

Abstract
Notch signaling is reported to regulate angiogenesis, interacting with vascular endothelial growth factor (VEGF) signaling. HMG CoA reductase inhibitors (statins) also alter Notch signaling in vascular cells, but the mechanism and involvement of Notch and VEGF signaling in statin-mediated angiogenesis remain unclear. Here, we examined how statins activate the endothelial Notch1, and promote angiogenesis and arteriogenesis. We examined blood flow recovery after hindlimb ischemia in wild-type (WT) and Notch1 mutant mice treated with or without pitavastatin (3 mg/kg/day, p.o.). Although VEGF induction was not altered in ischemic limbs, pitavastatin promoted blood flow recovery in ischemic limbs in control mice but not in Notch1 mutant mice. Furthermore, pitavastatin induced endothelial ephrinB2 downstream of Notch1 and increased the density of both capillaries and arterioles in the ischemic limbs of WT but not of Notch1 mutant mice. Pitavastatin (100 nmol/l) rapidly activated γ-secretase and Notch1 in human umbilical vein endothelial cells without VEGF induction, which was suppressed by pharmacological inhibition and knockdown of Akt. Pitavastatin also augmented endothelial proliferation and tube formation on Matrigel, which were suppressed by either γ-secretase inhibition or knockdown of Notch1. Pitavastatin-induced microvascular sprouting was also impaired in Notch1 mutant aortic explants. Taken together, pitavastatin activates Notch1 through Akt-dependent stimulation of γ-secretase in endothelial cells, and thereby increases vasculogenesis without VEGF induction.
Keywords: Akt, angio/arteriogenesis, endothelial function, Notch signal pathway, statins
Statins were developed to lower plasma cholesterol by inhibiting HMG-CoA reductase, the rate-limiting enzyme in the mevalonate pathway responsible for cholesterol synthesis. Statins also have cholesterol-independent or ‘pleiotropic’ effects, including direct inhibition of small GTP-binding proteins to activate various signaling cascades in vascular cells.1,2 It has been reported that low-dose statin administration promotes blood flow recovery in animal ischemia models, and that this mechanism is involved in the activation of phosphatidylinositol 3-kinase (PI3K)/Akt signaling pathway.3,4
Notch receptors are highly conserved membrane-bound receptors, which regulate binary cell fate during development and in the adult.5 These receptors directly transduce a signal from the cell surface to the nucleus through regulated intramembrane proteolysis. Binding of ligands promotes a terminal ectodomain-shedding extracellular cleavage followed by γ-secretase-mediated proteolysis within the trans-membrane domain. These events release the Notch intra-cellular domain into the nucleus, in which it interacts with RBP-J protein, and the complex functions as a transcription factor for downstream target genes such as hairy enhancer of split homolog-1 (Hes-1). Thus, γ-secretase strictly regulates the terminal cleavage event and Notch signal activity.6 The Notch signaling pathway regulates vasculogenesis downstream of vascular endothelial growth factor (VEGF) and upstream of ephrinB2.7,8 Endothelial ephrinB2 reverse signaling has been shown to promote embryonic and post-natal angio/arteriogenesis in vivo.9–11 Thus, Notch1 receptor is a key molecule involved in angio/arteriogenesis in adults.
Notch1 and 4 are highly expressed in endothelial cells,6 which have overlapping roles in vascular remodeling and morphogenesis during development.12 We have shown that endothelial Notch1 has a critical role in angiogenetic vascular remodeling during embryogenesis13 and angiogenesis in adult ischemic limbs, downstream the VEGF signaling pathway.14 Statins reportedly increase the expression of Notch-related proteins in neural progenitor cells and vascular cells of a rat stroke model,15,16 however; the mechanisms involved in the regulation of Notch and VEGF signaling are complicated. In the growing vessels of retina, Notch ligand, Delta-like 4 (Dll4) activates Notch signal to reduce VEGF receptor 2 and 3 expression, and suppress sprouting and branching, which was antagonized by Notch signal activation by Jagged-1.17,18 Meanwhile, a recent report showed that Notch1 signaling augments adult angiogenesis by induction of VEGF receptor 1.19
Using a hindlimb ischemia model of Notch1-deficient mice, we investigated the role of Notch1 in statin-mediated angio/arteriogenesis in ischemic tissues. Furthermore, we examined the interaction among statin, VEGF and its receptors, γ-secretase and Notch1 in vascular cells. The results showed that pitavastatin activates endothelial Notch1 downstream of PI3K/Akt pathway, resulting in enhanced angio/arteriogenesis in ischemic limbs without VEGF induction.
MATERIALS AND METHODS Animals
The investigation conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85–23, revised 1996). The experimental protocol was approved by the Animal Studies Committee of Nagoya University. Notch1 heterozygous-deficient mice (N1+/−) with C57BL/6J background (Jackson Laboratory) and wild-type mice (WT) were generated as described previously.14 Pitavastatin (1.0 and 3.0 mg/kg/day, a generous gift from Kowa Soyaku) or vehicle (0.5% carboxymethylcellulose) was administered orally to each 20 mice for 8 weeks.
Surgical Induction of Hindlimb Ischemia
Following surgical hindlimb ischemia,14,20 blood flow recovery was monitored in real-time with a Laser Doppler blood flow (LDBF) analyzer (Moor LDI; Moor Instruments). The left femoral artery and vein were then gently excised from the proximal portion of the femoral artery to the distal portion of the saphenous artery. The remaining arterial branches including the perforator arteries were also excised. Blood flow was monitored with a LDBF analyzer before and on post-operative days 0, 3, 7 and 14. To avoid data variability due to ambient light and temperature, the hindlimb blood flow was expressed as the ratio of left (ischemic) to right (non-ischemic) LDBF.
