

Abstract
Bovine herpesvirus 1 (BHV-1) establishes a lifelong latent infection in sensory neurons following acute infection. Increased corticosteroid levels, due to stress, increases the incidence of reactivation from latency. Within minutes, corticosteroids activate the glucocorticoid receptor and transcription of promoters containing a glucocorticoid receptor element. A single intravenous injection of the synthetic corticosteroid dexamethasone consistently induces reactivation from latency in calves. Lytic cycle viral gene expression is detected within 6 h after dexamethasone treatment of calves latently infected with BHV-1. Cellular transcription factors are induced by dexamethasone in trigeminal ganglionic neurons within 1.5 h after dexamethasone treatment, suggesting they promote viral gene expression during the early phases of reactivation from latency, which we operationally defined as the escape from latency. In this study, immunohistochemistry was utilized to examine viral protein expression during the escape from latency. Within 1.5 h after dexamethasone treatment, bICP0 and a late protein (VP16) were consistently detected in a subset of trigeminal ganglionic neurons. Most neurons expressing bICP0 also expressed VP16. Additional studies revealed that neurons expressing the glucocorticoid receptor also expressed bICP0 or VP16 at 1.5 h after dexamethasone treatment. Two other late proteins, glycoprotein C and D, were not detected until 6 h after dexamethasone treatment and were detected in only a few neurons. These studies provide evidence that VP16 and the promiscuous viral trans-activator (bICP0) are expressed during the escape from latency, suggesting they promote the production of infectious virus in a small subset of latently infected neurons.
INTRODUCTION
Bovine herpesvirus 1 (BHV-1) induces clinical disease in the upper respiratory tract, nasal cavity, or ocular cavity of cattle. BHV-1 establishes latency in sensory neurons but periodically reactivates from latency and consequently is widespread in cattle (1–4). Infection inhibits cell-mediated immunity (5–8) and CD8+ T cell recognition of infected cells (9–12) and induces apoptosis in CD4+ T cells (13, 14). Two viral regulatory proteins, bICP0 and bICP27, inhibit interferon-dependent transcription (3, 15–18). Infection also erodes mucosal surfaces within the upper respiratory tract, which promotes establishment of bacterial pathogens in the lower respiratory tract (19–21).
The incidence of BHV-1 reactivation from latency is increased following stressful stimuli that increase corticosteroid levels (reviewed in references 22, 23, and 24). Regardless of the reactivation stressor, lytic cycle viral gene expression, which is nearly undetectable during latency, must be activated. Administration of the synthetic corticosteroid dexamethasone (DEX) to latently infected calves or rabbits initiates BHV-1 reactivation from latency 100% of the time (1, 2, 4, 25–27). Six hours after DEX treatment, lytic cycle viral RNA expression is readily detected in a subset of trigeminal ganglionic neurons of latently infected calves (28, 29). DEX treatment of latently infected calves induces apoptosis of T cells that persist in trigeminal ganglia (TG) after infection (28). T cells also persist in TG of humans or mice latently infected with HSV-1 (30–36) and are proposed to promote the maintenance of latency (37–41).
Within 3 h after calves latently infected with BHV-1 are treated with DEX, 11 cellular genes are induced more than 10-fold in TG (42). Pentraxin 3, a regulator of innate immunity and neurodegeneration, is stimulated 35- to 63-fold at 3 or 6 h after DEX treatment. Two transcription factors, promyelocytic leukemia zinc finger (PLZF) and Slug, are induced more than 15-fold 3 h after DEX treatment. PLZF or Slug stimulates BHV-1 productive infection 20-fold or 5-fold, respectively, and Slug stimulates the late glycoprotein C promoter more than 10-fold. Additional DEX-induced transcription factors, sterile alpha motif (SAM) pointed domain-containing Ets transcription factor (SPDEF), Kruppel-like transcription factor 15 (KLF15), KLF4, KLF6, and GATA6, stimulate productive infection and certain key viral promoters. The DEX-inducible cellular transcription factors are predicted to mediate certain aspects of the early phases of reactivation from latency, including activation of lytic cycle viral gene expression. Latently infected neurons that express detectable levels of lytic cycle viral proteins within 6 h after DEX treatment are operationally defined as escaping latency (42).
In this study, we examined bICP0, VP16, gC, and gD protein expression during the escape from latency. bICP0- and VP16-positive TG neurons were detected at 1.5 h after DEX treatment but were not readily detected prior to DEX treatment. Consecutive sections were prepared, and subsequent studies demonstrated that bICP0 and VP16 were frequently detected in the same neuron. Conversely, gC- or gD-positive neurons were not detected until 6 h after DEX treatment, and only a few positive neurons were detected. At 1.5 h after DEX treatment, bICP0 or VP16 and the glucocorticoid receptor (GR) were detected in the same neuron. These studies suggest that activation of the GR by DEX directly stimulates lytic cycle viral gene expression during the escape from latency.
MATERIALS AND METHODS
Cells and virus.
Bovine kidney (CRIB) cells were grown in Earle's modified Eagle's medium (EMEM) supplemented with 10% fetal calf serum, penicillin (10 U/ml), and streptomycin (100 μg/ml).
The Cooper strain of BHV-1 was grown in CRIB cells and was used for all studies.
Calf studies.
All TG samples from calves used for this study were previously described (42). In brief, BHV-1-free crossbred calves (∼200 kg) were inoculated with 107 PFU of BHV-1 into each nostril and eye as described previously (25, 43–47). Calves were housed under strict isolation and given antibiotics before and after BHV-1 infection to prevent secondary bacterial infections. At 60 days postinoculation (dpi), calves were injected intravenously (jugular vein) with 100 mg of DEX. Calves were then transported to the veterinary diagnostic lab. Prior to euthanasia by electrocution, calves were heavily sedated with xylazine. After decapitation, TG were collected, and samples from each TG were formalin fixed and then paraffin embedded. The remainder of both TG was minced into small pieces and placed into a single 50-ml conical tube, and the tube was placed in a dry-ice ethanol bath. TG samples were then stored at −80°C. It took approximately 5 min to collect TG, mince the TG, place TG pieces in a 50-ml conical tube, and submerge the tube in a dry-ice ethanol bath after decapitation. One calf was decapitated at a time to ensure samples were processed in a timely manner. Calves were decapitated in the same order in which they were injected with DEX to ensure that the time points after DEX treatment were as close as possible to the designated time point. Three calves/time point were used for these studies. Experiments were performed in accordance with the American Association of Laboratory Animal Care guidelines and the University of Nebraska IACUC committee.
