

Abstract
Amantadine has been used for decades as an inhibitor of the influenza A virus M2 protein (AM2) in the prophylaxis and treatment of influenza A infections, but its clinical use has been limited by its central nervous system (CNS) side effects as well as emerging drug-resistant strains of the virus. With the goal of searching for new classes of M2 inhibitors, a structure–activity relation study based on 2-[3-azaspiro(5,5)undecanol]-2-imidazoline (BL-1743) was initiated. The first generation BL-1743 series of compounds has been synthesized and tested by two-electrode voltage-clamp (TEV) assays. The most active compound from this library, 3-azaspiro[5,5]undecane hydrochloride (9), showed an IC50 as low as 0.92 ± 0.11 μM against AM2, more than an order of magnitude more potent than amantadine (IC50 = 16 μM). 15N and 13C solid-state NMR was employed to determine the effect of compound 9 on the structure and dynamics of the transmembrane domain of AM2 (AM2-TM) in phospholipid bilayers. Compared to amantadine, spiro-piperidine 9 (1) induces a more homogeneous conformation of the peptide, (2) reduces the dynamic disorder of the G34-I35 backbone near the water-filled central cavity of the helical bundle, and (3) influences the dynamics and magnetic environment of more residues within the transmembrane helices. These data suggest that spiro-piperidine 9 binds more extensively with the AM2 channel, thus leading to stronger inhibitory potency.
Introduction
The influenza virus, especially the H5N1 strain of influenza A, is a serious threat to human health. The outbreak of highly pathogenic H5N1 avian influenza virus in 1997 and 2004/5 caused the death of millions of chickens and had a very high mortality rate among the limited number of infected humans. These cases cause great concern about the transmission of avian virus mutants among humans. The spread of influenza virus is mainly due to two factors: antigenic drift and antigenic shift.1 Antigenic drift due to point mutations of the virus’ negative-strand RNAs generates slightly modified antigens on the virus surface, while antigenic shift causes reassortment of RNAs between viruses that infect avian populations and viruses that infect humans. The latter is a more serious problem since it produces more radically different antigenic determinants, for which there is little pre-existing immunity. Thus there is a great need for anti-influenza therapeutics. Currently there are two classes of small-molecule drugs in use to treat influenza virus infection:2 amantadine and rimantadine (Figure 1), which target the virus′ M2 proton channel, and Tamiflu (oseltamivir), which targets the virus surface protein neuraminidase.
Figure 1.

Crystal structure of the amantadine-bound AM2-TM complex (PDB accession code: 3C9J).8
The AM2 proton channel plays an important role in viral replication by facilitating uncoating of the virus after its endocytosis into the host cells. The low pH of the endosome activates the AM2 channel, thus allowing proton flux into the viral interior. This acidification dissociates the viral RNA from its bound matrix proteins,3 a process that is required to release the viral genetic material to the cytoplasm for replication.4 In a later stage of viral replication, the M2 protein also equilibrates the pH gradient between the Golgi lumen and the cytoplasm, thus preventing premature conformational changes of hemagglutinin.5,6 AM2 is a homotetrameric, type III integral membrane protein containing a small N-terminal periplasmic domain (23 residues), a single transmembrane domain (20 residues), and a C-terminal cytoplasmic tail (53 residues).7
Structural studies of membrane proteins such as AM2 are often hampered by the difficulties associated with the production, functional characterization, and structure determination of this class of proteins. Structural analysis of AM2 has the additional challenge of pH-induced changes in the TM region. Recently one of our groups solved the structures of the AM2 TM domain (22–46) both with (pH 5.3) and without (pH 7.3) bound amantadine using X-ray crystallography.8 In the crystal structure of the amantadine-M2TM complex, one drug binds each M2 tetramer in the pore, and the drug is surrounded by residues whose mutations cause amantadine resistance. This finding is consistent with the stoichiometry previously inferred from fluorescence titrations9 and electrophysiological measurements,10 which showed the binding of one drug per tetramer with a Hill coefficient of 1.0. Moreover, the amantadine and rimantadine sensitivity of AM2 channels expressed either in Xenopus oocytes or in mammalian cells revealed the drug-binding region in the TM domain to encompass residues V27, A30, S31, and G34. No mutations from the C-terminus to G34 have been found to be amantadine resistant, except H37 and W41, which are functionally important for the channel activity. These results are less consistent with the solution NMR structure of AM2(18–60) at higher pH (7.5), solved in the presence of 40 mM rimantadine.11 The solution NMR structure showed four rimantadine molecules per AM2 tetramer channel bound on the outside of the helices toward the cytoplasmic side of the membrane, with the basic amine group of rimantadine interacting with D44. The authors suggested that rimantadine indirectly inhibits the channel by stabilizing the closed form of AM2 and that drug-resistant mutants escape inhibition by stabilizing the open state of the channel. However, this allosteric inhibition mechanism is not consistent with recent electrophysiological measurements of the amantadine-resistant mutations of AM2.12 Furthermore, mutations to the side chains that form the proposed surface site remained amantadine sensitive.8 Taken together, these data strongly suggest that the drug-binding site that is responsible for inhibition is located in the channel pore.
Amantadine targets AM2 by blocking the acidification of the virus entrapped in endosomes.13 Unfortunately the use of amantadine-related drugs is limited by central nervous system (CNS) side effects and the rapid emergence of drug-resistant viruses such as L26F, V27A, A30T, and S31N.14–16 Extensive structure–activity relationship (SAR) studies of adamantyl derivatives2,17–25 have been evaluated, leading to a series of potent adamantane analogues active against H2N2 and H3N2 viruses. However, few other molecular scaffolds have been explored, which led us to search for novel scaffolds that might provide new avenues for developing antagonists of AM2. The spirene guanidine analogue, 2-[3-azaspiro(5,5)undecanol]-2-imidazoline (BL-1743) (Figure 2), was previously discovered through a high-throughput screen based on the ability of inhibitors to reverse the toxicity associated with M2 channels expressed in the yeast Saccharomyces cerevisiae membranes.26 It is one of the few examples of nonadamantane-based M2 inhibitors reported in the literature.27–29
Figure 2.
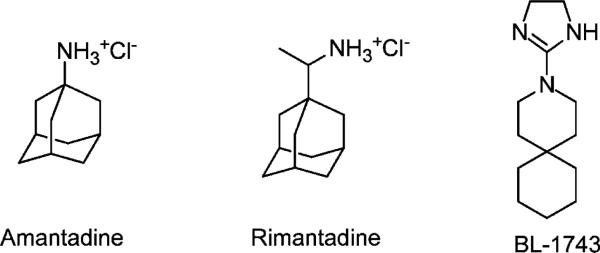
Chemical structures of AM2 channel inhibitors.
The M2 channel, due to its proton conductance properties, disrupts the electrochemical potential across the yeast cell membrane when expressed in high copy numbers. This disruption results in a retarded growth rate and eventually yeast cell death. The mechanism of AM2 channel inhibition by BL-1743 was further characterized by electrophysiological methods.30 BL-1743 is also able to inhibit the AM2 channel expressed in Xenopus oocytes, as determined using the two-electrode voltage clamp (TEV) technique. It was found that the majority of M2 sequences isolated from influenza viruses resistant to amantadine were also resistant to BL-1743, which suggests that BL-1743 binds competitively with amantadine. Interestingly, the kinetics of channel inhibition by BL-1743 were more rapid, showing a fast onset of inhibition as well as a reasonably rapid reversal of inhibition following removal of the compound.30,31 This behavior contrasts with that of amantadine, whose second-order rate constant for the onset of inhibition is much slower than the diffusion-controlled rate, and whose off-rate is essentially irreversible on the minute to hour time scale of the experiment. The Hill coefficient for inhibition was 1.0, which is consistent with the binding ratio of one BL-1743 per AM2 tetramer. Given the structural difference between BL-1743 and the amantadine class of compounds, and the lack of any published SAR studies for analogs of BL-1743, we have begun to explore this scaffold.
The goals of this study are to (1) explore the structure–activity relationship of BL-1743; (2) determine the binding specificity; and (3) characterize the effects of the most potent inhibitor on the conformation and dynamics of AM2-TM in the lipid bilayer using solid-state NMR (SSNMR) spectroscopy. SSNMR is ideally suited to atomic-level structural analysis of membrane proteins and their ligands in lipid bilayers, since it requires neither long-range order nor fast isotropic mobility. Previously, SSNMR has been used to investigate the orientation, dynamics, and conformation of key residues of the influenza M2 protein; 32–35 however, it is only recently beginning to be used to investigate drug–M2 interactions in lipid bilayers.36,37 We report herein the synthesis of our first generation BL-1743 series of inhibitors. Binding of the most potent compound, 3-azaspiro[5,5]undecane hydrochloride (9), to AM2 was characterized by 2D 13C and 15N NMR correlation experiments and line width analysis.