Immunohistochemistry
The thigh adductor skeletal muscles were processed for immunohistochemistry of CD31 and α-smooth muscle actin (αSMA) as described previously.14 The primary antibody for CD31 (1:500; BD Pharmingen, Franklin Lakes, NJ, USA) and αSMA (1:500; Sigma, St Louis, MO, USA) was applied to the sections overnight at 4°C to detect the endothelium and smooth muscle layer, respectively. After washing, the sections were sequentially incubated with proper fluorescein iso-thiocyanate (FITC)- or PE-conjugated secondary antibodies (Molecular Probes, Eugene, OR, USA; Vector Laboratories, Burlingame, CA, USA) for 1 h, counterstained with 4′,6′-diamidino-2-phenylindole (DAPI; Molecular Probes) and coverslipped with Antifade mounting medium (Vector Laboratories). Some samples were processed for immunohistochemical staining for biotinylated Griffonia simplicifolia isolectinB4 (Vector Laboratories) and αSMA (Dako, Carpinteria, CA, USA), and developed with Vector ABC Biotin Kit and Vectastain DAB Substrate Kit (Vector Laboratories) to detect capillaries and arterioles, respectively, followed by counterstaining with methyl green. Digital images of five microscopic fields from four different sections from each animal were stored. Capillary density was expressed as the number of isolectinB4-positive cells under high-power field (h.p.f.) (magnification, × 400). Arteriole density was expressed as the number of circumvented signals of αSMA-positive cells/h.p.f. (magnification, × 200). For arterial diameter measurement, the αSMA-stained thick wall vessels and short (10 μm) arterial diameters were measured with the software Scion image (Scion corporation) and averaged. The data are presented as averaged diameter and perimeter in eight regions.
Cell Culture and Reagents
Human umbilical vein endothelial cells (HUVECs; Clonetics, Lonza Cologne, Cologne, Germany) of passages 3 through 5 were cultured in EGM-2 (Lonza Cologne). Human aortic smooth muscle cells (HASMC; Lonza Cologne) of passages 3 through 5 were cultured in HuMedia (Lonza Cologne). In some experiments, the cells were pretreated with vehicle (DMSO), mevalonate (200 μmol/l; Sigma), inhibitors for PI3-kinase, (LY294002 at 10 μmol/l; Calbiochem), Akt (SH-5 at 10 μmol/l; Calbiochem), nitric oxide synthetase (NOS) (NG-nitro-L-arginine methyl ester or L-NAME at 1 mmol/l; Dojin Molecular Technologies, Kumamoto, Japan) and γ–secretase (N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester or DAPT at 20 μmol/l; Calbiochem), and stimulated with various concentrations of pitavastatin (kindly supplied by Kowa Pharmaceutical), atorvastatin (10 nmol/l; Sigma Aldrich) and rosuvastatin (10 nmol/l; Sigma Aldrich). Antibodies for VEGF, total Akt, phospho-Akt, total Jagged-1, total Notch1, cleaved Notch1 and presenilin1 (C-terminal antibody), ephrin B2 and β-actin were described previously.14 Rabbit anti-Hes-1 antibody was generously provided by Dr Tetsuo Sudo, (TORAY Corporation, Yokohama, Japan).14,21 Antibodies for Flk-1 and Flt-1 were purchased from Santa Cruz Biotechnology and Cell Signaling, respectively.
SiRNA Delivery to HUVECS
HUVECs were transfected with either a Qiagen small-interfering RNA (siRNA) against Akt (catalog #SI02757244) at 200 pmol, Notch1 (catalog #SI00119028) at 200 pmol or non-silencing control RNA (catalog #1027290) at 200 pmol, using Lipofectamine 2000 reagent (Invitrogen Life Technologies, Carlsbad, CA, USA), using the instructions provided by the manufacturer. Cells were used in the experiments at 24 h after transfection.
Western Blot Analysis
Western blot analysis was performed to examine in tissues and cells treated with the reagents as previously described.14
Assay for γ-Secretase Activity
The assay for γ-secretase activity was performed as described previously.22 Briefly, solubilized membrane preparations were incubated with 8 μmol/l intramolecularly quenched fluorogenic peptide probe, harboring the substrate for γ-secretase (Peptode International). Fluorescence was measured using a plate reader (Fluoroskan Ascent CF, Labsystems) with excitation wavelength at 355 nm and emission wavelength at 440 nm.14,22
Endothelial Cell Proliferation Assay
Cell proliferation assay was determined with cell count and 5-bromo-2′-deoxyuridine (BrdU) uptake as described previously.14 HUVECs (2500 cells) were pretreated with or without mevalonate (200 μmol/l), LY294002 (10 μmol/l), SH-5 (10 μmol/l) or DAPT (20 μmol/l), and then stimulated with pitavastatin (0–1.0 μmol/l). After 72-h incubation, the cells were fixed and stained with 0.1% crystal violet solution to quantify the number of cells. Cellular DNA synthesis was also assessed using the BrdU incorporation assay kit (Roche Applied Science, Indianapolis, IN, USA). HUVECs were pretreated as described above and then stimulated with pitavastatin (0–1.0 μmol/l, 16 h). The cells were properly labeled, stained and then incorporation of BrdU was measured according to the protocol. Experiments were performed for eight separate times for each group in triplicate.14
Assessment of Tube Formation
Cells were washed with Hank’s balanced salt solution, stained with Calcein AM (8 μg/ml in Hank’s balanced salt solution; Dojin Molecular Technologies), and seeded on Matrigel-coated plates at a density of 2 × 104 cells per well in Dulbecco’s modified Eagle medium (DMEM) with 1% fetal bovine serum (FBS), with and without pitavastatin (100 nmol/l) and DAPT (20 μmol/l). Some cells were incubated with pitavastatin (100 nmol/l) followed by siRNA transfection described above. The cells were incubated for 6 h at 37°C and photomicrographs were taken at × 40 magnification.14 Total tube length was measured using the Scion Image program and expressed as percentage of control.
Aortic Ring Assay
This ex vivo angiogenesis assay was performed essentially as described previously.14 Descending thoracic aortas from WT and N1 +/− mice were collected, the adventitia was dissected free and the aorta was cut into several 1-mm rings. Rings were then embedded on the growth factor-reduced Matrigel (BD Bioscience) supplemented with DMEM, 1% FBS, heparin (10 U/ml), antibiotics, pitavastatin (100 nmol/l) and DAPT (20 μmol/l). Experiments were performed using aortic rings from five mice per group.14 Sprout length was quantified using the software Scion image with a calibrated micrometer. The greatest distance from the aortic ring body to the end of the vascular sprouts was measured at three distinct points per ring and in three different rings per mice.
Statistical Analysis
All group data except arterial diameter and perimeter in the text and figures are reported as mean ± s.d. The data on arterial diameter and perimeter are expressed as mean ± s.e. Statistical analysis of the data was carried out using Student’s unpaired t-test. Values of P<0.05 were considered significant.
