

Abstract
Cancer-associated protein tyrosine kinase (PTK) mutations usually are gain-of-function (GOF) mutations that drive tumor growth and metastasis. We have found 50 JAK1 truncating mutations in 36 of 635 gynecologic tumors in the Total Cancer Care® (TCC®) tumor bank. Among cancer cell lines containing JAK1 truncating mutations in the Cancer Cell Line Encyclopedia databank, 68% are gynecologic cancer cells. Within JAK1 the K142, P430, and K860 frame-shift mutations were identified as hot spot mutation sites. Sanger sequencing of cancer cell lines, primary tumors, and matched normal tissues confirmed the JAK1 mutations and showed that these mutations are somatic. JAK1 mediates interferon (IFN)-γ-regulated tumor immune surveillance. Functional assays show that JAK1 deficient cancer cells are defective in IFN-γ-induced LMP2 and TAP1 expression, loss of which inhibits presentation of tumor antigens. These findings identify recurrent JAK1 truncating mutations that could contribute to tumor immune evasion in gynecologic cancers, especially in endometrial cancer.
Mutations in PTK genes usually result in GOF mutants that drive human cancer. Examples include EGFR mutations in non-small cell lung cancer1, FLT3 mutations in acute myeloid leukemia2 and the BCR-ABL fusion gene in chronic myeloid leukemia3. Mutations in these genes result in constitutively active PTKs and/or PTKs with altered substrate specificity. These mutant PTKs can transform immortalized model cells in cell culture and can induce tumors in animals. Mutant PTKs have been previously targeted for cancer therapy1,3,4.
JAK1 and JAK2 are PTKs that mediate the cytokine receptor signaling. GOF JAK2 mutations such as JAK2V617F have been linked to myeloproliferative neoplasms (MPNs)4,5. The JAK inhibitor Ruxolitinib is now used to treat myelofibrosis6. In a laboratory experiment, certain randomly generated JAK1 missense mutations were found to be able to transform the cytokine-dependent mouse BaF3 pro-B cells7.
However, JAK1 and JAK2 also mediate the interferon-γ (IFN-γ) signaling. IFN-γ plays important roles in tumor immune surveillance and has antiproliferative/apoptotic activity8,9,10. IFN-γ activates the receptor-associated JAK1/JAK2 to phosphorylate STAT1. STAT1 induces the primary IFN-γ response genes, such as IRF1, which then induce secondary response genes to mediate its biological effects11. The low molecular weight protein-2 (LMP2) of the proteosome and transporter associated with antigen processing-1 (TAP1) are IFN-γ-induced major histocompatibility complex (MHC) related genes12. LMP2 and TAP1 assist in the processing and transport of intracellular antigens for presentation by the MHC class I complex on the cell surface. MHC class I presentation allows for tumor recognition by cytotoxic T lymphocytes (CTLs)8,13 and is central to tumor immune surveillance8,10,12,13. Consistently, it was observed that IFN-γ receptor deficient mice are hypersensitive to the chemical carcinogen methylcholanthrene-induced tumors10. Moreover, LMP2 knockout female mice developed spontaneous uterine leiomyosarcoma, suggesting that LMP2 is obligatory for immune surveillance of this type of uterine neoplasm in the animals14. In a follow-up study of 101 human smooth muscle tumors of the uterus, 6 point mutations in the promoter of LMP2 gene in 6 tumors and 5 missense mutations in the JAK1 catalytic domain were found in 6 tumors that had impaired IFN-γ signaling in transfected cells15.
In this study, we searched for JAK1 mutations in 3,274 tumors among TCC® tumor samples from 48 different tissues16. We found JAK1 loss-of-function (LOF) mutations as identified by truncating mutations that lose the PTK domain, which occur predominantly in endometrial cancer. We also found that JAK1 truncating mutations in Cancer Cell Line Encyclopedia (CCLE) databank occur most often in endometrial cancer cell lines. Compilation of JAK1 truncating mutation events revealed three mutation hot spots. Re-sequencing of cancer cell line and tumor tissue samples available to us confirmed JAK1 mutations in these cancer cells and tumor tissues. Furthermore, cells containing JAK1 truncating mutations are defective in IFN-γ-induced antigen processing machinery proteins LMP2/TAP1 and cell surface expression of HLA molecules. These findings reveal recurrent JAK1 deficiency in gynecologic cancer that could contribute to tumor immune evasion.