Immunohistochemistry.
Immunohistochemistry was performed essentially as previously described (28, 48–50) using the ABC kit (Vector Laboratories). In brief, TG from calves were fixed in neutral-buffered formalin and then embedded in paraffin. Thin sections (4 to 5 um) were cut and mounted onto slides. Tissue sections were incubated 20 min at 65°C followed by two incubations of 10 min in xylene and rehydrated in graded alcohols. Tissue sections were then incubated with 0.03% hydrogen peroxide in phosphate-buffered saline (PBS; pH 7.4) for 20 min at room temperature to block endogenous peroxidase. After 3 washes in Tris-buffered saline (TBS; 5 min each) at room temperature, tissue sections were digested with 40 μl of a ready-to-use proteinase K solution (catalog no. 53020; Dako) for 20 min at 37°C to enhance antigen retrieval. Tissue sections were then blocked with 5% normal serum diluted in TBS containing 0.25% bovine serum albumin (BSA) for 45 min at room temperature in a humidified chamber.
A peptide-specific rabbit antibody was made that was directed against bICP0 (Affinity Bioreagents, Golden, CO), and the antibody was affinity purified. This antibody specifically recognizes bICP0 in infected or transfected cells. A VP16-specific rabbit polyclonal antibody was obtained from Vikram Misra (University of Saskatchewan, Saskatoon, CA). The gC- and gD-specific antibodies were obtained from Shafiqul Chowdhury (Louisiana State University Veterinary School, Baton Rouge, LA). The GR-specific rabbit polyclonal antibody was purchased from Santa Cruz Biotechnology (H-300) and was reported to detect the mouse, rat, and human protein. The designated rabbit polyclonal antibodies or the mouse monoclonal antibodies were used at a 1:500 dilution and incubated overnight in a humidified chamber at 4°C and were washed in TBS (pH 7.6) the next day. Biotinylated goat anti-rabbit IgG (Vector Laboratories; catalog no. PK-6101) or biotinylated donkey anti-mouse IgG (Vector Laboratories; catalog no. PK-6102) was then incubated with the section for 30 min at room temperature in a humidified chamber. Next, the avidin-biotinylated enzyme complex was added to slides for 30 min at room temperature in a humidified chamber. After 3 washes in TBS, slides were incubated with freshly prepared substrate (Vector Laboratories; catalog no. SK-4800), rinsed with distilled water, and counterstained with hematoxylin. Thin sections from mock infected or latently infected calves were used as a negative control.
Immunofluorescence to examine GR localization.
CRIB cells were incubated in EMEM supplemented with 2% charcoal-stripped fetal bovine serum (FBS) at 37°C for 24 h and then placed on a glass slide for an additional 24 h. After 2 h of DEX treatment (100 nM), cells were fixed in 4% paraformaldehyde for 10 min, and confocal microscopy was performed. The GR primary antibody (catalog no. 3660; Cell Signaling) was diluted 1:250 in PBS with 0.05% Tween 20 and 1% BSA and incubated on coverslips for 2 h at room temperature. After three washes, coverslips were incubated with Alexa Fluor 488 goat anti-rabbit IgG (H+L) (catalog no. A-11008; Invitrogen) at a dilution of 1:100 for 1 h in the dark. After the slides were washed, DAPI (4′,6-diamidino-2-phenylindole) staining was performed to visualize the nucleus. Coverslips were then mounted on slides by use of Gel Mount aqueous mounting medium (Electron Microscopy Sciences). Images were obtained with a Bio-Rad confocal laser-scanning microscope (MRC-1024ES).
Western blot analysis.
At the designated times after infection, whole-cell lysate was prepared. Cells were washed with PBS and suspended in NP-40 lysis buffer (100 mM Tris [pH 8.0], 1 mM EDTA, 100 mM NaCl, 1% NP-40, 1 mM phenylmethylsulfonyl fluoride, and one tablet of complete protease inhibitor [Roche Molecular Biochemicals] in 10 ml buffer). Cell lysate was incubated on ice for 30 min, sonicated, and then clarified by centrifugation at 10,000 × g at 4°C for 15 min. Protein concentrations were quantified by the Bradford assay. For SDS-PAGE, proteins were mixed with an equal amount of 1× sample loading buffer (62.5 mM Tris-HCl [pH 6.8], 2% sodium dodecyl sulfate, 50 mM dithiothreitol, 0.1% bromophenol blue, 10% glycerol) and boiled for 5 min. Proteins were separated in an 8 or 12% SDS-PAGE gel. After electrophoresis, proteins were transferred onto a polyvinylidene difluoride membrane (Immobilon-P; Millipore) and blocked for 4 h in 5% nonfat dry milk with Tris-buffered saline-0.1% Tween 20 (TBS-T). Membranes were then incubated with primary antibody overnight at 4°C. An antibody directed against β-actin (Santa Cruz Biotechnology, Santa Cruz, CA) was used as a loading control. After 45 min of being washed with TBS-T, blots were incubated with donkey anti-rabbit horseradish peroxidase-conjugated immunoglobulin G (Amersham Biosciences), which was diluted 1:2,000 in 5% nonfat milk in TBS-T. Blots were washed 45 min with TBS-T and exposed to Amersham ECL reagents, and then autoradiography was performed. Primary antibodies were described above. The GR (Cell Signaling; catalog no. 3660), bICP0, and VP16 antibodies were diluted 1:1,000. The gC and gD antibodies were diluted 1:500 in blocking buffer. The secondary donkey anti-rabbit and anti-mouse antibodies were purchased from GE Healthcare.
RESULTS
Viral protein expression during productive infection.
