

Abstract
During infection by herpes simplex virus 1 (HSV-1), the viral capsid is transported around the cytoplasm along the microtubule (MT) network. Although molecular motors have been implicated in this process, the composition of the molecular machinery required for efficient directional transport is unknown. We previously showed that dystonin (BPAG1) is recruited to HSV-1 capsids by the capsid-bound tegument protein pUL37 to promote efficient cytoplasmic transport of capsids during egress. Dystonin is a cytoskeleton cross-linker which localizes at MT plus ends and has roles in retrograde and anterograde transport in neurons. In this study, we investigated the role of dystonin during the entry stages of HSV-1 infection. Because of the way in which the MT network is organized, capsids are required to change their direction of motion along the MTs as they travel from the point of entry to the nucleus, where replication takes place. Thus, capsids first travel to the centrosome (the principal microtubule organizing center) by minus-end-directed transport and then switch polarity and travel to the nucleus by plus-end-directed transport. We observed that transport of capsids toward the centrosome was slowed, but not blocked, by dystonin depletion. However, transport of capsids away from the centrosome was significantly impaired, causing them to accumulate in the vicinity of the centrosome and reducing the numbers reaching the nucleus. We conclude that, during entry of HSV-1, dystonin has a specific role in plus-ended transport of capsids from the centrosome to the nucleus.
INTRODUCTION
A successful outcome of infection demands precise control of particle movement around the cell. The cell has a number of transport mechanisms available, but the most important for herpesviruses is the microtubule (MT) network (1, 2), which is the main route of movement between the cell surface, where virus entry and exit take place, and the nucleus, which is the site of virus transcription, DNA replication, and capsid assembly. The MT network is typically organized around one or more microtubule-organizing centers (MTOCs), with the MT minus ends anchored at the MTOC and the plus ends radiating outwards (3). Because of this arrangement, a herpesvirus capsid has to switch polarity in order to travel from the plasma membrane to the nucleus. Thus, the capsids travel from the plasma membrane to the centrosome (the principal MTOC in most cell types) by minus-end-directed transport but must then transfer to another MT to complete its journey by plus-end-directed transport. The direction of transport along MTs is determined by the molecular motors that transport the cargo. These are of two basic types, kinesins and dynein, which carry out plus-end- and minus-end-directed transport, respectively. Association of herpes simplex virus 1 (HSV-1) capsids with molecular motors, such as dynein or kinesins, has been reported in vitro (4), and kinesin 3 interaction with the viral membrane protein pUs9 was shown to be important for anterograde transport of pseudorabies virus (PrV) capsids in neurons (5). Two other viral proteins that are known to have important roles in herpesvirus capsid transport are the inner tegument proteins pUL36 and pUL37, two proteins interacting with each other (6) and essential for growth of HSV-1 (7, 8). Unlike most tegument proteins, these two remain attached to the capsid during transport to the nucleus (9–12). pUL36 has been shown to interact with the dynein/dynactin motor complex in transfected cells (13) and is required for active capsid transport and nuclear targeting (14–19). pUL37 was also found to have a role in efficient capsid transport during entry (20) and egress (16, 21). In previous studies, we showed that the MT-binding protein dystonin (BPAG1) is recruited to capsids via pUL37 and is required for efficient transport of HSV-1 capsids during virus egress (22). In this study, we extended our analysis to look at the role of dystonin during virus entry. Live-cell imaging of cells depleted of dystonin showed that dystonin is not required for minus-end-directed transport of capsids from the sites of entry to the centrosome. However, it plays an important role in plus-end-directed transport of capsids from the centrosome to the nucleus.
MATERIALS AND METHODS
Cells and viruses.
293T, baby hamster kidney (BHK), and human fetal foreskin fibroblast 2 (HFFF2) cells were grown at 37°C in Dulbecco's modified Eagle medium (DMEM; PAA Laboratories) supplemented with 8% fetal calf serum (FCS). For live-cell microscopy studies, cells were grown on 35-mm ibidi petri dishes.
Wild-type (WT) HSV-1 (strain 17+), vSR27-VP26GFP (expressing a green fluorescent protein [GFP]-tagged capsid protein), and tsK/luci (provided by C. Preston) were propagated on BHK cells infected at 0.01 PFU per cell, and virions were concentrated from the medium supernatant by centrifugation at 15,000 × g for 2 h. The tsK/luci virus was generated as described earlier (23). As the tsK virus has a temperature-sensitive lesion in the ICP4 protein that is not relevant to our studies, all experiments using this virus were performed at the permissive temperature for this mutant (31°C). vSR27-VP26GFP was generated as described in reference 22.
Antibodies.
The following antibodies were used. Mouse anti-alpha-tubulin clone DM1A and mouse anti-gamma-tubulin clone GTU-88 were obtained from Sigma. In Fig. 2 and 6, capsids were visualized using the rabbit purified HSV-1 nuclear C capsid (PTNC) antibody (23). Mouse anti-ICP0 11060 antibody was from Santa Cruz Biotechnology. Mouse DM165 antibody against VP5 was described previously (24); mouse monoclonal antibody (MAb) 4846 against glycoprotein D (gD) was a gift of A. Cross (University of Glasgow). Alexa Fluor 488- or Alexa Fluor 568-conjugated goat anti-mouse antibodies (GAM488 and GAM568, respectively) and Alexa Fluor 568-conjugated goat anti-rabbit antibody (GAR568) were obtained from Molecular Probes. Goat anti-mouse Dylight680 was obtained from Cell Signaling.
Fig 2.

Onset of viral protein production in dystonin-depleted and control cells assessed by Western blotting and immunofluorescence analysis. (A) shControl or shDyst HFFF2 cells were infected with 5 PFU/cell of WT HSV-1. Cells were harvested at 2 h, 4 h, 7 h, 12 h, or 16 h after infection, and the levels of the immediate early protein ICP0 and of the late capsid protein VP5 were monitored using antibodies 11060 and DM165, respectively. Alpha-tubulin was detected using MAb DM1A and was used as a loading control. Protein quantities were normalized to the amount of alpha-tubulin, and the relative levels of ICP0 and VP5 in shDyst and shLuc cells were calculated. (B) ShControl or shDyst HFFF2 cells were infected with 1 PFU/cell of WT HSV-1. At 3 h after infection, the cells were fixed and stained for ICP0 (green) or capsids (red) using antibodies 11060 and PTNC, respectively. The numbers indicate the percentages of cells that were ICP0 positive out of 69 shControl cells and 80 shDyst cells counted.
