

Abstract
Acute cerebral ischemia elicits an innate immune response that leads to a cascade of events that culminates in necrotic death of neurons and injury to their supportive structures in the neurovascular unit. Indeed, clinical studies have shown a close relationship between elevated levels of inflammatory markers and the risk for ischemic stroke. However, the signaling pathways that link these events are not well understood. A central regulator of inflammatory response is the transcription factor, nuclear factor-kappa B (NF-κB). The activation of NF-κB is required for the transcriptional induction of many proinflammatory mediators involved in innate immunity, such as cellular adhesion molecules, cytokines, and growth factors. Therefore, factors that modulate the activity of NF-κB could potentially regulate inflammatory processes in ischemic stroke. Here, we review the relationship between NF-κB and ischemic stroke, its role in the neurovascular unit, and discuss some animal models that suggest that this relationship is causal.
Keywords: NF-kappa B, stroke, innate immunity, inflammation, cerebral ischemia
Stroke is one of the most devastating manifestations of two common diseases, atherosclerosis and systemic hypertension. Stroke also occurs in less common syndromes (e.g., CADASIL, sickle cell anemia, and cerebral vasculitis). Regional cessation of cerebral blood flow is followed instantaneously by neurologic deficit that, at onset or by evolution over days, is frequently highly disabling or fatal. Recovery from stroke is highly variable, and long-term outlook is often poor and compounded by the possibility of further events. Other than preventing stroke by treating or preventing the causative vasculopathies, current treatment is aimed at modulating intravascular thrombotic pathways to achieve early recanalization. The window of opportunity for this approach is highly limited, so the foundation of stroke management is supportive therapy aimed at facilitating functional compensation for the deficit caused.
Strategies aimed at minimizing tissue damage in the ischemic region are yet to provide demonstrated benefits to patients. These are predicated on regulating of brain tissue response to ischemia on the cellular and molecular level. The most relevant cellular response pathways are believed to be neuronal excitotoxicity, the hypoxia response, the control of apoptosis, and inflammation. Inflammation in the context of ischemic stroke can be considered to have two elements: (1) the response of the resident cells in cerebral tissue to mediators of endogenous danger/injury signals and (2) the recruitment into tissue and subsequent activation of leukocytes (predominantly neutrophils and monocytes), enabled by disruption of the blood–brain barrier.
The nuclear factor-kappa B (NF-κB) transcriptional activation pathway is considered to be a “master regulator” of both these aspects of inflammation and indeed critical to the regulation of apoptosis.1 As such, descriptions of its role in ischemic stroke are of considerable relevance to an understanding of the relationship between ischemia and tissue damage in stroke. It should be borne in mind that there is a large amount of literature implicating NF-κB activation in atherosclerosis, the disease process that underlies stroke. This includes specific studies of atheroma at the carotid artery bifurcation, the lesion that is responsible for causing ischemic stroke.2,3 However, that literature lies outside of the scope of this review.
NF-κB and innate inflammation
The NF-κB family of Rel-homology domain (RHD) containing proteins subsumes the classical functions ascribed to gene transcription factors1 (Table 1). Furthermore, their interaction with the inhibitor κB (IκB) family of ankyrin repeat domain (ARD) proteins and its specific regulation by IκB kinase (IKK) produces an exquisitely regulated molecular switch, known collectively as the NF-κB pathway. The NF-κBs themselves (RelA [p65], cRel, RelB, p50 [NF-κB1], p52 [NF-κB2]) bear nuclear localization sequence (NLS) and homo/heterodimerization motifs that allow them to access and bind to κB sites in DNA promoter regions and drive transcription. Although combinatorial diversity is incomplete (p52 cannot homodimerize, for instance), it allows for some variation in functionality (e.g., p50 homodimers occupy κB sites but are transcriptionally inactive).
Table 1.
NF-κB/Rel family
| NF-κB | IκB | Species |
|---|---|---|
| Rel A (p65) | IκB-α | Mammalian |
| c-Rel | IκB-β | Mammalian |
| p50 (NF-κB1) | IκB-γ/p105 | Mammalian |
| p52 (NF-κB2) | IκB-δ/p100 | Mammalian |
| Rel B | IκB-ε | Mammalian |
| Dorsal | Cactus | Drosophila |
| v-Rel | ? | Viral |
A large number of genes are NF-κB dependent (Table 2), and the three most prominent gene transcription programs associated with this transcription factor are the following: (1) inflammation (e.g., IL-6, iNOS, ICAM1, MMP9, COX2); (2) regulation of apoptosis, typified by the Bcl2 family; and (3) IκBs (α, β, ε, γ, ζ, p105, p100). These functional inhibitors act as a classic negative feedback loop by binding via ankyrin repeats to the NLS of the NF-κBs and sequestering them in the cytoplasm, or, in the case of IκBα, exporting them from the nucleus also. Again, there is diversity within the family in the extent of inhibition (α, β, p105, p100 most critical) and predilection for NF-κB family members (e for RelA and cRel, Bcl3 for p50/p52 heterodimer). Inhibition of NF-κB by IκB is released by a series of events, starting with phosphorylation of specific serines (32 and 36 in IκBα) by the IKK complex. IKK is composed of two serine kinases (IKK1/α and IKK2/β) and a regulatory subunit (IKKγ/NF-κB essential modulator [NEMO]).
Table 2.