Results
JAK2 mutations have been linked to MPNs. Some of laboratory-generated JAK1 mutants also have transformation activity in BaF3 cells. We originally searched for potential GOF JAK1 and JAK2 mutations in the newly completed massively-parallel sequence data from the TCC® project (a targeted gene sequencing dataset) that contains >3000 cases of human cancer. Non-synonymous JAK1 mutations were found in 134 tumors among 3274 tumor samples (Supplementary Table 1). Gynecologic (GYN) cancer notably has the highest non-synonymous mutation rate (8.3%, 53 tumors carrying 78 non-synonymous mutations among 635 tumors analyzed) (Table 1). Strikingly, 64% of these mutations are frame shift or nonsense mutations (Fig. 1, Table 1). As shown in Fig. 1, these frame-shift or nonsense mutations result in truncated JAK1 lacking the essential PTK domain. Therefore, they are LOF mutations. The highest JAK1 truncating mutation rate (9.5%) was observed in corpus uterine cancer (tumors from endometrium or uterus tissue), followed by cervical uterine cancer (4.1%). In further examination of uterus cancer with JAK1 truncating mutations, it was found that all are endometrial adenocarcinoma except 1 case of which medical record is not available (sTable 1). No truncating mutation was found in brain, esophagus, hematologic, kidney, prostate, or soft tissue cancer in 595 tumors although non-synonymous mutations have been detected in these tumors, whereas truncating mutations were observed in breast, colorectal, lung, pancreas, skin, and stomach at much lower rates (Table 1).
Table 1. Samples with JAK1 mutations in the TCC project+.
| Case | |||
|---|---|---|---|
| Tissue | Total | Non-synonymous Mutation# | Truncating Mutation# |
| Brain | 79 | 1 | 0 |
| Breast | 427 | 10 | 1 |
| Colorectal* | 539 | 23 | 9 |
| Esophagus | 44 | 2 | 0 |
| Gallbladder | 2 | 1 | 0 |
| GYN** | 635 | 53 | 36 |
| (Corpus uterine***) | (348) | (43) | (33) |
| HEME-CLL | 94 | 2 | 0 |
| Kidney | 243 | 8 | 0 |
| Lung | 603 | 14 | 2 |
| Pancreas | 161 | 4 | 1 |
| Prostate | 52 | 1 | 0 |
| Skin | 209 | 10 | 1 |
| Soft tissue | 45 | 2 | 0 |
| Stomach | 55 | 4 | 2 |
+See supplementary Table 1 for the complete list.
*Large bowel, small intestine, and rectum-anus.
**Cervix, endometrial, gynecologic, ovary, uterus, vagina, and vulva.
***Endometrium and uterus.
#Some cases have more than one mutations, for examples, 14 cases of GYN tumors have two different JAK1 truncating mutations and thus the total JAK1 truncating mutation events detected in GYN cancer is 50.
Figure 1. JAK1 truncating mutations found in the TCC human tumors and CCLE cancer cells.

Schematic representation of JAK1 protein and the truncating mutation sites identified in the TCC tumors and CCLE cells. Vertical bars above JAK1 are proportional to the numbers of mutations found in the TTC and CCLE datasets. * indicates positions of nonsense mutations. Horizontal bars end at the position of stop codons.
To assess whether JAK1 mutations observed in cancer tissues occur in cancer cells, we examined the Cancer Cell Line Encyclopedia (CCLE) databank17. We reasoned that if JAK1 truncating mutations were detected in cancer cell lines, this would be indicative of JAK1 truncating mutations occurring within cancer cells in the tumor tissues. There are 72 non-synonymous JAK1 mutations in 57 different cancer cell lines in CCLE, including 25 truncating mutations in 19 cell lines (sTable 2, Fig. 1). Thirteen of these 19 cell lines (68.4%) are GYN cancer cells, which are represented by 11 endometrial and 2 ovarian cancer cell lines. Thus, JAK1 truncating mutations were detected mostly in endometrial cancer cell lines in CCLE. Manual examination of Binary Alignment/MAP (BAM) files of JAK1 truncating mutations from cases in TCC and CCLE17 confirmed the presence of point and insertion/deletion mutations. Compiling of JAK1 truncating mutation events in the TCC and CCLE samples identified K142fs, P430fs, and K860fs as the three hot spot mutation sites (Fig. 1).