Fig 6.

Effect of dystonin silencing on capsid localization during entry. ShControl or shDyst HFFF2 cells were infected with 25 PFU/cell of WT HSV-1 for 3 h at 37°C in the presence of 100 μg/ml cycloheximide before being fixed. (A) Capsids were visualized with the rabbit anticapsid antibody PTNC and GAR568 (red), and centrosomes were visualized with MAb GTU-88 against gamma-tubulin and GAM488 (green). Nuclei were visualized with DAPI (blue). z-stacks of the whole-cell thickness were collected and are shown here in projection. Arrowheads, centrosomes; white circles, an area of 9 μm2 centered on the centrosome excluding any overlapping nuclear area. The density of capsids (in number of capsids per μm2 of surface [c/μm2]; see below) in each circle is specified. (B) Nuclear capsids (defined as capsids present within the DAPI-labeled area) and centrosomal capsids (defined as capsids present within the area delineated by the white circles) were counted. A total of 2,102 capsids were counted in 19 shControl cells and 1,083 capsids were counted in 11 shDyst cells. The nuclear and centrosomal surface areas were calculated using Zeiss Axiovision software, and the number of capsids per μm2 of surface (capsid density) was calculated. Asterisks indicate statistical differences (t test; *, P < 0.01; **, P = 0.001).
shRNA.
The use of lentiviruses expressing short hairpin RNA (shRNA) directed against dystonin has been described previously (23, 25). Silencing of dystonin was done using the sequence GTGTTGAAAGCCATTTAGA (shDyst). This sequence (position 1897 on the murine open reading frame) corresponds to the plakin domain of human and murine dystonin and is common to all known isoforms of dystonin. It is not conserved within sequences of other known plakin proteins. The control corresponded to an shRNA sequence specific for the luciferase gene (shControl; GTGCGTTGCTAGTACCAAC) for all experiments except the luciferase experiments, where an shRNA construct specific for GFP (shGFP; GAGTACAACTACAACAGCC) was used. Silencing efficiency was routinely assessed by real-time reverse transcription-PCR from total RNA before every experiment, as described in reference 22.
Western blotting and quantification.
Cells transduced with the shControl or shDyst1 lentivirus (here referred to as shControl and shDyst cells, respectively) were infected with 5 PFU/cell of HSV-1 WT for 2 h, 4 h, 7 h, 12 h, or 16 h before being harvested and lysed directly with Laemmli buffer. Cell lysates were analyzed by Western blotting using antibodies 11060 and DM165 against ICP0 and VP5, respectively. Antibody DM1A against alpha-tubulin was used as a loading control. Western blots were visualized and quantified using the quantitative near-infrared fluorescent Dylight680-conjugated GAM secondary antibody and an Odyssey Imaging system (LI-COR). Quantification was performed using Image Studio software (version 1.1.7; LI-COR).
Virus penetration assay.
shControl and shDyst HFFF2 cells were incubated with gradient-purified vSR27-VP26GFP virions for 1 h at 4°C or for 2 h at 37°C. The cells were then fixed with 4% paraformaldehyde and labeled for gD with MAb 4846 and GAM568. Capsids were visualized through GFP fluorescence, and nuclei were visualized with DAPI (4′,6-diamidino-2-phenylindole). The total numbers of capsids present on randomly chosen cells were determined by counting the number of diffraction-limited green spots, and the proportion having envelopes was determined by colocalization between GFP (capsid) and Alexa Fluor 568 (envelope) signals. Counting was done using the Cell Counter plug-in of ImageJ software (version 1.47m).
Fluorescence microscopy.
Immunofluorescence that included tubulin staining was done as follows. Cells were fixed in a mix of 3.7% formaldehyde and 0.1% Triton X-100 in PEM buffer (100 mM PIPES [piperazine-N,N′-bis(2-ethanesulfonic acid)], 5 mM EGTA, 2 mM MgCl2, pH 6.8) for 5 min at room temperature, as described by Vielkind and Swierenga (26). Microtubules were stained using MAb DM1A against alpha-tubulin (Sigma) and GAM568 antibody (90 min for each incubation).
To visualize centrosomes, cells were fixed with −20°C methanol and incubated at −20°C for 5 min. The methanol was removed, and cells were left to dry before being rehydrated with phosphate-buffered saline. Centrosomes were stained using MAb GTU-88 against gamma-tubulin (Sigma) and GAM488. Capsids were stained using the rabbit antibody PTNC (23) and GAR568.
Samples were mounted in ImmuMount mounting agent (Thermo) containing 1 μg/ml DAPI dihydrochloride (Sigma) for DNA staining. All samples were visualized using a Zeiss Axio Observer Z1 microscope with a ×63 Plan-Apochromat oil-immersion objective (numerical aperture, 1.4; Zeiss).
Live-cell microscopy.
All live-cell microscopy was done at room temperature (∼23°C). Under these conditions, capsid motion is slower, thereby allowing more efficient tracking, as detailed previously (22). Movies were recorded at a rate of 1 frame/s for 55 to 60 s with an average exposure time of 200 ms, except for centrosome-based movies (see Movies S3 and S4 in the supplemental material), where acquisition was for 120 s at a 1-frame/s rate.
Capsid tracking.
Capsid tracking was done as described in reference 22. Briefly, movies were imported into ImageJ (version 1.47m). Capsids were tracked using the Particles Detector & Tracker plug-in (version 1.5) (27). Depending on the quality of each individual movie, detection parameters were set as follows: radius, 3 or 5; cutoff, 3.0 or 0.0; percentile, 0.6 to 1.5%. Linking parameters were a link range of 2 and a displacement of 10 to 20. Capsid motion was analyzed using the coordinates provided by the software to calculate the distance to the origin. Capsid tracking starts as the capsid enters the field of view and stops as it leaves it or when it moves out of focus. The directionality of each trajectory was estimated according to the position of the nucleus (see Fig. 5). A capsid which, over the period of the recording, moved away from the cell periphery and toward the nucleus was categorized as having retrograde motion, a capsid that moved away from the nucleus and toward the cell periphery was categorized as having anterograde motion, and capsids with no movement or no clear directionality were categorized as “none or both.”