Initial studies were performed to examine the specificity of BHV-1 antibodies that detect three late viral proteins (gC, gD, and VP16) and the promiscuous viral trans-activator (bICP0). The BHV-1 bICP0 mRNA is expressed as an immediate early (IE) and early (E) transcript because it contains an IE and E promoter (51, 52). For these studies, bovine kidney cells (CRIB) were infected with BHV-1 using a multiplicity of infection (MOI) of 2 PFU/cell. Total cell lysate was prepared at 3, 6, 12, and 24 h after infection in the absence or presence of the viral DNA polymerase inhibitor phosphonoacetic acid (PAA). As expected, bICP0 was expressed throughout productive infection, and PAA had no effect on expression (Fig. 1A, bICP0 panel). VP16 was not readily detected until 6 h after infection, and PAA reduced VP16 protein levels. Expression of the viral glycoprotein gC was not readily detected until 12 h after infection, and its expression was reduced by PAA treatment. Conversely, gD expression was detected earlier than VP16 or gC, and its expression was not affected by PAA treatment. These results are consistent with HSV-1 studies examining the kinetics of gC, gD, and VP16. For example, the HSV-1 gD gene is a prototype γ1 gene that is expressed early during infection, and its expression is not dramatically altered by PAA (53, 54). Conversely, the gC and VP16 genes are γ2 genes that are expressed relatively late during productive infection, and their expression is dramatically inhibited by PAA (53, 54).
Fig 1.

Analysis of BHV-1 protein expression in productively infected cells. (A) Western blot analysis comparing the expression of BHV-1 genes following infection of bovine CRIB cells. Monolayers of CRIB cells were mock infected or infected with BHV-1 (MOI = 2) in the presence or absence of 400 μg/ml of phosphonoacetic acid (PAA). At the designated times after infection (hours), whole-cell lysate was prepared. A total of 50 μg of protein was separated in an 8% SDS-PAGE gel, and viral proteins were detected by Western blotting using antibodies specific for the designed BHV-1 genes. β-Actin protein levels were analyzed in the respective samples as a loading control. The position of molecular weight markers for the respective panels is denoted. Molecular weight of the markers is shown on the right. The approximate molecular weight of bICP0 is 97, VP16 is 65, gC is 95, and gD is 77. (B) Subcellular localization of viral proteins in productively infected cells. Confocal microscopy was performed to detect BHV-1 proteins in CIRB cells infected with BHV-1 (MOI = 2) for 20 h and then fixed with formalin. Mock-infected CIRB cells served as a control. Fixed cells were incubated with antibodies specific for the designed viral genes. An Alexa Fluor 488 goat anti-rabbit secondary antibody (green) or Alexa Fluor 633 goat anti-mouse antibody (red) was used to detect viral protein expression. Images were obtained with a Bio-Rad confocal laser-scanning microscope (MRC-1024ES) with excitation/emission at 488/520 nm. Nuclear DNA (blue) was stained with DAPI (4′,6-diamidino-2-phenylindole). Merged images are shown, and they are representative of three independent experiments.
CRIB cells infected for 20 h were fixed in formalin, and then confocal microscopy was performed to determine if these antibodies recognized a viral protein in formalin-fixed cells. We chose a time late after infection because we knew the late proteins would be expressed at readily detectable levels. As expected, bICP0 was detected in the nucleus of infected cells, but the other three viral proteins were localized primarily to membranes in infected cells (Fig. 1B). None of the antisera directed against the respective viral proteins recognized a protein in mock-infected CRIB cells (Fig. 1B, mock panels, and data not shown). In summary, these studies demonstrated that the respective antibodies recognized the expected viral protein by Western blot analysis and by confocal microscopy using formalin-fixed cells.
Detection of bICP0 and VP16 during the escape from latency.
To test whether viral protein expression can be detected during the escape from latency, bICP0, VP16, gC, and gD protein expression was examined in TG sections by performing immunohistochemistry (IHC). The rationale for examining bICP0 protein expression is that bICP0 mRNA is consistently detected during DEX-induced reactivation from latency (55), and bICP0 stimulates productive infection and reactivation from latency (1–3, 23, 56, 57). BHV-1-encoded VP16 may promote the escape from latency because it activates expression of viral IE genes. HSV-1-encoded VP16, a late transcript, has been reported to be differentially expressed in neurons relative to cultured cells and consequently may stimulate reactivation from latency (58, 59). TG neurons were recognized by bICP0 or VP16 antiserum (Fig. 2) when calves latently infected with wild-type (wt) BHV-1 were treated with DEX for 1.5 h. Certain areas of TG sections contained bICP0- or VP16-positive neurons, whereas other areas did not. As expected, bICP0- or VP16-positive neurons were not readily detected during latency, and in general fewer bICP0- or VP16-positive neurons were detected at 3 or 6 h after DEX treatment relative to 1.5 h. The number of neurons stained by the gC or gD antiserum was low, and the intensity of staining was reduced relative to the results obtained with the VP16 or bICP0 antiserum (Fig. 3). We were unable to readily detect gC or gD prior to 6 h after DEX treatment (data not shown). In contrast to VP16+ or bICP0+ neurons, open reading frame 2-positive (ORF2+) neurons were readily detected during latency but not 6 h after DEX treatment (60).
Fig 2.

bICP0 and VP16 are expressed in sensory neurons following DEX treatment to induce reactivation from latency. IHC was performed using the bICP0 or VP16 antibody as described in Materials and Methods. Arrows denote neurons that were recognized by the respective antibody. Magnification, ×400.
Fig 3.

The glycoproteins gC and gD are weakly expressed in sensory neurons following DEX treatment to induce reactivation from latency. IHC was performed using the gC- or gD-specific antibody as described in Materials and Methods using TG sections from latently infected calves (latency) 6 h after DEX treatment. Arrows denote neurons that were recognized by the respective antibody. Magnification is ×400.
bICP0 and VP16 are frequently expressed in the same neuron during DEX-induced reactivation.
To determine whether bICP0 and VP16 were expressed in the same neuron following DEX treatment, consecutive sections were prepared from TG samples at 1.5, 3, and 6 h after DEX treatment, and each section was stained with the bICP0- or VP16-specific antiserum. At 3 h after DEX treatment, most VP16+ neurons were also bICP0+ (Fig. 4A; double-positive neurons denoted by arrows). We also examined sections 1.5 and 6 h after DEX treatment, and like 3 h after DEX treatment, nearly all VP16+ neurons were bICP0+ (Fig. 4B).
Fig 4.

bICP0 and VP16 are frequently expressed in the same neuron following DEX-induced reactivation from latency. (A) Consecutive sections were cut from formalin-fixed paraffin-embedded TG from calves latently infected with BHV-1 that were treated with DEX for 3 h. IHC was performed using the bICP0 antibody on one section, and the VP16 antibody was used to stain the consecutive section. Arrows denote neurons that were recognized by the respective antibody. Magnification is ×400. (B) The number of bICP0+ and VP16+ neurons (double positive) is shown for 1.5, 3, and 6 h after DEX treatment (black columns). The number of VP16+ neurons (white columns) or bICP0+ (gray columns) are also shown at the designated times after DEX-induced reactivation from latency.