Results and Discussion
Structure–Activity Relationship (SAR) Study of BL-1743
The activity of the inhibitors was measured using the TEV technique with full length Udorn AM2 protein in the Xenopus oocytes membrane.38 All inhibitors were initially tested at 100 μM; those that inhibited the AM2 channel activity by more than 70% (less than 30% remaining activity) were chosen for measurement of their IC50.
Synthesis of BL-1743 started with reduction of commercially available 3,3-pentamethylene glutarimide with LiAlH4 in refluxing THF to give 3-azaspiro[5,5]undecane hydrochloride (9) in 75% yield after treatment with HCl/ether. Subsequent nucleophilic substitution of 2-methylthio-2-imidazoline with 9 furnished the model compound BL-1743.39 Reductive amination of 9 with different aldehydes using NaBH(OAc)3/HCO2H in dichloroethane40 gave 1–8 with yields of 65% to 95%. The results showed that replacements of the imidazoline ring of BL-1743 with either hydrophobic substitutions or heterocycles lacking hydrogen-bond donors (HBD) led to complete loss of potency at 100 μM, as AM2 still retained >90% activity after inhibition. In contrast, inhibitors 7 and 8 with the imidazole headgroup retain moderate inhibition. This suggests that a hydrogen-bond donor may be necessary for the inhibitory activity.
Interestingly, the simple piperidine lacking the imidazoline group altogether, spiro-piperidine 9 (3-azaspiro[5,5]undecane hydrochloride), had an IC50 of 0.92 ± 0.11 μM (n = 6), representing a more than 45-fold increase in potency relative to BL-1743. Thus, the bulky imidazoline headgroup does not contribute to the affinity of BL-1743 to AM2. To determine whether the secondary amine is important for the activity of spiro-piperidine 9, it was converted to the corresponding N-methyl piperidine 10 by reductive amination. This modification decreased the activity approximately 20-fold (IC50 = 20.6 ± 2.14 μM; n = 6), indicating that the polar secondary amine might be functionally important for inhibitor binding. To assess the potential role of charge, spiro-piperidine 9 was modified to the corresponding unsubstituted guanidine (11), urea (12), thiourea (13), and sulfonamide (14). Syntheses of 11, 12, 13 were achieved using a standard procedure by treatment of 9 with 1,3-diBoc-2-methyl-2-thiopseudourea, trimethylsilyl isocyanate, and trityl thioisocyanate, respectively, followed by hydrolysis or acid deprotection (Scheme 2). The sulfonamide 14 was prepared using the literature reported sulfamoylating agent, N-(tert-Butoxycarbonyl)-N-[4-(dimethylazaniumylidene)-1,4-dihydropyridin-1-ylsulfonyl]azanide.41 The guanidine compound 11 had a potency similar to that of the parent BL-1743, indicating that the cyclic ring in the imidazoline was not essential for activity. The urea 12 and thiourea 13, by contrast, were not active. Interestingly, however, the sulfonamide 14 had a potency similar to that of BL-1743, indicating that a positively charged group was not a prerequisite for activity. Taken together, our results show that the most potent compound in this series was the simple piperidine, and conversion of the secondary amine in 9 to tertiary amines decreased the potency. Furthermore, while compound 9 is likely to be charged at neutral pH, an overall positive charge does not appear to be a prerequisite for activity, as long as appropriately placed polar groups were included as in 14.
Scheme 2.

Synthesis of Methylated Inhibitor 10, Guanidine (11), Urea (12), Thiourea (13), and Sulfonamide (14)
In our amantadine-complexed AM2-TM structure, the electron density map fits better with the positively charged amantadine amine pointing down toward H37, but the distance is too far (6.8 Å) for effective hydrogen bonding between the imidazoles and the amine (or ammonium group). However, more recent and higher-resolution AM2-TM crystal structures have shown two layers of well organized H2O above H37 (unpublished result). Thus, it is possible that the polar HBD group at the end of the inhibitor contributes to potency by forming hydrogen bonds with structural H2O molecules, while hydrophobic substituents at the same position would lead to energetically unfavorable dehydration of polar residues in the channel. This hypothesis might explain the reduced binding affinity of inhibitor 10 with methyl substitution. Similar structure–activity relations were also found in adamantane analogues, where methylation and ethylation of the amine group of all heterocycles caused a reduction of anti-influenza A virus activity.17,19–22,25
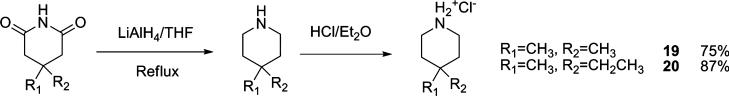
Spiro amines with different fused ring sizes have also been synthesized to determine which one best fits the AM2 channel. Reduction of 8-azaspiro[4,5]decane-7,9-dione by LiAlH4 gave 8-azaspiro[4,5]decane hydrochloride 15 after treatment with HCl/ether. 2-Azaspiro[4,5]decan-3-one was reduced and acidified to give 2-azaspiro[4,5]decane hydrochloride (16) under the same conditions. Ketal (17) and dithiene (18) versions of the spiro-piperidine 9 were synthesized by polyphosphoric acid catalyzed condensation.42
Compared to 9, the contraction of the bottom cyclohexane ring to cyclopentane (15) led to a significant loss of activity (IC50 = 8.14 ± 0.18 μM). Similarly, contraction of the piperidine ring to a pyrolidine (16) was also found to be unfavorable. These comparisons showed that the spiro[5,5]undecane was the most active. The ketal (17) and dithiene (18) retain the spiro six-membered ring of spiro-piperidine 9 but have increased hydrophilicity compared to 9 (clog P = 3.38). The introduction of a ketal in 17 completely abolished the binding affinity (IC50 > 100 μM), while dithiene 18 retained moderate binding (IC50 = 37.6 ± 2.67 μM). Presumably this finding reflects the energetics of dehydration of 17, which has a clog P of –0.30 as compared to that of 18 (clog P = 1.28).
To further test the role of the spiro system, the 6,6-fused decahydroisoquinoline (cis/trans mixture and trans isomer) was also tested and found to be less active than the corresponding spiro[5,5]undecane scaffold based inhibitors. Two 4,4-disubstituted piperidines (19 and 20) were also synthesized and found to be no more advantageous than the corresponding spiro-piperidine. Thus, the [5,5] spiro ring system appeared to provide the greatest activity.
Structural Basis for Activity
The most active compound, spiro-piperidine 9, was selected for docking experiments with AM2-TM. The minimum energy conformation of 9 was docked into the previously published high resolution AM2-TM structure (PDB code: 3BKD). As would be expected, the optimized docking conformation showed spiro-piperdine 9 located inside the channel with the amine group pointing either up or down the channel. The amine group of 9 could be hydrated in both orientations, but in neither case a specific interaction was formed between the amine and the AM2-TM protein.
When overlaying the optimized docking conformation of the 9-AM2-TM complex with the X-ray crystal structure of amantadine-bound AM2-TM, we found that spiro-piperidine 9 and amantadine occupied approximately the same region in the channel, spanning residues 27–34. A new structure was generated when overlaying the bottom cyclohexane ring of spiro-piperidie 9 with amantadine. Interestingly this hybrid molecule was recently reported in the literature to have highly potent anti-influenza activity, being 12-fold more active than amantadine and 2-fold more active than rimantadine when tested in virus infection assays. It is interesting to note that spiro-piperidine 9 is also 10- to 20-fold more active than amantadine.20 Thus, it is likely that the bulky, hydrophobic adamantyl group of the spiro adamantane compound is not required for potent activity.
