

Abstract
Background
Prior evidence suggests a link between inflammation and hypertension. Interleukin-6 (IL-6) has been implicated in animal studies to play an important role in angiotensin II (ANGII) mediated hypertension. The aim of this study was to examine the relationship of IL-6 and renin-angiotensin system (RAS) activity in human hypertension.
Methods
Data from 385 hypertensives and 196 normotensives are included in this report. Blood pressure and laboratory evaluation were performed on liberal and low sodium diets. IL-6 response to an ANGII infusion was evaluated to assess the effect of acute RAS activation.
Results
Hypertensives had higher baseline IL-6 and C-reactive protein (CRP) compared with normotensives on both diets. IL-6 increased in response to ANGII in hypertensives and normotensives (28% in hypertensives, 31% in normotensives, p = < 0.001 for change from baseline). In the setting of RAS activation by a low salt diet, multivariate regression analysis adjusted for age, BMI, gender, race and hypertension status demonstrated an independent positive association of PRA with CRP (beta = 0.199, p<0.0001). There was no significant difference in IL-6 or CRP levels between liberal and low sodium diets.
Conclusion
These findings confirm an association between hypertension and inflammation and provide human data supporting previous evidence from animal studies that IL-6 plays a role in ANGII mediated hypertension. Notably, compared to levels on a liberal sodium diet, neither IL-6 or CRP were higher with activation of the RAS by a low salt diet indicating that a low sodium diet is not inflammatory despite increased RAS activity.
Keywords: hypertension, inflammation, sodium, angiotensin, CRP, IL-6
Introduction
Accumulating evidence has established an important role of inflammation in the pathogenesis of cardiovascular diseases such as atherosclerosis and hypertension1–16. Elevated levels of interleukin-6 (IL-6), a key pro-inflammatory cytokine, has been shown to be associated with increased future risk of myocardial infarction in healthy men13 and a prognostic marker of future coronary events and mortality in patients with acute coronary syndrome14–17. In addition, evidence from animal studies strongly supports an important role for IL-6 in the pathogenesis of angiotensin II (ANGII)-mediated hypertension18–20. IL-6 levels increase in mice receiving ANGII infusions19, 20. In addition, it has been demonstrated that ANGII-mediated hypertension is attenuated or abolished in mice lacking a functional gene for IL-619, 20.
Evidence in humans evaluating the interplay of IL-6, ANGII and hypertension is limited. The majority of the human evidence supporting a link between inflammation and hypertension has been from epidemiological studies evaluating associations between blood pressure (BP) and biomarkers of inflammation, with C-reactive protein (CRP) being the most extensively studied marker of vascular inflammation 5, 6, 8, 10–12. Human studies evaluating the relationship between inflammation and renin-angiotensin system (RAS) activity are even more limited. A few clinical studies have suggested that angiotensin receptor blockers (ARBs) have anti-inflammatory effects, which may provide additional benefits beyond lowering of BP 21–23. The only human study that directly evaluated the relationship between ANGII and IL-6 was in a small cohort (N=11) of normotensives subjects and demonstrated an increase in IL-6 in response to ANGII infusion24.
The aim of this study was to examine systematically the relationship between IL-6, RAS activity and hypertension and determine if and how IL-6 and RAS activity are potentially linked with hypertension in humans. We hypothesized that inflammation in hypertension is associated with the level of activity of the RAS, and IL-6 is a key mediator of this inflammatory process. To provide a common ground with the majority of the previous studies evaluating the link between hypertension and inflammation, we also analyzed CRP levels.
Methods
Subjects and protocol
385 hypertensives and 196 normotensives studied by the International Hypertensive Pathotype (HyperPATH) group are included in this report. Subjects were recruited from the general population of the Brigham and Women’s Hospital, Boston (n = 239), Hospital Broussais in France (n = 96), University of Utah (n = 197) and Vanderbilt University (n = 49). The institutional review board of each institution approved the study. All subjects gave written informed consent before enrollment and underwent a standardized protocol. Some characteristics of subsets of this population have been reported previously25–28. However, the present analyses are original.
All subjects had a screening history, physical and laboratory examination. Subjects with known or suspected secondary hypertension, diabetes, coronary artery disease, stroke, overt renal insufficiency (serum creatinine > 1.5 mg/dl), current tobacco or illicit drug use, alcohol intake > 12 ounces per week, or other significant medical or psychiatric illnesses were excluded. Subjects with abnormal electrolytes or thyroid/liver function tests or electrocardiographic evidence of heart block, ischemia, or prior coronary events at the screening exam were excluded. Subjects were between ages 18 to 65. Race was self-defined.