Fig 5.

Impact of dystonin reduction on capsid transport during entry. shControl (A, B, and E) or shDyst (C, D, and F) HFFF2 cells were infected with 50 PFU/cell of vSR27-VP26GFP. Starting at 45 min after infection, cytoplasmic capsid movements were monitored by live-cell imaging at a rate of one frame per second. Results for each individual capsid tracked are plotted as the distance to the origin (A and C) or as the velocity (E and F) for each frame. The premature truncation of some lines is due to the capsids moving out of the field of view. A representative cell with all capsid trajectories plotted is shown for each condition (B and D). Dashed boxes, the area displayed in the corresponding movies (see Movies S1 and S2 in the supplemental material, respectively). Bars, 10 μm. (G) Summary of the maximum distances to the origin taken from the data shown in panels A and C as percentages of cells in categories of the distance to the origin. (H) Every moving capsid was tracked individually, and its directionality was estimated according to the position of the nucleus. Results are shown as the percentage of capsids having overall retrograde (away from the cell periphery and toward the nucleus) or anterograde (away from the nucleus and toward the cell periphery) motion. Both or none, either a capsid having opposite directionalities within the same run, a capsid that was not moving, or a capsid where the direction of movement relative to the nucleus could not be categorized.
Luciferase assay.
Replicate monolayers of HFFF2 cells were infected with 5 PFU/cell of tsK/luci and incubated at 31°C for the times required. Luciferase assays were performed using a luciferase assay system (Promega) according to the manufacturer's instructions. Luciferase activity was assessed on a Glomax 20/20 luminometer (Promega) and normalized to the cell count.
Fluorescence-activated cells sorter (FACS) analysis.
shControl or shDyst HFFF2 cells were infected with 5 PFU/cell of WT HSV-1 for 2 h, 4 h, or 6 h at 37°C. They were then harvested, fixed with 4% paraformaldehyde, and permeabilized with 0.1% Triton X-100. Cells were incubated with antibody 11060 against ICP0 and GAM488 for 1 h at room temperature and washed three times. Mock-infected shControl cells were similarly treated and were used as negative controls. Data were collected on an Accuri C6 flow cytometer (BD) mounted with a 20-mW 488-nm solid-state blue laser and a 533/30-nm optical filter. Data were analyzed using Accuri C6 software (version 1.0.264.21).
RESULTS
Initiation of infection is delayed in dystonin-depleted cells.
Using shRNA technology, we previously demonstrated that the levels of dystonin mRNA could be depleted by up to 80% in cells transduced with the shDyst1 lentivirus (shDyst cells) compared to the level in cells transduced with a control lentivirus (22). In shDyst cells, virus production was significantly delayed compared to that in control cells. Part of this delay could be explained by the severe defect in capsid egress observed in dystonin-depleted cells (22). To determine whether dystonin depletion also affected entry, shGFP (negative control) and shDyst cells were infected with tsK/luci, which encodes a luciferase gene under the control of a viral immediate early promoter. Cells were infected with 5 PFU/cell of tsK/luci for 2 h, 4 h, or 7 h postinfection (p.i.) and luciferase activity was monitored. As shown in Fig. 1, luciferase levels in shDyst cells (192 ± 9) were more than 15-fold lower than those in shGFP cells (3135 ± 230) at 2 h p.i. and corresponded to background levels (138 ± 97). At 4 h p.i., luciferase levels had increased only slightly in shDyst cells (365 ± 20) and were ∼330-fold lower than those in shGFP cells (120,639 ± 15,385). At 7 h p.i., luciferase levels in shDyst cells were higher (5,707 ± 1,113) but were still more than 2.5 log10 units lower than those in shGFP cells (1,522,604 ± 8,383). The rate of increase in luciferase levels at between 4 and 7 h p.i. in shDyst cells paralleled that in shGFP cells at between 0 and 2 h p.i. (Fig. 1B), suggesting that dystonin depletion affects the initiation of infection and not a later step.
Fig 1.
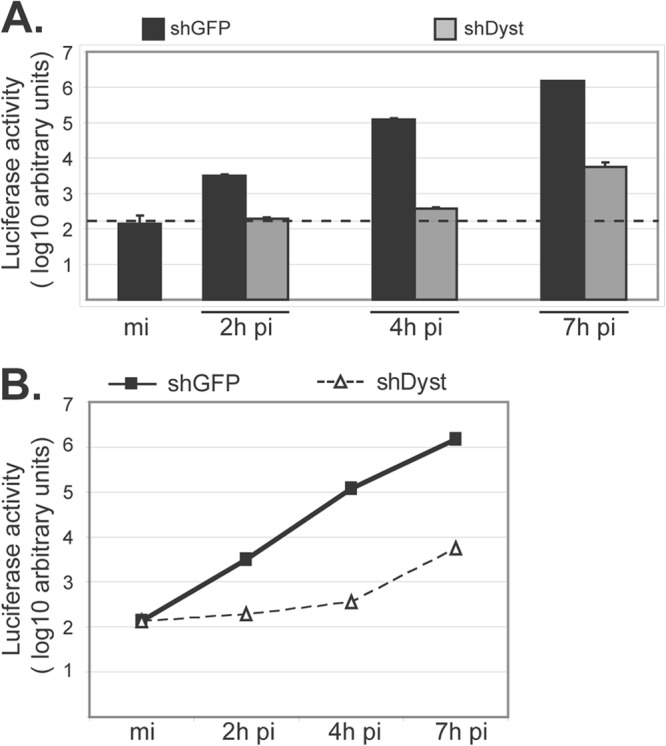
Initiation of HSV-1 replication in dystonin-depleted and control cells. (A) shGFP or shDyst HFFF2 cells were infected with 5 PFU/cell of tsK/luci and incubated for 2 h, 4 h, or 7 h at a permissive temperature (31°C). Cells were then harvested and lysed and luciferase activity was measured. Dashed line, background level. (B) Data shown in panel A are represented as a graph to show the rate of increase in luciferase levels with time after infection. mi, mock infected.
