

Abstract
Congenital human cytomegalovirus (HCMV) infection is the most frequent infectious cause of birth defects, primarily neurological disorders. Neural progenitor/stem cells (NPCs) are the major cell type in the subventricular zone and are susceptible to HCMV infection. In culture, the differentiation status of NPCs may change with passage, which in turn may alter susceptibility to virus infection. Previously, only early-passage (i.e., prior to passage 9) NPCs were studied and shown to be permissive to HCMV infection. In this study, NPC cultures derived at different gestational ages were evaluated after short (passages 3 to 6) and extended (passages 11 to 20) in vitro passages for biological and virological parameters (i.e., cell morphology, expression of NPC markers and HCMV receptors, viral entry efficiency, viral gene expression, virus-induced cytopathic effect, and release of infectious progeny). These parameters were not significantly influenced by the gestational age of the source tissues. However, extended-passage cultures showed evidence of initiation of differentiation, increased viral entry, and more efficient production of infectious progeny. These results confirm that NPCs are fully permissive for HCMV infection and that extended-passage NPCs initiate differentiation and are more permissive for HCMV infection. Later-passage NPCs being differentiated and more permissive for HCMV infection suggest that HCMV infection in fetal brain may cause more neural cell loss and give rise to severe neurological disabilities with advancing brain development.
INTRODUCTION
Human cytomegalovirus (HCMV), a beta-subfamily member of the Herpesviridae, is a ubiquitous pathogen. Congenital HCMV (cCMV) infection is the most common cause of birth defects and affects more children than other conditions such as Down syndrome and spina bifida (1). The prevalence of cCMV infection is ca. 1% in the United States (2–4), 2% in India (5), and even higher (2 to 3%) in China (6, 7). A total of 5 to 10% of congenital infections are symptomatic at birth and can present in the newborn period with microencephaly, cerebral ventriculomegaly, cerebral palsy, and intracranial calcification (4, 8, 9). The remainder are asymptomatic at birth, but of these 10 to 15% will develop late-onset sequelae, including sensorineural hearing loss (SNHL), mental retardation, visual impairment, learning disabilities (4, 10–14), and autism (15, 16). Infected infants can resolve the infection in visceral organs, such as the liver and lung, but can be left with sequelae in the brain and auditory system (17–19). The brain and auditory system are therefore the main targets of long-lasting and permanent end-organ disease. There are about 400,000 babies with cCMV infection among the annual 20 million newborns in China and an estimated 40,000 new cases suffering from the sequelae caused by cCMV infection every year. Of the long-lasting sequelae in infants with cCMV, SNHL can be expected to occur most frequently (20, 21).
Severe brain damage is most commonly observed when cCMV infection occurs at early gestation. It is characterized by ventriculo-encephalitis with loss of the normal cortical architecture and integrity of the ventricular epithelium layer (17, 22, 23). Based on these observations, we postulated that NPCs originating from early gestational age tissue might be more permissive to HCMV infection and in turn cause more severe damage.
Susceptibility of cell/tissue for CMV infection appears to increase during embryonic development, which is thought to be associated with the pathogenesis of neural cell loss and severe brain damage. In a mouse model, early embryos are nonpermissive for murine CMV (MCMV) infection, as judged by the lack of viral gene expression after injection of blastocysts (24), zygotes (25), or whole early embryos (E7.5) (26). MCMV injection of the yolk sac at midgestation (E8.5) results in developmental anomalies, and viral gene expression is detected in the eyes, neuroectoderm, and visceral organs (24, 27). It has been suggested that the main target of cCMV infection at midgestation is the mesenchyme, which is important for organogenesis, including the eyes and brain (15, 16, 28, 29). At late gestation (E15.5), the brain is a preferential site of infection, and obvious brain damage (porencephaly) and productive infection are observed when MCMV is injected into the cerebral ventricles (30). In vitro, murine embryo-derived pluripotent stem cells (ePSCs) are nonpermissive to MCMV infection (31, 32); however, they gain susceptibility and become permissive during differentiation (32). These results support the hypothesis that the stage of gestation and the differentiation status of progenitor/stem cells contribute to the susceptibility to CMV infection and clinical outcome.
Most cell types in the brain are susceptible to HCMV infection, but the extent of permissivity varies among different cell types. Using a mouse model, Tsutsui's group showed that the entire developing brain of infected animals exhibit viral antigen expression, but the viral genes are differentially expressed (33–37). The most susceptible area is the subventricular zone (SVZ), where the major cell type is the neural progenitor/stem cell (NPC) (37–39). Consistent with this, human NPCs cultured in vitro (prior to passage 9 [P9]) are fully permissive for HCMV infection (40–46). Typical virus-induced cytopathic effects (CPE) are observed with the full range of viral protein expression and production of infectious progeny (40, 42, 43, 45–48). In addition, viral infection disrupts NPC differentiation and downregulates expression of NPC markers GFAP, SOX2, Nestin, and DCX (42, 49).
Ex vivo expression of NPC-specific markers is maintained for longer than 20 weeks (50, 51). However, the capacity of NPCs to proliferate decreases with increasing culture time/number of passages, suggesting that extended passage in vitro may induce differentiation. The effect of extended passage or gestational age of the source tissues used to establish NPC cultures on HCMV replication has not been previously investigated. In the present study, NPCs were obtained from postmortem neonatal brain tissues at different gestational stages. NPC cultures were evaluated for cell morphology and expression of NPC markers at various passages and for the ability to support HCMV replication. Gestational age of source tissues did not impact NPC morphology, marker gene expression, or HCMV replication. However, regardless of gestational age, a clear change in morphology occurred between passages 9 and 11. NPCs with late-passage (P11 to P20) morphology were more efficiently infected by HCMV and produced higher titers of infectious progeny than early (P3 to P9)-passage NPCs. In addition to morphological changes, NPCs that triggered differentiation at later passages were more susceptible to HCMV infection. These findings imply a mechanism for permanent sequelae caused by cCMV infection.