Solid-State NMR Characterization of AM2-TM Bound to Spiro-piperidine 9
To investigate the interaction between AM2-TM and spiro-piperidine 9 as well as the effect of the drug on the peptide structure, we measured the 15N and 13C SSNMR spectra of bilayer-bound AM2-TM peptide complexed to the drug. The 15N cross-polarization (CP) spectra of the peptide were measured at both 313 and 243 K. The low-temperature spectra report the conformation and conformational distribution of AM2-TM, while the high-temperature spectra reflect the dynamics of the peptide channel. AM2-TM (22–46) was bound to DLPC bilayers at a peptide/lipid molar ratio of 1:15, where the peptide is known to form tetramers based on previous 19F spin diffusion NMR experiments.43 Figure 5a compares the low-temperature 15N spectra of the apo, amantadine-bound, and spiro-piperidine 9-bound AM2-TM. The spiro-piperidine-bound peptide shows significantly narrower line widths for all four labeled sites (L26, A29, G34, and I35) than either the apo or the amantadine-bound peptide. Since at low temperature all conformations are present, the narrower line widths suggest that spiro-piperidine binding induces a more homogeneous conformation of AM2-TM. To verify this possibility, we carried out 15N spin–echo measurements at 243 K. Residues L26, A29, and the upfield G34 peak (G34a) show very similar 15N T2 values of 10–11 ms for both the amantadine-bound peptide and the spiro-piperidine-bound peptide, confirming that the line narrowing by spiro-piperidine 9 indeed results from reduced conformational heterogeneity of these residues. However, the downfield G34 peak (G34b) increased its 15N T2 from 12 to 15 ms, while the I35 15N T2 increased from 13 to 22 ms in the presence of 9. These significant T2 increases indicate that the G34–I35 junction of the peptide is more immobilized by spiro-piperidine 9 than by amantadine; thus part of the line narrowing for these two sites is due to the reduction of dynamic disorder.
Figure 5.

15N CP-MAS spectra of LAGI-AM2-TM in DLPC bilayers at (a) 243 and (b) 313 K. Three states of the peptide are compared: apo (bottom), bound to amantadine (middle row), and bound to spiro-piperidine 9 (top row).
2D 15N–13C correlation spectra in Figure 6 confirm the increased conformational homogeneity of AM2-TM upon spiro-piperidine binding. The most significant line narrowing is found at I35 and L26, whereas more modest line narrowing is observed at A29 and G34 (Table 5). The line narrowing of L26 is interesting, because this residue is at the edge of the binding pocket of amantadine based on the crystal structure.8 Indeed, amantadine-bound AM2-TM did not show a narrower L26 15N line width than apo AM2-TM. This suggests that the bulkier spiro-piperidine 9 has a stronger rigidifying effect that extends over a larger area of the AM2 channel. Similarly, based on the 15N line width, I35 exhibits little conformational ordering by amantadine but is significantly ordered by spiro-piperidine 9. Taken together, the data indicate that spiro-piperidine 9, compared to amantadine, induces a more homogeneous structure of AM2-TM from L26 to I35.
Figure 6.

2D 15N–13C spectra of LAGI AM2-TM in DLPC bilayers at 243 K. (a) Apo peptide. (b) Amantadine-bound peptide. (c) Spiro-piperidine 9 bound peptide. (d) 1D 15N cross sections of G34. Note the separation of two G34 peaks in the spiro-piperidine-bound sample.
Table 5.
15N Chemical Shifts (δN) and Full Width at Half Maximum (δ1/2) of LAGI AM2-TM in DLPC Bilayers at 243 K
| Apo |
Amantadine-bound |
Spiro-piperidine 9 bound |
||||
|---|---|---|---|---|---|---|
| δN (ppm) | δ1/2 (Hz) | δN (ppm) | δ1/2 (Hz) | δN (ppm) | δ1/2 (Hz) | |
| L26 | 117.7 | 340 | 117.5 | 340 | 117.5 | 210 |
| A29 | 120.9 | 200 | 121.7 | 210 | 121.2 | 170 |
| G34a | 107.1 | 270 | 106.3 | 240 | 106.6 | 280 |
| G34b | - | - | 109.7 | 230 | 110.3 | 140 |
| I35 | 122.3 | 280 | 124.6 | 270 | 124.8 | 160 |
The 15N spectra in Figures 5 and 6 also show that the G34 chemical shifts are very sensitive to the state of AM2-TM. The apo peptide shows a broad G34 15N peak, indicating a distribution of conformations. Upon amantadine binding this broad 15N peak is partly resolved into two peaks with similar heights. Spiro-piperidine 9 shifts the conformational equilibrium to predominantly the downfield 15N conformer (G34b, 110.3 ppm), which also becomes narrower, indicating reduced conformational heterogeneity.
In addition to 15N chemical shifts, we measured the 13C chemical shifts of the peptide using the 2D INADEQUATE experiment. Figure 7 shows the 2D spectra of AM2-TM in the three states. The main line width reduction is found for L26 side chain Cγ and Cδ peaks, which became much better resolved in the spiro-piperidine-bound sample (Figure 7b). In addition, the other L26 Cδ peak (24 ppm) is shifted downfield compared to the apo and amantadine-bound peptide (Figure 7c). A recent study by London et al.44 and our own studies55 of Val and Leu side chains in protein NMR structures found that methyl 13C chemical shifts are sensitive to protein side chain conformation. Thus, these L26 chemical shift changes suggest perturbation and ordering of the L26 side chain by spiro-piperidine 9.
Figure 7.

(a) 2D 13C–13C INADEQUATE spectrum of spiro-piperidine-bound AM2-TM at 243 K. (b) The L26 Cγ/Cδ region of apo, amantadine-bound, and spiro-piperidine-bound peptide. (c) 1D cross sections at ω1 = 49 ppm shows the line narrowing and chemical shift changes induced by the two drugs.
In the liquid-crystalline phase of DLPC lipids, it is known that the apo and amantadine-bound AM2-TM undergoes fast (>120 kHz) uniaxial rotational diffusion around the membrane normal.32 This is also observed for the spiro-piperidine-bound peptide based on 15N spectra measured as a function of temperature (Figure 8). The lowest (243 K) and highest (313 K) temperatures give the greatest intensities, whereas at intermediate temperatures of 263 – 293 K most 15N signals are suppressed. This trend is characteristic of molecular motion, which interferes with 1H decoupling and 1H–15N CP when the motional rates are comparable to the 1H–15N dipolar couplings. When the motions are much slower or faster than the dipolar interaction, rigid-limit or motionally averaged spectra with high intensities are obtained.
Figure 8.

(a) Variable-temperature 15N CP-MAS spectra of LAGI AM2-TM in DLPC bilayers from 243 to 313 K. (b) I35 and G34 intensities as a function of temperature. G34a: 106.6 ppm. G34b: 110 ppm. Note the recovery of the G34a intensity at 313 K, in contrast to G34b and I35. (c) L26 and A29 15N intensities as a function of temperature.
Figure 8b and c plot the intensities of the four 15N peaks at different temperatures quantitatively. The L26, A29, and G34a intensities show the aforementioned minimum at intermediate temperature. In contrast, I35 and G34b do not exhibit intensity recovery at high temperature, indicating that these two residues are still in the intermediate motional regime at 313 K. Thus, the two G34 conformations have different dynamics, with the G34b conformer being more immobilized than G34a. The intermediate motion of G34b and I35 at 313 K is also consistent with their longer 15N T2 values at low temperature, indicating that this region of the peptide is more immobilized by spiro-piperidine 9 than are other residues.
Conclusion
In summary, a class of spirene-based AM2 inhibitors was identified from this study. The most potent compound from this series, 3-azaspiro[5,5]undecane hydrochloride (9), showed an IC50 of 0.92 ± 0.11 μM, which is more than an order of magnitude more potent than amantadine. Solid-state NMR data indicate that compound 9 interacts with AM2-TM differently from amantadine: it affects a longer stretch of the transmembrane helix, induces a more homogeneous conformation of the peptide, and immobilizes the G34-I35 region. These structural findings make compound 9 an attractive molecule for atomic-level structural investigation of drug-complexed AM2-TM. It is well-known that the apo-form of AM2-TM has extensive conformational and dynamic disorder in lipid bilayers32,36,45 and detergents.11 Amantadine was previously found to accelerate the peptide motion, which enhances the NMR spectral resolution at ambient temperature.36 However, the low temperature 15N spectra shown here indicate that substantial conformational heterogeneity remains in the amantadine-bound AM2-TM. Spiro-piperidine 9 significantly reduces this conformational heterogeneity and immobilizes the G34-I35 region of the peptide, which is a hot spot of conformational change in response to environments. These favorable properties make spiro-piperidine 9 a promising compound for examining the structure of the M2-channel complex in lipid bilayers. Continuing synthetic studies have been initiated to generate more diversity based on the spirene scaffold, with the aim of discovering inhibitors to target AM2 mutants, predominantly S31N.