RESULTS
Notch1 is Involved in Statin-Induced Blood Flow Recovery Following Ischemia
To examine the role of Notch1 in statin-induced angiogenesis in response to ischemia, we subjected WT and N1+/− mice to a hindlimb ischemia and examined the blood flow recovery (Figure 1a). With vehicle treatment, total cholesterol levels were significantly higher in N1+/− mice compared with WT mice, though the difference was small (WT vs N1+/−; 1.71 ± 0.07 vs 2.25 ± 0.07 mmol/l, respectively, P<0.001; n = 5 for each group; Table 1). Treatment with pitavastatin reduced total cholesterol levels to a similar extent in both groups (Table 1).
Figure 1.

Impaired blood flow recovery and angiogenesis in Notch1 heterozygous-deficient mice (N1 +/−) mice treated with pitavastatin (3 mg/kg/day). (a) LDBF showing high-perfusion signal (red to white) in ischemic hindlimb of wild-type (WT), which was augmented by pitavastatin treatment (top left), compared with low-perfusion signal (dark blue) in ischemic hindlimbs of N1 +/− mice, with or without pitavastatin treatment (bottom left). Quantitative analysis of blood flow recovery, following femoral artery ligation, expressed as ischemic to control LDBF ratio in WT and N1 +/− mice (right). *P<0.01 compared with N1 +/− with vehicle on day 7, **P<0.001 compared with N1 +/− with vehicle on day 14 and #P<0.001 compared with WT with vehicle on day 14, n = 8 in each group. (b) Immunostaining for isolectin B4 (brown-red color) in ischemic adductor muscle tissues of WT and N1 +/− mice (left), with and without pitavastatin treatment. The photomicrographs are representative data from eight individual mice from each group. Bar = 25 μm. Quantitative analysis of capillary density (five microscopic fields from four different sections per mice; eight mice each) in WT and N1 +/− mice with or without the pitavastatin treatment (number/h.p.f.; × 400 magnification) (right). *P<0.05 compared with WT mice with vehicle. **P<0.01 compared with WT mice with pitavastatin.
Table 1.
Serum lipid profiles of WT and N1+/− mice treated with vehicle or pitavastatin (mmol/l)
| Treatment | T-CHO | HDL-C | TG |
|---|---|---|---|
| WT(Vehicle) | 1.71 ± 0.07 | 0.83 ± 0.02 | 1.11 ± 0.15 |
| N1+/− (Vehicle) | 2.25 ± 0.07* | 1.37 ± 0.10 | 1.05 ± 0.06 |
| WT (Pitavastatin 3 mg/kg per day) | 1.29 ± 0.14 | 0.54 ± 0.03 | 0.56 ± 0.01 |
| N1+/− (Pitavastatin 3 mg/kg per day) | 1.34 ± 0.12 | 0.67 ± 0.03 | 0.51 ± 0.09 |
Abbreviations: HDL-C, high-density lipoprotein cholesterol; T-CHO, total cholesterol; TG, triglycerides.
Data are shown as mean ± s.d.
P<0.001, compared with WT with vehicle. n = 5 for each group.
As reported previously,14 blood flow recovery in N1+/− was impaired as early as 7 post-operative day (ratio of ischemic to control limb: WT vs N1+/−; 0.34 ± 0.12 and 0.14 ± 0.05, respectively, P<0.01) and reduced up to 47.3% of that of WT at 14 post-operative days (WT vs N1+/−; 0.56 ± 0.04 and 0.29 ± 0.03, respectively, P<0.001; n = 8 for each group). We also examined the effect of oral pitavastatin at two doses on blood flow recovery in WT mice. Pitavastatin at 1.0 mg/kg/day hardly improved the recovery in WT mice (data not shown). However, at 3.0 mg/kg/day, pitavastatin significantly augmented the flow recovery in WT mice by about 50% at 14 post-operative days (WT with vehicle and WT with pitavastatin; 0.56 ± 0.04 and 0.73 ± 0.07, respectively, P<0.001; n = 8 for each group). However, pitavastatin did not result in the recovery of impaired blood flow in N1+/− mice (N1 +/− with vehicle and N1 +/− with pitavastatin; 0.29 ± 0.03 and 0.34 ± 0.02, respectively, P = NS; n = 8 for each group). Four out of eight N1 +/− mice treated with the vehicle and three out of eight N1+/− mice treated with the pitavastatin, but none of WT mice, showed limb necrosis.
As the improvement in blood flow recovery corresponds to increased tissue capillary density, we examined the capillary densities in ischemic adductor muscles of WT and N1 +/− mice. As reported previously,14 there was no difference in capillary density in the control limbs between WT and N1 +/− mice with or without the treatment (data not shown). Notch1 haploinsufficiency was associated with reduced capillary density in the ischemic limbs, which was not restored by the statin treatment (Figure 1b). The capillary density in WT mice was significantly increased by the treatment, and was remarkably higher than that of N1+/− after the statin treatment (62 ± 5.2 and 39 ± 4.1 capillaries/h.p.f., respectively; n = 8 **P<0.01 compared with WT mice).
Statins Accelerate Arteriogenesis via Notch1 Signaling
To determine the effects of Notch1 haploinsufficiency in arteriogenesis, we examined the formation of small arteries by immunohistochemistry using CD31 (endothelial cell layer) and αSMA. As shown in Figure 2a, there was no difference in the arterioles of the control limbs between WT and N1 +/− mice following treatment. However, the αSMA-positive vessel walls of collateral arteries in the ischemic limb were thicker in WT than in N1+/− mice. Furthermore, the vessel walls were thicker and larger in WT mice treated with pitavastatin compared with those with the vehicle-treated WT mice. Pitavastatin had no such effect in N1 +/− mice. The new arterioles of pitavastatin-treated and -untreated N1 +/− mice had smaller vessel areas (Figure 2a). Indeed the pitavastatin treatment significantly enlarged perimeter and diameter of new arterioles in WT mice but not in N1 +/− mice. We also examined the density of αSMA-positive arterioles in the ischemic adductor muscles of WT and N1+/− mice, with or without the treatment. Haploinsufficiency of Notch1 reduced arteriole density in the ischemic limbs. After the statin treatment, which was concordant with the data of blood flow recovery (Figure 1a), the density of arterioles was significantly higher in WT mice (5.0 ± 0.65/h.p.f.) than in N1 +/− mice (2.4 ± 0.50/h.p.f., n = 8 each, **P<0.001 compared with WT mice, Figure 2b).
Figure 2.