Hypertension was defined as seated diastolic BP ≥100 mmHg off antihypertensive medication, ≥90 mmHg with 1 medication, or treatment with ≥2 medications. Subjects with hypertension requiring more than four medications were excluded. Normotensives, in addition to having BP less than 140/90 mmHg, reported no first degree relatives diagnosed with hypertension before age 60. Subjects taking an angiotensin converting enzyme inhibitor, ARBs, or mineralocorticoid receptor antagonist were transitioned to amlodipine three months prior to study to minimize interference with assessment of the RAS. If needed, hydrochlorothiazide was added to control BP. All antihypertensive medications were discontinued two weeks before initiating the diet phase of the study and three weeks before protocol initiation.
Subjects then completed two dietary phases, a five to seven day course of liberal sodium (200 mmol/day) and seven days of low sodium (10 mmol/day) diets. Each diet was isocaloric, contained 100 mmol/day potassium, 20 mmol/day calcium, and was caffeine and alcohol-free. On the final day of each diet, subjects were admitted to the General Clinical Research Center (GCRC) and remained fasting and supine overnight. Baseline BP and laboratory assessments were performed the following morning with subjects in the supine position followed by an ANGII infusion at 3 ng/kg/min × 55 minutes and repeat laboratory evaluation was performed 45–55 minutes after the start of the ANGII infusion. The ANGII infusion was performed to evaluate IL-6 response to an acute increase in RAS activity. Sodium balance was confirmed by measuring sodium and creatinine in a 24-hour urine collection (≥ 150 mmol urinary sodium for liberal sodium and ≤ 30 mmol for low-sodium). BP was measured at five-minute intervals using an automated device (DINAMAP; Critikon, Tampa, FL). Three consecutive readings were averaged for analysis.
Laboratory analysis
Blood samples were collected on ice and centrifuged for 20 minutes. All samples (serum, plasma and urine) were frozen without preservatives and stored at −80 degrees centigrade until assayed. Aldosterone and renin levels were measured by radioimmunoassay techniques, as previously described28–30. CRP levels were measured using a high-sensitivity CRP enzyme-linked immunoassay (ALPCO, NH). IL-6 levels were measured using a quantitative enzyme-linked immunoassay (R&D Systems, Inc., Minneapolis, MN).
Statistics
Statistical analyses were performed using SPSS version 15.0 (SPSS, Chicago, IL). Subject groups were compared using a two-sample t-test for normally distributed data and non-parametric tests when the raw data was not normally distributed. Paired samples t-test was used for comparison of repeated measures including comparison of liberal and low salt CRP and IL-6 levels and IL-6 levels before and after ANGII infusion. Data were analyzed for determinants of CRP and IL-6 levels using univariate and multivariate regression. The natural-log of CRP, IL-6, plasma renin activity (PRA) and aldosterone were used to meet normality assumptions for the regression analyses. Data are reported as mean ± standard error. Statistical significance was indicated by p < 0.05.
Results
Baseline Characteristics
The baseline characteristics of the hypertensive and normotensives subjects are shown in Table 1. The hypertensives were older and had higher BMI. There were minor differences in the race and gender distributions between the two groups, which were not statistically significant. Baseline BP was higher in hypertensives compared to normotensives and on liberal salt compared to low salt diet. Serum aldosterone and PRA were higher on a low salt diet than on a liberal salt diet in hypertensives and normotensives. There were mild differences in baseline aldosterone and PRA levels between normotensives and hypertensives as shown in Table 1.
Table 1.
