ZH 231/1-1) to Q.Zhou.
References
- 1.Palinski W. New evidence for beneficial effects of statins unrelated to lipid lowering. Arterioscler Thromb Vasc Biol. 2001;21:3–5. doi: 10.1161/01.atv.21.1.3. [DOI] [PubMed] [Google Scholar]
- 2.Wang CY, Liu PY, Liao JK. Pleiotropic effects of statin therapy: molecular mechanisms and clinical results. Trends Mol Med. 2008;14:37–44. doi: 10.1016/j.molmed.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhou Q, Liao JK. Statins and cardiovascular diseases: from cholesterol lowering to pleiotropy. Curr Pharm Des. 2009;15:467–478. doi: 10.2174/138161209787315684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alberts AW, Chen J, Kuron G, Hunt V, Huff J, Hoffman C, Rothrock J, Lopez M, Joshua H, Harris E, Patchett A, Monaghan R, Currie S, Stapley E, Albers-Schonberg G, Hensens O, Hirshfield J, Hoogsteen K, Liesch J, Springer J. Mevinolin: a highly potent competitive inhibitor of hydroxymethylglutaryl-coenzyme A reductase and a cholesterol-lowering agent. Proc Natl Acad Sci U S A. 1980;77:3957–3961. doi: 10.1073/pnas.77.7.3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alberts AW. Discovery, biochemistry and biology of lovastatin. Am J Cardiol. 1988;62:10J–15J. doi: 10.1016/0002-9149(88)90002-1. [DOI] [PubMed] [Google Scholar]
- 6.Istvan ES, Deisenhofer J. Structural mechanism for statin inhibition of HMG-CoA reductase. Science. 2001;292:1160–1164. doi: 10.1126/science.1059344. [DOI] [PubMed] [Google Scholar]
- 7.Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature. 1990;343:425–430. doi: 10.1038/343425a0. [DOI] [PubMed] [Google Scholar]
- 8.Van Aelst L, D’Souza-Schorey C. Rho GTPases and signaling networks. Genes Dev. 1997;11:2295–2322. doi: 10.1101/gad.11.18.2295. [DOI] [PubMed] [Google Scholar]
- 9.Illingworth DR, Tobert JA. HMG-CoA reductase inhibitors. Adv Protein Chem. 2001;56:77–114. doi: 10.1016/s0065-3233(01)56003-9. [DOI] [PubMed] [Google Scholar]
- 10.Germershausen JI, Hunt VM, Bostedor RG, Bailey PJ, Karkas JD, Alberts AW. Tissue selectivity of the cholesterol-lowering agents lovastatin, simvastatin and pravastatin in rats in vivo. Biochem Biophys Res Commun. 1989;158:667–675. doi: 10.1016/0006-291x(89)92773-3. [DOI] [PubMed] [Google Scholar]
- 11.Corsini A, Bellosta S, Baetta R, Fumagalli R, Paoletti R, Bernini F. New insights into the pharmacodynamic and pharmacokinetic properties of statins. Pharmacol Ther. 1999;84:413–428. doi: 10.1016/s0163-7258(99)00045-5. [DOI] [PubMed] [Google Scholar]
- 12.Hall A. G proteins and small GTPases: distant relatives keep in touch. Science. 1998;280:2074–2075. doi: 10.1126/science.280.5372.2074. [DOI] [PubMed] [Google Scholar]
- 13.Laufs U, Kilter H, Konkol C, Wassmann S, Bohm M, Nickenig G. Impact of HMG CoA reductase inhibition on small GTPases in the heart. Cardiovasc Res. 2002;53:911–920. doi: 10.1016/s0008-6363(01)00540-5. [DOI] [PubMed] [Google Scholar]
- 14.Cicha I, Schneiderhan-Marra N, Yilmaz A, Garlichs CD, Goppelt-Struebe M. Monitoring the cellular effects of HMG-CoA reductase inhibitors in vitro and ex vivo. Arterioscler Thromb Vasc Biol. 2004;24:2046–2050. doi: 10.1161/01.ATV.0000145943.19099.a3. [DOI] [PubMed] [Google Scholar]
- 15.Takemoto M, Sun J, Hiroki J, Shimokawa H, Liao JK. Rho-kinase mediates hypoxia-induced downregulation of endothelial nitric oxide synthase. Circulation. 2002;106:57–62. doi: 10.1161/01.cir.0000020682.73694.ab. [DOI] [PubMed] [Google Scholar]