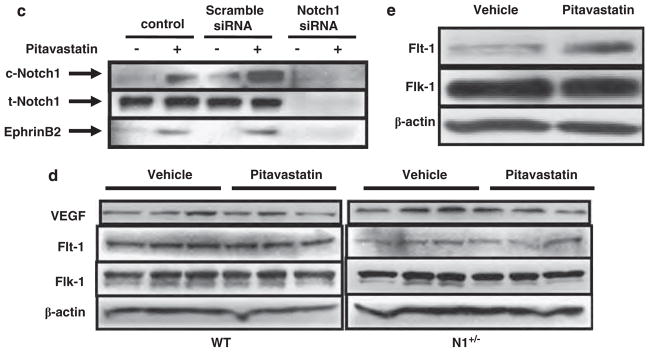
Impaired post-natal arteriogenesis in Notch1 heterozygous-deficient mice (N1 +/−) mice treated with pitavastatin. (a) Top: representative pictures of immunostaining for α-smooth muscle actin (SMA) and CD31 for new vessels from eight individual mice from each group. Bar = 10 μm. Bottom: arterial perimeter and diameter quantitative data in ischemic limbs from wild-type (WT) and N1 +/− mice with the vehicle and pitavastatin treatment and control limbs. *P<0.05 compared WT mice with the vehicle treatment. **P<0.05 compared WT mice with the pitavastatin treatment. (b) Left: representative photomicrographs of αSMA-positive arterioles in the ischemic adductor muscles of WT and N1 +/− mice, with the vehicle or pitavastatin treatment from eight individual mice from each group. Bar = 50 μm. Right: quantitative analysis of arteriole density (five microscopic fields from four different sections per mice; eight mice each; number/h.p.f.; ×200 magnification). *P<0.05, compared with WT with the vehicle treatment. **P<0.001 compared with WT mice with the pitavastatin treatment. (c) Immunoblots showing total and cleaved Notch1 (t- and c-Notch1), and Ephrin-B2 expression in human umbilical vein endothelial cells (HUVECs) transfected with scramble or Notch1 small-interfering RNA (siRNA) in the absence or presence of pitavastatin (100 nmol/l, 2 h). Representative data of three independent experiments with similar results. (d) Immunoblots showing VEGF, Flt-1 and Flk-1 expression in ischemic limbs from WT and N1 +/− mice in the absence or presence of pitavastatin at 3 post-operative days. Representative data of three independent experiments with similar results. (e) Immunoblots showing Flt-1 and Flk-1 expression in HUVECs in the absence or presence of pitavastatin. Representative data of three independent experiments with similar results.
To investigate whether pitavastatin induces ephrinB2, downstream of Notch1 activation, HUVECs were incubated in the presence of pitavastatin followed by transfection with scramble or Notch1 siRNA (Figure 2c). Pitavastatin increased cleaved or active Notch1 in control and scramble-transfected HUVECs within 2 h, resulting in the induction of ephrinB2, but had no effect on the expression of total Notch1. Knockdown of Notch1 markedly reduced Notch1 expression in HUVECs. The induction of ephrinB2 was diminished by Notch1 knockdown (Figure 2c).
Pitavastatin Unchanged VEGF, Flt-1 and Flk-1 Expression in Ischemic Limbs
As VEGF signaling is involved in the activation of endothelial Notch signaling,14 we examined the expression of VEGF in control and ischemic limbs (Figure 2d) at 3 days after the procedure. As reported previously,14 similar VEGF expression was noted in the control and ischemic limbs in WT and N1 +/− mice (Figure 2d, data on control limbs not shown). Pitavastatin neither increased VEGF, Flt-1 or Flk-1 expression in both groups of mice (Figure 2c) nor altered VEGF expression in cultured HUVECs (Figure 3a). However, the expression of Flt-1 was decreased in ischemic limbs in N1 +/− mice with or without the pitavastatin treatment (Figure 3d). Meanwhile, pitavastatin increased the expression of Flt-1 in cultured HUVECs (Figure 3e).
Figure 3.

Pitavastatin induces Notch1 cleavage and Hes-1 expression through Akt. (a) Immunoblots showing the time course of pitavastatin (100 nmol/l)-induced Akt and Notch1 activation, VEGF, Notch1 ligand (Jagged-1) and Hes-1 expression in human umbilical vein endothelial cells (HUVECs). Activation of Akt was assessed by Ser473 phosphorylation of Akt (p-Akt) relative to the total Akt (t-Akt). Activation of Notch1 was determined by the amount of cleaved Notch1 (c-Notch1) relative to total Notch1 (t-Notch1). Representative blots of three independent experiments with similar results. (b) Quantitative data based on three independent experiments. *P<0.001 and #P<0.05, compared with the control (0 time point).
Pitavastatin Activates Endothelial Notch1 and γ-Secretase Activity
To test whether statins activate Notch1 in endothelial cells, HUVECs were treated with pitavastatin at 100 nmol/l for up to 120 min. Pitavastatin rapidly increased the cleavage of Notch1 in HUVECs, reaching a peak level at 30 min (Figure 3). This corresponded to an increase in Hes-1. These changes were also associated with Akt phosphorylation. Short-term treatment with pitavastatin did not change the levels of total Notch1 or Jagged-1 in endothelial cells. Pitavastatin-induced Notch1 activation reached a peak level at a lower concentration (as low as 100 nmol/l, Figure 4a). Stimulation of HASMCs with various concentrations of pitavastatin (0–10 μM) resulted in little or no change in activation of Akt and Notch1 (Figure 4b). Treatment with atorvastatin (10 nmol/l) also activated endothelial Notch1, but this was not observed with rosuvastatin (10 nmol/l) (Figure 4a). This tendency was concordant with Akt activation (data not shown) and γ-secretase activity (Figure 4c). Treatment of HUVECs with pitavastatin significantly increased γ-secretase activity in a dose-dependent manner (control vs pitavastatin at 10 nmol/l and 100 nmol/l; 100 ± 6.0%, 188 ± 23%, 263 ± 18%, respectively. Figure 4c).
Figure 4.