To confirm these results, shControl and shDyst cells were infected with 1 PFU/cell of WT virus, and the levels of expression of the immediate early gene RL2, encoding the protein ICP0, and the late gene UL19, encoding the major capsid protein VP5, were monitored by Western blotting (Fig. 2A). ICP0 could be clearly detected in shControl cells as early as 4 h p.i. and at increasing levels until the latest time (16 h) analyzed. In contrast, ICP0 was first detected in small amounts in shDyst cells at 7 h p.i., and its level then increased rapidly to reach a level similar to that in shControl cells by 16 h. Expression of VP5 presented a similar pattern, although with late gene expression kinetics. Thus, VP5 was present from 7 h p.i. in shControl cells but was first detected at 12 h p.i. in shDyst cells. As with ICP0, the amounts of VP5 in shDyst cells had reached the level of that in shControl cells by 16 h p.i. Similar results were obtained with cells infected with 3 or 5 PFU/cell, but only a very moderate effect was observed with 50 PFU/cell (data not shown).
Immunofluorescence analysis of cells infected with 1 PFU/cell of HSV-1 WT showed behavior similar to that described above, where only ∼2% of shDyst cells but nearly half of shControl cells were assessed to be positive for ICP0 by 3 h after infection (Fig. 2B). To quantify this effect, we carried out FACS analysis. ShControl or shDyst cells were mock infected or infected with HSV-1 WT for 2 h, 4 h, or 6 h. Cells were then fixed, permeabilized, stained for ICP0, and analyzed by flow cytometry. At 2 h, ∼13% of shControl cells had a detectable amount of ICP0, while at 4 h and 7 h, the percentages had increased to 38% and 47%, respectively. In comparison, ICP0 expression was greatly reduced in shDyst cells, where 0%, 4%, and 7% of cells were ICP0 positive at 2 h, 4 h, and 7 h, respectively (Fig. 3A). In keeping with these results, the fluorescence intensity was also markedly reduced in shDyst cells compared to shControl cells (Fig. 3B).
Fig 3.
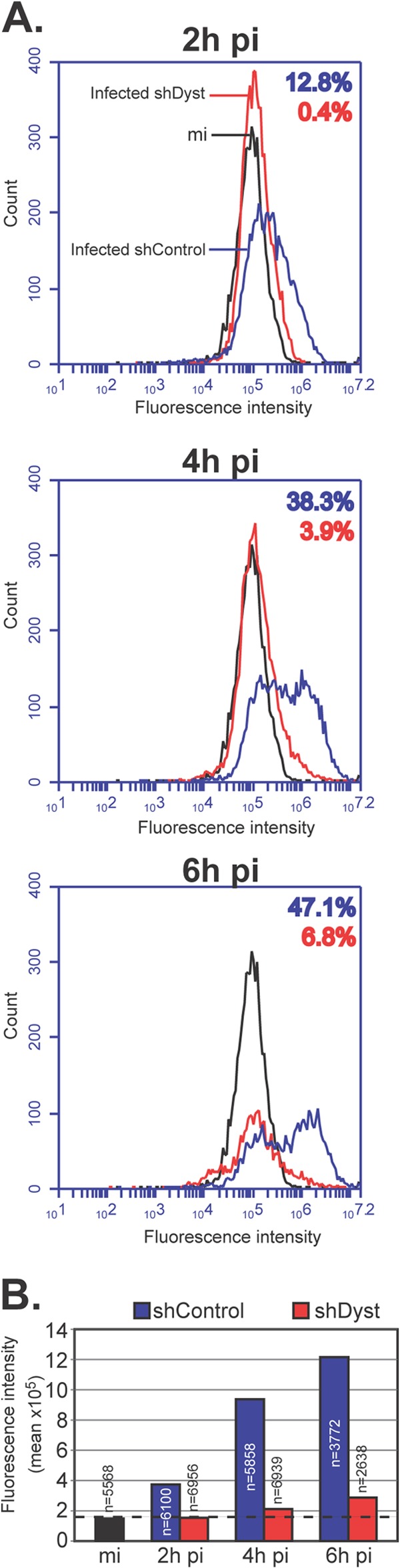
Onset of viral protein production in dystonin-depleted and control cells assessed by FACS analysis. (A) Mock-infected shControl cells or shControl or shDyst HFFF2 cells infected with 5 PFU/cell of WT HSV-1 were harvested at 2 h, 4 h, or 6 h after infection, fixed, permeabilized, and incubated with antibody 11060 against ICP0 and GAM488. Cells were then analyzed by flow cytometry. The numbers of fluorescing cells and their fluorescence intensities were determined for mock-infected shControl cells (black line), infected shControl cells (blue line), or infected shDyst cells (red line). The percentages of the total cell population identified as positive (i.e., above the background level, as defined by the fluorescence intensity of the mock-infected shControl cells) are indicated using the same color coding. (B) Summary of mean fluorescence intensities indicated for panel A. The number of cells analyzed is given for each condition. Dashed line, background level defined by the fluorescence intensity of mock-infected shControl cells.
Taken together, these data provide strong support for a mechanism in which a lack of dystonin delays the onset of virus expression but has no effect on expression once it has started.
Dystonin is dispensable for virus entry into the cell.