JAK2 truncating mutations were found in 17 TCC tumors (sTable 3). Ten of these are GYN cancer, including 9 uterine cancer. IFN-γ-activated JAK1/JAK2 mainly phosphorylate STAT1 to regulate IFN-γ response genes8,18. STAT1 truncating mutations were found in 6 TCC tumors (sTable 4). Four of these are corpus uterine cancer. Thus, JAK2 and STAT1 truncating mutations were also detected predominantly in GYN cancer but at lower rates than that of JAK1 mutations.
To confirm JAK1 truncating mutations in cancer cells, we sampled 6 CCLE GYN cell lines. CCLE data indicate that Hela and OVCAR3 cells have wildtype JAK1 whereas HEC1B, IGROV1, TOV21G, and MFE296 cells have truncating mutations. Consistently, our sequencing data showed no JAK1 mutation in Hela and OVCAR3 (Fig. 2a, sFig. 1a). IGROV1 cells have homozygous K860fs*43 mutation (2580delA) (Fig. 2b–c). TOV21G cells have heterozygous K860fs*43 mutation (2580delA) and heterozygous N339fs*3 mutation (1016delA) (sFig. 1c–d). Besides 1289delC reported in CCLE, HEC1B cells have 1290–1291insC, resulting in homozygous P430fs mutations (P430fs*2; P430fs*63) (sFig. 1b). MFE296 cells have homozygous K860fs*43 mutation (2580delA) (sFig. 1e). Consistently, immunoblotting analysis detected the full-length 130-kDa JAK1 in Hela and OVCAR3 cells but not in HEC1B, IGROV1, TOV21G, or MFE296 cells (Fig. 3a). In comparison, all 6 cell lines express the full-length JAK2 protein (sFig. 3). To assess whether the mutated JAK1 gene is expressed in the 4 cell lines, we examined the JAK1 transcript in the cells by RT-PCR. Our data showed that the JAK1 mRNA was readily detectable in all four GYN cell lines that contain the JAK1 gene mutations (sFig. 4), suggesting that the truncated JAK1 is expressed in these cells.
Figure 2. Sanger DNA sequencing to examine JAK1 frame shift mutation at K860.

Genomic DNA of Hela cells (a), IGROV1 cells (c), an endometrial tumor (2T) or the matched normal endometrial tissue (2N) from the same patient (d) were prepared and sequenced across the K860 region. (b) CCLE igv_snapshot of the sequence alignment that identifies the K860fs mutation in IGROV1 cells.
Figure 3. JAK1 deficient cancer cells are defective in the IFN-γ-stimulated LMP2/TAP1 antigen-processing machinery pathway.

(a) Cell lysates from 6 cell lines were analyzed by immunoblotting with an anti-JAK1 antibody. Position of the 130-kDa full-length JAK1 is indicated. Other bands appear on the immunoblot are unknown non-specific immunoreactivities. (b–c) Cells were treated with IFNγ for the indicated time. Cell lysates were analyzed by immunoblotting for IFN-γ-induced pSTAT1, IRF1, LMP2, and TAP1.
We obtained frozen tissues from 7 TCC corpus uterine tumors that contain 9 JAK1 truncating mutations in the TCC Mutation dataset and 6 matched normal tissues (Table 2). DNA was sequenced across the mutation sites identified in the TCC data. Eight of 9 mutations were detected in these tumor samples (Table 2, Fig. 2d, and sFig. 2). The TCC data show that the DS-55004 tumor has double K142fs and K860fs mutations with mutation allele fractions of 0.19 and 0.60, respectively. The K142fs mutation was not detected by our Sanger-sequencing, although it was possible the mutation allele fraction was below our detection limit. However, the K860fs mutation was detected (Table 2 and sFig. 2). Thus, all 7 tumors have confirmed JAK1 mutations. None of the matched normal tissues has the corresponding JAK1 mutations (Table 2, Fig. 2d, and sFig. 2). Thus, JAK1 mutations detected in these tumors are somatic mutations.