- 16.Brown JH, Del Re DP, Sussman MA. The Rac and Rho hall of fame: a decade of hypertrophic signaling hits. Circ Res. 2006;98:730–742. doi: 10.1161/01.RES.0000216039.75913.9e. [DOI] [PubMed] [Google Scholar]
- 17.Takemoto M, Node K, Nakagami H, Liao Y, Grimm M, Takemoto Y, Kitakaze M, Liao JK. Statins as antioxidant therapy for preventing cardiac myocyte hypertrophy. J Clin Invest. 2001;108:1429–1437. doi: 10.1172/JCI13350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wassmann S, Laufs U, Baumer AT, Muller K, Konkol C, Sauer H, Bohm M, Nickenig G. Inhibition of geranylgeranylation reduces angiotensin II-mediated free radical production in vascular smooth muscle cells: involvement of angiotensin AT1 receptor expression and Rac1 GTPase. Mol Pharmacol. 2001;59:646–654. doi: 10.1124/mol.59.3.646. [DOI] [PubMed] [Google Scholar]
- 19.Maack C, Kartes T, Kilter H, Schafers HJ, Nickenig G, Bohm M, Laufs U. Oxygen free radical release in human failing myocardium is associated with increased activity of rac1-GTPase and represents a target for statin treatment. Circulation. 2003;108:1567–1574. doi: 10.1161/01.CIR.0000091084.46500.BB. [DOI] [PubMed] [Google Scholar]
- 20.Rashid M, Tawara S, Fukumoto Y, Seto M, Yano K, Shimokawa H. Importance of Rac1 signaling pathway inhibition in the pleiotropic effects of HMG-CoA reductase inhibitors. Circ J. 2009;73:361–370. doi: 10.1253/circj.cj-08-0817. [DOI] [PubMed] [Google Scholar]
- 21.Yano M, Matsumura T, Senokuchi T, Ishii N, Murata Y, Taketa K, Motoshima H, Taguchi T, Sonoda K, Kukidome D, Takuwa Y, Kawada T, Brownlee M, Nishikawa T, Araki E. Statins activate peroxisome proliferator-activated receptor gamma through extracellular signal-regulated kinase 1/2 and p38 mitogen-activated protein kinase-dependent cyclooxygenase-2 expression in macrophages. Circ Res. 2007;100:1442–1451. doi: 10.1161/01.RES.0000268411.49545.9c. [DOI] [PubMed] [Google Scholar]
- 22.Sugamura K, Sugiyama S, Matsuzawa Y, Nozaki T, Horibata Y, Ogawa H. Benefit of adding pioglitazone to successful statin therapy in nondiabetic patients with coronary artery disease. Circ J. 2008;72:1193–1197. doi: 10.1253/circj.72.1193. [DOI] [PubMed] [Google Scholar]
- 23.Zelvyte I, Dominaitiene R, Crisby M, Janciauskiene S. Modulation of inflammatory mediators and PPARgamma and NFkappaB expression by pravastatin in response to lipoproteins in human monocytes in vitro. Pharmacol Res. 2002;45:147–154. doi: 10.1006/phrs.2001.0922. [DOI] [PubMed] [Google Scholar]
- 24.Corti R, Osende JI, Fallon JT, Fuster V, Mizsei G, Jneid H, Wright SD, Chaplin WF, Badimon JJ. The selective peroxisomal proliferator-activated receptor-gamma agonist has an additive effect on plaque regression in combination with simvastatin in experimental atherosclerosis: in vivo study by high-resolution magnetic resonance imaging. J Am Coll Cardiol. 2004;43:464–473. doi: 10.1016/j.jacc.2003.08.048. [DOI] [PubMed] [Google Scholar]