(a) Immunoblots showing Notch1 activation by 30-min treatment with pitavastatin (0–1.0 μmol/l), atorvastatin (10 nmol/l) and rosuvastatin (10 nmol/l). Experiments were performed three times with similar results. (b) Immunoblots showing the lack of effect of 30-min treatment with various concentrations of pitavastatin (0–10 μmol/l) on the expression of active Akt and Notch1 in HASMCs. (c) Effect of mevalonate (Mev) (200 μmol/l), PI3K inhibitor LY294002 (Ly) (10 μmol/l) and DAPT (20 μmol/l) in the absence or presence of pitavastatin (10 or 100 nmol/l, 30 min), atorvastatin (10 nmol/l, 30 min) and rosuvastatin (10 nmol/l, 30 min) on γ-secretase activity in human umbilical vein endothelial cells. *P<0.01 compared with control, **P<0.001 compared with control and #P<0.001 compared with pitavastatin 100 nmol/l alone. n = 3 for each group.
Akt Mediates Pitavastatin-Induced Notch1 Activation
Excess mevalonate (200 μmol/l) reversed the Notch1 activation effect of pitavastatin (Figure 5a). Co-treatment with 10 μmol/l of LY294002, a PI3K inhibitor, or 10 μmol/l of SH-5, an Akt inhibitor, suppressed the statin-induced Notch1 cleavage and Hes-1 expression. γ-Secretase inhibitor, DAPT (20 μmol/l), efficiently blocked the statin-induced Notch1 cleavage and Hes-1 expression, but NOS inhibitor, L-NAME (1 mmol/l), had no effect on Notch1 activation. These results were concordant with γ-secretase activity. Co-treatment with mevalonate, LY294002 and DAPT reduced γ-secretase activity down to the control level (Figure 4c). To analyze whether Akt mediates statin-induced Notch1 activation, HUVECs were transfected with scramble and Akt siRNA in the presence or absence of pitavastatin (Figure 5b). Similar to Figure 3, pitavastatin increased active Notch1 and Hes-1 in control and scramble siRNA-transfected HUVECs. Pitavastatin also increased the C-terminal fragment of presenilin1 in control and scramble siRNA-transfected HUVECs, which was concordant with activation of γ-secretase (Figure 4c). Transfection with Akt siRNA efficiently decreased total and phospho-Akt (Figure 5b). Indeed transfection with Akt siRNA reduced the cleavage of presenilin1 and Notch1, and the induction of Hes-1.
Figure 5.

(a) Effects of mevalonate (200 μmol/l), LY294002 (10 μmol/l), Akt inhibitor SH-5 (10 μmol/l), γ-secretase inhibitor DAPT (20 μmol/l) and NOS inhibitor NG-nitro-L-arginine methyl ester (L-NAME) (1 mmol/l) on pitavastatin (100 nmol/l) 30 min-induced Akt activation (p-Akt), Notch1 cleavage (c-Notch1) and Hes-1 expression. Experiments were performed three times with similar results. (b) Representative immunoblots showing total and phosphorylated Akt (t- and p-Akt), C-terminal fragment of presenilin 1 (c-Presenilin 1), Jagged-1, total and cleaved Notch (t- and c-Notch1) and Hes-1 expression in human umbilical vein endothelial cells transfected with Akt small-interfering RNA (siRNA) in the absence or presence of pitavastatin (100 nmol/l, 30 min). Three independent experiments provided similar results.
Notch1 Mediates Statin-Induced Endothelial Proliferation
The effect of pitavastatin on endothelial proliferation was determined by determining the cell counts and BrdU incorporation because endothelial proliferation reflects pro-angiogenetic activity (Figure 6). Pitavastatin significantly increased the cell count in a dose-dependent manner, which decreased at 1 μM (Figure 6a). This was reversed by excess mevalonate and suppressed by inhibitors of PI3K, Akt and γ-secretase similar to endothelial Notch1 activation (Figures 4a and 6a). Pitavastatin also increased BrdU incorporation in HUVECs, whereas excess mevalonate and inhibitors of PI3K, Akt and γ-secretase reduced the incorporation, in agreement with the changes in the cell count (Figure 6b).
Figure 6.
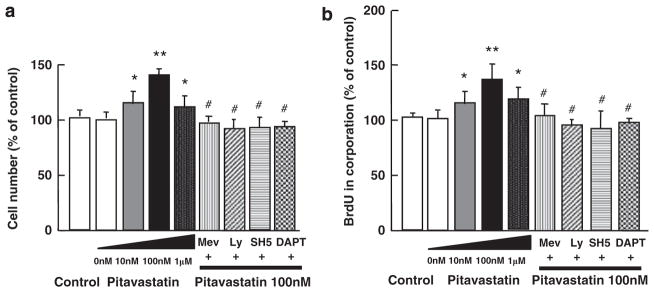
Pitavastatin induces endothelial cell proliferation through γ-secretase activation. (a) Cell counts. (b) 5-bromo-2′-deoxyuridine (BrdU) incorporation in human umbilical vein endothelial cells in the absence or presence of pitavastatin (10 nmol/l–1.0 μmol/l). Effects of mevalonate (200 μmol/l), LY294002 (Ly)(10 μmol/l), Akt inhibitor SH-5 (10 μmol/l), NOS inhibitor NG-nitro-L-arginine methyl ester (L-NAME) (1 mmol/l) and γ-secretase inhibitor DAPT (20 μmol/l) on proliferation were also determined. *P<0.007 compared with control and **P<0.001 compared with control and #P<0.001 compared with pitavastatin (100 nmol/l). n = 8 in each group.
Notch1 Mediates Statin-Induced In Vitro Tube Formation and Ex Vivo Microvascular Sprouting
Matrigel-based in vitro angiogenesis assay was performed to determine whether Notch1 contributes to the statin-induced angiogenesis (Figure 7). Compared with basal conditions, endothelial cells formed tubular networks progressively following exposure to pitavastatin (Figure 7a). Notch signal inhibition combined with a γ-secretase inhibitor blocked pitavastatin-induced tubular formation (Figure 7a). To determine the involvement of Notch1 in the pitavastatin-induced tubular formation, tube-forming assay was performed after transfection of scramble and Notch1 siRNA. Knockdown of Notch1 efficiently suppressed the expression of Notch1, resulting in suppression of tube formation (Figure 7b).
Figure 7.