Experimental Details
In Vitro cRNA Transcription, Heterologous Expression, and Electrophysiological Recordings
The cDNA encoding to the influenza virus A/Udorn/72 a.m.2 protein was inserted into pGEM-HJ (a gift from N.Dascal Tel-Aviv University, Israel) for expression on Xenopus oocytes. Plasmid was linearized with HindIII, and capped cRNA was transcribed in vitro using T7 RNA polymerase (mMessage mMachine; Ambion, Austin, TX). The quality of transcripts was assessed by agarose gel electrophoresis and ethidium bromide staining and analytical UV spectroscopy. Stage V–VI Xenopus laevis oocytes were prepared as described previously.46 Oocytes were injected with 5–10 ng of cRNA in 50 nL/oocyte and assayed 2–3 days later. Two electrode voltage clamp recordings were carried out using TEV-200 (Dagan, Minneapolis, MN) connected to DIGIDATA 1440A and pCLAMP10 (Axon Instruments, Foster City, CA). Oocytes were superfused with Barth's solution containing 88 mM NaCl, 1 mM KCl, 2.4 mM NaHCO3, 0.3 mM NaNO3, 0.71 mM CaCl2, 0.82 mM MgCl2, and 15 mM HEPES for pH 8.5 or 15 mM MES for pH 5.5. Currents were recorded at –20 mV. Dose–inhibition curves were usually constructed by applying 1–3 concentrations per oocyte of antagonist mixed in recording pH 5.5 barth's solution, and currents were normalized to the steady-state current obtained with pH 5.5 barth solution alone. Data were analyzed using the ORIGIN 8.0 software (OriginLab, Northampton, MA).
Preparation of M2 Membrane Samples for Solid-State NMR
13C- and 15N-labeled AM2-TM (22–46) of the Udorn strain47 (SSDPL VVAASII GILHLIL WILDRL) was synthesized by PrimmBiotech (Cambridge, MA) and purified to >95%. 13C and 15N-labeled amino acids were placed at L26, A29, G34, and I35 in one sample and V27, A30, I33, and L38 in another sample.
AM2-TM was reconstituted into 1,2-dilauroyl-sn-glycero-3-phosphatidylcholine (DLPC) bilayers by detergent dialysis as described before.43,48 The peptide structure and dynamics have been extensively characterized in this bilayer.32,36 AM2-TM was first dissolved in octyl-β-d-glucopyranoside (OG) solution and then mixed with the lipid vesicle solution at pH 7.5. OG was then removed by dialysis for 3 days. The peptide–lipid suspension was ultracentrifuged to obtain the membrane pellet. The peptide/lipid molar ratio was 1:15.3; azaspiro[5,5]undecane was added to the membrane by either direct titration to the pellet at a peptide/drug molar ratio of 1:2 or through a 10 mM solution before ultracentrifugation. For the latter, the percent of spiro-piperidine 9 remaining in the supernatant was quantified by 1H solution NMR to be 60%, giving a peptide/drug molar ratio of 1:8 in the membrane pellet.
Solid-State NMR Spectroscopy
SSNMR experiments were carried out on Bruker wide-bore AVANCE and DSX spectrometers at 14.1 and 9.4 T (Karlsruhe, Germany) using 4 mm magic-angle-spinning (MAS) probes. Radio-frequency pulse lengths were typically 5 μs for 13C and 15N and 3.5–4.0 μs for 1H. 1H TPPM49 or SPINAL50 decoupling field strengths of 60–70 kHz were applied. 13C and 15N chemical shifts were referenced, respectively, to the α-Gly C′ signal at 176.49 ppm on the TMS scale and the 15N signal of N-acetyl-valine at 122.0 ppm on the liquid ammonia scale. 2D 13C–13C correlation was carried out using the dipolar INADEQUATE sequence51,52 which better resolves side chain methyl signals than the standard spin diffusion experiments. 2D 15N–13C correlation was carried out using an HSQC-type sequence53 incorporating a REDOR pulse train for 15N–13C coherence transfer.54 The 2D spectra were measured at 243 K to freeze the peptide motion.
The numbers of scans for the 1D 15N CP spectra of the apo-, amantadine-bound, and spiro-piperidine-bound AM2-TM were 3000–6000 at low temperatures (<263 K) and 30 000–40 000 at high temperatures (>263 K). For the 2D 15N–13C correlation spectra, 352 scans were coadded per t1 slice and a maximum t1 evolution time of 8.8 ms was used, resulting in a measuring time of 21 h for each experiment. For the 2D 13C–13C INADEQUATE spectra, the number of scans per t1 slice was 192 and the maximum t1 evolution time was 6.4 ms. The measuring time was 20 h for each experiment.
Chemical Synthesis
All chemicals were purchased from commercial vendors and used without further purification unless otherwise noted. 3,3-Pentamethylene glutarimide was purchased from Bosche, NJ. 1H and 13C NMR spectra were recorded on a DMX-360 NMR spectrometer. Chemical shifts are reported in parts per million referenced with respect to residual solvent (CHCl3 = 7.26 ppm and DMSO-d6 = 2.50 ppm) or from internal standard tetramethylsilane (TMS = 0.00 ppm). The following abbreviations were used in reporting spectra: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, dd = doublet of doublets. All reactions were carried out under a N2 atmosphere, unless otherwise stated. HPLC grade solvents were used for all the reactions. Column chromatography was performed using silica gel (230–400 mesh). Low-resolution mass spectra were obtained on an LC platform from Micromass using an ESI technique.
3-Azaspiro[5,5]undecane Chloride (9)
To a solution of the LiAlH4 (0.38 g, 10 mmol) in THF (20 mL) at 0 °C was added 3,3-pentamethylene glutarimide (0.36 g, 2 mmol) in THF (10 mL) dropwise under a nitrogen atmosphere. The reaction was allowed to warm to RT over 30 min and then refluxed overnight. The solution was cooled to 0 °C and quenched with H2O (0.38 mL), 15% NaOH (0.38 mL), and H2O (1.14 mL) sequentially. The resulting slurry was filtered. The filtrate was diluted with CH2Cl2, washed with brine, dried over MgSO4, filtered, concentrated in vacuo, and purified by flash chromatography to give 3-azaspiro[5,5]undecane as a yellow oil. The hydrochloride salt was prepared by passing HCl gas into the ether solution of 3-azaspiro[5,5]undecane at 0 °C, followed by decanting the ether supernatant. The final white solid (0.28 g, 75% yield) was dried in vacuo. 1H NMR (360 MHz, CDCl3) δ 9.29 (br s, 2H), 3.14 (t, J = 5.87 Hz, 4H), 1.74 (t, J = 5.87 Hz, 4H), 1.43 (m, 10H); 13C NMR (90 MHz, CDCl3) δ 39.93, 35.85, 32.76, 30.60, 26.53, 21.37; ESI-MS: Calculated for C10H20ClN (M – Cl)+ 154.2, Found: 154.5.
3-(4,5-Dihydro-1H-imidazol-2-yl)-3-azaspiro[5,5]undecane Hydroiodide (BL-1743)
3-Azaspiro[5,5]undecane (0.38 g, 2 mmol) and 2-methylthio-2-imidazoline hydroiodide (0.44 g, 1.8 mmol) were refluxed in CH3OH overnight, concentrated in vacuo, and purified by flash chromatography to give a yellow oil (0.26 g, 65% yield). 1H NMR (360 MHz, CDCl3) δ 8.27 (br s, 2H), 3.72 (s, 4H), 3.39 (t, J = 5.83 Hz, 4H), 1.50 (t, J = 5.94 Hz, 4H), 1.47–1.44 (m, 10H); 13C NMR (90 MHz, CDCl3) δ 159.48, 43.39, 42.73, 36.02, 35.26, 30.83, 26.65, 21.61. 21.49; ESI-MS: Calculated for C13H24IN3 (M – I)+ 222.3, Found: 222.5.
3-(Cyclopropylmethyl)-3-azaspiro[5,5]undecane (1)
To a solution of 3-azaspiro[5,5]undecane (0.34 g, 2.2 mmol), cyclopropanecarbaldehyde (0.14 g, 2 mmol), and NaBH(OAc)3 (0.64 g, 3 mmol) in dichloroethane (10 mL) was added AcOH (0.12 mL, 2 mmol), and the resulting suspension was stirred overnight, quenched with NaHCO3, and extracted with CH2Cl2 twice. The combined organic layer was washed with brine and dried with MgSO4. The solution was filtered, concentrated in vacuo, and purified by flash chromatography. The compound was converted to the hydrochloride by passing HCl gas into the ether solution of 3-(cyclopropylmethyl)-3-azoniaspiro[5,5]undecane at 0 °C, followed by decanting the ether supernatant, the final white solid (0.39 g, 80% yield) was dried in vacuo. 1H NMR (360 MHz, CDCl3) δ 2.45 (t, J = 5.66 Hz, 4H), 2.23 (d, J = 6.51 Hz 2H), 1.49 (t, J = 5.66 Hz, 4H), 1.48–1.40 (m, 6H), 1.38–1.30 (m, 4H), 0.92–0.82 (m, 1H), 0.52–0.47 (m, 2H), 0.11–0.17 (m, 2H); 13C NMR (90 MHz, CDCl3) δ 64.55, 49.67, 36.47, 35.46, 31.00, 27.12, 21.73, 8.74, 4.19; ESI-MS: Calculated for C14H25N (M + H)+ 208.4, Found: 208.4.