Baseline Characteristics of the Hypertensive and Normotensive Study Cohorts
| Hypertensives (N=385) | Normotensives (N=196) | p-value | |
|---|---|---|---|
| Age (years) | 49 ± 8 | 38 ± 12 | < 0.001 |
| Gender (female%) | 53% | 46% | 0.106 |
| Race (white%) | 85.5% | 79% | 0.052 |
| BMI (kg/m2) | |||
| HS | 28.2 ± 3.8 | 25.2 ± 3.9 | < 0.001 |
| LS | 27.4 ± 3.8 | 24.6 ± 3.9 | < 0.001 |
| SBP (mmHg) | |||
| HS | 147 ± 20 | 112 ± 12 | < 0.001 |
| LS | 132 ± 17 | 106 ± 10 | < 0.001 |
| DBP (mmHg) | |||
| HS | 87 ± 11 | 67 ± 8 | < 0.001 |
| LS | 80 ± 10 | 64 ± 7 | < 0.001 |
| PRA (ng/ml/hr) | |||
| HS | 0.48 ± 0.42 | 0.44 ± 0.38 | 0.38 |
| LS | 2.3 ± 2 | 3.2 ± 2 | < 0.001 |
| Serum Aldo (ng/dl) | |||
| HS | 5.2 ± 3.3 | 3.6 ± 1.7 | < 0.001 |
| LS | 17.6 ± 12 | 19.8 ± 12 | 0.008 |
Mean ± SD for continuous variables; percentages for categorical variables.
Abbreviations: HS –> liberal salt diet; LS –> low salt diet
BMI -> body mass index; SBP -> Systolic blood pressure; DBP -> diastolic blood pressure.
Relationship between Hypertension and Inflammation on Baseline Liberal Salt Diet
Hypertensives had significantly higher (p = < 0.001 for IL-6 and p = 0.008 for CRP) unadjusted baseline IL-6 and CRP levels compared with normotensives (Fig 1). Given the differences in baseline characteristics of our hypertensive and normotensive cohorts and potential for confounding related to these differences, we evaluated the relationship between hypertension status and the levels of CRP and IL-6 adjusting for the effects of age, BMI, gender and race. The relationship between hypertension status and CRP was no longer statistically significant (p = 0.58) when adjusted for the above covariates, with BMI (standardized beta 0.41, p < 0.001) and age (standardized beta 0.197, p < 0.001) being significant confounders and independent predictors of CRP. However, the association between hypertension status and IL-6 remained significant (p = 0.018) indicating an independent association between hypertension and IL-6 even after controlling for relevant confounders.
Figure 1. Plasma CRP and IL-6 in Hypertensives versus Normotensives on Liberal Salt Diet.
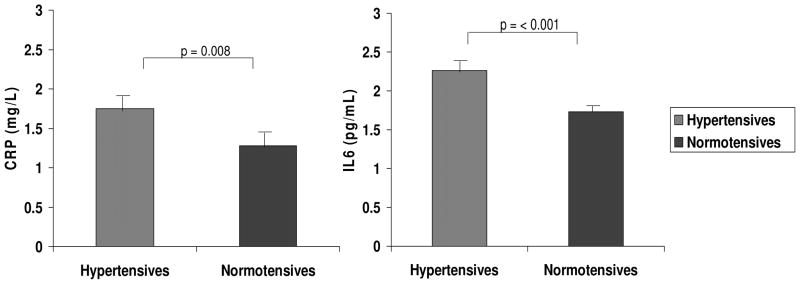
Hypertensives had significantly higher unadjusted baseline mean CRP and IL-6 levels. Error bars = SEM.
RAS Activity and Inflammation
1) IL-6 Response to ANGII infusion
To evaluate the inflammatory response to an acute increase in RAS activity, we studied the change in IL-6 levels in response to ANGII infusion at 3 ng/kg/min for 45–55 minutes. IL-6 levels increased significantly in response to ANGII infusion in both hypertensives and normotensives (28% in HT and 31% in NT, p = < 0.001 for change from baseline in both groups; Fig 2). There was no significant difference in the magnitude of change in IL-6 in response to ANGII between normotensives and hypertensives.
Figure 2. IL6 response to Angiotensin II infusion.

IL6 increased significantly in response to ANGII in both hypertensives and normotensives. Error bars = SEM. Analysis was performed using paired samples.
2) IL-6 Response to Low Salt Diet
Having noted that IL-6 levels increase in response to ANGII infusion on a liberal salt diet, we evaluated IL-6 response to endogenous RAS activation by a low-sodium (< 10 mmol sodium/day) diet for 1 week. PRA levels were significantly higher on a low sodium diet compared to the levels on a liberal sodium diet in hypertensives and normotensives (Fig 3) consistent with activation of the RAS by the low sodium diet. However, IL-6 levels were not significantly different on the low salt diet compared to levels on the liberal salt diet in either hypertensives or normotensives (Fig 3). To test if there may be a ceiling effect on a low salt diet whereby the IL-6 levels cannot increase further than the baseline threshold achieved on the liberal salt diet, we again evaluated the IL-6 response to ANGII, now on the low salt diet. IL-6 levels increased significantly in response to an ANGII infusion on the low salt diet in both normotensives and hypertensives (data not shown; p < 0.001 in both groups) similar to the response on the liberal salt diet, thus ruling out a ceiling effect. Similar to IL-6, CRP levels were also not significantly different between low and liberal salt diets. Comparing hypertensives to normotensives, baseline IL-6 (Figure 3) and CRP levels on the low salt diet were again significantly higher in hypertensives. Hypertension status remained significantly associated with both CRP (p = 0.025) and IL-6 (p = 0.006) after controlling for age, BMI, gender and race.