Notch1 mediates tube formation and microvascular sprouting in vitro. (a) Top: representative photomicrographs (× 400 magnification) showing Matrigel-based, capillary-like tube formation in human umbilical vein endothelial cells (HUVECs) treated with pitavastatin (100 nmol/l) and γ-secretase inhibitor DAPT (20 μmol/l). Bottom: quantitative data of capillary-like tube formation. Data shown are mean ± s.d., n = 5 in each group. *P<0.01 compared with the absence of pitavastatin and **P<0.01 compared with pitavastatin only. (b) Top: immunoblots showing the expression of Notch1 and β-actin in HUVECs transfected with scramble or Notch1 small-interfering RNA (siRNA). Center: representative photomicrographs (× 400 magnification) showing Matrigel-based, capillary-like tube formation in HUVECs transfected with scramble or Notch1 siRNA in the presence of pitavastatin (100 nmol/l). Bottom: quantitative data of capillary-like tube formation. Data shown are mean ± s.d., n = 5 in each group. *P<0.01 compared with the absence of pitavastatin and **P<0.01 compared with pitavastatin plus scramble siRNA. (c) Top: representative fields (× 40 magnification) showing microvessel outgrowth in response to pitavastatin (100 nmol/l) and DAPT (20 μmol/l) in aortic explants from wild-type (WT) and Notch1 heterozygous-deficient mice (N1+/−) mice. Bottom: microvessel outgrowth was quantified as percentage of that in WT in the absence of pitavastatin. Data shown are mean ± s.d., n = 5 in each group, *P<0.001 compared with WT in the absence of pitavastatin ,**P<0.001 compared with WT in the absence of pitavastatin and #P<0.001 compared with WT with pitavastatin.
Further microvascular sprouting was examined in aortic rings from WT and N1 +/− cultured with or without pitavastatin (Figure 7c). Microvessel sprouting was significantly impaired in aortic rings from N1 +/− mice (100 ± 8.9 vs 26.6 ± 1.3% of control, WT vs N1+/−, n = 5, **P<0.001), as reported previously.14 Pitavastatin increased sprouting in aortic rings of WT mice compared with the control (100 ± 8.9 vs 205 ± 24% of control, n = 5, *P< 0.001), but did not affect that in N1+/− mice (26.6 ± 1.3 vs 28.5 ± 2.3% of control, n = 5, P = NS). This effect was diminished by pharmacological Notch signal inhibition in aortic rings from WT mice (205 ± 24 vs 68 ± 6.4% of control, respectively; n = 5, #P<0.001 compared with aortic rings from WT mice with the treatment).
DISCUSSION
We demonstrated in the present study that pitavastatin promoted angio/arteriogenesis in ischemic limbs of WT mice but not of N1+/− mice (Figures 1 and 2). The aortic explant assay highlighted the importance of endothelial Notch1 for pitavastatin-enhanced microvessel outgrowth (Figure 7c). The results also showed that pitavastatin activated endothelial γ-secretase and Notch1 signaling downstream of the PI3K/Akt pathway (Figures 3–5). Furthermore, pitavastatin augmented the expression of its downstream targets, Hes-1, and the arterial marker, ephrinB2, to promote arteriogenesis (Figures 2 and 3). Notch1 has a critical role in endothelial proliferation and migration activity.14,23 Indeed, pitavastatin increased the pro-angiogenic activity of endothelial cells, including proliferation and tube formation, which was ameliorated by Notch1 inhibition or knockdown (Figures 6 and 7).
Recent reports have demonstrated that statins induce Notch-related proteins in neural progenitor cells and vascular cells in a rat stroke model,15,16 and cultured bone marrow stromal cells.24 However, it has not been clear whether Notch1 activation in vessels contributes to statin-induced angio/arteriogenesis in vivo, and how statins activate endothelial Notch signal in detail. Accordingly, we compared statin-induced angiogenic effects in WT and N1 +/− mice, and have shown the direct evidence that Notch1 is indeed involved in statin-mediated angio/arteriogenesis in vivo for the first time. We also addressed the more in-depth mechanism of statin-mediated Notch signal activation through Akt and γ-secrtase activation (Figure 3), according to our previous findings.14 It was hardly discussed how the statin treatment promotes arteriogenesis apart from endothelial activation and increased blood flow recovery, but we showed endothelial ephrin B2 induction by pitavastatin in the downstream of Notch1 activation, resulting in statin-mediated arteriogenesis (Figure 2).
In the present study, pitavastatin did not alter VEGF expression in ischemic limbs and cultured endothelial cells, suggesting that VEGF induction is not involved in pitavastatin-induced Notch signal activation (Figures 2 and 3). The pitavastatin treatment did not change the expression of VEGF receptors in ischemic limbs of both mice. On the other hand, decrease in Flt-1 expression was observed in ischemic limbs of N1+/− mice. This would be concordant to the recent reports that Notch1 signal inhibition reduces the expression of Flt-1.19,25 Funahashi et al19 showed that Notch1 decoy treatment reduced tumor angiogenesis and the expression of endothelial Flt-1 without alteration of Flk-1. Thus, haplo-insufficiency of Notch1 might decline the responsiveness to VEGF and placental growth factor derived from ischemic skeletal muscle. Meanwhile, the pitavastatin treatment increased Flt-1 expression in cultured HUVECs (Figure 2e). This would suggest that the pleiotropic effects of statins would be closely involved in Notch and VEGF signaling without VEGF induction.
It is reported that statins improve endothelial function by reducing cholesterol and through other cholesterol-independent pleiotropic effects.2 One of the potent pleiotropic effects is believed to be activation of the PI3K/protein kinase Akt/eNOS axis in endothelial cells.3 As pitavastatin reduced total cholesterol levels in both groups to the same level (Table 1), Notch signal pathway is unlikely to be involved in the cholesterol-dependent effect of pitavastatin. Akt and Notch1 activation by pitavastatin peaked at 100 nmol/l and decreased at 1 μM, and pharmacological inhibition and knockdown of Akt diminished pitavastatin-mediated Notch1 activation in HUVECs. Furthermore, we also demonstrated that other statins also activated Notch1 in cultured HUVECs and observed the same effects for atorvastatin. However, Notch1 activation in vitro was hardly detected in rosuvastatin-treated HUVECs. The reason for this finding is likely because of the fact that rosuvastatin is a hydrophilic agent with poor penetration in HUVECs and difficult to activate Akt in an in vitro system.26 Indeed, pitavastatin activated endothelial Akt and Notch1 in a similar manner, which reflects dual concentration-dependent effects on endothelial proliferation (Figure 6). Thus, Notch1, a critical angiogenesis regulator, could contribute at least in part to the Akt-mediated effects of pitavastatin. Interestingly, L-NAME, a NOS inhibitor, had little or no effect on pitavastatin-induced Notch1 activation in agreement with our previous report that L-NAME did not change VEGF-mediated Notch1 activation (Figure 4a).14 These results indicate a strong relationship between Akt and Notch signal activation. We reported previously that Notch 1 activation in SMCs promotes neointimal formation.27 The present results also confirmed that pitavastatin did not change Akt and Notch1 activation in SMCs and, thus, does not cause exacerbation of arteriosclerosis (Figure 4b).