3-(Cyclopentylmethyl)-3-azaspiro[5,5]undecane (2)
This compound was prepared by the same procedure described for the synthesis of 1. 95% yield. 1H NMR (360 MHz, CDCl3) δ 2.35 (t, J = 5.56 Hz, 4H), 2.25 (d, J = 7.08 Hz, 2H), 2.06–2.02 (m, 1H), 1.76–1.73 (m, 2H), 1.58–1.52 (m, 4H), 1.45 (t, J = 5.56 Hz, 4H), 1.43–1.39 (m, 6H), 1.39–1.31 (m, 4H), 1.20–1.14 (m, 2H); 13C NMR (90 MHz, CDCl3) δ 65.52, 49.93, 37.80, 36.51, 35.47, 32.08, 31.00, 27.14, 25.44, 21.74; ESI-MS: Calculated for C16H29N (M + H)+ 236.4, Found: 236.6.
3-(Thiophen-3-ylmethyl)-3-azaspiro[5,5]undecane (3)
This compound was prepared by the same procedure described for the synthesis of 1. 92% yield. 1H NMR (360 MHz, CDCl3) δ 7.24 (d, J = 4.88 Hz, 1H), 7.09 (s, 1H), 7.04 (d, J = 4.88 Hz, 1H), 3.35 (s, 2H), 2.37 (t, J = 5.52 Hz, 4H), 1.46 (t, J = 5.52 Hz, 4H), 1.37–1.34 (m, 6H), 1.30–1.26 (m, 4H); 13C NMR (90 MHz, CDCl3) δ 129.02, 125.39, 123.11, 58.14, 49.37, 36.77, 36.35, 30.92, 27.08, 21.72; ESI-MS: Calculated for C15H23NS (M + H)+ 250.4, Found: 250.4.
3-((1-Methyl-1H-pyrazol-4-yl)methyl)-3-azaspiro[5,5]undecane (4)
This compound was prepared by the same procedure described for the synthesis of 1. 82% yield. 1H NMR (360 MHz, CDCl3) δ 7.93 (s, 1H), 7.28 (s, 1H), 3.87 (s, 3H), 3.41 (s, 2H), 2.38 (t, J = 5.38 Hz, 4H), 1.46 (t, J = 5.38 Hz, 4H), 1.39–1.34 (m, 6H), 1.30–1.24 (m, 4H); 13C NMR (90 MHz, CDCl3) δ 141.15, 140.30, 130.18, 52.93, 48.97, 39.03, 36.29, 35.45, 30.90, 27.05, 21.70; ESI-MS: Calculated for C15H25N3 (M + H)+ 248.4, Found: 248.6.
3-(3-Azaspiro[5,5]undecan-3-ylmethyl)-5-methylisoxazole (5)
This compound was prepared by the same procedure described for the synthesis of 1. 75% yield. 1H NMR (360 MHz, CDCl3) δ 6.00 (s, 1H), 3.54 (s, 2H), 2.41 (t, J = 5.73, 4H), 2.39 (s, 3H), 1.46 (t, J = 5.73 Hz, 4H), 1.39–1.35 (m, 6H), 1.30–1.25 (m, 4H); 13C NMR (90 MHz, CDCl3) δ 169.41, 161.90, 102.05, 53.94, 49.49, 36.74, 30.75, 27.05, 21.68, 12.47; ESI-MS: Calculated for C15H24N2O (M + H)+ 249.4, Found: 249.6.
3-(Pyridin-2-ylmethyl)-3-azaspiro[5,5]undecane (6)
This compound was prepared by the same procedure described for the synthesis of 1. 84% yield. 1H NMR (360 MHz, CDCl3) δ 8.56 (d, J = 4.8 Hz, 1H), 7.63 (td, J = 7.6, 1.6 Hz, 1H), 7.40 (d, J = 7.2 Hz, 1H), 7.15–7.13 (m, 1H), 3.65 (s, 2H), 2.44 (t, J = 5.58 Hz, 4H), 1.48 (t, J = 5.58 Hz, 4H), 1.39–1.36 (m, 6H), 1.30–1.25 (m, 4H); 13C NMR (90 MHz, CDCl3) δ 149.36, 136.47, 130.58, 123.42, 122.03, 65.32, 49.79, 36.44, 35.46, 30.87, 27.10, 21.72; ESI-MS: Calculated for C16H24N2 (M + H)+ 245.4, Found: 245.6.
3-((1H-Imidazol-4-yl)methyl)-3-azaspiro[5.5]undecane (7)
This compound was prepared by the same procedure as described for synthesis of 1. 68% yield. 1H NMR (360 MHz, CD3OD) δ 8.97 (s, 1H), 7.81 (s, 1H), 4.56 (s, 2H), 3.34 (br s, 4H), 1.77 (br s, 4H), 1.54–1.48 (m, 10H); 13C NMR (90 MHz, CDCl3) δ 137.47, 124.34, 123.60, 50.18, 49.86, 34.31, 31.21, 27.58, 22.50; ESI-MS: Calculated for C14H23N3 (M + H)+ 234.4, Found: 234.6.
3-((1H-Imidazol-2-yl)methyl)-3-azaspiro[5.5]undecane (8)
This compound was prepared by the same procedure described for the synthesis of 1. 65% yield. 1H NMR (360 MHz, CD3OD) δ 7.51 (s, 2H), 4.55 (s, 2H), 3.22 (t, J = 5.94 Hz 4H), 1.74 (t, J = 6.01 Hz, 4H), 1.47–1.45 (m, 10H); 13C NMR (90 MHz, CDCl3) δ 123.49, 51.45, 50.37, 34.64, 31.16, 27.62, 22.51; ESI-MS: Calculated for C14H23N3 (M + H)+ 234.4, Found: 234.6.
3-Methyl-3-azoniaspiro[5,5]undecane Chloride (10)
To a solution of 3-azaspiro[5,5]undecane (0.38 g, 2 mmol), formaldehyde (0.15 g 5 mmol), and NaBH(OAc)3 (0.42 g, 2 mmol) in dichloroethane (10 mL) was added AcOH (0.12 g, 2 mmol); the resulting suspension was stirred overnight, diluted with NaHCO3, extracted with CH2Cl2 twice, washed with brine, and dried with MgSO4. The solution was filtered, concentrated in vacuo, and purified by flash chromatography. The hydrochloride salt was prepared by passing HCl gas into the ether solution of 3-methyl-3-azoniaspiro[5,5]undecane at 0 °C, followed by decanting the ether supernatant; the final white solid (0.33 g, 80% yield) was dried in vacuo. 1H NMR (360 MHz, CDCl3) δ 3.35–3.31 (m, 2H), 2.83–2.79 (m, 2H), 2.75 (d, J = 4.89 Hz, 3H), 2.10–2.00 (m, 2H), 1.77–1.74 (m, 2H), 1.45–1.39 (m, 10H); 13C NMR (90 MHz, CDCl3) δ 50.56, 43.69, 39.69, 32.91, 31.43, 26.49, 21.55; ESI-MS: Calculated for C11H22ClN (M – Cl)+ 168.4, Found: 168.6.
3-Azaspiro[5,5]undecane-3-carboximidamide (11)
3-Azaspiro-[5,5]undecane (0.38 g, 2 mmol), HgCl2 (0.54 g, 2 mmol), and 1,3-bis(tert-butoxycarbonyl)-2-methyl-2-thiopseudourea (0.58 g, 2 mmol) were stirred in DMF (10 mL) under a N2 atmosphere for 24 h, H2O was added, and the white precipitate was removed by filtration. The precipitate was washed with CH2Cl2 twice, and the combined filtrate was extracted with CH2Cl2, washed with brine, dried with MgSO4, concentrated in vacuo, and purified by flash chromatography. The Boc protecting group was removed by 20% TFA/CH2Cl2 for 2 h at RT. Excess TFA was removed by passing N2 through the solution to give a yellow solid which was subsequently purified by flash chromatography (0.28 g, 71% over two steps). 1H NMR (360 MHz, CD3CN) δ 5.11 (br s 2H), 3.12 (t, J = 5.74 Hz, 4H), 1.57 (t, J = 5.74 Hz, 4H), 1.46–1.35 (m, 10H); 13C NMR (90 MHz, CD3CN) δ 157.20, 41.78, 35.64, 35.06, 26.48, 21.26, 20.94; ESI-MS: Calculated for C11H21N3 (M + H)+ 196.3, Found: 196.3.