Table 2. JAK1 frame-shift truncating mutation in TCC tumors and matched normal tissues+.
| Barcode | JAK1 mutation (TCC) | Mutation allele fraction (TCC) | Primary tissue | ID | JAK1 mutation in tumor (Sanger Sequencing) | Matched normal tissue | ID | JAK1 mutation in normal tissue (Sanger Sequencing) |
|---|---|---|---|---|---|---|---|---|
| DS-58859 | K860fs | 0.85 | Endometrium | 1T | mut | N/A | N/A | N/A |
| DS-57828 | K860fs | 0.53 | Endometrium | 2T | mut | Endometrium | 2N | wt |
| DS-55004 | K142fs; K860fs | 0.19; 0.60 | Uterus** | 3T | K142wt; K860mut | Uterus | 3N | wt |
| DS-60386 | K860fs | 0.45 | Endometrium* | 4T | mut | Large Bowel | 4N | wt |
| DS-55027 | R174X | 0.42 | Uterus** | 5T | mut | Uterus | 5N | wt |
| DS-55352 | P430fs | 0.31 | Uterus** | 6T | mut | Uterus | 6N | wt |
| DS-80370 | K142fs; P430fs | 0.37; 0.26 | Endometrium | 7T | K142mut; P430mut | Endometrium | 7N | wt |
+Mut, mutation; wt, wildtype.
*Tumor tissue collected from metastatic tumor in vagina.
**Adenocarcinoma.
To determine if the IFN-γ-stimulated LMP2/TAP1 pathway is defective in GYN cancer cell lines carrying JAK1 truncating mutations, we compared IFN-γ-induced STAT1 tyrosine phosphorylation (pSTAT1), IRF1, LMP2, and TAP1 in the above six GYN cancer cell lines. IFN-γ induced pSTAT1 and expression of IRF1, LMP2, and TAP1 in Hela and OVCAR3 cells but these IFN-γ-induced responses were defective in HEC1B, IGROV1, TOV21G, and MFE296 cells (Fig. 3b–c). LMP2 and TAP1 are induced by IFN-γ via a shared bidirectional promoter activated by the IFN-γ-induced IRF112. As shown in Fig. 4. IRF1-dependent LMP2- and TAP1-promoter activities were readily induced by IFN-γ in Hela and OVCAR3 cells but not in other cell lines. This defect in the expression of the antigen-processing machinery proteins was not due to a non-specific cytotoxic effect of IFN-γ in these JAK1 mutation cells because these cells were resistant to growth inhibition induced by IFN-γ (Fig. 5a). Consistently, p27 and p21 levels were not changed by IFN-γ treatment in JAK1 mutation cell lines whereas they were induced by IFN-γ in Hela and OVCAR3 cells (Fig. 5b).
Figure 4. Analysis of IFN-γ-induced LMP2- and TAP1-promoter activities.

(a) Comparison of IFN-γ-induced, IRF1-dependent LMP2 and TAP1-promoter activities in GYN cancer cell lines. The basal LMP2-Luc or TAP1-Luc activity, respectively, in Hela cells was set as 1 for comparison of Luc activities. Data were from two independent experiments performed in duplicate (n = 4). (b) JAK1 mutation cells were transfected with pCMV-JAK1 or the control vector and LMP2/TAP1-promoter reporter plasmids as in (a). IFN-γ-stimulated LMP2- and TAP1-promoter luciferase activities were assayed. *, p < 0.05 by the Mann-Whitney test. Differences in the basal LMP2- and TAP1-luciferase activities between cells transfected with pCMV-JAK1 and the control vector were p < 0.05 in all cell lines.
Figure 5. Effects of IFN-γ on cell proliferation/viability of GYN cancer cells.

(a) Cells were treated with IFN-γ for 4 days and viable cells were measured. Data were from two independent experiments performed in quadruple. *, p <0.05 by the Mann-Whitney test. (b) Cells were treated with IFN-γ for the indicated time and cell lysates were analyzed by immunoblotting with indicated antibodies.
We next co-transfected a wildtype JAK1 expression vector with the LMP2- or TAP1-promoter luciferase reporter to determine whether the exogenous JAK1 could rescue the defect in the IFN-γ-stimulated LMP2 and TAP1-promoter activities in JAK1 mutation cell lines. As shown in Fig. 4b, the basal LMP2- and TAP1-promoter activities were elevated significantly in JAK1-transfected cells. The LMP2- and TAP1 promoter activities were further increased in response to IFN-γ stimulation. The levels of IFN-γ-stimulated LMP2- and TAP1-promoter activities in these JAK1-transfected cells were not as high as those observed in Hela or OVCAR3 cells. This may be due to differences in unknown genetic attributes between cell lines. However, the JAK1 truncation mutants in these cell lines retain the protein-protein interacting FERM domain (HEC1B cells) or the FERM-SH2-pseudokinase domains (MFE296, IGROV1, and TOV21G cells). Conceivably, these truncated JAK1 proteins may exert a dominant-negative effect in the cells to prevent a full restoration of the IFN-γ-stimulated activities by the exogenous JAK1.