We also investigated the mechanism of γ-secretase-dependent Notch signal activation. The multiprotein enzyme complex γ-secretase, which includes presenilin, nicastrin, Aph-1 and Pen-2, cleaves Notch receptors.28 γ-Secretase is a critical target to regulate angiogenesis and tumorigenesis through Notch signal activation.29 However, regulation of presenilin, which comprises the active catalytic core of γ-secretase, is still unknown in detail. As shown in Figures 4c and 5b, pitavastatin increased presenilin1 cleavage and γ-secretase activity in HUVECs downstream Akt activation, resulting in endothelial Notch1 activation. Indeed, knockdown of Akt and pharmacological inhibition of PI3K and Akt decreased γ-secretase activity, resulting in reduced statin-mediated Notch1 activation. These findings are quite consistent with our previous study that VEGF increases endothelial presenilin processing, γ-secretase activity and Notch1 activation through the PI3K/Akt pathway.14
Growing evidence indicates that Notch signaling promotes angio/arteriogenesis not only in developmental state but also in ischemia-induced angiogenesis in adults.6–8,13,14,19 Meanwhile, Notch signal activation regulates VEGF receptor expression and angiogenic activity in retinal endothelial cells in a ligand-dependent manner. Dll4-mediated Notch signaling suppresses tip cells sprouting in retina, which was antagonized by Notch signal activation by Jagged-1.17,18 Thus, negative and positive roles for Notch signaling in endothelial sprouting and angiogenetic activity have been discussed. In the present study, we observed abundant Jagged-1 expression in HUVECs (Figure 3a). However, we could not detect the expression of Dll4 in HUVECs and vessels in ischemic limbs with western blotting and immunohistochemistry (antibody; R&D systems, AF1389), even after the statin treatment. This might suggest that the results in the present study are mainly dependent on Jagged-1-mediated Notch activation to promote angio/arteriogenesis. Furthermore, statins would activate endothelial Notch1 signal in ischemic limbs in a manner distinct from retinal model. For instance, retinal endothelial tip cell filopodia are guided by a gradient of VEGF. However, in hindlimb ischemia model, the source of VEGF is likely to be more diffused.
The ephrin B2/EphB4 signaling has a critical role during development to determine the fate of blood vessel identity downstream of the Notch1 in endothelium.12,13,30 Arterial and venous specification in the developing embryo is genetically identified through the distinctive expression of ephrinB2 in cells that form future arteries.31 Recent studies also reported that EphB4/ephrinB2 regulates tumor vessel remodeling and promotes post-natal angiogenesis in a myocardial ischemia model.32,33 Thus, the ephrin B2/EphB4 signaling is a critical target to modulate proper angio/arterio genesis downstream of Notch1. Indeed, we showed that pitavastatin increased ephrinB2 in endothelial cells by Notch1 activation (Figure 2c), and promoted the growth of collateral arteries with larger vessel areas in WT but not in N1+/− mice. These findings suggest that Notch1 is an important downstream target of pleiotropic effects in vascular endothelial cells, consistent with the pro-angiogenic effects of statin.3
In conclusion, we have demonstrated in the present study that statin-mediated angio/arteriogenesis were reduced in Notch1 mutant mice. We also investigated the mechanisms of pitavastatin-induced activation of endothelial Notch1. Our study provided direct evidence for the involvement of Notch signaling pathway in angio/arteriogenesis in vivo by statins.
Acknowledgments
We thank Dr Yoshiyuki Rikitake (Department of Cardiology, Kobe University Graduate School of Medicine) for careful reading and suggestion. We also thank Kowa Soyaku for providing pitavastatin. This work was supported by research grants from the Kanae Foundation (to KT), the Kowa life Science Foundation (to KT), the Mitsubishi Pharma Research Foundation (to KT) and Grant-in-Aid for Scientific Research (Kakenhi 21590950 to KT, Kakenhi 20249045 to TM).
Footnotes
DISCLOSURE/CONFLICT OF INTEREST
The authors declare no conflict of interest.
References
- 1.Van Aelst L, D’Souza-Schorey C. Rho GTPases and signaling networks. Genes Dev. 1997;11:2295–2322. doi: 10.1101/gad.11.18.2295. [DOI] [PubMed] [Google Scholar]
- 2.Wang CY, Liu PY, Liao JK. Pleiotropic effects of statin therapy: molecular mechanisms and clinical results. Trends Mol Med. 2008;14:37–44. doi: 10.1016/j.molmed.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kureishi Y, Luo Z, Shiojima I, et al. The HMG-CoA reductase inhibitor simvastatin activates the protein kinase Akt and promotes angiogenesis in normocholesterolemic animals. Nat Med. 2000;6:1004–1010. doi: 10.1038/79510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sata M, Nishimatsu H, Suzuki E, et al. Endothelial nitric oxide synthase is essential for the HMG-CoA reductase inhibitor cerivastatin to promote collateral growth in response to ischemia. Faseb J. 2001;15:2530–2532. doi: 10.1096/fj.01-0415fje. [DOI] [PubMed] [Google Scholar]
- 5.Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science. 1999;284:770–776. doi: 10.1126/science.284.5415.770. [DOI] [PubMed] [Google Scholar]
- 6.Iso T, Kedes L, Hamamori Y. HES and HERP families: multiple effectors of the Notch signaling pathway. J Cell Physiol. 2003;194:237–255. doi: 10.1002/jcp.10208. [DOI] [PubMed] [Google Scholar]
- 7.Lawson ND, Scheer N, Pham VN, et al. Notch signaling is required for arterial-venous differentiation during embryonic vascular development. Development. 2001;128:3675–3683. doi: 10.1242/dev.128.19.3675. [DOI] [PubMed] [Google Scholar]
- 8.Lawson ND, Vogel AM, Weinstein BM. sonic hedgehog and vascular endothelial growth factor act upstream of the Notch pathway during arterial endothelial differentiation. Dev Cell. 2002;3:127–136. doi: 10.1016/s1534-5807(02)00198-3. [DOI] [PubMed] [Google Scholar]