Sensory neurons that express bICP0 and VP16 frequently express the glucocorticoid receptor.
DEX, like the natural corticosteroids, binds and activates the glucocorticoid receptor (GR) (reviewed in references 61 and 62). Since GR is expressed in rat sensory neurons (63), we predicted that bICP0 and VP16 expression may be stimulated directly by DEX in neurons that express the GR. Support for this prediction comes from the finding that the bICP0 E promoter is stimulated by DEX in transient-transfection studies (55). Thus, it was of interest to determine (i) if the GR is expressed in a subset of bovine TG neurons and (ii) whether VP16+ and bICP0+ neurons express the GR.
Initial studies tested whether commercially available antiserum recognizes the bovine GR. Antiserum directed against GR (MR-20; Santa Cruz Biotechnology) recognizing mouse, rat, and human proteins was used for this study. Western blots revealed that the GR antisera specifically recognized a protein with an approximate molecular weight of 90 in bovine kidney cells (CRIB) (Fig. 5A, B lanes) and mouse cells (M lanes), which is the expected size of the GR (61, 62). Confocal microscopy demonstrated that the antisera recognized a nuclear protein in CRIB cells following treatment with DEX (Fig. 5B), which occurs when the GR binds DEX. Prior to DEX treatment, the GR was localized throughout CRIB cells. The GR antisera also recognized a subset of TG neurons after treatment with DEX for 3 h (Fig. 5C). In general, the signal localized to the nucleus, suggesting the GR was activated after DEX treatment (denoted by arrows). In uninfected bovine TG, the signal was generally disperse and not readily detected compared to TG sections prepared from calves after DEX treatment. The signal appeared to be more concentrated in a few neurons from mock-infected calves (denoted by arrows), which may have been the result of stress during transportation of calves to the necropsy room prior to euthanasia.
Fig 5.

Detection of GR in bovine kidney cells (CRIB). (A) Bovine cells (CRIB) or mouse neuroblastoma cells (Neuro-2A) (30 μg protein/lane) were electrophoresed in an SDS-PAGE gel, and Western blot analysis was performed using the commercially available GR antibody (Santa Cruz; H-300). Lanes denoted B are cell lysate prepared from bovine kidney cells (CRIB), and lanes denoted M are cell lysate from mouse neuroblastoma cells (Neuro-2A). (B) Cells were treated with DEX, and immunofluorescence was performed as described in Materials and Methods. As controls, certain cultures were not treated with DEX. Localization of the GR was examined by immunofluorescence. Nuclear DNA was identified by DAPI staining. (C) IHC was performed with TG samples from uninfected calves and 3 h after DEX treatment using procedures described in Materials and Methods. Arrows denote the GR+ neurons. Magnification, ×400.
Consecutive sections were prepared from calves treated with DEX, and studies were performed to determine if the GR was expressed in sensory neurons expressing bICP0 or VP16. At 1.5 h after DEX treatment, GR+ and bICP0+ neurons were detected in certain areas of TG sections (Fig. 6A, double-positive neurons denoted by arrows). We detected 71 GR+ and bICP0+ neurons but only 2 that were GR+ but not bICP0+ and no bICP0+ neurons that were GR negative. VP16+ and GR+ neurons were also readily detected at 1.5 h after DEX treatment (Fig. 6B, double-positive neurons denoted by arrows). GR+ neurons that were not VP16+ were occasionally detected at 1.5 h after DEX treatment (Fig. 6B, neurons denoted by closed circles). Seventy-two GR+ and VP16+ neurons were detected, but only two were identified in the same field that were just GR+. At 1.5 h after DEX treatment, no bICP0+ or VP16+ neurons were detected that were GR negative.
Fig 6.
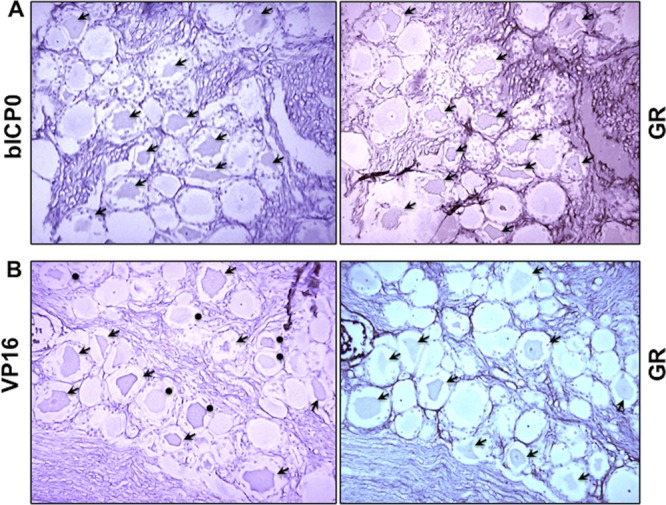
GR+ neurons frequently express bICP0 or VP16 during the escape from latency. Consecutive sections were cut from formalin-fixed paraffin-embedded TG from calves latently infected with BHV-1 that were treated with DEX for 1.5 h. (A) IHC was performed using the bICP0 antibody on one section and the GR antibody on the adjacent section. (B) IHC was performed using the VP16 antibody on one section and the GR antibody on the adjacent section. Arrows denote neurons that were recognized by the respective antibody. Closed circles denote GR+ neurons that were VP16 negative. Magnification is ×400.
DISCUSSION
In this study, two viral proteins that regulate viral transcription (VP16 and bICP0) were readily detected in sensory neurons within 1.5 h after reactivation from latency was initiated with the synthetic corticosteroid DEX. These two viral proteins were frequently detected in the same neuron. In addition, neurons that expressed bICP0 or VP16 also frequently expressed the GR. Conversely, fewer neurons appeared to express the late proteins, gC and gD, before or after DEX treatment. Although one could argue that we were unable to detect gC or gD in TG neurons because the antibodies were not as good as the VP16 or bICP0 antibodies used, the gC and gD antibodies clearly recognized a virus-specific protein in formalin-fixed cells prepared from productively infected bovine cells. Furthermore, the gC and gD antibodies specifically recognized abundant levels of a virus-specific protein during productive infection in a Western blot.