3-Azaspiro[5,5]undecane-3-carboxamide (12)
Triethylamine (0.42 mL, 3 mmol) was added to 3-azaspiro[5,5]undecane (0.38 g, 2 mmol) in CH2Cl2 (10 mL) at 0 °C, and then trimethylsilyl isocyanate (0.35 g, 3 mmol) was added dropwise to the solution under N2. The solution was allowed to warm to RT and stirred for 7 h. H2O was added, and the solution was extracted with CHCl3 twice; the combined organic layer was dried with MgSO4, filtered, concentrated in vacuo, and purified by flash chromatography to give a white powder (0.32 g, 82% yield). 1H NMR (360 MHz, CD3CN) δ 5.23 (br s, 2H), 3.31 (t, J = 5.87 Hz, 4H), 1.46–1.36 (m, 14H); 13C NMR (90 MHz, CD3CN) δ 160.23, 41.09, 37.21, 36.95, 27.87. 22.57, 21.83; ESI-MS: Calculated for C11H20N2O (M – H)– 195.3, Found: 195.1.
3-Azaspiro[5,5]undecane-3-carbothioamide(13)
3-Azaspiro[5,5]-undecane (0.38 g, 2 mmol) and trityl isothiocyanate (0.54 g, 1.8 mmol) were stirred in THF at RT for 8 h until the disappearance of trityl isothiocyanate (based on TLC), and then the mixture was concentrated in vacuo and purified by flash chromatography (hexane/CHCl3 = 1:1 to CHCl3) to give a white foam. This was followed by deprotection of the trityl group using 20% TFA/CH2Cl2 for 2 h at RT. TFA was removed by passing N2 through the solution to give a yellow solid which was subsequently purified by flash chromatography (0.29 g, 76% yield over two steps). 1H NMR (360 MHz, CD3CN) δ 6.04 (br s 2H), 3.70 (t, J = 5.76 Hz, 4H), 1.48–1.37 (m, 14H), 1.46–1.36 (m, 14H); 13C NMR (90 MHz, CD3CN) δ 181.49, 45.25, 37.46, 37.08, 32.16, 27.89, 22.73; ESI-MS: Calculated for C11H20N2S (M + Na)+ 236.4, Found: 236.8.
3-Azaspiro[5,5]undecane-3-sulfonamide (14)
The sulfamoylating agent, N-(tert-butoxycarbonyl)-N-[4-(dimethylazaniumylidene)-1,4-dihydropyridin-1-ylsulfonyl]azanide, was synthesized according to literature protocols.41 3-Azaspiro[5,5]undecane (0.38 g, 2 mmol) and the sulfamoylating agent (0.602 g, 2 mmol) were stirred in CH2Cl2 (10 mL) at RT overnight. Solvent was removed by rotary evaporation, and the residue was purified by flash chromatography to give tert-butyl 3-azaspiro[5,5]undecan-3-ylsulfonylcarbamate. The Boc protecting group was removed by 20% TFA/CH2Cl2 for 2 h at RT. Excess TFA was removed by passing N2 through the solution to give a yellow solid which was subsequently purified by flash chromatography (0.29 g, 62% yield for three steps). 1H NMR (360 MHz, CD3CN) δ 5.01 (br s 2H), 3.03 (t, J = 5.78 Hz, 4H), 1.53 (t, J = 5.78 Hz, 4H), 1.46–1.35 (m, 10H); 13C NMR (90 MHz, CD3CN) δ 41.78, 35.62, 35.06, 26.41, 21.06, 20.84; ESI-MS: Calculated for C10H20N2O2S (M + H)+ 233.3, Found: 233.1.
8-Azoniaspiro[4,5]decane Chloride (15)
Synthesis of 15 was accomplished following the same procedure described for 9. 72% yield. 1H NMR (360 MHz, CDCl3) δ 2.80 (t, J = 5.62, 4H), 1.61 (t, J = 5.62 Hz, 4H), 1.43–1.40 (m, 8H); 13C NMR (90 MHz, CDCl3) δ 44.18, 41.43, 38.70, 35.45, 24.46; ESI-MS: Calculated for C9H18ClN (M – Cl)+ 140.4, Found: 140.6.
2-Azoniaspiro[4,5]decane Chloride (16)
Synthesis of 16 was accomplished following the same procedure described for 9. 85% yield. 1H NMR (360 MHz, CDCl3) δ 9.67 (br s, 2H), 3.39 (Quintet, J = 7.10 Hz, 2H), 3.07 (t, J = 5.75 Hz, 2H), 1.85 (t, J = 7.49 Hz, 2H), 1.52–1.46 (m, 10H); 13C NMR (90 MHz, CDCl3) δ 54.68, 43.97, 43.05, 35.86, 35.44, 25.72, 23.47; ESI-MS: Calculated for C9H18ClN (M – Cl)+ 140.4, Found: 140.2.
1,5-Dioxa-9-azaspiro[5,5]undecane (17)
4-Piperidone hydrochloride (0.31 g, 2 mmol), 1,3-propanediol (0.76 g, 10 mmol), and phosphoric acid (0.5 g) were stirred at 65 °C for 6 h. Saturated Na2CO3 was added, and the solution was extracted with CHCl3 twice; the combined CHCl3 was washed with DI H2O, dried over Mg2SO4, and concentrated in vacuo to give a yellow oil (0.23 g, 73% yield). 1H NMR (360 MHz, CDCl3) δ 3.91 (t, J = 5.62, 4H), 2.86 (t, J = 5.58, 4H), 2.50 (s br, 1H), 1.84 (t, J = 5.58 Hz, 4H), 1.73 (quintet, J = 3.56, 2H); 13C NMR (90 MHz, CDCl3) δ 108.62, 59.41, 43.26, 34.55, 25.87; ESI-MS: Calculated for C8H15NO2 (M + H)+ 158.2, Found: 158.5.
1,5-Dithia-9-azaspiro[5,5]undecane (18)
Synthesis of 18 was accomplished using the same procedure described for 9. 75% yield. 1H NMR (360 MHz, CDCl3) δ 2.96 (t, J = 5.39, 4H), 2.83 (t, J = 5.39, 4H), 2.06–1.97 (m, 6H), 1.56 (s, 1H); 13C NMR (90 MHz, CDCl3) δ 48.97, 42.49, 38.87, 26.20, 25.87; ESI-MS: Calculated for C8H15NS2 (M + H)+ 190.3, Found: 190.4.
4,4-Dimethylpiperidinium Chloride (19)
Synthesis of 19 was accomplished using the same procedure described for 9. 87% yield. 1H NMR (360 MHz, CDCl3) δ 9.37 (br s, 2H), 3.15 (t, J = 5.97, 4H), 1.68 (t, J = 5.97, 4H), 1.04 (s, 6H), 1.56 (s, 1H); 13C NMR (90 MHz, CDCl3) δ 40.64, 35.21, 28.29, 27.64; ESI-MS: Calculated for C7H16ClN (M – Cl)+ 114.4, Found: 114.6.
4-Ethyl-4-methylpiperidinium Chloride (20)
Synthesis of 20 was accomplished using the same procedure described for 9. 82% yield. 1H NMR (360 MHz, CDCl3) δ 9.38 (br s, 2H), 3.19–3.09 (m, 4H), 1.77–1.73 (m, 2H), 1.71–1.62 (m, 2H), 1.37 (q, J = 7.34 Hz, 2H), 0.97 (s, 3H), 0.85 (t, J = 7.45 Hz, 2H); 13C NMR (90 MHz, CDCl3) δ 40.37, 33.41, 33.24, 30.70, 22.88, 7.52; ESI-MS: Calculated for C8H18ClN (M – Cl)+ 128.2, Found: 128.4.
Figure 3.

Docked conformation of spiro-piperidine 9 in AM2-TM channel (Only three of four helices are shown to enhance viewing of the inhibitor). (a) Crystal structure of the amantadine-AM2 complex (PDB: 3C9J). Minimized conformation of spiro-piperidine 9 inside the AM2 channel with the amine group pointing down (b) and up (c). Docking experiments are performed using Autodoc. Side chains of drug-surrounding residues V27, A30, S31, A34 and functionally essential residues H37 and W41 are shown.
Figure 4.