A possible explanation for the delay in the onset of infection observed in dystonin-depleted cells is a defect in the ability of the virus to enter the cell. To determine whether this was the case, we carried out experiments to examine virus entry at the cell surface, as assessed by the presence of the virion envelope. shControl or shDyst cells were incubated with 5 PFU/cell of vSR27-VP26GFP (expressing a GFP-tagged capsid protein) and then fixed, permeabilized, and stained for glycoprotein D (gD) (Fig. 4A). Intact virions should appear as both GFP- and gD-positive spots, as their envelope and capsid are tightly associated. This was the case when infection was carried out at 4°C, where virus particles can attach to the cell but membrane fusion is inhibited (Fig. 4A). Under these conditions, almost all capsids seen on cells were associated with gD (97% ± 3%) (Fig. 4B). However, in cells infected at 37°C, membrane fusion can occur, leading to dissociation of the capsid and envelope proteins. Under these conditions, 88% of the capsids (±4%) in shControl cells were gD negative, indicating successful viral entry. Similarly, 84% of the capsids (±4%) in shDyst cells were gD negative (Fig. 4). In addition, the absolute numbers of cytosolic capsids per cell were similar in shControl and shDyst cells (Fig. 4C). This clearly demonstrates that entry occurs normally in cells depleted for dystonin.
Fig 4.

Virus penetration in dystonin-depleted and control cells. (A) shControl and shDyst HFFF2 cells were incubated with vSR27-VP26GFP virions for 1 h at 4°C or for 2 h at 37°C. The cells were fixed and labeled for gD with MAb 4846 and GAM568 (red). Capsids were visualized through GFP fluorescence (green). Nuclei were visualized with DAPI (blue). Bars, 20 μm. (B) The total numbers of capsids present on randomly chosen cells from the experiment whose results are shown in panel A were counted, and the proportion having envelopes was determined by colocalization between GFP (capsid) and Alexa Fluor 568 (envelope) signals. Results are shown as the numbers of gD-positive (yellow) and gD-negative (green) capsids expressed as a percentage of the total number of capsids counted. (C) Average number of cytosolic (gD-negative) capsids per cell at 37°C. A total of 270 (ShControl, 4°C), 1,870 (shControl, 37°C), and 1,836 (shDyst, 37°C) particles were analyzed in 27, 43, and 46 cells, respectively.
Capsid transport in dystonin-depleted cells.
Given the demonstrated role of dystonin in capsid transport during egress, an obvious early step that might be affected by depletion of dystonin would be the MT-based retrograde transport of capsids from the cell periphery to the nucleus. To test this hypothesis, we monitored infected shControl or shDyst cells by live-cell microscopy. These cells were infected at 37°C with 50 PFU/cell of vSR27-VP26GFP, and capsid movements were monitored by live-cell microscopy at 30 min postinfection. Live-cell imaging was carried out at room temperature (∼25°C) because capsids moved too fast to be tracked efficiently at 37°C with our equipment (see Materials and Methods and reference 22).
On initial examination, capsid movement was observed in both shControl and shDyst cells (see Movies S1 and S2 in the supplemental material). This indicated that dystonin silencing did not have as dramatic an influence on overall capsid transport during entry as that seen during egress, when capsid movement was almost completely blocked (22). Capsid movement was measured and plotted as the distance moved from the point of origin at the start of imaging against time (Fig. 5A and C). The capsid velocity (Fig. 5E and F), the maximum distance moved (Fig. 5G), and the direction of movement (Fig. 5H) were also measured. The histogram in Fig. 5H shows the overall direction of capsid movement (retrograde for movement from the cell periphery to the nucleus; anterograde for movement from the nucleus to the cell periphery) rather than the time spent moving in one or the other direction. Analysis of a total of 185 capsid trajectories revealed differences in capsid trafficking between infected shControl and shDyst cells (Fig. 5A and C). Capsid velocities were significantly lower in shDyst cells (average of maxima = 0.5 ± 0.2 μm/s) than in shControl cells (average of maxima = 1.4 ± 0.8 μm/s) (Fig. 5E and F), although the effect was not as great as that seen at late times after infection (22). As a consequence, capsids tended to travel longer distances under control conditions than in shDyst cells (Fig. 5G). However, the direction of motion was similar for the two conditions, with 45% of capsids showing retrograde movement (i.e., from the cell periphery to the nucleus) in shControl cells and 39% showing retrograde movement in shDyst cells (Fig. 5H).
Thus, the overall pattern of capsid transport was not disrupted in dystonin-depleted cells, but the rate of movement was reduced.
Effect of dystonin silencing on nuclear targeting of capsids.
Despite the limited effects of dystonin depletion on overall capsid movement at early times of infection (Fig. 5), the observed delay in immediate early gene expression when dystonin levels were reduced (Fig. 1 to 3) suggested that an early, prenuclear step was affected. Given the usual close proximity of the centrosome to the nucleus, the majority of capsid movement during entry would be expected to be minus-end directed along the MTs toward the centrosome, with only a small amount of plus-end-directed transport required to reach the nucleus, while the reverse should be true during egress, when plus-end-directed transport would predominate. Therefore, because blocking the limited amount of plus-end-directed transport during entry would explain why capsid transport was less dramatically affected than during egress (22) (Fig. 5), we hypothesized that plus-end-directed transport of capsids was involved. If dystonin is required for plus-end-directed transport, depleting it might be expected to have no effect on capsids moving toward the centrosome but would prevent them from moving away from it, leading to an accumulation of capsids in the vicinity of the centrosome with a concomitant decrease in the number of capsids reaching the nucleus.
To test whether this was the case, shControl or shDyst HFFF2 cells were infected with 25 PFU/cell of WT HSV-1 for 3 h at 37°C in the presence of cycloheximide, an inhibitor of de novo protein synthesis commonly used in studies of HSV capsid accumulation at the nucleus (1, 2, 28–31). Addition of cycloheximide increases capsid retention at the nucleus and prevents confusion between input capsids and newly formed nuclear capsids.
Numerous capsids were seen in the area of the nucleus in shControl cells and frequently formed a distinct ring around the nuclear periphery (Fig. 6A). In contrast, capsids never formed rings around the nuclei of shDyst cells, and overall, there were fewer capsids associated with the nuclei. Visual comparison gave the impression that capsids were present at a higher density in the vicinity of the centrosome in shDyst cells (Fig. 6A). To quantify these observations, we compared the number of capsids present over the nuclear area and at the nuclear rim (as estimated by DAPI staining) to the number of capsids present within an area of 9 μm2 centered on the centrosome (minus any overlapping nuclear area). The number of capsids counted in each region was divided by its area to measure capsid density (number of capsids per μm2). This showed that in shControl cells (n = 19 cells), 0.22 capsid/μm2 was present in nuclear areas and 0.07 capsid/μm2 was present in centrosomal areas (Fig. 6B). These proportions were inverted in shDyst cells (n = 11 cells), where capsid density was lower in nuclear areas (0.09 capsid/μm2) than in centrosomal areas (0.24 capsid/μm2). These results show that reducing the levels of dystonin limits the ability of capsids to reach the nucleus and causes them to concentrate in the vicinity of the centrosome.