In general, stress increases corticosteroid levels and activates the GR, which we predict is a molecular switch that can consistently initiate the escape from latency (see Fig. 7 for a schematic of the events that are necessary for reactivation from latency). During the escape from latency, several key events occur that result in lytic cycle viral gene expression. First, specific DEX-induced cellular transcription factors stimulate certain viral promoters and productive infection (42). Second, lytic cycle viral gene expression, including that of the bICP0 and VP16 genes, may also be stimulated directly by an activated GR. Third, DEX represses expression of latency-related (LR) gene products (27, 60, 64), which is important because certain LR gene products can inhibit productive infection (56, 64–66). Finally, apoptosis of T cells in TG that persist during latency occurs following DEX treatment of latently infected calves (28), which may enhance the incidence of reactivation from latency because CD8+ T cells are proposed to maintain latency (37–40, 67). We predict that the pleiotropic effects of increased corticosteroids levels and activation of the GR lead to the escape from latency.
Fig 7.

Putative steps that occur during DEX-induced escape from latency. For details, see Discussion.
DEX, as with other natural corticosteroids, specifically bind the GR and mineralocorticoid receptor (MR) (reviewed in reference 61), resulting in nuclear localization of the GR or MR (68). Nuclear GR or MR dimers stimulate transcription by binding consensus glucocorticoid response elements (GRE; 5′-GGTACANNNTGTTCT-3′) (69, 70). A GR or MR monomer can also stimulate transcription by binding to a GR (1/2) binding site, of which the consensus 1/2 binding site is TGTTTCT or GGTACA, reviewed in reference 62. Although it is possible that MR+ TG neurons promote reactivation from latency, we were unable to identify a commercially available antibody that recognizes the MR receptor in bovine cells (data not shown). Within 5 min of glucocorticoid treatment, GR enters the nucleus, binds to glucocorticoid response elements, and induces rapid changes in chromatin conformation and transcriptional activation (reviewed in references 71 and 72). The BHV-1 genome contains 58 GR (1/2) binding sites in 24 BHV-1 promoters (data not shown), and a previous study demonstrated that DEX stimulates the bICP0 early promoter in transient-transfection assays (55). It will be of interest to directly test whether viral promoters are actually bound by a GR during the escape from latency.
In a small subset of latently infected neurons, DEX-induced expression of bICP0 and/or VP16 is predicted to stimulate lytic cycle viral gene expression, productive infection, and production of infectious virus in a small subset of latently infected neurons (Fig. 7). Most neurons that escape latency do not produce infectious virus because they apparently lack factors necessary for productive infection; consequently, these neurons reestablish latency (27). This study suggested that expression of late genes is one bottleneck that must be overcome to produce infectious virus. It may not be necessary to produce high levels of late proteins; however, expression of all late proteins necessary for producing infectious viruses would appear to be necessary. Several subtypes of sensory neurons exist in TG (73), and certain subtypes of TG neurons are more permissive for HSV-1 and HSV-2 productive infection (73–75). Consequently, we predict that only certain neuronal subtypes, which are latently infected, can escape latency and have the necessary factors to activate expression of viral genes necessary to produce infectious virus. An unanswered question that is raised as a result of these studies is, “What is the threshold of corticosteroids that must be achieved to induce successful reactivation from latency?” Every day, mammals encounter increases in corticosteroids, but those that are latently infected with their respective Alphaherpesvirinae subfamily member do not always successfully reactivate from latency, as judged by shedding of infectious virus.
Published studies concluded that the normal cascade of viral gene expression in cultured cells is different than what occurs during the escape from latency. For example, a late viral promoter (the gC gene) is trans-activated by DEX-inducible cellular transcription factors (SPDEF and Slug) (42) and Notch1 or Notch3 (66). Furthermore, the bICP0 E promoter, but not the bICP0 and bICP4 IE promoter (the IEtu1 promoter), is activated during reactivation from latency (55). Finally, this study demonstrated that the VP16 gene (a late viral gene), but not the gC or gD gene, is detected in TG neurons within 1.5 h after latently infected calves are treated with DEX. Several studies also suggest that the normal cascade of HSV-1 gene expression is different during reactivation from latency. For example, E gene expression and DNA replication are proposed to occur prior to IE gene expression (76–79). Another study concluded that expression of a late HSV-1 gene (the VP16 gene) promotes the exit from latency (59). During explant-induced reactivation from latency, viral gene expression was reported to be “disorganized” (80, 81). It does not appear that BHV-1 reactivation from latency is a random process in cattle, because we consistently detected VP16 and bICP0 but not gC or gD protein expression in TG neurons within 1.5 h after DEX treatment.
ACKNOWLEDGMENTS
This research was supported by grants from the Agriculture and Food Research Initiative Competitive Grants Program (09-01653 and 2013-01041) from the USDA National Institute of Food and Agriculture. A PHS grant to the Nebraska Center for Virology (1P20RR15635) has also supported certain aspects of these studies.