Figure 3. PRA and IL6 levels on Low Salt Diet versus Liberal Salt Diet in Hypertensives and Normotensives.

PRA was significantly higher on low salt diet. IL-6 levels were similar on low salt compared with the levels on liberal salt diet in hypertensives and normotensives. Analysis was performed using paired samples only (N = 191 hypertensives; 142 normotensives). Error bars = SEM.
3) Relationship between RAS Components and Inflammatory Markers
In multivariate regression models controlling for age, BMI, race, gender and hypertension status, PRA was positively associated with CRP on the low salt diet (standardized beta = 0.2; p <0.001). There was no significant association between serum aldosterone and either CRP or IL-6. When normotensives and hypertensives were analyzed separately, the significant positive association between PRA and CRP levels was observed in both groups (p = 0.008 in hypertensives and p = 0.04 in normotensives). No significant associations were detected between either PRA or aldosterone and the levels of CRP or IL6 under conditions of endogenous RAAS suppression by a liberal salt diet.
Discussion
In this study, we have demonstrated a clear relationship between IL-6 and hypertension by comparing hypertensives to normotensives and adjusting for relevant confounders,. Consistent with the IL-6 results and similar to multiple previous reports, we have also confirmed an association between higher CRP levels and hypertension. In addition, we have systematically examined, in normotensive and hypertensive human subjects, the relationship between RAS activity and IL-6, under conditions of controlled dietary sodium. This is the first study to directly evaluate this relationship in human hypertensives. We have demonstrated a significant rise in IL-6 in response to infused ANGII in both hypertensives and normotensives, supporting an inflammatory effect of acute RAS activation. Consistent with the idea that an activated RAS is associated with inflammation, endogenous activation of RAS by a low salt diet reflected by a higher PRA, as a surrogate for ANGII, showed a positive association with CRP in both hypertensives and normotensives.
We were unable to demonstrate an association between PRA and IL-6 on a low salt diet which could potentially be due to differences in the time course of release of these biomarkers. IL-6, a proinflammatory cytokine produced by various cells including macrophages, endothelial cells and vascular smooth muscle cells, is an early regulator of inflammation, which plays a critical role in driving the chronic inflammatory process including triggering hepatic synthesis of acute phase reactants such as CRP 31. Therefore, it is conceivable that circulating levels of IL-6 may vary depending on whether they are measured during acute versus chronic stimulation. Serum aldosterone did not show a positive association with CRP or IL-6. Not surprisingly, we were unable to detect an association between PRA or aldosterone with CRP or IL-6 on the liberal salt diet given the narrow range of PRA and aldosterone levels under conditions of endogenous RAS suppression by liberal salt intake,. Interestingly, despite the higher PRA levels on the low salt diet, the absolute levels of IL-6 or CRP were not higher on low salt compared with a liberal salt diet in either hypertensives or normotensives suggesting that a low sodium diet may have an independent protective effect in reducing inflammation.
The role of inflammation in the pathogenesis of cardiovascular disease is now widely accepted. However, the details of the inflammatory mechanisms and precise inflammatory mediators are yet to be fully understood. CRP has been shown to be a predictor of future cardiovascular risk32. Several lines of evidence suggest a key role for IL-6 in coronary heart disease. In apparently healthy men, elevated IL6 levels are associated with increased risk of future myocardial infarction13. In patients with unstable angina, high IL-6 levels are associated with an increased risk of coronary events14, 15. In addition, IL-6 has been shown to be a predictor of future mortality in patients with myocardial infarction17 as well as identify patients who will benefit from early invasive treatment strategies16.