Genes requiring NF-κB activation
| Cellular adhesion molecules |
| Intercellular adhesion molecule-1 (ICAM-1) |
| Vascular cell adhesion molecule-1 (VCAM-1) |
| Endothelial leukocyte adhesion molecule-1 (ELAM-1 or E-selectin) |
| Inflammatory cytokines |
| Interleukin (IL)-2, -6, and -8 |
| Tumor necrosis factor (TNF)-α and -β |
| Macrophage colony stimulating factor (M-CSF or CSF-1) |
| Granulocyte colony stimulating factor (G-CSF) |
| Granulocyte/macrophage-colony stimulating factor (GM-CSF) |
| Interferon-β |
| Tissue factor |
| Macrophage chemotactic protein-1 (MCP-1) |
| Immunologic mediators |
| Immunoglobulin (IgG) κ light chain |
| T cell receptor α and β chain |
| Major histocompatability complex (MHC) class I |
| Major histocompatability complex (MHC) class II |
| Invariant chain (Ii) |
| β2-Microglobulin |
| Type II inducible nitric oxide synthase (iNOS) |
| Viral enhancers |
| Human immunodeficiency virus-1 (HIV-1) |
| Cytomegalovirus (CMV) |
| Adenovirus |
| Simian virus 40 (SV40) |
| Transcription factors |
| IκB-α |
| c-Rel |
| NF-κB/p105 |
| c-Myc |
| Interferon regulatory factor-1 (IRF-1) |
Phosphorylation of IκBs triggers their polyubiquitination by the ubiquitin ligase SCFTrCP, which is followed by their degradation in the 26S proteasome, thereby allowing for NF-κB nuclear translocation and DNA binding (Fig. 1). The exception to this is p105 and p100, IκB proteins, which contain both Rel homology and ankyrin repeat domains, and that upon phosphorylation are cleaved to produce NF-κB p50 and NF-κB p52, respectively. The canonical NF-κB switch associated with inflammation is dependent on NEMO to integrate upstream stimuli, IKK2 to degrade IκBα, and activation of RelA/p50 heterodimers. In the resting state, this switch is in the “off” position, and upon activation, the de novo synthesis of IκBs produces a negative feedback loop that results in a series of damped oscillations, with two to four “on” periods over then next 72 h, each one with less amplitude than the last. A noncanonical pathway has been described in B-lymphocytes, in which the BAFF-BAFF-R ligand–receptor interaction drives IKK2/NF-κB inducing kinase (NIK) phosphorylation of p100 and formation of RelB/p52 dimers, which transcribe antiapoptotic genes. This noncanonical pathway does not act as a molecular switch but rather as a constitutive homeostatic regulator of B cell survival.1
Figure 1.
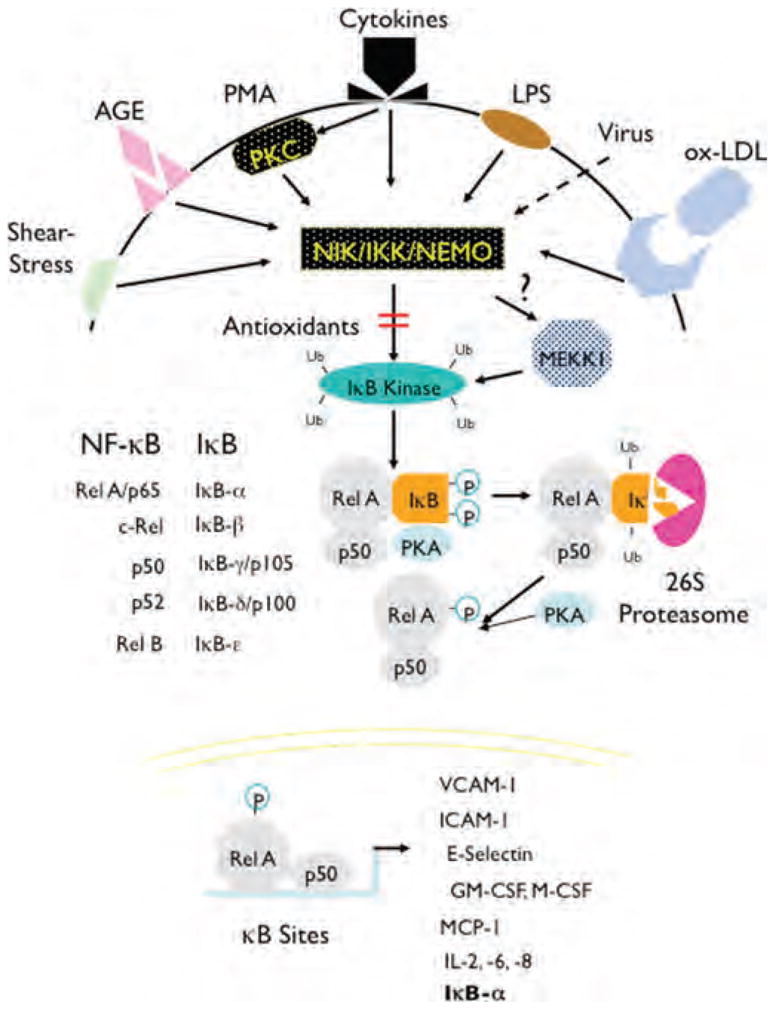
Activation of NF-κB and induction of proinflammatory genes. The activation of NF-κB requires serine 32 and 36 phosphorylation of IκB by IKK complex, leading to ubiquitination and 26S proteosome degradation of IκB. This allows for the nuclear translocation of NF-κB, where it transactivates genes with κB cis-acting elements. VCAM-1, vascular cell adhesion molecule-1; ICAM-1, intercellular adhesion molecule-1; GM-CSF, guanulocyte/monocyte-colony stimulating factor; M-CSF, macrophage-colony stimulating factor; MCP-1, monocyte chemotactic protein-1; IL, interleukin.