Effect of dystonin silencing on capsid trafficking in the vicinity of the centrosome.
To directly ascertain the effect of dystonin silencing on plus-end-directed MT transport, capsid trafficking to and from the centrosome was imaged by live-cell microscopy of shControl and shDyst HFFF2 cells infected with 50 PFU/cell of vSR27-VP26GFP. Capsid monitoring started once the centrosome became distinguishable as a result of capsid accumulation in its vicinity (see Movies S3 and S4 in the supplemental material). As early as 45 min after infection, capsids could be seen accumulating close to the nucleus. Subsequent staining of fixed cells for tubulin confirmed that this area of capsid accumulation was an MTOC, most likely the centrosome (arrowhead in Fig. 7A). As can be seen in Movies S3 and S4 in the supplemental material, capsids were clearly moving both toward and away from the MTOC in shControl cells (see Movie S3 in the supplemental material). In contrast, although capsids were also seen moving toward the MTOC in shDyst cells, very few were seen leaving it (see Movie S4 in the supplemental material). To quantify this effect, capsids were classified as trafficking either toward (marked as red lines in Fig. 7B and C) or away from (marked as yellow lines) the MTOC. A total of 1,219 moving capsids and their directions were monitored in shControl cells (n = 32 cells) and shDyst cells (n = 32 cells). This revealed that in shControl cells, 45% of moving capsids were traveling away from the MTOC, with 55% moving in the opposite direction, whereas in shDyst cells, only 21% of capsids were moving away from the MTOC and 79% were moving toward it. As each capsid that moves to the centrosome would subsequently be expected to move away from it, the expected ratio of capsids moving to and from the MTOC would be approximately 50:50. That 45% of capsids were identified as moving away from the MTOC in control cells means that this is the case for 45/55, or 82%, of capsids. This drops to 21/79, or 26.5%, in shDyst cells, which, although not indicative of a complete block, does suggest that a capsid is more likely to become trapped in the vicinity of the centrosome. Therefore, the ability of capsids to be redirected from the MTOC to the nucleus is at least three times less efficient in shDyst cells than in shControl cells.
Fig 7.

Capsid trafficking in the vicinity of the centrosome. shControl or shDyst HFFF2 cells were infected with 50 PFU/cell of vSR27-VP26GFP at 37°C. (A) shControl cells were fixed at 3 h postinfection and stained for microtubules with an antibody directed against alpha-tubulin and GAM568 (red). Capsids were visualized through direct GFP fluorescence (green). White arrowhead, MTOC. Bar, 20 μm. (B, C) shControl or shDyst cells were infected for 45 min at 37°C, and capsid trafficking was monitored by time-lapse microscopy at room temperature (see Movies S3 and S4 in the supplemental material, respectively). Capsid movement was visualized through a maximum-intensity projection of the whole time-lapse stack and interpreted as moving to (red) or from (yellow) the centrosome. (D) A total of 1,219 capsids from 114 different movies were tracked in shControl (n = 32) and shDyst (n = 32) HFFF2 cells. Their motion relative to the centrosome was determined and plotted as a percentage of the total number of capsids tracked.
This supports a role for dystonin in the transport of HSV-1 capsids by plus-end-directed MT transport, which is in accordance with the known functions of pUL37.
DISCUSSION
In an earlier paper, we reported that the tegument protein pUL37 of HSV-1 can bind the cellular protein dystonin and that capsids colocalized with endogenous dystonin in infected cells in a UL37-dependent manner (22). Dystonin had previously been suggested to act as a linker between MT motor proteins and their cargoes during retrograde transport (32, 33), to be involved in anterograde transport (34), and to localize to the plus ends of MTs (22, 35). When dystonin expression was inhibited, we observed a dramatic decrease in capsid movement at late times of infection and interpreted this as indicating a role for dystonin in capsid transport during egress. In view of these observations, it was rather surprising that depletion of dystonin did not appear to have a dramatic effect on overall capsid transport during entry, while it strongly delayed the onset of virus expression at high multiplicities of infection (MOIs; 1, 3, or 5 PFU/cell) but not at the very high MOI of 50 PFU/cell (data not shown). However, closer analysis revealed that although transport of capsids to the MTOC was not strongly affected, redirection of capsids from the MTOC to the nucleus was significantly impaired. To make this observation, cells were infected with 50 PFU/cell to have a sufficient number of capsids to track. Under these conditions, the delay in the onset of infection was very moderate (data not shown), which can be explained by the remaining number of capsids that leave from the MTOC in the absence of dystonin (Fig. 7). Since the onset of infection was strongly delayed at an MOI of 1, 3, or 5, it is possible that the ratio of capsids arriving at the MTOC that are redirected to the nucleus is actually much lower under these conditions than that observed in our live-cell experiments.
The typical perinuclear location of the centrosome ensures that during entry, most of the movement will be between the plasma membrane and the centrosome. This transport involves minus-end-directed motors (13), and although capsid movement was slower in the absence of dystonin, it was not prevented (Fig. 5). However, the shorter distance between the centrosome and the nucleus is expected to be crossed by plus-end-directed transport, and this was strongly impaired in dystonin-depleted cells. Based on these observations, we postulate that the pUL37-dystonin interaction has a specific role in plus-end-directed transport of incoming capsids. Interestingly, although pUL37 is important for efficient nuclear targeting of incoming PrV capsids (20), it is not essential for nuclear binding of HSV-1 capsids and viral DNA release (17). A role in plus-end-directed transport is in accordance with the reported role of its binding partner dystonin in anterograde transport of cellular vesicles (34) and its localization to the plus ends of MTs (22, 35) and provides the first evidence that transport directionality can be conferred on the capsid by recruitment of cellular nonmotor proteins.