Hypertension, a well established risk factor for cardiovascular disease, has been previously linked with chronic mild inflammation5–10. Experimental evidence has suggested that the RAS, which is known to play an important role in the pathogenesis of hypertension and hypertensive end-organ damage, is also linked with inflammation. There is substantial data supporting the role of ANGII as a pro-inflammatory mediator33–37. A growing body of evidence from animal studies strongly implicate that IL6 plays a significant role in mediating both inflammatory and BP responses to ANGII. Experimental evidence has shown that IL-6 is released from vascular tissue in response to ANGII and the effect of ANGII on IL-6 release is blocked by an ARB18. Animal studies have shown that ANGII infusion induces an increase in plasma IL-6 and BP in wild-type mice, whereas in IL-6 knockout mice, the blood pressure effects of ANGII are attenuated or ameliorated19, 20.
The results of our study provide evidence in humans supporting the association between ANGII and IL-6. This relationship has never been evaluated before in human hypertensives. The only previous human data evaluating this relationship was in a small cohort of 11 normotensive individuals that showed an increase in IL-6 in response to aldosterone and ANGII infusions24. This previous study24 had also suggested that ANGII induced IL-6 through a mineralocorticoid receptor-dependent mechanism. However, more recent evidence from mouse models suggests that stimulation of IL-6 by aldosterone is transient and BP effects of IL-6 are likely linked to ANGII rather than aldosterone38. In our study, we found no significant associations between serum aldosterone levels and either IL-6 or CRP.
Of particular interest is the effect of dietary sodium on inflammation. It has been shown previously in animal models that RAS-mediated end-organ damage is dependent on the presence of high sodium intake39, 40. A low salt diet alone was shown to be effective in preventing cardiac damage in a Nω-nitro-L-arginine methyl ester (L-NAME)/ANGII rat model of cardiac injury without reducing BP and despite a 10-fold increase in plasma aldosterone levels40. In our study, we did not find an increase in levels of IL-6 or CRP on a low salt diet compared to the levels on a liberal salt diet despite activation of the RAS by the low salt diet and the observed positive association between PRA and CRP levels. These findings are consistent with the evidence from animal studies of a complex role of dietary sodium in modulating the deleterious effects of increased RAS activity with a potential protective effect of a very low sodium diet. It has been suggested previously, based on the results of animal studies, that the RAS may exert its deleterious effects only when the levels are inappropriate for the dietary salt status.
Our findings should be interpreted in the context of the study design. They confirm an association between hypertension and inflammation and further demonstrate an association between RAS activity and inflammation. The cross-sectional nature of the study limits the ability to establish causal relationships and the underlying mechanisms remain to be established. Hypertensives in our database had mild to moderate hypertension. Therefore results may not be applicable to severe hypertensives. Our subjects were under age 66. Hence results may not apply to older individuals. The strengths of this study are the large sample size, ability to control for relevant confounders, carefully controlled experimental conditions, and elimination of potential confounding effects of drugs in that hypertensives were off medications when studied. Finally, our findings confirmed that factors such as BMI are significant mediators of inflammation in humans and added new ones to the list, i.e. RAS activity and dietary salt.
Conclusion
Our study provides confirmatory evidence that individuals with hypertension have greater background inflammation, as indicated by higher levels of IL-6 and CRP, compared to normotensives. In addition, we have demonstrated that increased levels of activity of the RAS are associated with inflammation as demonstrated by a significant rise in IL-6 in response to exogenous ANGII infusion and a positive association of PRA with CRP levels when the RAS is endogenously activated by a low salt diet. The increase in IL-6 levels in response to ANGII infusion is consistent with and provides human data supporting evidence from animal studies that IL-6 plays a role in the pathogenesis of ANGII hypertension. Serum aldosterone levels showed no significant relationship with either IL-6 or CRP, which may suggest that the ANGII portion of the RAS is the stronger factor driving the inflammatory effects of RAS in hypertension, which is also consistent with recent evidence from animal models of ANGII hypertension38. These findings may have implications for the choice of therapeutic agents for treatment of hypertension. Also of particular note is the role of dietary sodium as a mediator of inflammation. Despite activation of the RAS, IL-6 and CRP levels were not higher on a low salt diet compared to levels on a liberal salt diet suggesting that a low sodium diet is not inflammatory despite the increase in activity of the RAS.
Acknowledgments
Acknowledgements and Sources of Funding
We gratefully acknowledge the support of the dietary, nursing, administrative, and laboratory staff of the General Clinical Research Centers in which these studies were performed, three of which were supported by grants from the National Center for Research Resources, NIH (M01RR02635, M01RR00095, M01RR00064). We also acknowledge the numerous investigators, fellows, nurses and research coordinators at each of the study sites, who have participated in the HyperPATH study group. We gratefully acknowledge their contribution to the study of these subjects.