- 9.Adams RH, Wilkinson GA, Weiss C, et al. Roles of ephrinB ligands and EphB receptors in cardiovascular development: demarcation of arterial/venous domains, vascular morphogenesis, and sprouting angiogenesis. Genes Dev. 1999;13:295–306. doi: 10.1101/gad.13.3.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hayashi S, Asahara T, Masuda H, et al. Functional ephrin-B2 expression for promotive interaction between arterial and venous vessels in postnatal neovascularization. Circulation. 2005;111:2210–2218. doi: 10.1161/01.CIR.0000163566.07427.73. [DOI] [PubMed] [Google Scholar]
- 11.Roca C, Adams RH. Regulation of vascular morphogenesis by Notch signaling. Genes Dev. 2007;21:2511–2524. doi: 10.1101/gad.1589207. [DOI] [PubMed] [Google Scholar]
- 12.Krebs LT, Xue Y, Norton CR, et al. Notch signaling is essential for vascular morphogenesis in mice. Genes Dev. 2000;14:1343–1352. [PMC free article] [PubMed] [Google Scholar]
- 13.Limbourg FP, Takeshita K, Radtke F, et al. Essential role of endothelial Notch1 in angiogenesis. Circulation. 2005;111:1826–1832. doi: 10.1161/01.CIR.0000160870.93058.DD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Takeshita K, Satoh M, Li M, et al. Critical role of endothelial Notch1 signaling in postnatal angiogenesis. Circ Res. 2007;100:70–78. doi: 10.1161/01.RES.0000254788.47304.6e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen J, Zacharek A, Li A, et al. Atorvastatin promotes presenilin-1 expression and Notch1 activity and increases neural progenitor cell proliferation after stroke. Stroke. 2008;39:220–226. doi: 10.1161/STROKEAHA.107.490946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zacharek A, Chen J, Cui X, et al. Simvastatin increases notch signaling activity and promotes arteriogenesis after stroke. Stroke. 2009;40:254–260. doi: 10.1161/STROKEAHA.108.524116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Benedito R, Roca C, Sorensen I, et al. The notch ligands Dll4 and Jagged1 have opposing effects on angiogenesis. Cell. 2009;137:1124–1135. doi: 10.1016/j.cell.2009.03.025. [DOI] [PubMed] [Google Scholar]
- 18.Suchting S, Freitas C, le Noble F, et al. The Notch ligand Delta-like 4 negatively regulates endothelial tip cell formation and vessel branching. Proc Natl Acad Sci USA. 2007;104:3225–3230. doi: 10.1073/pnas.0611177104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Funahashi Y, Shawber CJ, Vorontchikhina M, et al. Notch regulates the angiogenic response via induction of VEGFR-1. J Angiogenes Res. 2010;2:3. doi: 10.1186/2040-2384-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murohara T, Asahara T, Silver M, et al. Nitric oxide synthase modulates angiogenesis in response to tissue ischemia. J Clin Invest. 1998;101:2567–2578. doi: 10.1172/JCI1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ito T, Udaka N, Yazawa T, et al. Basic helix-loop-helix transcription factors regulate the neuroendocrine differentiation of fetal mouse pulmonary epithelium. Development. 2000;127:3913–3921. doi: 10.1242/dev.127.18.3913. [DOI] [PubMed] [Google Scholar]
- 22.Farmery MR, Tjernberg LO, Pursglove SE, et al. Partial purification and characterization of gamma-secretase from post-mortem human brain. J Biol Chem. 2003;278:24277–24284. doi: 10.1074/jbc.M211992200. [DOI] [PubMed] [Google Scholar]
- 23.Morrow D, Cullen JP, Cahill PA, et al. Ethanol stimulates endothelial cell angiogenic activity via a Notch- and angiopoietin-1-dependent pathway. Cardiovasc Res. 2008;79:313–321. doi: 10.1093/cvr/cvn108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu J, Liu X, Chen J, et al. Simvastatin enhances bone marrow stromal cell differentiation into endothelial cells via notch signaling pathway. Am J Physiol Cell Physiol. 2009;296:C535–C543. doi: 10.1152/ajpcell.00310.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hamilton Outtz H, Wu JK, Wang X, et al. Notch1 deficiency results in decreased inflammation during wound healing and regulates vascular endothelial growth factor receptor-1 and inflammatory cytokine expression in macrophages. J Immunol. 2010;185:4363–4373. doi: 10.4049/jimmunol.1000720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.White CM. A review of the pharmacologic and pharmacokinetic aspects of rosuvastatin. J Clin Pharmacol. 2002;42:963–970. [PubMed] [Google Scholar]
- 27.Li Y, Takeshita K, Liu PY, et al. Smooth muscle Notch1 mediates neointimal formation after vascular injury. Circulation. 2009;119:2686–2692. doi: 10.1161/CIRCULATIONAHA.108.790485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.De Strooper B. Aph-1, Pen-2, and Nicastrin with Presenilin generate an active gamma-secretase complex. Neuron. 2003;38:9–12. doi: 10.1016/s0896-6273(03)00205-8. [DOI] [PubMed] [Google Scholar]
- 29.Miele L, Miao H, Nickoloff BJ. NOTCH signaling as a novel cancer therapeutic target. Curr Cancer Drug Targets. 2006;6:313–323. doi: 10.2174/156800906777441771. [DOI] [PubMed] [Google Scholar]
- 30.Swift MR, Weinstein BM. Arterial-venous specification during development. Circ Res. 2009;104:576–588. doi: 10.1161/CIRCRESAHA.108.188805. [DOI] [PubMed] [Google Scholar]
- 31.Aitsebaomo J, Portbury AL, Schisler JC, et al. Brothers and sisters: molecular insights into arterial-venous heterogeneity. Circ Res. 2008;103:929–939. doi: 10.1161/CIRCRESAHA.108.184937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hainaud P, Contreres JO, Villemain A, et al. The role of the vascular endothelial growth factor-Delta-like 4 ligand/Notch4-ephrin B2 cascade in tumor vessel remodeling and endothelial cell functions. Cancer Res. 2006;66:8501–8510. doi: 10.1158/0008-5472.CAN-05-4226. [DOI] [PubMed] [Google Scholar]
- 33.Mansson-Broberg A, Siddiqui AJ, Genander M, et al. Modulation of ephrinB2 leads to increased angiogenesis in ischemic myocardium and endothelial cell proliferation. Biochem Biophys Res Commun. 2008;373:355–359. doi: 10.1016/j.bbrc.2008.06.036. [DOI] [PubMed] [Google Scholar]