- 25.Liu PY, Liu YW, Lin LJ, Chen JH, Liao JK. Evidence for statin pleiotropy in humans: differential effects of statins and ezetimibe on rho-associated coiled-coil containing protein kinase activity, endothelial function, and inflammation. Circulation. 2009;119:131–138. doi: 10.1161/CIRCULATIONAHA.108.813311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Robinson JG, Smith B, Maheshwari N, Schrott H. Pleiotropic effects of statins: benefit beyond cholesterol reduction? A meta-regression analysis. J Am Coll Cardiol. 2005;46:1855–1862. doi: 10.1016/j.jacc.2005.05.085. [DOI] [PubMed] [Google Scholar]
- 27.Landmesser U, Bahlmann F, Mueller M, Spiekermann S, Kirchhoff N, Schulz S, Manes C, Fischer D, de Groot K, Fliser D, Fauler G, Marz W, Drexler H. Simvastatin versus ezetimibe: pleiotropic and lipid-lowering effects on endothelial function in humans. Circulation. 2005;111:2356–2363. doi: 10.1161/01.CIR.0000164260.82417.3F. [DOI] [PubMed] [Google Scholar]
- 28.Laufs U, La Fata V, Plutzky J, Liao JK. Upregulation of endothelial nitric oxide synthase by HMG CoA reductase inhibitors. Circulation. 1998;97:1129–1135. doi: 10.1161/01.cir.97.12.1129. [DOI] [PubMed] [Google Scholar]
- 29.Laufs U, Liao JK. Post-transcriptional regulation of endothelial nitric oxide synthase mRNA stability by Rho GTPase. J Biol Chem. 1998;273:24266–24271. doi: 10.1074/jbc.273.37.24266. [DOI] [PubMed] [Google Scholar]
- 30.Rikitake Y, Kim HH, Huang Z, Seto M, Yano K, Asano T, Moskowitz MA, Liao JK. Inhibition of Rho kinase (ROCK) leads to increased cerebral blood flow and stroke protection. Stroke. 2005;36:2251–2257. doi: 10.1161/01.STR.0000181077.84981.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wolfrum S, Dendorfer A, Rikitake Y, Stalker TJ, Gong Y, Scalia R, Dominiak P, Liao JK. Inhibition of Rho-kinase leads to rapid activation of phosphatidylinositol 3-kinase/protein kinase Akt and cardiovascular protection. Arterioscler Thromb Vasc Biol. 2004;24:1842–1847. doi: 10.1161/01.ATV.0000142813.33538.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kureishi Y, Luo Z, Shiojima I, Bialik A, Fulton D, Lefer DJ, Sessa WC, Walsh K. The HMG-CoA reductase inhibitor simvastatin activates the protein kinase Akt and promotes angiogenesis in normocholesterolemic animals. Nat Med. 2000;6:1004–1010. doi: 10.1038/79510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Plenz GA, Hofnagel O, Robenek H. Differential modulation of caveolin-1 expression in cells of the vasculature by statins. Circulation. 2004:109:e7–8. doi: 10.1161/01.CIR.0000111128.83347.7A. author reply e7–8. [DOI] [PubMed] [Google Scholar]
- 34.Michel JB, Feron O, Sacks D, Michel T. Reciprocal regulation of endothelial nitric-oxide synthase by Ca2+-calmodulin and caveolin. J Biol Chem. 1997;272:15583–15586. doi: 10.1074/jbc.272.25.15583. [DOI] [PubMed] [Google Scholar]
- 35.Michel JB, Feron O, Sase K, Prabhakar P, Michel T. Caveolin versus calmodulin. Counterbalancing allosteric modulators of endothelial nitric oxide synthase. J Biol Chem. 1997;272:25907–25912. doi: 10.1074/jbc.272.41.25907. [DOI] [PubMed] [Google Scholar]
- 36.Feron O, Dessy C, Desager JP, Balligand JL. Hydroxy-methylglutaryl-coenzyme A reductase inhibition promotes endothelial nitric oxide synthase activation through a decrease in caveolin abundance. Circulation. 2001;103:113–118. doi: 10.1161/01.cir.103.1.113. [DOI] [PubMed] [Google Scholar]
- 37.Vasa M, Fichtlscherer S, Adler K, Aicher A, Martin H, Zeiher AM, Dimmeler S. Increase in circulating endothelial progenitor cells by statin therapy in patients with stable coronary artery disease. Circulation. 2001;103:2885–2890. doi: 10.1161/hc2401.092816. [DOI] [PubMed] [Google Scholar]