This research was supported by the following grants: The National Institutes of Health (NIH) grants HL47651, HL59424, DK63214, HL094452, a Specialized Center of Research (SCOR) in Molecular Genetics of Hypertension (P50HL055000) and a K30 Grant (NCRR) RR02229207. Dr. Chamarthi was supported by the following grants: T32 HL007609 (NIH) and 5 KL2 RR025757 (NIH). Dr. Thomas was funded by a K23 award from the NIH (5K23RR017394).
Footnotes
Disclosure: Authors have no conflicts of interest to declare
Conflict(s) of Interest/Disclosure(s): None
References
- 1.Ross R. Atherosclerosis--an inflammatory disease. N Engl J Med. 1999;340(2):115–26. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 2.Ridker PM. Intrinsic fibrinolytic capacity and systemic inflammation: novel risk factors for arterial thrombotic disease. Haemostasis. 1997;27 (Suppl 1):2–11. doi: 10.1159/000217475. [DOI] [PubMed] [Google Scholar]
- 3.Ridker PM, Cushman M, Stampfer MJ, Tracy RP, Hennekens CH. Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. N Engl J Med. 1997;336(14):973–9. doi: 10.1056/NEJM199704033361401. [DOI] [PubMed] [Google Scholar]
- 4.Sinisalo J, Paronen J, Mattila KJ, et al. Relation of inflammation to vascular function in patients with coronary heart disease. Atherosclerosis. 2000;149(2):403–11. doi: 10.1016/s0021-9150(99)00333-0. [DOI] [PubMed] [Google Scholar]
- 5.Bautista LE, Lopez-Jaramillo P, Vera LM, Casas JP, Otero AP, Guaracao AI. Is C-reactive protein an independent risk factor for essential hypertension? J Hypertens. 2001;19(5):857–61. doi: 10.1097/00004872-200105000-00004. [DOI] [PubMed] [Google Scholar]
- 6.Bautista LE. Inflammation, endothelial dysfunction, and the risk of high blood pressure: epidemiologic and biological evidence. J Hum Hypertens. 2003;17(4):223–30. doi: 10.1038/sj.jhh.1001537. [DOI] [PubMed] [Google Scholar]
- 7.Bautista LE, Vera LM, Arenas IA, Gamarra G. Independent association between inflammatory markers (C-reactive protein, interleukin-6, and TNF-alpha) and essential hypertension. J Hum Hypertens. 2005;19(2):149–54. doi: 10.1038/sj.jhh.1001785. [DOI] [PubMed] [Google Scholar]
- 8.Bautista LE, Atwood JE, O’Malley PG, Taylor AJ. Association between C-reactive protein and hypertension in healthy middle-aged men and women. Coron Artery Dis. 2004;15(6):331–6. doi: 10.1097/00019501-200409000-00006. [DOI] [PubMed] [Google Scholar]
- 9.Chae CU, Lee RT, Rifai N, Ridker PM. Blood pressure and inflammation in apparently healthy men. Hypertension. 2001;38(3):399–403. doi: 10.1161/01.hyp.38.3.399. [DOI] [PubMed] [Google Scholar]
- 10.Sesso HD, Buring JE, Rifai N, Blake GJ, Gaziano JM, Ridker PM. C-reactive protein and the risk of developing hypertension. JAMA. 2003;290(22):2945–51. doi: 10.1001/jama.290.22.2945. [DOI] [PubMed] [Google Scholar]
- 11.Lakoski SG, Cushman M, Palmas W, Blumenthal R, D’Agostino RB, Jr, Herrington DM. The relationship between blood pressure and C-reactive protein in the Multi-Ethnic Study of Atherosclerosis (MESA) J Am Coll Cardiol. 2005;46(10):1869–74. doi: 10.1016/j.jacc.2005.07.050. [DOI] [PubMed] [Google Scholar]
- 12.Lakoski SG, Herrington DM, Siscovick DM, Hulley SB. C-reactive protein concentration and incident hypertension in young adults: the CARDIA study. Arch Intern Med. 2006;166(3):345–9. doi: 10.1001/archinte.166.3.345. [DOI] [PubMed] [Google Scholar]
- 13.Ridker PM, Rifai N, Stampfer MJ, Hennekens CH. Plasma concentration of interleukin-6 and the risk of future myocardial infarction among apparently healthy men. Circulation. 2000;101(15):1767–72. doi: 10.1161/01.cir.101.15.1767. [DOI] [PubMed] [Google Scholar]