The classical “molecular switch” function of NF-κB is pivotal in the control of innate inflammation. The IKK signaling complex serves as an integrator of a wide variety of xenogeneic and endogenous danger signals, both extracellular (via toll-like receptors [TLRs], and receptors of advanced glycation end products [RAGE]) and intracellular (via noninflammasome NOD-like receptors) (Table 3). Mediators of inflammation also principally use this pathway, including the proinflammatory cytokines (e.g., TNFα, IL-1β, TWEAK), the chemokine family, and complement proteins. The promiscuity of the NF-κB pathway is counterbalanced by tight control of signal strength and duration by the IκBs. This has been shown to be dependent on the duration of the inducing stimulus, rather than its strength. A stimulus of short duration will result in a rather stereotyped activation-downregulation over 1 h. Prolonged stimulation of the pathway leads to more prolonged and variable activation.1 This enables sensitivity in responding to an inflammatory trigger without leading to a potentially damaging exaggerated inflammation cascade.
Table 3.
Activators of NF-κB
| Inflammatory mediators |
| Tumor necrosis factor-α |
| Interleukin-1 and -2 |
| Lymphotoxin |
| Leukotriene B4 |
| Growth factors |
| Platelet-derived growth factor (PDGF) |
| Transforming growth factor-β1 (TGF-β1) |
| Viral mediators |
| Viral infection (HIV, EBV, CMV) |
| Double-stranded RNA |
| Epstein–Barr nuclear antigen-2 |
| Bacterial mediators |
| Lipopolysaccharide (LPS) |
| Muramyl proteins |
| Exotoxin B |
| Oxidants |
| Hydrogen peroxide |
| Ultraviolet light |
| Drugs |
| Phorbolesters |
| Okadaic acid |
| Cycloheximide |
| Anisomycin |
| Pervanadate |
| Physical stress |
| Laminar shear stress |
| Stretch |
| Cyclic strain |
Assessment and modulation of NF-κB activation in vivo
The description of the role of the NF-κB pathway in vivo has been underscored by the development of a number of assays that measure this linear series of molecular events in an overlapping fashion. These multiple techniques have been used together to cross-validate each other. Starting downstream, we have the development of gene transcription reporters (κB-β-globin and κB-lacZ) that have been expressed as germline transgenes in mice. Gene array experiments using classical NF-κB stimuli have been used to develop low-density gene arrays describing κB activity. Moving upstream, electrophoretic mobility shift assays (EMSA; gel shifts) have been used to quantify NF-κB DNA binding and to identify (using antibody supershift) the composition of the functional dimers. Immunofluorescent and confocal microscopy techniques have been used to quantify and localize NF-κB (particularly RelA) as it shuttles in and out of the nucleus. At the most upstream end, we have IκB abundance assays (Western blot or immunofluorescence), IκB phoshporylation assays, and IKK kinase activity assays.
To probe the function of NF-κB in vivo, a number of predominantly loss-of-function approaches have been used. Germline deletions of cRel, p50, and IKK2 have been established and are viable.4,5 RelA deletion is developmentally lethal, so an alternative approach has been the tissue specific overexpression of a mutant IκBα with serines 32 and 36 replaced by alanines (referred to as a dominant negative [DN] or super-repressor [IκBαSR]).6,7 IKK2 and NIK dominant negative transgenic mice are also available. A number of small molecule NF-κB inhibitors (e.g., pyrrolidinedithiocarbamate [PDTC]) have been used, and while these are often nonspecific, they have been useful in establishing an association between NF-κB inhibition and biological outcomes. The most specific reagents are the IKK inhibitors, which were developed as part of therapeutic programs and have been used as loss-of-function interventions to link NF-κB to biology in a causative relationship.8
NF-κB and ischemic stroke
The association of NF-κB with stroke was first suggested by a study examining the expression of RelA and p50 by immunohistochemistry on brain tissue obtained from six patients who died after stroke.9 RelA and p50 were detected in macrophages throughout the infarcts, and in astroglia, predominantly in the ischemic penumbra (the zone between necrotic and healthy brain tissue). This initial observation was reinforced by a subsequent study in three patients who died within 2 days of stroke showing nuclear translocation of RelA, again in the penumbra, but this time predominantly in neurons.10 The paradigm that inflammation and apoptosis are contributory processes to irreversible tissue loss in stroke, combined with early observations of NF-κB expression in stroke, led to the hypothesis that NF-κB is centrally implicated in the evolution of cerebral infarction.