Footnotes
Published ahead of print 7 August 2013
REFERENCES
- 1.Jones C. 1998. Alphaherpesvirus latency: its role in disease and survival of the virus in nature. Adv. Virus Res. 51:81–133. [DOI] [PubMed] [Google Scholar]
- 2.Jones C. 2003. Herpes simplex virus type 1 and bovine herpesvirus 1 latency. Clin. Microbiol. Rev. 16:79–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jones C. 2009. Regulation of innate immune responses by bovine herpesvirus 1 and infected cell protein 0. Viruses 1:255–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jones C, Geiser V, Henderson G, Jiang Y, Meyer F, Perez S, Zhang Y. 2006. Functional analysis of bovine herpesvirus 1 (BHV-1) genes expressed during latency. Vet. Microbiol. 113:199–210. [DOI] [PubMed] [Google Scholar]
- 5.Carter JJ, Weinberg AD, Pollard A, Reeves R, Magnuson JA, Magnuson NS. 1989. Inhibition of T-lymphocyte mitogenic responses and effects on cell functions by bovine herpesvirus 1. J. Virol. 63:1525–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Griebel P, Ohmann HB, Lawman MJ, Babiuk LA. 1990. The interaction between bovine herpesvirus type 1 and activated bovine T lymphocytes. J. Gen. Virol. 71:369–377. [DOI] [PubMed] [Google Scholar]
- 7.Griebel P, Qualtiere L, Davis WC, Gee A, Bielefeldt Ohmann H, Lawman MJ, Babiuk LA. 1987. T lymphocyte population dynamics and function following a primary bovine herpesvirus type-1 infection. Viral Immunol. 1:287–304. [DOI] [PubMed] [Google Scholar]
- 8.Griebel PJ, Qualtiere L, Davis WC, Lawman MJ, Babiuk LA. 1987. Bovine peripheral blood leukocyte subpopulation dynamics following a primary bovine herpesvirus-1 infection. Viral Immunol. 1:267–286. [DOI] [PubMed] [Google Scholar]
- 9.Hariharan MJ, Nataraj C, Srikumaran S. 1993. Down regulation of murine MHC class I expression by bovine herpesvirus 1. Viral Immunol. 6:273–284. [DOI] [PubMed] [Google Scholar]
- 10.Hinkley S, Hill AB, Srikumaran S. 1998. Bovine herpesvirus-1 infection affects the peptide transport activity in bovine cells. Virus Res. 53:91–96. [DOI] [PubMed] [Google Scholar]
- 11.Koppers-Lalic EA, Reits EAJ, Ressing ME, Lipinska AD, Abele R, Koch J, Rezende MM, Admiraal P, van Leeuwen D, Bienkowsaka-Szewczyc K, Mettenleiter TC, Rijsewijk FAM, Tampe R, Neefjes J, Wiertz EJHJ. 2005. Varicelloviruses avoid T cell recognition by UL49.5-mediated inactivation of the transporter associated with antigen processing. Proc. Natl. Acad. Sci. U. S. A. 102:5144–5149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nataraj C, Eidmann S, Hariharan MJ, Sur JH, Perry GA, Srikumaran S. 1997. Bovine herpesvirus 1 downregulates the expression of bovine MHC class I molecules. Viral Immunol. 10:21–34. [DOI] [PubMed] [Google Scholar]
- 13.Eskra L, Splitter GA. 1997. Bovine herpesvirus-1 infects activated CD4+ lymphocytes. J. Gen. Virol. 78:2159–2166. [DOI] [PubMed] [Google Scholar]
- 14.Winkler MT, Doster A, Jones C. 1999. Bovine herpesvirus 1 can infect CD4(+) T lymphocytes and induce programmed cell death during acute infection of cattle. J. Virol. 73:8657–8668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.da Silva LF, Jones C. 2012. The ICP27 protein encoded by bovine herpesvirus type 1 (bICP27) interferes with promoter activity of the bovine genes encoding beta interferon 1 (IFN-β1) and IFN-β3. Virus Res. 169:162–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Henderson G, Zhang Y, Jones C. 2005. The bovine herpesvirus 1 gene encoding infected cell protein 0 (bICP0) can inhibit interferon-dependent transcription in the absence of other viral genes. J. Gen. Virol. 86:2697–2702. [DOI] [PubMed] [Google Scholar]
- 17.Saira K, Zhou Y, Jones C. 2007. The infected cell protein 0 encoded by bovine herpesvirus 1 (bICP0) induces degradation of interferon response factor 3 (IRF3), and consequently inhibits beta interferon promoter activity. J. Virol. 81:3077–3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saira K, Jones C. 2009. The infected cell protein 0 encoded by bovine herpesvirus 1 (bICP0) associates with interferon regulatory factor 7 (IRF7), and consequently inhibits beta interferon promoter activity. J. Virol. 83:3977–3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Highlander SK. 2001. Molecular genetic analysis of virulence in Mannheimia (Pasteurella) haemolytica. Front. Biosci. 6:D1128–D1150. [DOI] [PubMed] [Google Scholar]
- 20.Highlander SK, Fedorova ND, Dusek DM, Panciera R, Alvarez LE, Renehart C. 2000. Inactivation of Pasteurella (Mannheimia) haemolytica leukotoxin causes partial attenuation of virulence in a calf challenge model. Infect. Immun. 68:3916–3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zecchinon L, Fett T, Desmecht D. 2005. How Mannheimia haemolytica defeats host defense through a kiss of death mechanism. Vet. Res. 36:133–156. [DOI] [PubMed] [Google Scholar]
- 22.Jones C, da Silva LF, Sinani D. 2011. Regulation of the latency-reactivation cycle by products encoded by the bovine herpesvirus 1 (BHV-1) latency-related gene. J. Neurovirol. 17:535–545. [DOI] [PubMed] [Google Scholar]
- 23.Jones C, Chowdhury S. 2007. A review of the biology of bovine herpesvirus type 1 (BHV-1), its role as a cofactor in the bovine respiratory disease complex, and development of improved vaccines. Adv. Anim. Health 8:187–205. [DOI] [PubMed] [Google Scholar]
- 24.Perng G-C, Jones C. 2010. Towards an understanding of the herpes simplex virus type 1 latency-reactivation cycle. Interdiscip. Perspect. Infect. Dis. 2010:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Inman M, Lovato L, Doster A, Jones C. 2002. A mutation in the latency related gene of bovine herpesvirus 1 interferes with the latency-reactivation cycle of latency in calves. J. Virol. 76:6771–6779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jones C, Newby TJ, Holt T, Doster A, Stone M, Ciacci-Zanella J, Webster CJ, Jackwood MW. 2000. Analysis of latency in cattle after inoculation with a temperature sensitive mutant of bovine herpesvirus 1 (RLB106). Vaccine 18:3185–3195. [DOI] [PubMed] [Google Scholar]
- 27.Rock D, Lokensgard J, Lewis T, Kutish G. 1992. Characterization of dexamethasone-induced reactivation of latent bovine herpesvirus 1. J. Virol. 66:2484–2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Winkler MT, Doster A, Sur JH, Jones C. 2002. Analysis of bovine trigeminal ganglia following infection with bovine herpesvirus 1. Vet. Microbiol. 86:139–155. [DOI] [PubMed] [Google Scholar]