- 14.Biasucci LM, Liuzzo G, Fantuzzi G, et al. Increasing levels of interleukin (IL)-1Ra and IL-6 during the first 2 days of hospitalization in unstable angina are associated with increased risk of in-hospital coronary events. Circulation. 1999;99(16):2079–84. doi: 10.1161/01.cir.99.16.2079. [DOI] [PubMed] [Google Scholar]
- 15.Koukkunen H, Penttila K, Kemppainen A, et al. C-reactive protein, fibrinogen, interleukin-6 and tumour necrosis factor-alpha in the prognostic classification of unstable angina pectoris. Ann Med. 2001;33(1):37–47. doi: 10.3109/07853890109002058. [DOI] [PubMed] [Google Scholar]
- 16.Lindmark E, Diderholm E, Wallentin L, Siegbahn A. Relationship between interleukin 6 and mortality in patients with unstable coronary artery disease: effects of an early invasive or noninvasive strategy. Jama. 2001;286(17):2107–13. doi: 10.1001/jama.286.17.2107. [DOI] [PubMed] [Google Scholar]
- 17.Tan J, Hua Q, Li J, Fan Z. Prognostic value of interleukin-6 during a 3-year follow-up in patients with acute ST-segment elevation myocardial infarction. Heart Vessels. 2009;24(5):329–34. doi: 10.1007/s00380-008-1128-8. [DOI] [PubMed] [Google Scholar]
- 18.Funakoshi Y, Ichiki T, Ito K, Takeshita A. Induction of interleukin-6 expression by angiotensin II in rat vascular smooth muscle cells. Hypertension. 1999;34(1):118–25. doi: 10.1161/01.hyp.34.1.118. [DOI] [PubMed] [Google Scholar]
- 19.Lee DL, Sturgis LC, Labazi H, et al. Angiotensin II hypertension is attenuated in interleukin-6 knockout mice. Am J Physiol Heart Circ Physiol. 2006;290(3):H935–40. doi: 10.1152/ajpheart.00708.2005. [DOI] [PubMed] [Google Scholar]
- 20.Brands MW, Banes-Berceli AK, Inscho EW, Al-Azawi H, Allen AJ, Labazi H. Interleukin 6 knockout prevents angiotensin II hypertension: role of renal vasoconstriction and janus kinase 2/signal transducer and activator of transcription 3 activation. Hypertension. 56(5):879–84. doi: 10.1161/HYPERTENSIONAHA.110.158071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ridker PM, Danielson E, Rifai N, Glynn RJ. Valsartan, blood pressure reduction, and C-reactive protein: primary report of the Val-MARC trial. Hypertension. 2006;48(1):73–9. doi: 10.1161/01.HYP.0000226046.58883.32. [DOI] [PubMed] [Google Scholar]
- 22.Dandona P, Kumar V, Aljada A, et al. Angiotensin II receptor blocker valsartan suppresses reactive oxygen species generation in leukocytes, nuclear factor-kappa B, in mononuclear cells of normal subjects: evidence of an antiinflammatory action. J Clin Endocrinol Metab. 2003;88(9):4496–501. doi: 10.1210/jc.2002-021836. [DOI] [PubMed] [Google Scholar]
- 23.Fliser D, Buchholz K, Haller H. Antiinflammatory effects of angiotensin II subtype 1 receptor blockade in hypertensive patients with microinflammation. Circulation. 2004;110(9):1103–7. doi: 10.1161/01.CIR.0000140265.21608.8E. [DOI] [PubMed] [Google Scholar]
- 24.Luther JM, Gainer JV, Murphey LJ, et al. Angiotensin II induces interleukin-6 in humans through a mineralocorticoid receptor-dependent mechanism. Hypertension. 2006;48(6):1050–7. doi: 10.1161/01.HYP.0000248135.97380.76. [DOI] [PubMed] [Google Scholar]
- 25.Grant FD, Romero JR, Jeunemaitre X, et al. Low-renin hypertension, altered sodium homeostasis, and an alpha-adducin polymorphism. Hypertension. 2002;39(2):191–6. doi: 10.1161/hy0202.104273. [DOI] [PubMed] [Google Scholar]