It is not clear whether the directional specificity seen during entry would account for the observed behavior during egress. The proximity of the centrosome to the nucleus might seem to imply that outward transport would be predominantly plus ended, given the relatively greater distance between the centrosome and the sites of envelopment. However, there is increasing evidence that functional centrosomes are no longer present in HSV-infected cells by the time that newly formed capsids are entering the cytoplasm (36–38). Capsid transport must therefore be along MTs originating from noncentrosomal sites. The polarity of these MTs is not known, and it remains unclear whether outgoing capsids are directed to the sites of envelopment by plus- or minus-end-directed transport.
Herpesvirus intracellular transport is a complex process that is likely to involve different viral and cellular proteins, depending on the cell type and the stage of the viral cycle. This is of particular relevance in differentiated neurons, a highly asymmetric cell type, where the distances traveled are typically much longer than those in other cells. The state of the capsid during transport from the neuronal cell body to the tip of the axon is still a matter of debate. Two models have been proposed, with one suggesting that capsids are transported to the axon tips for envelopment and the other suggesting that capsids are enveloped in the cell body and mature virions are transported to the axon tips (39). Recent studies have shown that the viral membrane protein pUs9 interacts with kinesin 3 to transport enveloped virions from the cell body to the axonal terminus (5). As for the alternative mechanism, it would be interesting to determine whether the pUL37-dystonin interaction plays any role in transport of unenveloped capsids to the axon tips. The diverse requirements of transporting virus capsids around cells makes it clear that many of the factors involved in the complex intracellular trafficking of herpesviruses, together with the mechanisms associated with them, are still to be unraveled.
Interaction studies have failed to identify a direct interaction between pUL37 and molecular motors (22, 40), as has been done for pUL36 (13) and pUs9 (5). Since pUL36 appears to play a central role in capsid transport along microtubules (16) and given the apparent involvement of the pUL37-dystonin interaction in controlling transport polarity, it seems plausible that pUL37 may act as a regulatory protein modulating the outcome of the interaction between pUL36 and the motor proteins.
Supplementary Material
ACKNOWLEDGMENTS
We thank Y. Gaudin for his support of this project. We are grateful to C. Preston (CVR, Glasgow, Scotland) for his gift of the tsK/Luci virus and to R. Everett (CVR, Glasgow, Scotland) for his gift of silencing plasmids.
This work was funded by the CNRS and the MRC.
Footnotes
Published ahead of print 31 July 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.01633-13.
REFERENCES
- 1.Mabit H, Nakano MY, Prank U, Saam B, Dohner K, Sodeik B, Greber UF. 2002. Intact microtubules support adenovirus and herpes simplex virus infections. J. Virol. 76:9962–9971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sodeik B, Ebersold MW, Helenius A. 1997. Microtubule-mediated transport of incoming herpes simplex virus 1 capsids to the nucleus. J. Cell Biol. 136:1007–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bartolini F, Gundersen GG. 2006. Generation of noncentrosomal microtubule arrays. J. Cell Sci. 119:4155–4163. [DOI] [PubMed] [Google Scholar]
- 4.Radtke K, Kieneke D, Wolfstein A, Michael K, Steffen W, Scholz T, Karger A, Sodeik B. 2010. Plus- and minus-end directed microtubule motors bind simultaneously to herpes simplex virus capsids using different inner tegument structures. PLoS Pathog. 6:e1000991. 10.1371/journal.ppat.1000991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kramer T, Greco TM, Taylor MP, Ambrosini AE, Cristea IM, Enquist LW. 2012. Kinesin-3 mediates axonal sorting and directional transport of alphaherpesvirus particles in neurons. Cell Host Microbe 12:806–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klupp BG, Fuchs W, Granzow H, Nixdorf R, Mettenleiter TC. 2002. Pseudorabies virus UL36 tegument protein physically interacts with the UL37 protein. J. Virol. 76:3065–3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Desai P, Sexton GL, McCaffery JM, Person S. 2001. A null mutation in the gene encoding the herpes simplex virus type 1 UL37 polypeptide abrogates virus maturation. J. Virol. 75:10259–10271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Desai PJ. 2000. A null mutation in the UL36 gene of herpes simplex virus type 1 results in accumulation of unenveloped DNA-filled capsids in the cytoplasm of infected cells. J. Virol. 74:11608–11618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aggarwal A, Miranda-Saksena M, Boadle RA, Kelly BJ, Diefenbach RJ, Alam W, Cunningham AL. 2012. Ultrastructural visualization of individual tegument protein dissociation during entry of herpes simplex virus 1 into human and rat dorsal root ganglion neurons. J. Virol. 86:6123–6137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Antinone SE, Smith GA. 2010. Retrograde axon transport of herpes simplex virus and pseudorabies virus: a live-cell comparative analysis. J. Virol. 84:1504–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Granzow H, Klupp BG, Mettenleiter TC. 2005. Entry of pseudorabies virus: an immunogold-labeling study. J. Virol. 79:3200–3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Luxton GW, Haverlock S, Coller KE, Antinone SE, Pincetic A, Smith GA. 2005. Targeting of herpesvirus capsid transport in axons is coupled to association with specific sets of tegument proteins. Proc. Natl. Acad. Sci. U. S. A. 102:5832–5837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zaichick SV, Bohannon KP, Hughes A, Sollars PJ, Pickard GE, Smith GA. 2013. The herpesvirus VP1/2 protein is an effector of dynein-mediated capsid transport and neuroinvasion. Cell Host Microbe 13:193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abaitua F, Hollinshead M, Bolstad M, Crump CM, O'Hare P. 2012. A nuclear localization signal in herpesvirus protein VP1-2 is essential for infection via capsid routing to the nuclear pore. J. Virol. 