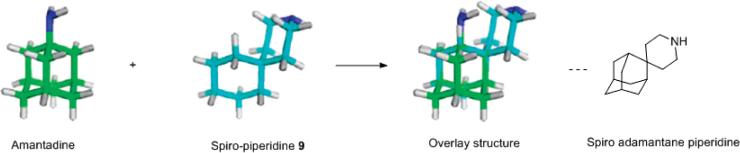
Conformational analysis of spiro-piperidine 9. 3D conformations of amantadine and spiro-piperidine 9 are minimized using MOPAC in Chem3D. Models are generated in Pymol.
Scheme 1.

Synthesis of BL-1743 and N-Substituted Azaspiro[5,5]undecane 1–8
Scheme 3.

Synthesis of Spiro-Amines 15, 16, 17, and 18 with Different Ring Sizes and Substitutions
Scheme 4.
Synthesis 4,4-Disubstituted Piperidines
Table 1.
AM2 Channel Proton Conductivity after Applying 100 Mm Inhibitors with Cyclic Hydrophobic and Heterocyclic Head Groups

| |||
|---|---|---|---|
| Compound | R | AM2 remaing Activity after 100uM inhibition | IC50 (uM) |
| BL-1743 |
|
25% | 45.3±3.9 |
| 1 |
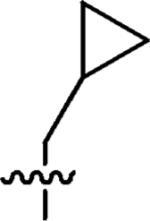
|
100% | n.a |
| 2 |
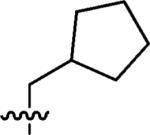
|
100% | n.a |
| 3 |
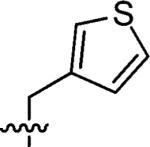
|
90% | n.a |
| 4 |
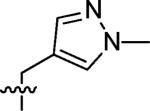
|
100% | n.a |
| 5 |
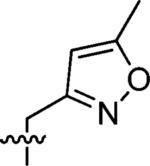
|
100% | n.a |
| 6 |
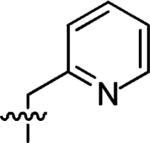
|
100% | n.a |
| 7 |
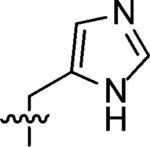
|
70% | n.a |
| 8 |
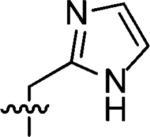
|
49% | n.a |
a As a positive control, the AM2 channel showed 6% remaining activity after 100 μM amantadine inhibition. The IC50 of amantadine was determined to be 16 μM.
Table 2.
AM2 Channel Activities after Applying 100 μM Inhibitors with HBD Acyclic Head Groups
| |||
|---|---|---|---|
| Compound | R | AM2 remaining activity after 100uM inhibition | IC50 (uM) |
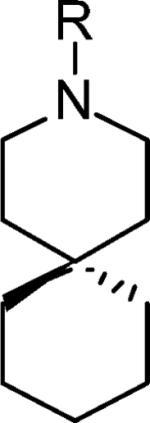
| 9 | H | 5% | 0.92 ± 0.11 |
| 10 |
|
7% | 20.6 ± 2.14 |
| 11 |
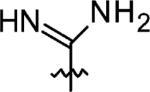
|
13% | 22.8 ± 1.82 |
| 12 |
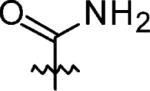
|
30% | n.d |
| 13 |
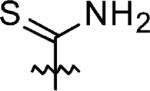
|
60% | n.d |
| 14 |
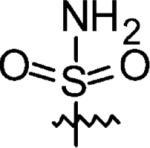
|
14% | 15.3 ± 2.28 |
Table 3.
AM2 Channel Activities after Applying 100 μM Inhibitors of 3-Azaspiro[5,5]undecane Analogues
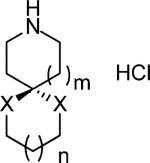
| |||
|---|---|---|---|
| Compound | n, X | Activity remaining activity after 100uM inhibition | IC50 (μM) |
| 15 | m = 1 | 16.2% | 8.14 ± 1.08 |
| n = 0 | |||
| X = CH2 | |||
| 16 | m = 0 | 6% | 12.0 ± 1.6 |
| n = 1 | |||
| X = CH2 | |||
| 17 | m = 1 | 100% | n.a |
| n = 1 | |||
| X = O | |||
| 18 | m = 1 | 12% | 37.6 ± 2.67 |
| n = 1 | |||
| X = S | |||
Table 4.
Potency of Decahydroisoquinoline and 4,4-Disubstituted Acyclic Piperidines
| Compound | AM2 remaining Activity after 100uM inhibition |
|---|---|
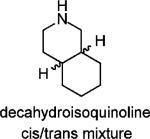
|
45.0% |
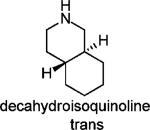
|
15% |
| 19 | 55% |
| 20 | 22% |
Acknowledgment
Support for this work was provided by an NSF Grant MCB-543473 to M.H. and GM56416 and AI74517 to W.F.D. J.W. thanks Dr. Jonathan G. Rudick for kindly proofreading the manuscript.
References
- 1.De Clercq E. Nat. Rev. Drug Discovery. 2006;5:1015–1025. doi: 10.1038/nrd2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lagoja IM, De Clercq E. Med. Res. Rev. 2008;28:1–38. doi: 10.1002/med.20096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhirnov OP. Virology. 1990;176:274–279. doi: 10.1016/0042-6822(90)90253-n. [DOI] [PubMed] [Google Scholar]
- 4.Martin K, Helenius A. Cell. 1991;67:117–130. doi: 10.1016/0092-8674(91)90576-k. [DOI] [PubMed] [Google Scholar]
- 5.Ciampor F, Bayley PM, Nermut MV, Hirst EMA, Sugrue RJ, Hay AJ. Virology. 1992;188:14–24. doi: 10.1016/0042-6822(92)90730-d. [DOI] [PubMed] [Google Scholar]
- 6.Grambas S, Hay AJ. Virology. 1992;190:11–18. doi: 10.1016/0042-6822(92)91187-y. [DOI] [PubMed] [Google Scholar]
- 7.Lamb RA, Zebedee SL, Richardson CD. Cell. 1985;40:627–633. doi: 10.1016/0092-8674(85)90211-9. [DOI] [PubMed] [Google Scholar]
- 8.Stouffer AL, Acharya R, Salom D, Levine AS, Di Costanzo L, Soto CS, Tereshko V, Nanda V, Stayrook S, DeGrado WF. Nature. 2008;451:596–599. doi: 10.1038/nature06528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Czabotar PE, Martin SR, Hay AJ. Virus Res. 2004;99:57–61. doi: 10.1016/j.virusres.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 10.Wang C, Takeuchi K, Pinto LH, Lamb RA. J. Virol. 1993;67:5585–5594. doi: 10.1128/jvi.67.9.5585-5594.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schnell JR, Chou JJ. Nature. 2008;451:591–595. doi: 10.1038/nature06531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jing XH, Ma CL, Ohigashi Y, Oliveira FA, Jardetzky TS, Pinto LH, Lamb RA. Proc. Natl. Acad. Sci. U.S.A. 2008;105:10967–10972. doi: 10.1073/pnas.0804958105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hay AJ, Wolstenholme AJ, Skehel JJ, Smith MH. EMBO J. 1985;4:3021–3024. doi: 10.1002/j.1460-2075.1985.tb04038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bright RA, Medina MJ, Xu XY, Perez-Oronoz G, Wallis TR, Davis XHM, Povinelli L, Cox NJ, Klimov AI. Lancet. 2005;366:1175–1181. doi: 10.1016/S0140-6736(05)67338-2. [DOI] [PubMed] [Google Scholar]
- 15.Bright RA, Shay DK, Shu B, Cox NJ, Klimov AI. JAMA J. Am. Med. Assoc. 2006;295:891–894. doi: 10.1001/jama.295.8.joc60020. [DOI] [PubMed] [Google Scholar]
- 16.Deyde VM, Xu XY, Bright RA, Shaw M, Smith CB, Zhang Y, Shu YL, Gubareva LV, Cox NJ, Klimov AI. J. Infect. Dis. 2007;196:249–257. doi: 10.1086/518936. [DOI] [PubMed] [Google Scholar]
- 17.Stamatiou G, Foscolos GB, Fytas G, Kolocouris A, Kolocouris N, Pannecouque C, Witvrouw M, Padalko E, Neyts J, De Clercq E. Bioorg. Med. Chem. 2003;11:5485–5492. doi: 10.1016/j.bmc.2003.09.024. [DOI] [PubMed] [Google Scholar]
- 18.Zoidis G, Fytas C, Papanastasiou L, Foscolos GB, Fytas G, Padalko E, de Clercq E, Naesens L, Neyts J, Kolocouris N. Bioorg. Med. Chem. 2006;14:3341–3348. doi: 10.1016/j.bmc.2005.12.056. [DOI] [PubMed] [Google Scholar]