- 38.Spiel AO, Mayr FB, Leitner JM, Firbas C, Sieghart W, Jilma B. Simvastatin and rosuvastatin mobilize Endothelial Progenitor Cells but do not prevent their acute decrease during systemic inflammation. Thromb Res. 2008 doi: 10.1016/j.thromres.2008.03.007. [DOI] [PubMed] [Google Scholar]
- 39.Dimmeler S, Aicher A, Vasa M, Mildner-Rihm C, Adler K, Tiemann M, Rutten H, Fichtlscherer S, Martin H, Zeiher AM. HMG-CoA reductase inhibitors (statins) increase endothelial progenitor cells via the PI 3-kinase/Akt pathway. J Clin Invest. 2001;108:391–397. doi: 10.1172/JCI13152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Llevadot J, Murasawa S, Kureishi Y, Uchida S, Masuda H, Kawamoto A, Walsh K, Isner JM, Asahara T. HMG-CoA reductase inhibitor mobilizes bone marrow--derived endothelial progenitor cells. J Clin Invest. 2001;108:399–405. doi: 10.1172/JCI13131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weis M, Heeschen C, Glassford AJ, Cooke JP. Statins have biphasic effects on angiogenesis. Circulation. 2002;105:739–745. doi: 10.1161/hc0602.103393. [DOI] [PubMed] [Google Scholar]
- 42.Chandrasekar B, Mummidi S, Mahimainathan L, Patel DN, Bailey SR, Imam SZ, Greene WC, Valente AJ. Interleukin-18-induced human coronary artery smooth muscle cell migration is dependent on NF-kappaB- and AP-1-mediated matrix metalloproteinase-9 expression and is inhibited by atorvastatin. J Biol Chem. 2006;281:15099–15109. doi: 10.1074/jbc.M600200200. [DOI] [PubMed] [Google Scholar]
- 43.Shimizu K, Aikawa M, Takayama K, Libby P, Mitchell RN. Direct anti-inflammatory mechanisms contribute to attenuation of experimental allograft arteriosclerosis by statins. Circulation. 2003;108:2113–2120. doi: 10.1161/01.CIR.0000092949.67153.74. [DOI] [PubMed] [Google Scholar]
- 44.Jakobisiak M, Bruno S, Skierski JS, Darzynkiewicz Z. Cell cycle-specific effects of lovastatin. Proc Natl Acad Sci U S A. 1991;88:3628–3632. doi: 10.1073/pnas.88.9.3628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang Z, Kozai T, van der Loo B, Viswambharan H, Lachat M, Turina MI, Malinski T, Luscher TF. HMG-CoA reductase inhibition improves endothelial cell function and inhibits smooth muscle cell proliferation in human saphenous veins. J Am Coll Cardiol. 2000;36:1691–1697. doi: 10.1016/s0735-1097(00)00924-4. [DOI] [PubMed] [Google Scholar]
- 46.Laufs U, Marra D, Node K, Liao JK. 3-Hydroxy-3-methylglutaryl-CoA reductase inhibitors attenuate vascular smooth muscle proliferation by preventing rho GTPase-induced down-regulation of p27(Kip1) J Biol Chem. 1999;274:21926–21931. doi: 10.1074/jbc.274.31.21926. [DOI] [PubMed] [Google Scholar]
- 47.Li M, Liu Y, Dutt P, Fanburg BL, Toksoz D. Inhibition of serotonin-induced mitogenesis, migration, and ERK MAPK nuclear translocation in vascular smooth muscle cells by atorvastatin. Am J Physiol Lung Cell Mol Physiol. 2007;293:L463–471. doi: 10.1152/ajplung.00133.2007. [DOI] [PubMed] [Google Scholar]
- 48.Hauck L, Harms C, Grothe D, An J, Gertz K, Kronenberg G, Dietz R, Endres M, von Harsdorf R. Critical role for FoxO3a-dependent regulation of p21CIP1/WAF1 in response to statin signaling in cardiac myocytes. Circ Res. 2007;100:50–60. doi: 10.1161/01.RES.0000254704.92532.b9. [DOI] [PubMed] [Google Scholar]