Using a rodent model of focal cerebral ischemia induced by middle cerebral artery occlusion (MCAO), several investigators tested the hypothesis that NF-κB contributes to ischemic stroke.11 MCAO is achieved by the insertion of an occlusive monofilament into the MCA, either permanently or transiently, over 60–120 min, and followed by reperfusion. The MCAO model shows similarity to human MCAO with respect to the development of focal neurodeficit, the regional nature of the infarct, and its evolution through acute necrosis in the core, edema, inflammation, and cell death in the penumbra.12 Transient MCAO in the rat is followed over hours by an acute NF-κB activation, as demonstrated by increased p65 and p50 expression in ischemic region neurons, and gel shifts of homogenized lesional tissue.13
There are several studies where severity of stroke has been modulated by various interventions; NF-κB activation has been detected and the covariation between stroke severity and NF-κB activity has established an association. Atorvastatin was used in the rat permanent MCAO model to show that a reduction in stroke size (using triphenyltetrazolium chloride staining and corrected for cerebral edema), edema, and neurodeficit was associated with reduced NF-κB expression, as well as that of other factors potentially upstream of NF-κB, such as high mobility group box 1 (HMGB1), receptor of advanced glycation end products (RAGE), and toll-like receptor 4 (TLR4).14 In the rat transient MCAO model, a neuroprotective lipid, palmitoylethanolamine, reduced infarct size and neurodeficit, and also reduced NF-κB expression and lesional apoptosis as determined by TUNEL staining.15 Adiponectin was shown to have similar effects on stroke in the rat transient MCAO model, in association with a reduction in NF-κB activity, as shown by reduced nuclear translocation of RelA within the ischemic lesion.16 Pyruvate, a reactive oxygen species (ROS) scavenger, had similar effects, and in this study, the improvement in stroke severity was paralleled by a reduction in RelA expression and binding to a κB consensus DNA sequence. There was evidence of reduced inflammation, with less neutrophil, macrophage and microglial activity within the lesion. Matrix metalloproteinase 9 (MMP-9) expression was reduced.17 MMP-9 expression is NF-κB-dependent and MMP-9 mediates the disruption of the blood–brain barrier that causes brain edema in stroke.18
In an impressive feat of microsurgical technique, the transient MCAO model has been scaled down for use in the mouse, which allows for the evaluation of the impact of gene deletions and overexpression. CD36 is a scavenger receptor upstream of NF-κB, and CD36 knockout mice exhibit milder stroke after MCAO, with less neutrophilia and glial reaction. Gel shift on lesional tissue shows that this amelioration is associated with less NF-κB (RelA) activity and a reduced expression of NF-κB-dependent genes (iNOS, COX-2, ICAM-1, Nox-2).19,20 Gene deletion of IL-1 converting enzyme, which results in IL-1b deficiency, generated similar findings.21,22 To establish a causative relationship between NF-κB and stoke severity, loss-of-function experiments have been conducted. Nonspecific pharmacologic approaches to inhibit NF-κB, using n-acetylcysteine and PDTC, have caused a reduction in stroke size in the rat and mouse, respectively. This effect was associated with reduced RelA and p50 expression and reduced NF-κB activity using both gel shift and RelA nuclear localization techniques.23–25 Similar effects in rat transient MCAO were observed using the 26S proteasome inhibitor, MLN519, which inhibits NF-κB by preserving IκB-mediated sequestration.26,27 Further specificity in in vivo loss-of-function experiments has been achieved using genetic manipulation. In one study, rats were subjected to four cortical injections of adenovirus expressing IκBαSR prior to permanent MCAO. NF-κB inhibition caused a reduction in stroke size and neurodeficit.28
This body of data has created a strong paradigm whereby NF-κB activation takes place as an acute response to injury in stroke, participates in blood–brain barrier disruption, inflammation, and contributes to neuronal cell death. However, further data in similar experimental systems appear conflicting and hint at more complexity than allowed by the “NF-κB as deleterious in stroke” model. The application of transient or permanent MCAO to p50 deficient mice has indeed shown a reduction in stroke size, as might be expected.10,29 Although p50 is often heterodimerized with RelA or cRel in active NF-κB, p50 homodimers are inactive and may even block κB transcription by occupying DNA binding sites. However, these descriptions of MCAO in p50 knockouts confirmed that NF-κB activity, by gel shift and κB-b-globin reporter assay, was indeed reduced by the absence of p50. Nevertheless, subsequent studies have shown an increase in neurodegeneration after transient MCAO in p50−/− mice, using the Fluoro-Jade marker, and in permanent MCAO, p50 deletion increased infarct volume and penumbral apoptosis on the basis of TUNEL and caspase 3 staining, and reduced the proliferative response postinfarction in neurons, astrocytes, and microglia by BrdU staining.30,31 This controversy is not restricted to p50 loss of function. Stroke size is also increased, rather than reduced, in the cRel deficient mouse.32 This variability can also be seen in the IKK-targeted approach. BMS345541 is an IKK1 and 2 inhibitor with potency in the low micromolar range.33 This agent reduced stroke size after permanent MCAO using extrinsic microbipolar coagulation in mice, albeit after intracerebroventricular administration (as systemic administration did not lead to CNS penetration). However, when an NEMO-binding peptide was used to inhibit IKK and NF-κB activation in a transient carotid occlusion model in neonatal rat (a model of focal perinatal hypoxia/ischemia), successful suppression of NF-κB activation produced an exacerbation in focal neurodeficit over chronic phase.34 A less specific NF-κB inhibitor, diethyldithiocarbamate, produced an increase in postreperfusion apoptosis and stroke size in the rat MCAO model, while effectively inhibiting NF-κB in lesional tissue as shown by gel shift.35
Potential role of NF-κB in the neurovascular unit