- 19.Kolocouris A, Tataridis D, Fytas G, Mavromoustakos T, Foscolos GB, Kolocouris N, De Clercq E. Bioorg. Med. Chem. Lett. 1999;9:3465–3470. doi: 10.1016/s0960-894x(99)00631-9. [DOI] [PubMed] [Google Scholar]
- 20.Kolocouris N, Zoidis G, Foscolos GB, Fytas G, Prathalingham SR, Kelly JM, Naesens L, De Clercq E. Bioorg. Med. Chem. Lett. 2007;17:4358–4362. doi: 10.1016/j.bmcl.2007.04.108. [DOI] [PubMed] [Google Scholar]
- 21.Stamatiou G, Kolocouris A, Kolocouris N, Fytas G, Foscolos GB, Neyts J, De Clercq E. Bioorg. Med. Chem. Lett. 2001;11:2137–2142. doi: 10.1016/s0960-894x(01)00388-2. [DOI] [PubMed] [Google Scholar]
- 22.Tataridis D, Fytas G, Kolocouris A, Fytas C, Kolocouris N, Foscolos GB, Padalko E, Neyts J, De Clercq E. Bioorg. Med. Chem. Lett. 2007;17:692–696. doi: 10.1016/j.bmcl.2006.10.092. [DOI] [PubMed] [Google Scholar]
- 23.Kolocouris N, Foscolos GB, Kolocouris A, Marakos P, Pouli N, Fytas G, Ikeda S, Declercq E. J. Med. Chem. 1994;37:2896–2902. doi: 10.1021/jm00044a010. [DOI] [PubMed] [Google Scholar]
- 24.Kolocouris N, Kolocouris A, Foscolos GB, Fytas G, Neyts J, Padalko E, Balzarini J, Snoeck R, Andrei G, DeClercq E. J. Med. Chem. 1996;39:3307–3318. doi: 10.1021/jm950891z. [DOI] [PubMed] [Google Scholar]
- 25.Zoidis G, Tsotinis A, Kolocouris N, Kelly JM, Prathalingam SR, Naesens L, De Clercq E. Org. Biomol. Chem. 2008;6:3177–3185. doi: 10.1039/b804907f. [DOI] [PubMed] [Google Scholar]
- 26.Kurtz S, Luo GX, Hahnenberger KM, Brooks C, Gecha O, Ingalls K, Numata KI, Krystal M. Antimicrob. Agents Chemother. 1995;39:2204–2209. doi: 10.1128/aac.39.10.2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ochiai H, Sakai S, Hirabayashi T, Shimizu Y, Terasawa K. Antiviral Res. 1995;27:425–430. doi: 10.1016/0166-3542(95)00040-s. [DOI] [PubMed] [Google Scholar]
- 28.Alnakib W, Higgins PG, Willman J, Tyrrell DAJ, Swallow DL, Hurst BC, Rushton A. J. Antimicrob. Chemother. 1986;18:119–129. doi: 10.1093/jac/18.1.119. [DOI] [PubMed] [Google Scholar]
- 29.Garcia Martinez A, Teso Vilar E, Garcia Fraile A, de la Cerero S, Rodriguez Herrero ME, Martinez Ruiz P, Subramanian LR, Garcia Gancedo A. J. Med. Chem. 1995;38:4474–4477. doi: 10.1021/jm00022a012. [DOI] [PubMed] [Google Scholar]
- 30.Tu Q, Pinto LH, Luo GX, Shaughnessy MA, Mullaney D, Kurtz S, Krystal M, Lamb RA. J. Virol. 1996;70:4246–4252. doi: 10.1128/jvi.70.7.4246-4252.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gandhi CS, Shuck K, Lear JD, Dieckmann GR, DeGrado WF, Lamb RA, Pinto LH. J. Biol. Chem. 1999;274:5474–5482. doi: 10.1074/jbc.274.9.5474. [DOI] [PubMed] [Google Scholar]
- 32.Cady SD, Goodman C, Tatko CD, DeGrado WF, Hong M. J. Am. Chem. Soc. 2007;129:5719–5729. doi: 10.1021/ja070305e. [DOI] [PubMed] [Google Scholar]
- 33.Wang J, Kim S, Kovacs F, Cross TA. Protein Sci. 2001;10:2241–2250. doi: 10.1110/ps.17901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hu J, Fu R, Nishimura K, Zhang L, Zhou HX, Busath DD, Vijayvergiya V, Cross TA. Proc. Natl. Acad. Sci. U.S.A. 2006;103:6865–6870. doi: 10.1073/pnas.0601944103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nishimura K, Kim S, Zhang L, Cross TA. Biochemistry. 2002;41:13170–13177. doi: 10.1021/bi0262799. [DOI] [PubMed] [Google Scholar]
- 36.Cady SD, Hong M. Proc. Natl. Acad. Sci. U.S.A. 2008;105:1483–1488. doi: 10.1073/pnas.0711500105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hu J, Asbury T, Achuthan S, Li C, Bertram R, Quine JR, Fu R, Cross TA. Biophys. J. 2007;92:4335–4343. doi: 10.1529/biophysj.106.090183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Giffin K, Rader RK, Marino MH, Forgey RW. FEBS J. 1995;357:269–274. doi: 10.1016/0014-5793(94)01369-c. [DOI] [PubMed] [Google Scholar]
- 39.Aspinall SR, Bianco EJ. J. Am. Chem. Soc. 1951;73:602–603. [Google Scholar]
- 40.AbdelMagid AF, Carson KG, Harris BD, Maryanoff CA, Shah RD. J. Org. Chem. 1996;61:3849–3862. doi: 10.1021/jo960057x. [DOI] [PubMed] [Google Scholar]
- 41.Winum JY, Toupet L, Barragan V, Dewynter G, Montero JL. Org. Lett. 2001;3:2241–2243. doi: 10.1021/ol0161312. [DOI] [PubMed] [Google Scholar]
- 42.Weerawarna SA, Jewell RA. Preparation of piperidone ketal used as primary oxidant of cellulose fiber, involves condensing alcohol with piperidone in presence of polyphosphoric acid. EP1457491-A1; US2004192920-A1; CA2458736-A1; JP2004307478-A; US6852860-B2.
- 43.Luo W, Hong M. J. Am. Chem. Soc. 2006;128:7242–7251. doi: 10.1021/ja0603406. [DOI] [PubMed] [Google Scholar]
- 44.London RE, Wingad BD, Mueller GA. J. Am. Chem. Soc. 2008;130:11097–11105. doi: 10.1021/ja802729t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li C, Qin H, Gao FP, Cross TA. Biochim. Biophys. Acta. 2007;1768:3162–3170. doi: 10.1016/j.bbamem.2007.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shimbo K, Brassard DL, Lamb RA, Pinto LH. Biophys. J. 1996;70:1335–1346. doi: 10.1016/S0006-3495(96)79690-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ito T, Gorman OT, Kawaoka Y, Bean WJ, Webster RG. J. Virol. 1991;65:5491–5498. doi: 10.1128/jvi.65.10.5491-5498.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cady SD, Mishanina TV, Hong M. J. Mol. Biol. 2009;385:1127–1141. doi: 10.1016/j.jmb.2008.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bennett AE, Rienstra CM, Auger M, Lakshmi KV, Griffin RG. J. Chem. Phys. 1995;103:6951–6958. [Google Scholar]
- 50.Fung BM, Khitrin AK, Ermolaev K. J. Magn. Reson. 2000;142:97–101. doi: 10.1006/jmre.1999.1896. [DOI] [PubMed] [Google Scholar]
- 51.Bax A, Freeman R, Kempsell SP. J. Am. Chem. Soc. 1980;102:4849–4851. [Google Scholar]
- 52.Hong M. J. Magn. Reson. 1999;136:86–91. doi: 10.1006/jmre.1998.1631. [DOI] [PubMed] [Google Scholar]
- 53.Hong M, Griffin RG. J. Am. Chem. Soc. 1998;120:7113–7114. [Google Scholar]
- 54.Gullion T, Schaefer J. J. Magn. Reson. 1989;81:196–200. [Google Scholar]
- 55.Hong M, Mishanina TV, Cady SD. J. Am. Chem. Soc. 2009 doi: 10.1021/ja901550q. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]