- 29.Winkler MTC, Doster A, Jones C. 2000. Persistence and reactivation of bovine herpesvirus 1 in the tonsil of latently infected calves. J. Virol. 74:5337–5346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cantin EM, Hinton DR, Chen J, Openshaw H. 1995. Gamma interferon expression during acute and latent nervous system infection by herpes simplex virus type 1. J. Virol. 69:4898–4905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Halford WP, Gebhardt BM, Carr DJ. 1996. Persistent cytokine expression in trigeminal ganglion latently infected with herpes simplex virus type 1. J. Immunol. 157:3542–3549. [PubMed] [Google Scholar]
- 32.Liu T, Tang Q, Hendricks RL. 1996. Inflammatory infiltration of the trigeminal ganglion after herpes simplex virus type 1 corneal infection. J. Virol. 70:264–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shimeld C, Whiteland JL, Nicholls SM, Grinfeld E, Easty DL, Gao H, Hill TJ. 1995. Immune cell infiltration and persistence in the mouse trigeminal ganglion after infection of the cornea with herpes simplex virus type 1. J. Neuroimmunol. 61:7–16. [DOI] [PubMed] [Google Scholar]
- 34.Shimeld C, Whiteland JL, Williams NA, Easty DL, Hill TJ. 1997. Cytokine production in the nervous system of mice during acute and latent infection with herpes simplex virus type 1. J. Gen. Virol. 78:3317–3325. [DOI] [PubMed] [Google Scholar]
- 35.Shimeld C, Whiteland JL, Williams NA, Easty DL, Hill TJ. 1996. Reactivation of herpes simplex virus type 1 in the mouse trigeminal ganglion: an in vivo study of virus antigen and immune cell infiltration. J. Gen. Virol. 77:2583–2590. [DOI] [PubMed] [Google Scholar]
- 36.Theil D, Derfuss T, Paripovic I, Herberger S, Meinl E, Schueler O, Strupp M, Arbusow V, Brandt T. 2003. Latent herpesvirus infection in human trigeminal ganglia causes chronic immune response. Am. J. Pathol. 163:2179–2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Khanna KM, Bonneau RH, Kinchington PR, Hendricks RL. 2003. Herpes simplex virus-specific memory CD8+ T cells are selectively activated and retained in latently infected sensory ganglia. Immunity 18:593–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Knickelbein JE, Khanna KM, Yee MB, Baty CJ, Kinchington PR, Hendricks RL. 2008. Noncytotoxic lytic granule-mediated CD8+ T cell inhibition of HSV-1 reactivation from neuronal latency. Science 322:268–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu T, Khanna KM, Carriere BN, Hendricks RL. 2001. Gamma interferon can prevent herpes simplex virus type 1 reactivation from latency in sensory neurons. J. Virol. 75:11178–11184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu T, Khanna KM, Chen X, Fink DJ, Hendricks RL. 2000. CD8(+) T cells can block herpes simplex virus type 1 (HSV-1) reactivation from latency in sensory neurons. J. Exp. Med. 191:1459–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Prbhakaran K, Sheridan BS, Kinchington PR, Khanna KM, Decman V, Lathrop K, Hendricks RL. 2005. Sensory neurons regulate the effector functions of CD8+ T cells in controlling HSV-1 latency ex vivo. Immunity 23:515–523. [DOI] [PubMed] [Google Scholar]
- 42.Workman A, Eudy J, Smith L, Frizzo da Silva L, Sinani D, Bricker H, Cook E, Doster A, Jones C. 2012. Cellular transcription factors induced in trigeminal ganglia during dexamethasone-induced reactivation from latency stimulate bovine herpesvirus 1 productive infection and certain viral promoters. J. Virol. 86:2459–2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Inman M, Lovato L, Doster A, Jones C. 2001. A mutation in the latency-related gene of bovine herpesvirus 1 leads to impaired ocular shedding in acutely infected calves. J. Virol. 75:8507–8515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lovato L, Inman M, Henderson G, Doster A, Jones C. 2003. Infection of cattle with a bovine herpesvirus 1 (BHV-1) strain that contains a mutation in the latency related gene leads to increased apoptosis in trigeminal ganglia during the transition from acute infection to latency. J. Virol. 77:4848–4857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Perez S, Meyer F, Saira K, Doster A, Jones C. 2008. Premature expression of the latency-related RNA encoded by bovine herpesvirus 1 correlates with higher levels of beta interferon RNA expression in productively infected cells. J. Gen. Virol. 89:1338–1345. [DOI] [PubMed] [Google Scholar]
- 46.Perez S, Lovato L, Zhou J, Doster A, Jones C. 2006. Comparison of inflammatory infiltrates in trigeminal ganglia of cattle infected with wild type BHV-1 versus a virus strain containing a mutation in the LR (latency-related) gene. J. Neurovirol. 12:392–397. [DOI] [PubMed] [Google Scholar]
- 47.Perez S, Inman M, Doster A, Jones C. 2005. Latency-related gene encoded by bovine herpesvirus 1 promotes virus growth and reactivation from latency in tonsils of infected calves. J. Clin. Microbiol. 43:393–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Meyer F, Perez S, Jiang Y, Zhou Y, Henderson G, Jones C. 2007. Identification of a novel protein encoded by the latency-related gene of bovine herpesvirus 1. J. Neurovirol. 13:569–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Meyer F, Perez S, Geiser V, Sintek M, Inman M, Jones C. 2007. A protein encoded by the bovine herpes virus 1 (BHV-1) latency related gene interacts with specific cellular regulatory proteins, including the CCAAT enhancer binding protein alpha (C/EBP-a). J. Virol. 81:59–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Winkler MT, Schang LS, Doster A, Holt T, Jones C. 2000. Analysis of cyclins in trigeminal ganglia of calves infected with bovine herpesvirus-1. J. Gen. Virol. 81:2993–2998. [DOI] [PubMed] [Google Scholar]
- 51.Wirth UV, Fraefel C, Vogt B, Vlcek C, Paces V, Schwyzer M. 1992. Immediate-early RNA 2.9 and early RNA 2.6 of bovine herpesvirus 1 are 3′ coterminal and encode a putative zinc finger transactivator protein. J. Virol. 66:2763–2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wirth UV, Vogt B, Schwyzer M. 1991. The three major immediate-early transcripts of bovine herpesvirus 1 arise from two divergent and spliced transcription units. J. Virol. 65:195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Holland LE, Anderson KP, Shipman C, Wagner EK. 1980. Viral DNA synthesis is required for the efficient expression of specific herpes virus type 1 mRNA species. Virology 101:10–24. [DOI] [PubMed] [Google Scholar]