To try to resolve these inconsistencies, newer and more complex models of the role of NF-κB in stroke have been proposed.36,37 These have stressed the potential impact of regional/organotypic/molecular and temporal variation in NF-κB activation on whole-organ downstream effect in stroke. Perhaps the most significant duality in NF-κB function of relevance here is between the proinflammatory effects on the one hand, and the antiapoptotic effects on the other. Bcl2, TRAF-1, and XIAP are examples of κB-dependent induction of antiapoptotic regulators, which could limit stroke size. At the same time, proinflammatory mediators downstream of NF-κB (e.g., iNOS, COX-2, ICAM-1, IL-6, MMP9) could contribute to ischemic tissue damage. In regional terms, a cerebral infarct can be divided into core and penumbra. It is possible that NF-κB could be exerting opposite effects in the two regions. The regulator of the balance between neurodegeneration and neuroprotection for NF-κB may be the availability of ROS in each locality, as MCAO in superoxide dismutase 1 (SOD1) transgenic mice, that have reduced ROS, was associated with increased Bcl2, TRAF-1, and XIAP in the penumbral zone.38 Furthermore, this balance may be regulated differently in the different cells present in the ischemic cerebral region, at different times. Using a κB-lacZ reporter mouse, neuronal but not astroglial NF-κB has been shown to be constitutively active (by nuclear β-gal staining) in various parts of the adult brain, and in primary neuronal cultures gain and loss of function perturbation of constitutive NF-κB showed this to subsume an antiapoptotic function.39
In an in vivo model of neuronal survival (ischemia/reperfusion in the retina), astroglial-specific NF-κB inhibition, by means of the IκBαSR transgene under the control of the glial fibrillary acidic protein (GFAP) promoter, reduced inflammatory gene transcription and neuronal cell death in the ganglion cell layer.40 So while constitutive neuronal NF-κB can be neuroprotective, ischemia-induced astroglial NF-κB can be neurodegenerative. In the MCAO model, astroglial, but not neuronal, NF-κB activation is downstream of TLR2 and 4 activation, and mice deficient in these TLRs exhibit reduction in stroke size and neurodeficit, again imputing a deleterious role for astroglial NF-κB.41 However, when the relative contributions of neuronal and astroglial NF-κB were formally tested by comparing MCAO in the GFAP- IκBaSR mouse with that in the neuron-specific enolase (NSE)- IκBaSR mouse, it was reported that neuronal NF-κB inhibition reduced stroke size and apoptosis by TUNEL staining, whereas astroglial NF-κB inhibition had no effect.42 Clearly, then, although constitutive neuronal NF-κB may protect neurons from apoptosis, in the context of ischemic stroke, the overall effect of neuronal NF-κB may be deleterious. Furthermore, although astroglial NF-κB seems to be deleterious, the contribution of this cell type to the overall picture is indeterminate. Considerably less work has been done regarding the contribution of other cell types.
An intriguing study showed that in blood outgrowth endothelial cell cultures increased RelA expression and increased NF-κB activation on stimulus could be observed in sickle cell patients with cerebral occlusive vasculopathy (Circle of Willis abnormalities on angiography) compared with those without.43 We have investigated the effect of the IκBaSR transgene under control of the Tie2 promoter (endothelial cells and leukocytes) in the MCAO model. NF-κB inhibition in these cell types results in reduced edema but an increase in stroke size. Using bone marrow transplants with wild-type mice, it appears that the NF-κB effect on edema works primarily though endothelial MMP-9 expression and blood–brain permeability, whereas the protective effect on stroke size is mediated though leukocyte NF-κB.44 It is feasible that inflammatory gene programs orchestrated by NF-κB in leukocytes facilitate the clearance from ischemic tissue of mediators of tissue injury, thereby reducing stroke size. The overall contribution of this mechanism, given the apparent converse function of NF-κB in resident cells of the nervous system, remains to be assessed.
Because a significant body of data imputing a protective role for NF-κB in stroke comes from deletions of specific NF-κB family members in mice subjected to MCAO, it has been suggested that the balance of NF-κB activation as neuroprotective or deleterious is mediated through diversity in homo-and heterodimer composition. Good evidence to support this notion comes from another MCAO study, in which RelA, cRel, and p50 gene-deleted mice were directly compared. Because RelA deletion is embryonic lethal, RelA was deleted only in neurons and glia using a Cre-lox approach under control of the Nestin promoter. The results showed that infarct size was reduced in the RelA knockouts, whereas cRel and p50 deficiency had no effect.45
Current understanding limits our ability to resolve the apparent discrepancies in results across this literature, but it appears that the deleterious effect of NF-κB in stroke is mediated predominantly by the activation of RelA in resident cells of the central nervous system and is mediated via apoptosis. An in vitro study of astroglial cultures showed that κB-dependent gene transcription downstream of TNF-α could be potentially deleterious (e.g., ICAM-1) or protective (e.g., Mn-SOD), depending on the presence or absence of the p300 adaptor, but how this would be regulated in vivo is unknown.46
Another variable that could impact the balance of NF-κB effect in stroke is time. As would be expected from the inherent kinetic of the classical NF-κB pathway, MCAO studies in rat and mouse have shown by nuclear immunolocalization, phospho-RelA staining and gel shift, that an initial induction of NF-κB in ischemic brain at 2–4 h is followed by a downregulation.21,47 Another study tracks the kinetic further out and suggests a second peak in activation over days. This study ties this observation in with spatial/organotypic considerations to show that the early peak is in the core infarct and predominantly neuronal, whereas the late peak is in the penumbra and predominantly astroglial. In the carotid ischemia/reperfusion neonatal rat model, in which a peptide inhibitor of NEMO was used to demonstrate that NF-κB activation is overall neuroprotective, the timing of administration of the inhibitor produced opposite effects, so that treatment over the first 3 h after reperfusion reduced apoptotic neuronal loss, whereas treatment over the whole reperfusion period, or only over the later period (from 18 h), produced more neuronal loss through inhibition of the Bcl2 and Bcl-xL antiapoptotic pathways.48 However, timing of intervention has not been assessed in MCAO models in which NF-κB inhibition was overall favorable in outcome.