- 49.Lefer AM, Campbell B, Shin YK, Scalia R, Hayward R, Lefer DJ. Simvastatin preserves the ischemic-reperfused myocardium in normocholesterolemic rat hearts. Circulation. 1999;100:178–184. doi: 10.1161/01.cir.100.2.178. [DOI] [PubMed] [Google Scholar]
- 50.Matsuki A, Igawa A, Nozawa T, Nakadate T, Igarashi N, Nonomura M, Inoue H. Early administration of fluvastatin, but not at the onset of ischemia or reperfusion, attenuates myocardial ischemia-reperfusion injury through the nitric oxide pathway rather than its antioxidant property. Circ J. 2006;70:1643–1649. doi: 10.1253/circj.70.1643. [DOI] [PubMed] [Google Scholar]
- 51.Bulhak A, Roy J, Hedin U, Sjoquist PO, Pernow J. Cardioprotective effect of rosuvastatin in vivo is dependent on inhibition of geranylgeranyl pyrophosphate and altered RhoA membrane translocation. Am J Physiol Heart Circ Physiol. 2007;292:H3158–3163. doi: 10.1152/ajpheart.01354.2006. [DOI] [PubMed] [Google Scholar]
- 52.D’Annunzio V, Donato M, Erni L, Miksztowicz V, Buchholz B, Carrion CL, Schreier L, Wikinski R, Gelpi RJ, Berg G, Basso N. Rosuvastatin given during reperfusion decreases infarct size and inhibits matrix metalloproteinase-2 activity in normocholesterolemic and hypercholesterolemic rabbits. J Cardiovasc Pharmacol. 2009;53:137–144. doi: 10.1097/FJC.0b013e318197c5e9. [DOI] [PubMed] [Google Scholar]
- 53.Laufs U, Fata VL, Liao JK. Inhibition of 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase blocks hypoxia-mediated down-regulation of endothelial nitric oxide synthase. J Biol Chem. 1997;272:31725–31729. doi: 10.1074/jbc.272.50.31725. [DOI] [PubMed] [Google Scholar]
- 54.Di Napoli P, Taccardi AA, Grilli A, De Lutiis MA, Barsotti A, Felaco M, De Caterina R. Chronic treatment with rosuvastatin modulates nitric oxide synthase expression and reduces ischemia-reperfusion injury in rat hearts. Cardiovasc Res. 2005;66:462–471. doi: 10.1016/j.cardiores.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 55.De Caterina R, Libby P, Peng HB, Thannickal VJ, Rajavashisth TB, Gimbrone MA, Jr, Shin WS, Liao JK. Nitric oxide decreases cytokine-induced endothelial activation. Nitric oxide selectively reduces endothelial expression of adhesion molecules and proinflammatory cytokines. J Clin Invest. 1995;96:60–68. doi: 10.1172/JCI118074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jones SP, Teshima Y, Akao M, Marban E. Simvastatin attenuates oxidant-induced mitochondrial dysfunction in cardiac myocytes. Circ Res. 2003;93:697–699. doi: 10.1161/01.RES.0000097262.21507.DF. [DOI] [PubMed] [Google Scholar]
- 57.Libby P. Molecular bases of the acute coronary syndromes. Circulation. 1995;91:2844–2850. doi: 10.1161/01.cir.91.11.2844. [DOI] [PubMed] [Google Scholar]
- 58.Opper C, Clement C, Schwarz H, Krappe J, Steinmetz A, Schneider J, Wesemann W. Increased number of high sensitive platelets in hypercholesterolemia, cardiovascular diseases, and after incubation with cholesterol. Atherosclerosis. 1995;113:211–217. doi: 10.1016/0021-9150(94)05448-r. [DOI] [PubMed] [Google Scholar]