- 54.Holland LE, Anderson KP, Stringer JR, Wagner EK. 1979. Isolation and localization of herpes simplex virus type 1 mRNA abundant before viral DNA synthesis. J. Virol. 31:447–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Workman A, Perez S, Doster A, Jones C. 2009. Dexamethasone treatment of calves latently infected with bovine herpesvirus 1 (BHV-1) leads to activation of the bICP0 early promoter, in part by the cellular transcription factor C/EBP-alpha. J. Virol. 83:8800–8809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Geiser V, Inman M, Zhang Y, Jones C. 2002. The latency related (LR) gene of bovine herpes virus 1 (BHV-1) can inhibit the ability of bICP0 to activate productive infection. J. Gen. Virol. 83:2965–2971. [DOI] [PubMed] [Google Scholar]
- 57.Geiser V, Zhang Y, Jones C. 2005. Characterization of a BHV-1 strain that does not express the major regulatory protein, bICP0. J. Gen. Virol. 86:1987–1996. [DOI] [PubMed] [Google Scholar]
- 58.Kim JY, Mandarino A, Chao MV, Mohr I, Wilson AC. 2012. Transient reversal of episome silencing precedes VP16-dependent transcription during reactivation of HSV-1 in neurons. PLoS Pathog. 8:e1002540. 10.1371/journal.ppat.1002540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Thompson RL, Preston CM, Sawtell NM. 2009. De novo synthesis of VP16 coordinates the exit form HSV latency in vivo. PLoS Pathog. 5:1–12. 10.1371/journal.ppat.1000352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sinani D, Frizzo da Silva L, Jones C. 2013. A bovine herpesvirus 1 protein expressed in latently infected neurons (ORF2) promotes neurite sprouting in the presence of activated Notch1 or Notch3. J. Virol. 87:1183–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Funder JW. 1997. Glucocorticoids and mineralocorticoid receptors: biology and clinical relevance. Annu. Rev. Med. 48:231–240. [DOI] [PubMed] [Google Scholar]
- 62.Schonevild OJLM, Gaemers IC, Lamers WH. 2004. Mechanisms of glucocorticoid signalling. Biochim. Biophys. Acta 1680:114–128. [DOI] [PubMed] [Google Scholar]
- 63.DeLeon M, Covenas R, Chadi G, Narvaez JA, Fuxe K, Cintra A. 1994. Subpopulations of primary sensory neurons show coexistence of neuropeptides and glucocorticoid receptors in the rat spinal and trigeminal ganglia. Brain Res. 14:338–342. [DOI] [PubMed] [Google Scholar]
- 64.Jaber T, Workman A, Jones C. 2010. Small noncoding RNAs encoded within the bovine herpesvirus 1 latency-related gene can reduce steady-state levels of infected cell protein 0 (bICP0). J. Virol. 84:6297–6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bratanich AC, Hanson ND, Jones C. 1992. The latency-related gene of bovine herpesvirus 1 inhibits the activity of immediate-early transcription unit 1. Virology 191:988–991. [DOI] [PubMed] [Google Scholar]
- 66.Workman A, Sinani D, Pittayakhajonwut D, Jones C. 2011. A protein (ORF2) encoded by the latency related gene of bovine herpesvirus 1 interacts with Notch1 and Notch3. J. Virol. 85:2536–2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Decman V, Freeman ML, Kinchington PR, Hendricks RL. 2005. Immune control of HSV-1 latency. Viral Immunol. 18:466–473. [DOI] [PubMed] [Google Scholar]
- 68.Pratt WB, Toft DO. 1997. Steroid receptor interactions with heat shock protein and immunophillin chaperones. Endocr. Rev. 18:306–360. [DOI] [PubMed] [Google Scholar]
- 69.Giguere V, Hollenberg SM, Rosenfeld MG, Evans RM. 1986. Functional domains of the human glucocorticoid receptor. Cell 46:645–652. [DOI] [PubMed] [Google Scholar]
- 70.Wang JC, Derynck MK, Nonaka DF, Khodabakhsh DB, Haqq C, Yammamoto KR. 2004. Chromatin immunoprecipitation (ChIP) scanning identifies primary glucocorticoid receptor target genes. Proc. Natl. Acad. Sci. U. S. A. 101:15603–15608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chen J, Kinyamu HK, Archer TK. 2006. Changes in attitudes, changes in latitude: nuclear receptors remodeling chromatin to regulate transcription. Mol. Endocrinol. 20:1–13. [DOI] [PubMed] [Google Scholar]
- 72.Deroo BJ, Archer TK. 2001. Glucocorticoid receptor-mediated chromatin remodeling in vivo. Oncogene 20:3039–3046. [DOI] [PubMed] [Google Scholar]
- 73.Yang L, Voytek CC, Margolis TP. 2000. Immunohistochemical analysis of primary sensory neurons latently infected with herpes simplex virus type 1. J. Virol. 74:209–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bertke AS, Swanson SM, Chen J, Imai Y, Kinchingotn PR, Margolis TP. 2011. A5-positive primary sensory neurons are nonpermissive for productive infection with herpes simplex virus 1 in vitro. J. Virol. 85:6669–6677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Margolis TP, Imai Y, Yang L, Vallas V, Krause PR. 2007. Herpes simplex virus type 2 (HSV-2) establishes latent infection in a different population of ganglionic neurons than HSV-1: role of latency-associated transcripts. J. Virol. 81:1872–1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.KosZ-Vnenchak JJ, Coen DM, Knipe DM. 1993. Evidence for a novel regulatory pathway for herpes simplex virus gene expression in trigeminal ganglion neurons. J. Virol. 67:5383–5393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nichol PF, Chang JY, Johnson EM, Jr, Olivo PD. 1996. Herpes simplex virus gene expression in neurons: viral DNA synthesis is a critical regulatory event in the branch point between lytic and latent pathways. J. Virol. 70:5476–5486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pesola JM, Zhu J, Knipe DM, Coen DM. 2005. Herpes simplex virus 1 immediate-early and early gene expression during reactivation from latency under conditions that prevent infectious virus production. J. Virol. 79:14516–14525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tal-Singer R, Lasner TM, Podrzucki W, Skokotas A, Leary JJ, Berger SL, Frazer NW. 1997. Gene expression during reactivation of herpes simplex virus type 1 from latency in the peripheral nervous system is different from that during lytic infection of tissue cultures. J. Virol. 71:5268–5276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Du T, Zhou G, Roizman B. 2011. HSV-1 gene expression from reactivated ganglia is disordered and concurrent with suppression of latency-associated transcript and miRNAs. Proc. Natl. Acad. Sci. U. S. A. 108:18820–18824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Du T, Zhou G, Roizman B. 2012. Induction of apoptosis accelerates reactivation from latent HSV-1 in ganglionic organ cultures and replication in cell cultures. Proc. Natl. Acad. Sci. U. S. A. 109:14616–14621. [DOI] [PMC free article] [PubMed] [Google Scholar]