The timing of NF-κB has been further investigated in the context of ischemic preconditioning. In this paradigm, stroke severity after cerebral arterial occlusion can be lessened by transient ischemia prior to occlusion. For example, the death of CA1 hippocampal from a four-vessel (carotid and vertebral) 6-min occlusion was smaller if preceded by a 4-vessel 3-min occlusion (this is a model of hypoxic brain injury). The preconditioning ischemia was associated with NF-κB activation, and inhibition of NF-κB with PDTC showed that it is required for the preconditioning effect.49 Similar findings were observed in a two-vessel (bilateral common carotid artery) occlusion model (BCCAO), with the preconditioning effect (10 second ischemia/reperfusion events repeated five times) was abolished by diethyldithiocarbamic acid, which also inhibited NF-κB activation, and by TLR4 deficiency, which reduced κB-mediated expression of TNF-a, COX2 and iNOS.50,51 A simple explanation would be to posit that NF-κB activation during preconditioning causes induction of IκB, so that when the subsequent occlusive event occurs, NF-κB activity is reduced and therefore so is neuronal loss.
Summary
The contribution of NF-κB activation in stroke now appears far from simple, and the idea that clinical inhibition of the pathway might provide a new paradigm of therapy in stroke appears uncertain, and at this point, unlikely to be tested in patients. Nonetheless, as understanding of mechanisms of tissue loss in stroke increases, the likelihood of a future therapy for stroke that can be administered after the event and that reduces tissue loss and neurodeficit is growing.
Footnotes
Conflicts of interest
The authors declare no conflicts of interest.
References
- 1.Hoffmann A, Baltimore D. Circuitry of nuclear factor kappaB signaling. Immunol Rev. 2006;210:171–186. doi: 10.1111/j.0105-2896.2006.00375.x. [DOI] [PubMed] [Google Scholar]
- 2.Monaco C, Andreakos E, Kiriakidis S, et al. Canonical pathway of nuclear factor kappa B activation selectively regulates proinflammatory and prothrombotic responses in human atherosclerosis. Proc Natl Acad Sci USA. 2004;101:5634–5639. doi: 10.1073/pnas.0401060101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martin-Ventura JL, Blanco-Colio LM, Munoz-Garcia B, et al. NF-kappaB activation and Fas ligand overexpression in blood and plaques of patients with carotid atherosclerosis: potential implication in plaque instability. Stroke. 2004;35:458–463. doi: 10.1161/01.STR.0000114876.51656.7A. [DOI] [PubMed] [Google Scholar]
- 4.Snapper CM, Zelazowski P, Rosas FR, et al. B cells from p50/NF-kappa B knockout mice have selective defects in proliferation, differentiation, germ-line CH transcription, and Ig class switching. J Immunol. 1996;156:183–191. [PubMed] [Google Scholar]
- 5.Pasparakis M, Courtois G, Hafner M, et al. TNF-mediated inflammatory skin disease in mice with epidermis-specific deletion of IKK2. Nature. 2002;417:861–866. doi: 10.1038/nature00820. [DOI] [PubMed] [Google Scholar]
- 6.Beg AA, Sha WC, Bronson RT, et al. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-kappa B. Nature. 1995;376:167–170. doi: 10.1038/376167a0. [DOI] [PubMed] [Google Scholar]
- 7.Attar RM, Macdonald-Bravo H, Raventos-Suarez C, et al. Expression of constitutively active IkappaB beta in T cells of transgenic mice: persistent NF-kappaB activity is required for T cell immune responses. Mol Cell Biol. 1998;18:477–487. doi: 10.1128/mcb.18.1.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Strnad J, Burke JR. IkappaB kinase inhibitors for treating autoimmune and inflammatory disorders: potential and challenges. Trends Pharmacol Sci. 2007;28:142–148. doi: 10.1016/j.tips.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 9.Terai K, Matsuo A, Mcgeer EG, Mcgeer PL. Enhancement of immunoreactivity for NF-kappa B in human cerebral infarctions. Brain Res. 1996;739:343–349. doi: 10.1016/s0006-8993(96)01073-6. [DOI] [PubMed] [Google Scholar]
- 10.Nurmi A, Lindsberg PJ, Koistinaho M, et al. Nuclear factor-kappaB contributes to infarction after permanent focal ischemia. Stroke. 2004;35:987–991. doi: 10.1161/01.STR.0000120732.45951.26. [DOI] [PubMed] [Google Scholar]
- 11.Young W, Rappaport ZH, Chalif DJ, Flamm ES. Regional brain sodium, potassium, and water changes in the rat middle cerebral artery occlusion model of ischemia. Stroke. 1987;18:751–759. doi: 10.1161/01.str.18.4.751. [DOI] [PubMed] [Google Scholar]
- 12.Carmichael ST. Rodent models of focal stroke: size, mechanism, and purpose. NeuroRx. 2005;2:396–409. doi: 10.1602/neurorx.2.3.396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stephenson D, Yin T, Smalstig EB, et al. Transcription factor nuclear factor-kappa B is activated in neurons after focal cerebral ischemia. J Cereb Blood Flow Metab. 2000;20:592–603. doi: 10.1097/00004647-200003000-00017. [DOI] [PubMed] [Google Scholar]