To further assess if the deficiency in IFN-γ-regulated antigen processing machinery proteins LMP2 and TAP1 compromises MHC class I antigen presentation, we compared IFN-γ-induced cell surface expression of the MHC class I molecules HLA-ABC on Hela, MFE296 and HEC1B cells. These cells were randomly chosen to represent JAK1 wildtype and JAK1 mutation cell lines. MHC class I antigens are normally loaded into HLA molecules and transported to cell surface for presentation to CD8+ CTLs. When HLA molecules are not loaded with antigens, they become unstable and are degraded more rapidly in the cells. This results in a lower level of HLA molecules on the cell surface19,20. As shown in Fig. 6. HLA-ABC proteins on the cell surface of Hela cells increased 2.3 ± 0.4 fold (mean ± range from 2 independent experiments) after IFN-γ stimulation. In contrast, the HLA-ABC levels did not change on the surface of MFE296 or HEC1B cells after IFN-γ stimulation. These data indicate that MFE296 and HEC1B cells are defective in MHC class I antigen presentation in response to IFN-γ.
Figure 6. Analysis of cell surface HLA molecules.
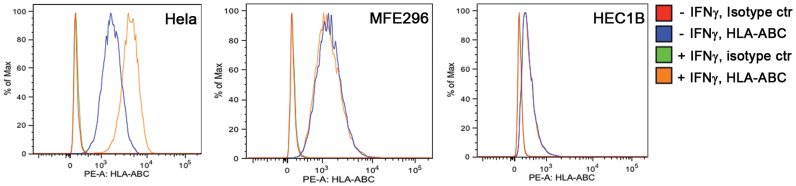
The amounts of HLA-ABC on the Hela, MFE296, and HEC1B cell surface were analyzed by flow cytometry after immunostaining with a PE-conjugated anti-HLA-ABC antibody as described in the Method. Data were from a representative experiment.
Discussion
Utilizing the newly completed massively-parallel sequence dataset of the TCC® project that contains >3,000 tumors from various human tissues, we found recurrent JAK1 LOF truncating mutations. The JAK1 deficiency is linked most often to endometrial cancer. Examination of JAK1 mutations in CCLE cancer cell lines lends support to the finding in the TCC data that JAK1 truncating mutations occur predominantly in endometrial cancer cells. Our analysis of the TCC and CCLE data further identified K142, P430, and K860 of JAK1 as three hot spot truncating mutation sites.
In addition to the truncating mutations, other non-synonymous JAK1 mutations were also observed in the TCC tumors. Effects of these missense mutations are not as readily predictable as truncating mutations and thus require further analysis individually by experiments. Conceivably, some of these mutations could be LOF. Consistent with this notion, five JAK1 missense mutations defective in JAK1 kinase activity were found in uterine leiomyosarcoma15. Nevertheless, mutation in none of these five residues (G871, G876, C881, Y987, R995) or a partial loss of function mutation site Q986 was found in TCC tumors or in CCLE cells.
RT-PCR data suggest that the mutated JAK1 genes are expressed in the cancer cell lines that harbor these mutations. These truncated forms of JAK1 retain protein-protein interaction domains. Although it remains to be analyzed, it is predicted that they may exert an inhibitory effect on IFN-γ signaling by sequestering JAK1 binding partners, such as the IFN-γ receptor, to a non-functional state. This might have prevented the full-restoration of IFN-γ signaling when a wildtype JAK1 was expressed in these cells in our experiments.
Recently, POLE mutations were reported in a subset of endometrial cancer21. Mutations in the N-terminal exonuclease domain of POLE may contribute to higher mutation rates. However, only 3 of the 36 TCC GYN tumors that carry JAK1 truncating mutations have POLE mutations in the N-terminal exonuclease domain (one P286R, P400T, and A465V mutation each). Thus, POLE defect-associated mutations may contribute but is not sufficient to explain high JAK1 truncating mutation rate that we observed in endometrial cancer.