- 59.Glynn RJ, Danielson E, Fonseca FA, Genest J, Gotto AM, Jr, Kastelein JJ, Koenig W, Libby P, Lorenzatti AJ, MacFadyen JG, Nordestgaard BG, Shepherd J, Willerson JT, Ridker PM. A randomized trial of rosuvastatin in the prevention of venous thromboembolism. N Engl J Med. 2009;360:1851–1861. doi: 10.1056/NEJMoa0900241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Laufs U, Gertz K, Huang P, Nickenig G, Bohm M, Dirnagl U, Endres M. Atorvastatin upregulates type III nitric oxide synthase in thrombocytes, decreases platelet activation, and protects from cerebral ischemia in normocholesterolemic mice. Stroke. 2000;31:2442–2449. doi: 10.1161/01.str.31.10.2442. [DOI] [PubMed] [Google Scholar]
- 61.Haramaki N, Ikeda H, Takenaka K, Katoh A, Sugano R, Yamagishi S, Matsuoka H, Imaizumi T. Fluvastatin alters platelet aggregability in patients with hypercholesterolemia: possible improvement of intraplatelet redox imbalance via HMG-CoA reductase. Arterioscler Thromb Vasc Biol. 2007;27:1471–1477. doi: 10.1161/ATVBAHA.106.128793. [DOI] [PubMed] [Google Scholar]
- 62.Aikawa M, Rabkin E, Sugiyama S, Voglic SJ, Fukumoto Y, Furukawa Y, Shiomi M, Schoen FJ, Libby P. An HMG-CoA reductase inhibitor, cerivastatin, suppresses growth of macrophages expressing matrix metalloproteinases and tissue factor in vivo and in vitro. Circulation. 2001;103:276–283. doi: 10.1161/01.cir.103.2.276. [DOI] [PubMed] [Google Scholar]
- 63.Ali FY, Armstrong PC, Dhanji AR, Tucker AT, Paul-Clark MJ, Mitchell JA, Warner TD. Antiplatelet actions of statins and fibrates are mediated by PPARs. Arterioscler Thromb Vasc Biol. 2009;29:706–711. doi: 10.1161/ATVBAHA.108.183160. [DOI] [PubMed] [Google Scholar]
- 64.Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–874. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 65.Vaughan CJ, Gotto AM, Jr, Basson CT. The evolving role of statins in the management of atherosclerosis. J Am Coll Cardiol. 2000;35:1–10. doi: 10.1016/s0735-1097(99)00525-2. [DOI] [PubMed] [Google Scholar]
- 66.Rezaie-Majd A, Prager GW, Bucek RA, Schernthaner GH, Maca T, Kress HG, Valent P, Binder BR, Minar E, Baghestanian M. Simvastatin reduces the expression of adhesion molecules in circulating monocytes from hypercholesterolemic patients. Arterioscler Thromb Vasc Biol. 2003;23:397–403. doi: 10.1161/01.ATV.0000059384.34874.F0. [DOI] [PubMed] [Google Scholar]
- 67.Rasmussen LM, Hansen PR, Nabipour MT, Olesen P, Kristiansen MT, Ledet T. Diverse effects of inhibition of 3-hydroxy-3-methylglutaryl-CoA reductase on the expression of VCAM-1 and E-selectin in endothelial cells. Biochem J. 2001;360:363–370. doi: 10.1042/0264-6021:3600363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Scalia R, Gooszen ME, Jones SP, Hoffmeyer M, Rimmer DM, 3rd, Trocha SD, Huang PL, Smith MB, Lefer AM, Lefer DJ. Simvastatin exerts both anti-inflammatory and cardioprotective effects in apolipoprotein E-deficient mice. Circulation. 2001;103:2598–2603. doi: 10.1161/01.cir.103.21.2598. [DOI] [PubMed] [Google Scholar]
- 69.Stalker TJ, Lefer AM, Scalia R. A new HMG-CoA reductase inhibitor, rosuvastatin, exerts anti-inflammatory effects on the microvascular endothelium: the role of mevalonic acid. Br J Pharmacol. 2001;133:406–412. doi: 10.1038/sj.bjp.0704070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kwak B, Mulhaupt F, Myit S, Mach F. Statins as a newly recognized type of immunomodulator. Nat Med. 2000;6:1399–1402. doi: 10.1038/82219. [DOI] [PubMed] [Google Scholar]