- 26.Kosachunhanun N, Hunt SC, Hopkins PN, et al. Genetic determinants of nonmodulating hypertension. Hypertension. 2003;42(5):901–8. doi: 10.1161/01.HYP.0000095615.83724.82. [DOI] [PubMed] [Google Scholar]
- 27.Chamarthi B, Kolatkar NS, Hunt SC, et al. Urinary free cortisol: an intermediate phenotype and a potential genetic marker for a salt-resistant subset of essential hypertension. J Clin Endocrinol Metab. 2007;92(4):1340–6. doi: 10.1210/jc.2006-2093. [DOI] [PubMed] [Google Scholar]
- 28.Hopkins PN, Lifton RP, Hollenberg NK, et al. Blunted renal vascular response to angiotensin II is associated with a common variant of the angiotensinogen gene and obesity. J Hypertens. 1996;14(2):199–207. doi: 10.1097/00004872-199602000-00008. [DOI] [PubMed] [Google Scholar]
- 29.Underwood RH, Williams GH. The simultaneous measurement of aldosterone, cortisol, and corticosterone in human peripheral plasma by displacement analysis. J Lab Clin Med. 1972;79(5):848–62. [PubMed] [Google Scholar]
- 30.Emanuel RL, Cain JP, Williams GH. Double antibody radioimmunoassay of renin activity and angiotensin II in human peripheral plasma. J Lab Clin Med. 1973;81(4):632–40. [PubMed] [Google Scholar]
- 31.Schuett H, Luchtefeld M, Grothusen C, Grote K, Schieffer B. How much is too much? Interleukin-6 and its signalling in atherosclerosis. Thromb Haemost. 2009;102(2):215–22. doi: 10.1160/TH09-05-0297. [DOI] [PubMed] [Google Scholar]
- 32.Ridker PM. High-sensitivity C-reactive protein: potential adjunct for global risk assessment in the primary prevention of cardiovascular disease. Circulation. 2001;103(13):1813–8. doi: 10.1161/01.cir.103.13.1813. [DOI] [PubMed] [Google Scholar]
- 33.Brasier AR, Recinos A, 3rd, Eledrisi MS. Vascular inflammation and the renin-angiotensin system. Arterioscler Thromb Vasc Biol. 2002;22(8):1257–66. doi: 10.1161/01.atv.0000021412.56621.a2. [DOI] [PubMed] [Google Scholar]
- 34.Kranzhofer R, Schmidt J, Pfeiffer CA, Hagl S, Libby P, Kubler W. Angiotensin induces inflammatory activation of human vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 1999;19(7):1623–9. doi: 10.1161/01.atv.19.7.1623. [DOI] [PubMed] [Google Scholar]
- 35.Kranzhofer R, Browatzki M, Schmidt J, Kubler W. Angiotensin II activates the proinflammatory transcription factor nuclear factor-kappaB in human monocytes. Biochem Biophys Res Commun. 1999;257(3):826–8. doi: 10.1006/bbrc.1999.0543. [DOI] [PubMed] [Google Scholar]
- 36.Mehta PK, Griendling KK. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol. 2007;292(1):C82–97. doi: 10.1152/ajpcell.00287.2006. [DOI] [PubMed] [Google Scholar]
- 37.Pastore L, Tessitore A, Martinotti S, et al. Angiotensin II stimulates intercellular adhesion molecule-1 (ICAM-1) expression by human vascular endothelial cells and increases soluble ICAM-1 release in vivo. Circulation. 1999;100(15):1646–52. doi: 10.1161/01.cir.100.15.1646. [DOI] [PubMed] [Google Scholar]
- 38.Sturgis LC, Cannon JG, Schreihofer DA, Brands MW. The role of aldosterone in mediating the dependence of angiotensin hypertension on IL-6. Am J Physiol Regul Integr Comp Physiol. 2009;297(6):R1742–8. doi: 10.1152/ajpregu.90995.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Young M, Head G, Funder J. Determinants of cardiac fibrosis in experimental hypermineralocorticoid states. Am J Physiol. 1995;269(4 Pt 1):E657–62. doi: 10.1152/ajpendo.1995.269.4.E657. [DOI] [PubMed] [Google Scholar]
- 40.Martinez DV, Rocha R, Matsumura M, et al. Cardiac damage prevention by eplerenone: comparison with low sodium diet or potassium loading. Hypertension. 2002;39(2 Pt 2):614–8. [PubMed] [Google Scholar]