Cancer-associated PTK gene mutations mostly result in GOF PTK mutants with deregulated protein kinase activity that drives malignant phenotypes. In contrast, JAK1 gene truncating mutations identified in this study give rise to truncated JAK1 lacking the essential kinase domain. Cancer cells containing these JAK1 LOF mutations are defective in the IFN-γ-regulated tumor antigen processing machinery. Thus, rather than directly driving tumor growth via a constitutively activated PTK, our findings suggest that JAK1 mutations in endometrial cancer facilitates tumorigenesis involving the mechanism of escaping CTL-mediated tumor immune surveillance that requires the MHC class I antigens.
Methods
Massively-parallel sequence data from the TCC project and BAM files examination
Frozen tumor samples from the TCC® project16 were subjected to genomic capture (performed by BGI, Shenzhen using SureSelect custom designs targeting 1,321 genes, Agilent Technologies, Inc., Santa Clara, CA) and massively parallel sequencing (90 base pair, paired end reads, performed by BGI, Shenzhen using GAIIx, Illumina, Inc., San Diego, CA). Sequences were aligned to the hs37d5 human reference with the Burrows-Wheeler Aligner (BWA)22. Insertion/deletion realignment, quality score recalibration, and variant identification were performed with the Genome Analysis ToolKit (GATK)23. Sequence variants were annotated with ANNOVAR24 and then analyzed using an in-house web-based display application. Alignments using BWA and Stampy25 were manually inspected with samtools tview26 to visually confirm mutation status.
Cell cultures
Hela cells27 were cultured in DMEM/10% fetal bovine serum (FBS). HEC1B cells were obtained from American Type Culture Collection and cultured in EMEM/10%FBS. MFE296 cells were from the European Collection of Cell Cultures and cultured in DMEM/10%FCS. OVCAR3 cells28 were cultured in RPMI1640/10%FBS. IGROV1 and TOV21G cells were provided by Dr. Patricia Kruk and cultured in Medium 199/MDCB105 (1:1)/10%FBS.
Sanger DNA sequencing
Genomic DNA was prepared from cultured cells or frozen tissue samples using DNAeasy Blood & Tissue Kit (Qiagen). Malignant or non-malignant state of each tissue was verified by histological examination by a pathologist in the Moffitt Histology Core. Genomic DNA regions containing the mutation sites identified in TCC or CCLE were amplified by PCR using GoTag Hot Start Green Master Mix reagent (Promega). The PCR primer pairs used were: K142U (5′-GGACGCCCTGCCCACATCTG)/K142L (5′-TGGGCCCAAACTTCCTACCTGAGC), annealing temperature: 58°C; R174U (5′-GGCGGCCCCTCCCTGTGTAG)/R174L (5′-TTCTGGCAACTGCATCTTCTTCATCAT), annealing temperature: 56°C; N339U (5′-AGCAGCACTGACCTTTTACCA)/N339L (5′-CCACTCTTCCCGGATCTTGTTTT), annealing temperature: 51°C; P430U (5′-TTGGGTTATAAAAATCTTCTCACTG)/P430L (5′-TGCCCCATCACTAAAACACG), annealing temperature: 46°C; K860U (5′-CAGCTTGGGGAGAAACAGGAG)/K860L (5′-TGGCCCCAGAATGAAAAAGAAT), annealing temperature: 50°C. PCR were performed using ~80 ng genomic DNA and the following parameters: 1 cycle at 95°C for 2 min; 30 cycles at the 95°C for 45 sec, annealing temperature for 45 sec, 72°C for 30 sec; and 1 cycle at 72°C for 5 min. PCR products were cleaned with ExoSAP-IT PCR product cleanup reagent (USB) and sequenced in both strands using BigDye Terminator v3.1 cycle sequencing kit (Life Technologies) and a 3130X Genetic Analyzer (Applied Biosystems). Sequencing primers were the same as the PCR primers.
IFN-γ treatment, immunoblotting, luciferase report, and cell proliferation assays
For immunoblot analysis, cells were treated with 20 ng/ml IFN-γ (Peprotech) for the duration indicated in the figures. Cell lysate preparation and immunoblot analysis were performed essentially as described previously29,30. Antibodies against JAK1, JAK2, pSTAT1, and cleaved PARP were from Cell Signaling Technologies. Antibodies to STAT1, IRF1, and LMP2 were from Santa Cruz Biotech. Anti-TAP1 antibdoy was from Enzo. Anti-p21 antibody was from NeoMarkers. Anti-p27 antibody was from BD Pharmingen. Anti-β-Actin antibody was from Sigma.