- 14.Wang L, Zhang X, Liu L, et al. Atorvastatin protects rat brains against permanent focal ischemia and down-regulates HMGB1, HMGB1 receptors (RAGE and TLR4), NF-kappaB expression. Neurosci Lett. 2010;471:152–156. doi: 10.1016/j.neulet.2010.01.030. [DOI] [PubMed] [Google Scholar]
- 15.Garg P, Duncan RS, Kaja S, Koulen P. Intracellular mechanisms of N-acylethanolamine-mediated neuroprotection in a rat model of stroke. Neuroscience. 2010;166:252–262. doi: 10.1016/j.neuroscience.2009.11.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen B, Liao WQ, Xu N, et al. Adiponectin protects against cerebral ischemia-reperfusion injury through anti-inflammatory action. Brain Res. 2009;1273:129–137. doi: 10.1016/j.brainres.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 17.Wang Q, Van HM, Tang XN, et al. Pyruvate protects against experimental stroke via an anti-inflammatory mechanism. Neurobiol Dis. 2009;36:223–231. doi: 10.1016/j.nbd.2009.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang G, Guo Q, Hossain M, et al. Bone marrow-derived cells are the major source of MMP-9 contributing to blood-brain barrier dysfunction and infarct formation after ischemic stroke in mice. Brain Res. 2009;1294:183–192. doi: 10.1016/j.brainres.2009.07.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kunz A, Abe T, Hochrainer K, et al. Nuclear factor-kappaB activation and postischemic inflammation are suppressed in CD36-null mice after middle cerebral artery occlusion. J Neurosci. 2008;28:1649–1658. doi: 10.1523/JNEUROSCI.5205-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cho S, Park EM, Febbraio M, et al. The class B scavenger receptor CD36 mediates free radical production and tissue injury in cerebral ischemia. J Neurosci. 2005;25:2504–2512. doi: 10.1523/JNEUROSCI.0035-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang FP, Wang ZQ, Wu DC, et al. Early NF-kappaB activation is inhibited during focal cerebral ischemia in interleukin-1beta-converting enzyme deficient mice. J Neurosci Res. 2003;73:698–707. doi: 10.1002/jnr.10654. [DOI] [PubMed] [Google Scholar]
- 22.Yang GY, Schielke GP, Gong C, et al. Expression of tumor necrosis factor-alpha and intercellular adhesion molecule-1 after focal cerebral ischemia in interleukin-1beta converting enzyme deficient mice. J Cereb Blood Flow Metab. 1999;19:1109–1117. doi: 10.1097/00004647-199910000-00007. [DOI] [PubMed] [Google Scholar]
- 23.Carroll JE, Howard EF, Hess DC, et al. Nuclear factor-kappa B activation during cerebral reperfusion: effect of attenuation with N-acetylcysteine treatment. Brain Res Mol Brain Res. 1998;56:186–191. doi: 10.1016/s0169-328x(98)00045-x. [DOI] [PubMed] [Google Scholar]
- 24.Crack PJ, Taylor JM, Ali U, et al. Potential contribution of NF-kappaB in neuronal cell death in the glutathione peroxidase-1 knockout mouse in response to ischemia-reperfusion injury. Stroke. 2006;37:1533–1538. doi: 10.1161/01.STR.0000221708.17159.64. [DOI] [PubMed] [Google Scholar]
- 25.Nurmi A, Vartiainen N, Pihlaja R, et al. Pyrrolidine dithiocarbamate inhibits translocation of nuclear factor kappa-B in neurons and protects against brain ischaemia with a wide therapeutic time window. J Neurochem. 2004;91:755–765. doi: 10.1111/j.1471-4159.2004.02756.x. [DOI] [PubMed] [Google Scholar]
- 26.Williams AJ, Hale SL, Moffett JR, et al. Delayed treatment with MLN519 reduces infarction and associated neurologic deficit caused by focal ischemic brain injury in rats via antiinflammatory mechanisms involving nuclear factor-kappaB activation, gliosis, and leukocyte infiltration. J Cereb Blood Flow Metab. 2003;23:75–87. doi: 10.1097/01.WCB.0000039285.37737.C2. [DOI] [PubMed] [Google Scholar]
- 27.Williams AJ, Dave JR, Tortella FC. Neuroprotection with the proteasome inhibitor MLN519 in focal ischemic brain injury: relation to nuclear factor kappaB (NF-kappaB), inflammatory gene expression, and leukocyte infiltration. Neurochem Int. 2006;49:106–112. doi: 10.1016/j.neuint.2006.03.018. [DOI] [PubMed] [Google Scholar]
- 28.Xu L, Zhan Y, Wang Y, et al. Recombinant adenoviral expression of dominant negative IkappaBalpha protects brain from cerebral ischemic injury. Biochem Biophys Res Commun. 2002;299:14–17. doi: 10.1016/s0006-291x(02)02573-1. [DOI] [PubMed] [Google Scholar]
- 29.Schneider A, Martin-Villalba A, Weih F, et al. NF-kappaB is activated and promotes cell death in focal cerebral ischemia. Nat Med. 1999;5:554–559. doi: 10.1038/8432. [DOI] [PubMed] [Google Scholar]
- 30.Li J, Lu Z, Li WL, et al. Cell death and proliferation in NF-kappaB p50 knockout mouse after cerebral ischemia. Brain Res. 2008;1230:281–289. doi: 10.1016/j.brainres.2008.06.130. [DOI] [PubMed] [Google Scholar]
- 31.Duckworth EA, Butler T, Collier L, et al. NF-kappaB protects neurons from ischemic injury after middle cerebral artery occlusion in mice. Brain Res. 2006;1088:167–175. doi: 10.1016/j.brainres.2006.02.103. [DOI] [PubMed] [Google Scholar]
- 32.Valerio A, Dossena M, Bertolotti P, et al. Leptin is induced in the ischemic cerebral cortex and exerts neuroprotection through NF-kappaB/c-Rel-dependent transcription. Stroke. 2009;40:610–617. doi: 10.1161/STROKEAHA.108.528588. [DOI] [PubMed] [Google Scholar]