- 71.Mulhaupt F, Matter CM, Kwak BR, Pelli G, Veillard NR, Burger F, Graber P, Luscher TF, Mach F. Statins (HMG-CoA reductase inhibitors) reduce CD40 expression in human vascular cells. Cardiovasc Res. 2003;59:755–766. doi: 10.1016/s0008-6363(03)00515-7. [DOI] [PubMed] [Google Scholar]
- 72.Wagner AH, Gebauer M, Guldenzoph B, Hecker M. 3-hydroxy-3-methylglutaryl coenzyme A reductase-independent inhibition of CD40 expression by atorvastatin in human endothelial cells. Arterioscler Thromb Vasc Biol. 2002;22:1784–1789. doi: 10.1161/01.atv.0000037098.20829.31. [DOI] [PubMed] [Google Scholar]
- 73.Weitz-Schmidt G, Welzenbach K, Brinkmann V, Kamata T, Kallen J, Bruns C, Cottens S, Takada Y, Hommel U. Statins selectively inhibit leukocyte function antigen-1 by binding to a novel regulatory integrin site. Nat Med. 2001;7:687–692. doi: 10.1038/89058. [DOI] [PubMed] [Google Scholar]
- 74.Weitz-Schmidt G, Welzenbach K, Dawson J, Kallen J. Improved lymphocyte function-associated antigen-1 (LFA-1) inhibition by statin derivatives: molecular basis determined by x-ray analysis and monitoring of LFA-1 conformational changes in vitro and ex vivo. J Biol Chem. 2004;279:46764–46771. doi: 10.1074/jbc.M407951200. [DOI] [PubMed] [Google Scholar]
- 75.Nassief A, Marsh JD. Statin therapy for stroke prevention. Stroke. 2008;39:1042–1048. doi: 10.1161/STROKEAHA.107.501361. [DOI] [PubMed] [Google Scholar]
- 76.Cholesteroldiastolic blood pressure, and stroke: 13,000 strokes in 450,000 people in 45 prospective cohorts. Prospective studies collaboration. Lancet. 1995;346:1647–1653. [PubMed] [Google Scholar]
- 77.Tanne D, Koren-Morag N, Graff E, Goldbourt U. Blood lipids and first-ever ischemic stroke/transient ischemic attack in the Bezafibrate Infarction Prevention (BIP) Registry: high triglycerides constitute an independent risk factor. Circulation. 2001;104:2892–2897. doi: 10.1161/hc4901.100384. [DOI] [PubMed] [Google Scholar]
- 78.Bloomfield Rubins H, Davenport J, Babikian V, Brass LM, Collins D, Wexler L, Wagner S, Papademetriou V, Rutan G, Robins SJ. Reduction in stroke with gemfibrozil in men with coronary heart disease and low HDL cholesterol: The Veterans Affairs HDL Intervention Trial (VA-HIT) Circulation. 2001;103:2828–2833. doi: 10.1161/01.cir.103.23.2828. [DOI] [PubMed] [Google Scholar]
- 79.Huang PL, Huang Z, Mashimo H, Bloch KD, Moskowitz MA, Bevan JA, Fishman MC. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature. 1995;377:239–242. doi: 10.1038/377239a0. [DOI] [PubMed] [Google Scholar]
- 80.Huang Z, Huang PL, Ma J, Meng W, Ayata C, Fishman MC, Moskowitz MA. Enlarged infarcts in endothelial nitric oxide synthase knockout mice are attenuated by nitro-L-arginine. J Cereb Blood Flow Metab. 1996;16:981–987. doi: 10.1097/00004647-199609000-00023. [DOI] [PubMed] [Google Scholar]
- 81.Endres M, Laufs U, Huang Z, Nakamura T, Huang P, Moskowitz MA, Liao JK. Stroke protection by 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase inhibitors mediated by endothelial nitric oxide synthase. Proc Natl Acad Sci U S A. 1998;95:8880–8885. doi: 10.1073/pnas.95.15.8880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ridker PM. Rosuvastatin in the primary prevention of cardiovascular disease among patients with low levels of low-density lipoprotein cholesterol and elevated high-sensitivity C-reactive protein: rationale and design of the JUPITER trial. Circulation. 2003;108:2292–2297. doi: 10.1161/01.CIR.0000100688.17280.E6. [DOI] [PubMed] [Google Scholar]