86:8998–9014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Batterson W, Furlong D, Roizman B. 1983. Molecular genetics of herpes simplex virus. VIII. Further characterization of a temperature-sensitive mutant defective in release of viral DNA and in other stages of the viral reproductive cycle. J. Virol. 45:397–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luxton GW, Lee JI, Haverlock-Moyns S, Schober JM, Smith GA. 2006. The pseudorabies virus VP1/2 tegument protein is required for intracellular capsid transport. J. Virol. 80:201–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roberts AP, Abaitua F, O'Hare P, McNab D, Rixon FJ, Pasdeloup D. 2009. Differing roles of inner tegument proteins pUL36 and pUL37 during entry of herpes simplex virus type 1. J. Virol. 83:105–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jovasevic V, Liang L, Roizman B. 2008. Proteolytic cleavage of VP1-2 is required for release of herpes simplex virus 1 DNA into the nucleus. J. Virol. 82:3311–3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schipke J, Pohlmann A, Diestel R, Binz A, Rudolph K, Nagel CH, Bauerfeind R, Sodeik B. 2012. The C terminus of the large tegument protein pUL36 contains multiple capsid binding sites that function differently during assembly and cell entry of herpes simplex virus. J. Virol. 86:3682–3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krautwald M, Fuchs W, Klupp BG, Mettenleiter TC. 2009. Translocation of incoming pseudorabies virus capsids to the cell nucleus is delayed in the absence of tegument protein pUL37. J. Virol. 83:3389–3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sandbaumhuter M, Dohner K, Schipke J, Binz A, Pohlmann A, Sodeik B, Bauerfeind R. 2013. Cytosolic herpes simplex virus capsids not only require binding inner tegument protein pUL36 but also pUL37 for active transport prior to secondary envelopment. Cell. Microbiol. 15:248–269. [DOI] [PubMed] [Google Scholar]
- 22.Pasdeloup D, McElwee M, Beilstein F, Labetoulle M, Rixon FJ. 2013. Herpesvirus tegument protein pUL37 interacts with dystonin/BPAG1 to promote capsid transport on microtubules during egress. J. Virol. 87:2857–2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pasdeloup D, Blondel D, Isidro AL, Rixon FJ. 2009. Herpesvirus capsid association with the nuclear pore complex and viral DNA release involve the nucleoporin CAN/Nup214 and the capsid protein pUL25. J. Virol. 83:6610–6623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McClelland DA, Aitken JD, Bhella D, McNab D, Mitchell J, Kelly SM, Price NC, Rixon FJ. 2002. pH reduction as a trigger for dissociation of herpes simplex virus type 1 scaffolds. J. Virol. 76:7407–7417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Everett RD, Rechter S, Papior P, Tavalai N, Stamminger T, Orr A. 2006. PML contributes to a cellular mechanism of repression of herpes simplex virus type 1 infection that is inactivated by ICP0. J. Virol. 80:7995–8005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vielkind U, Swierenga SH. 1989. A simple fixation procedure for immunofluorescent detection of different cytoskeletal components within the same cell. Histochemistry 91:81–88. [DOI] [PubMed] [Google Scholar]
- 27.Sbalzarini IF, Koumoutsakos P. 2005. Feature point tracking and trajectory analysis for video imaging in cell biology. J. Struct. Biol. 151:182–195. [DOI] [PubMed] [Google Scholar]
- 28.Dohner K, Wolfstein A, Prank U, Echeverri C, Dujardin D, Vallee R, Sodeik B. 2002. Function of dynein and dynactin in herpes simplex virus capsid transport. Mol. Biol. Cell 13:2795–2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ojala PM, Sodeik B, Ebersold MW, Kutay U, Helenius A. 2000. Herpes simplex virus type 1 entry into host cells: reconstitution of capsid binding and uncoating at the nuclear pore complex in vitro. Mol. Cell. Biol. 20:4922–4931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rode K, Dohner K, Binz A, Glass M, Strive T, Bauerfeind R, Sodeik B. 2011. Uncoupling uncoating of herpes simplex virus genomes from their nuclear import and gene expression. J. Virol. 85:4271–4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Turcotte S, Letellier J, Lippe R. 2005. Herpes simplex virus type 1 capsids transit by the trans-Golgi network, where viral glycoproteins accumulate independently of capsid egress. J. Virol. 79:8847–8860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu JJ, Ding J, Kowal AS, Nardine T, Allen E, Delcroix JD, Wu C, Mobley W, Fuchs E, Yang Y. 2003. BPAG1n4 is essential for retrograde axonal transport in sensory neurons. J. Cell Biol. 163:223–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu JJ, Ding J, Wu C, Bhagavatula P, Cui B, Chu S, Mobley WC, Yang Y. 2007. Retrolinkin, a membrane protein, plays an important role in retrograde axonal transport. Proc. Natl. Acad. Sci. U. S. A. 104:2223–2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ryan SD, Bhanot K, Ferrier A, De Repentigny Y, Chu A, Blais A, Kothary R. 2012. Microtubule stability, Golgi organization, and transport flux require dystonin-a2-MAP1B interaction. J. Cell Biol. 196:727–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kapur M, Wang W, Maloney MT, Millan I, Lundin VF, Tran TA, Yang Y. 2012. Calcium tips the balance: a microtubule plus end to lattice binding switch operates in the carboxyl terminus of BPAG1n4. EMBO Rep. 13:1021–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Avitabile E, Di Gaeta S, Torrisi MR, Ward PL, Roizman B, Campadelli-Fiume G. 1995. Redistribution of microtubules and Golgi apparatus in herpes simplex virus-infected cells and their role in viral exocytosis. J. Virol. 69:7472–7482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kotsakis A, Pomeranz LE, Blouin A, Blaho JA. 2001. Microtubule reorganization during herpes simplex virus type 1 infection facilitates the nuclear localization of VP22, a major virion tegument protein. J. Virol. 75:8697–8711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pasdeloup D, Labetoulle M, Rixon FJ. 2013. Differing effects of herpes simplex virus 1 and pseudorabies virus infection on centrosomal function. J. Virol. 87:7102–7112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Johnson DC, Baines JD. 2011. Herpesviruses remodel host membranes for virus egress. Nat. Rev. Microbiol. 9:382–394. [DOI] [PubMed] [Google Scholar]
- 40.Kelly BJ, Diefenbach E, Fraefel C, Diefenbach RJ. 2012. Identification of host cell proteins which interact with herpes simplex virus type 1 tegument protein pUL37. Biochem. Biophys. Res. Commun. 417:961–965. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.