For LMP2 and TAP1 promoter luciferate reporter assay, 3 × 105 cells in 6-well plates were co-transfected with either 1.8 μg LMP2-Luc, LMP2-IRF1mut-Luc, TAP1-Luc, or TAP1-IRF1mut-Luc12 and 0.2 μg pCMV-βgal30 plasmids for 24 h. Transfected cells were incubated in serum-free medium with or without 20 ng/ml IFN-γ for 24 h. Cell lysates were prepared for determination of luciferase and β-galactosidase activities as described30. Luciferase activity was normalized with the β-galactosidase activity. The wildtype JAK1 expression plasmid was obtained from GeneCopoeia. The JAK1 rescue experiment was performed by transfecting cells with a 1:1 ratio of 0.9 μg JAK1:LMP2-Luc (or TAP1-Luc) plus control plasmids as described above.
For cell proliferation/viability assay, cells were plated in black 96-well plates, allowed to attach overnight before addition of 300 ng/ml IFN-γ or left untreated for 4 days. Relative viable cell number was measured using CellTiterGlo reagent (Promega).
RT-PCR analysis of JAK1 mRNA
Total RNA was isolated from cells using the Trizol reagent (Life Technologies). RT-PCR was performed using the SuperScript One-Step RT-PCR with Platinum Tag reagents (Life Technologies). Two pairs of RT-PCR primers were used: 1) JAK1-F1 (5′-CCGGCTGGGCAGTGGAGAGT)/JAK1-R1 (5′-GGTAGTGGAGCCGGAGGGACAT) and 2) JAK1-F2 (5′-AGATGGCTACTTCCGGCTCACAG)/JAK1-R2 (5′- CGTACATCCCCTCCTCGCTTCCTT). The product of the JAK1-F1/R1 pairs is 178 bp while that of the JAK1-F2/R2 pairs is 163 bp. The reverse transcription step was performed at 53°C for 30 min using ~800 ng total RNA. The PCR step was performed using the following conditions: 1 cycle at 94°C for 2 min; 30 cycles at the 94°C for 30 sec, annealing temperature (53°C for F1/R1 primers, 55°C for F2/R2 primers) for 30 sec, 72°C for 20 sec; and 1 cycle at 72°C for 5 min.
Flow cytometry
Cells were treated with or without 50 ng/ml IFN-γ for 24 h, detached from plates using Accutase (BD Biosciences), and suspended in PBS containing 0.5% BSA. Cells were immunostained with a PE-conjugated mouse anti-human HLA-ABC antibody (clone G46-2.6) or the isotype control antibody (BD Pharmingen) according to the supplier's instruction. The stained cells were fixed with 4% paraformaldehyde. Data were acquired on a FACSCanto II flow cytometer with DIVA software (BD Biosciences.). A blue laser (488 nm) with a 585/42 bandpass (BP) filter and a 566 longpass (LP) filter were used to excite and collect the emission from the PE fluorochrome. The data were analyzed with the Flowjo (Treestar) software.
Author Contributions
Y.R. performed experiments. Y.Z. analyzed the genomic data. R.Z.L. developed the mutation library browser. D.A.F. reviewed the genomic data. K.L.W. advised on tumor immunology and provided reagents. J.K.T. analyzed genomic data. J.W. discovered the JAK1 truncating mutations initially, directed the project, performed experiments, and analyzed data. D.A.F., K.L.W., J.K.T., and J.W. wrote the manuscript.
Supplementary Material
Supplementary Information
Acknowledgments
We thank the TCC and Moffitt core facility staffs for assistance, Drs. Jin Cheng and Patricia Kruk for reagents, and Dr. Bill Dalton for facilitating the study. Funding for the TCC® sequencing project was provided by Merck (Whitehouse Station, NJ). This work was supported by the Dr. Tsai-fan Yu Cancer Research Fund and in part by the NIH grant R01CA178456.