- 83.Ridker PM, Danielson E, Fonseca FA, Genest J, Gotto AM, Jr, Kastelein JJ, Koenig W, Libby P, Lorenzatti AJ, MacFadyen JG, Nordestgaard BG, Shepherd J, Willerson JT, Glynn RJ. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med. 2008;359:2195–2207. doi: 10.1056/NEJMoa0807646. [DOI] [PubMed] [Google Scholar]
- 84.Ridker PM, Danielson E, Fonseca FA, Genest J, Gotto AM, Jr, Kastelein JJ, Koenig W, Libby P, Lorenzatti AJ, Macfadyen JG, Nordestgaard BG, Shepherd J, Willerson JT, Glynn RJ. Reduction in C-reactive protein and LDL cholesterol and cardiovascular event rates after initiation of rosuvastatin: a prospective study of the JUPITER trial. Lancet. 2009;373:1175–1182. doi: 10.1016/S0140-6736(09)60447-5. [DOI] [PubMed] [Google Scholar]
- 85.Takayama T, Hiro T, Yamagishi M, Daida H, Hirayama A, Saito S, Yamaguchi T, Matsuzaki M. Effect of rosuvastatin on coronary atheroma in stable coronary artery disease: multicenter coronary atherosclerosis study measuring effects of rosuvastatin using intravascular ultrasound in Japanese subjects (COSMOS) Circ J. 2009;73:2110–2117. doi: 10.1253/circj.cj-09-0358. [DOI] [PubMed] [Google Scholar]
- 86.Sudhop T, Lutjohann D, Kodal A, Igel M, Tribble DL, Shah S, Perevozskaya I, von Bergmann K. Inhibition of intestinal cholesterol absorption by ezetimibe in humans. Circulation. 2002;106:1943–1948. doi: 10.1161/01.cir.0000034044.95911.dc. [DOI] [PubMed] [Google Scholar]
- 87.Bruckert E, Giral P, Tellier P. Perspectives in cholesterol-lowering therapy: the role of ezetimibe, a new selective inhibitor of intestinal cholesterol absorption. Circulation. 2003;107:3124–3128. doi: 10.1161/01.CIR.0000072345.98581.24. [DOI] [PubMed] [Google Scholar]
- 88.Fichtlscherer S, Schmidt-Lucke C, Bojunga S, Rossig L, Heeschen C, Dimmeler S, Zeiher AM. Differential effects of short-term lipid lowering with ezetimibe and statins on endothelial function in patients with CAD: clinical evidence for ‘pleiotropic’ functions of statin therapy. Eur Heart J. 2006;27:1182–1190. doi: 10.1093/eurheartj/ehi881. [DOI] [PubMed] [Google Scholar]
- 89.Kastelein JJ, Akdim F, Stroes ES, Zwinderman AH, Bots ML, Stalenhoef AF, Visseren FL, Sijbrands EJ, Trip MD, Stein EA, Gaudet D, Duivenvoorden R, Veltri EP, Marais AD, de Groot E. Simvastatin with or without ezetimibe in familial hypercholesterolemia. N Engl J Med. 2008;358:1431–1443. doi: 10.1056/NEJMoa0800742. [DOI] [PubMed] [Google Scholar]
- 90.Ostad MA, Eggeling S, Tschentscher P, Schwedhelm E, Boger R, Wenzel P, Meinertz T, Munzel T, Warnholtz A. Flow-mediated dilation in patients with coronary artery disease is enhanced by high dose atorvastatin compared to combined low dose atorvastatin and ezetimibe: results of the CEZAR study. Atherosclerosis. 2009;205:227–232. doi: 10.1016/j.atherosclerosis.2008.11.032. [DOI] [PubMed] [Google Scholar]
- 91.Liu PY, Chen JH, Lin LJ, Liao JK. Increased Rho kinase activity in a Taiwanese population with metabolic syndrome. J Am Coll Cardiol. 2007;49:1619–1624. doi: 10.1016/j.jacc.2006.12.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Liu PY, Liao JK. A method for measuring Rho kinase activity in tissues and cells. Methods Enzymol. 2008;439:181–189. doi: 10.1016/S0076-6879(07)00414-4. [DOI] [PMC free article] [PubMed] [Google Scholar]