- 33.Herrmann O, Baumann B, De LR, et al. IKK mediates ischemia-induced neuronal death. Nat Med. 2005;11:1322–1329. doi: 10.1038/nm1323. [DOI] [PubMed] [Google Scholar]
- 34.Van Den Tweel ER, Kavelaars A, Lombardi MS, et al. Selective inhibition of nuclear factor-kappaB activation after hypoxia/ischemia in neonatal rats is not neuroprotective. Pediatr Res. 2006;59:232–236. doi: 10.1203/01.pdr.0000196807.10122.5f. [DOI] [PubMed] [Google Scholar]
- 35.Hill WD, Hess DC, Carroll JE, et al. The NF-kappaB inhibitor diethyldithiocarbamate (DDTC) increases brain cell death in a transient middle cerebral artery occlusion model of ischemia. Brain Res Bull. 2001;55:375–386. doi: 10.1016/s0361-9230(01)00503-2. [DOI] [PubMed] [Google Scholar]
- 36.Ridder DA, Schwaninger M. NF-kappaB signaling in cerebral ischemia. Neuroscience. 2009;158:995–1006. doi: 10.1016/j.neuroscience.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 37.Pizzi M, Sarnico I, Lanzillotta A, et al. Post-ischemic brain damage: NF-kappaB dimer heterogeneity as a molecular determinant of neuron vulnerability. FEBS J. 2009;276:27–35. doi: 10.1111/j.1742-4658.2008.06767.x. [DOI] [PubMed] [Google Scholar]
- 38.Song YS, Lee YS, Narasimhan P, Chan PH. Reduced oxidative stress promotes NF-kappaB-mediated neuroprotective gene expression after transient focal cerebral ischemia: lymphocytotrophic cytokines and antiapoptotic factors. J Cereb Blood Flow Metab. 2007;27:764–775. doi: 10.1038/sj.jcbfm.9600379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bhakar AL, Tannis LL, Zeindler C, et al. Constitutive nuclear factor-kappa B activity is required for central neuron survival. J Neurosci. 2002;22:8466–8475. doi: 10.1523/JNEUROSCI.22-19-08466.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dvoriantchikova G, Barakat D, Brambilla R, et al. Inactivation of astroglial NF-kappa B promotes survival of retinal neurons following ischemic injury. Eur J Neurosci. 2009;30:175–185. doi: 10.1111/j.1460-9568.2009.06814.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tang SC, Arumugam TV, Xu X, et al. Pivotal role for neuronal Toll-like receptors in ischemic brain injury and functional deficits. Proc Natl Acad Sci USA. 2007;104:13798–13803. doi: 10.1073/pnas.0702553104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang W, Potrovita I, Tarabin V, et al. Neuronal activation of NF-kappaB contributes to cell death in cerebral ischemia. J Cereb Blood Flow Metab. 2005;25:30–40. doi: 10.1038/sj.jcbfm.9600004. [DOI] [PubMed] [Google Scholar]
- 43.Enenstein J, Milbauer L, Domingo E, et al. Proinflammatory phenotype with imbalance of KLF2 and RelA: risk of childhood stroke with sickle cell anemia. Am J Hematol. 2010;85:18–23. doi: 10.1002/ajh.21558. [DOI] [PubMed] [Google Scholar]
- 44.Ahl D, Kim HH, Harari OA, et al. Increased infarct size and decreased vascular leakage following transient focal cerebral ischemia in mice with inhibition of endothelial and leukocyte NF-κB. Circulation. 2006;114:182. [Google Scholar]
- 45.Inta I, Paxian S, Maegele I, et al. Bim and Noxa are candidates to mediate the deleterious effect of the NF-kappa B subunit RelA in cerebral ischemia. J Neurosci. 2006;26:12896–12903. doi: 10.1523/JNEUROSCI.3670-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ginis I, Jaiswal R, Klimanis D, et al. TNF-alpha-induced tolerance to ischemic injury involves differential control of NF-kappaB transactivation: the role of NF-kappaB association with p300 adaptor. J Cereb Blood Flow Metab. 2002;22:142–152. doi: 10.1097/00004647-200202000-00002. [DOI] [PubMed] [Google Scholar]
- 47.Irving EA, Hadingham SJ, Roberts J, et al. Decreased nuclear factor-kappaB DNA binding activity following permanent focal cerebral ischaemia in the rat. Neurosci Lett. 2000;288:45–48. doi: 10.1016/s0304-3940(00)01203-9. [DOI] [PubMed] [Google Scholar]
- 48.Nijboer CH, Heijnen CJ, Groenendaal F, et al. Strong neuroprotection by inhibition of NF-kappaB after neonatal hypoxia-ischemia involves apoptotic mechanisms but is independent of cytokines. Stroke. 2008;39:2129–2137. doi: 10.1161/STROKEAHA.107.504175. [DOI] [PubMed] [Google Scholar]
- 49.Blondeau N, Widmann C, Lazdunski M, Heurteaux C. Activation of the nuclear factor-kappaB is a key event in brain tolerance. J Neurosci. 2001;21:4668–4677. doi: 10.1523/JNEUROSCI.21-13-04668.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rehni AK, Bhateja P, Singh N. Diethyl dithiocar-bamic acid, a possible nuclear factor kappa B inhibitor, attenuates ischemic postconditioning-induced attenuation of cerebral ischemia-reperfusion injury in mice. Can J Physiol Pharmacol. 2009;87:63–68. doi: 10.1139/Y08-100. [DOI] [PubMed] [Google Scholar]
- 51.Pradillo JM, Fernandez-Lopez D, Garcia-Yebenes I, et al. Toll-like receptor 4 is involved in neuroprotection afforded by ischemic preconditioning. J Neurochem. 2009;109:287–294. doi: 10.1111/j.1471-4159.2009.05972.x. [DOI] [PubMed] [Google Scholar]