References
- Pao W. & Girard N. New driver mutations in non-small-cell lung cancer. Lancet Oncol 12, 175–80 (2011). [DOI] [PubMed] [Google Scholar]
- Gilliland D. G., Jordan C. T. & Felix C. A. The molecular basis of leukemia. Hematology (Am Soc Hematol Educ Program), 80–97 (2004). [DOI] [PubMed] [Google Scholar]
- Deininger M., Buchdunger E. & Druker B. J. The development of imatinib as a therapeutic agent for chronic myeloid leukemia. Blood 105, 2640–53 (2005). [DOI] [PubMed] [Google Scholar]
- Tefferi A. & Gilliland D. G. Oncogenes in myeloproliferative disorders. Cell Cycle 6, 550–66 (2007). [DOI] [PubMed] [Google Scholar]
- Jatiani S. S., Baker S. J., Silverman L. R. & Reddy E. P. Jak/STAT pathways in cytokine signaling and myeloproliferative disorders: approaches for targeted therapies. Genes Cancer 1, 979–93 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tefferi A. JAK inhibitors for myeloproliferative neoplasms: clarifying facts from myths. Blood 119, 2721–30 (2012). [DOI] [PubMed] [Google Scholar]
- Gordon G. M., Lambert Q. T., Daniel K. G. & Reuther G. W. Transforming JAK1 mutations exhibit differential signalling, FERM domain requirements and growth responses to interferon-gamma. Biochem J 432, 255–65 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seliger B., Ruiz-Cabello F. & Garrido F. IFN inducibility of major histocompatibility antigens in tumors. Adv Cancer Res 101, 249–76 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall L., Burke F., Barton C., Smyth J. & Balkwill F. IFN-gamma induces apoptosis in ovarian cancer cells in vivo and in vitro. Clin Cancer Res 9, 2487–96 (2003). [PubMed] [Google Scholar]
- Kaplan D. H. et al. Demonstration of an interferon gamma-dependent tumor surveillance system in immunocompetent mice. Proc Natl Acad Sci U S A 95, 7556–61 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder K., Hertzog P. J., Ravasi T. & Hume D. A. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol 75, 163–89 (2004). [DOI] [PubMed] [Google Scholar]
- Wright K. L. et al. Coordinate regulation of the human TAP1 and LMP2 genes from a shared bidirectional promoter. J Exp Med 181, 1459–71 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaczynska M., Rock K. L. & Goldberg A. L. Gamma-interferon and expression of MHC genes regulate peptide hydrolysis by proteasomes. Nature 365, 264–7 (1993). [DOI] [PubMed] [Google Scholar]
- Hayashi T. & Faustman D. L. Development of spontaneous uterine tumors in low molecular mass polypeptide-2 knockout mice. Cancer Res 62, 24–7 (2002). [PubMed] [Google Scholar]
- Hayashi T. et al. Potential role of LMP2 as tumor-suppressor defines new targets for uterine leiomyosarcoma therapy. Sci Rep 1, 180 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenstermacher D. A., Wenham R. M., Rollison D. E. & Dalton W. S. Implementing personalized medicine in a cancer center. Cancer J 17, 528–36 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretina J. et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–7 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H. & Jove R. The STATs of cancer--new molecular targets come of age. Nat Rev Cancer 4, 97–105 (2004). [DOI] [PubMed] [Google Scholar]
- de la Salle H. et al. Homozygous human TAP peptide transporter mutation in HLA class I deficiency. Science 265, 237–41 (1994). [DOI] [PubMed] [Google Scholar]
- Van Kaer L., Ashton-Rickardt P. G., Ploegh H. L. & Tonegawa S. TAP1 mutant mice are deficient in antigen presentation, surface class I molecules, and CD4-8+ T cells. Cell 71, 1205–14 (1992). [DOI] [PubMed] [Google Scholar]
- Kandoth C. et al. Integrated genomic characterization of endometrial carcinoma. Nature 497, 67–73 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. & Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–60 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo M. A. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43, 491–8 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K., Li M. & Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38, e164 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunter G. & Goodson M. Stampy: a statistical algorithm for sensitive and fast mapping of Illumina sequence reads. Genome Res 21, 936–9 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–9 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunnick J. M. et al. Role of tyrosine kinase activity of epidermal growth factor receptor in the lysophosphatidic acid-stimulated mitogen-activated protein kinase pathway. J Biol Chem 273, 14468–75 (1998). [DOI] [PubMed] [Google Scholar]
- Xu C. X. et al. MicroRNA miR-214 regulates ovarian cancer cell stemness by targeting p53/Nanog. J Biol Chem 287, 34970–8 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Ren Y. et al. Critical role of Shp2 in tumor growth involving regulation of c-Myc. Genes Cancer 1, 994–1007 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Y. et al. Shp2E76K mutant confers cytokine-independent survival of TF-1 myeloid cells by up-regulating Bcl-XL. J Biol Chem 282, 36463–73 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information