MATERIALS AND METHODS
Tissue.
Postmortem fetal brain tissues from different gestational age cases were obtained according to the approval notice from the Institutional Review Board (WIVH10201202) and the Guidelines for Biomedical Research Involving Human Subjects at Wuhan Institute of Virology, Chinese Academy of Sciences. The tissues were from three different gestations: early gestation (10 to 12 weeks [n = 3], termed NPC-E1, -E2, and -E3), midgestation (20 to 23 weeks [n = 5], termed NPC-M1 to -M5), and late gestation (28 to 30 weeks [n = 4], termed NPC-L1 to -L4). All of the investigated cases died of causes unrelated to HCMV infection, the brain tissues and isolated NPCs were negative for HCMV DNA by PCR as described previously (52), and representative results are shown in Fig. S1 in the supplemental material.
NPC isolation and culture.
NPCs were isolated following a protocol modified from one described previously (53). Autopsy was conducted in a sterile field. The brain was removed with cerebellum and brainstem intact. Hippocampus and bilateral ventricular and subventricular zone tissues were acquired separately, and blood residue was removed with Hanks' buffer supplemented with high-dose antibiotics (1,000 U of penicillin/ml, 1,000 μg of streptomycin/ml), followed by a wash in basal medium (DGA) comprised of a 1:1 mixture of Dulbecco modified Eagle medium (DMEM)–F-12 containing GlutaMAX (2 mM; Gibco-BRL), penicillin-streptomycin (100 U/ml and 100 μg/ml; Gibco-BRL), gentamicin (50 μg/ml; Gibco-BRL), and amphotericin B (Fungizone; 1.5 μg/ml; Gibco-BRL). Tissues were diced with scalpel blades and eye scissors, followed by digestion with PPD solution (papain [2.5 U/ml], dispase II [40 U/ml], and DNase I [1 U/ml] in DMEM–F-12) at 37°C for 20 min. Red blood cells and tissue remnants were removed by centrifugation at 1,500 × g for 15 min in 90% Percoll in phosphate-buffered saline (PBS; Sigma).
Cells in the top Percoll layer were recovered, washed twice with DGA, and then cultured using uncoated or fibronectin-coated (14 μg/ml; Prospect) dishes in growth medium (GM) consisting of DGA supplemented with 10% BIT9500 (5 mg of bovine serum albumin/ml, 5 μg of recombinant human insulin/ml, 100 μg of human transferrin/ml; Stem Cell Technologies), human basic fibroblast growth factor (20 ng/ml; Prospect), and human epithelial growth factor (EGF; 20 ng/ml; Prospect). The culture medium was half replaced with fresh GM every day for the first 7 days or until neurospheres or colonies formed. After 7 days, the culture medium was half replaced every other day until NPCs became confluent. At this stage, cultures were defined as passage 1 (P1). Culture medium that was removed was clarified by centrifugation and stored at −20°C to be used later as conditioned medium (CM). Confluent NPC monolayers and neurospheres were digested with Accutase (1× Accutase enzyme; Millipore) and then passed to uncoated or fibronectin-coated dishes at 1:3 for subculture, and the medium was half replaced every other day. Serial subcultures were maintained in uncoated or fibronectin-coated dishes and defined as P2, P3, etc. (Fig. 1), until cells were used for infection. Cultures were maintained at 37°C in a humidified atmosphere containing 5% CO2.
Fig 1.
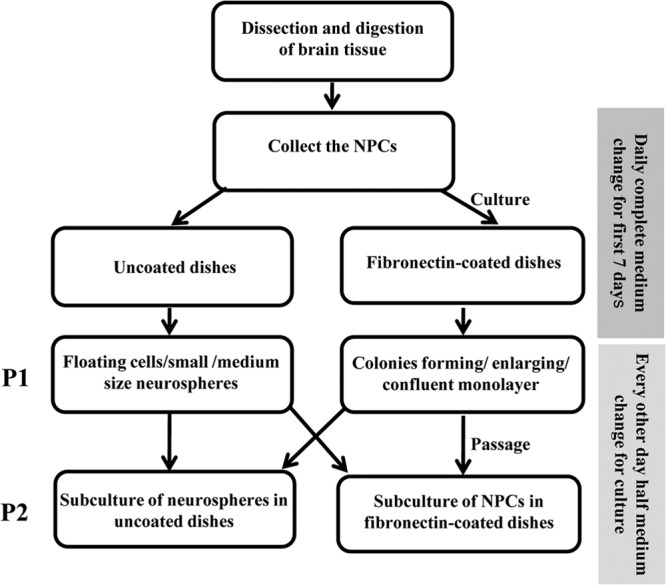
Chart flow for NPCs culture. See Materials and Methods for details.
Virus culture and infection.
HCMV Towne strain (ATCC-VR977) was propagated in human foreskin fibroblasts (HFFs). Viruses in infected cell culture supernatants were concentrated by ultracentrifugation (46,000 × g for 4 h). Pellets were resuspended in GM containing 1% dimethyl sulfoxide (DMSO), stored at −80°C, and titered by plaque assay on HFFs following 1:10 serial dilutions as described previously (54). Since the concentrated HCMV was resuspended in GM containing 1% DMSO, the mock infection was inoculated with equal volume of GM containing 1% DMSO without virus. Confluent NPC monolayers on fibronectin-coated dishes were lifted and dissociated with Accutase. NPCs were infected as described previously (42, 43). Briefly, cells were counted and reseeded onto poly-d-lysine-coated (50 μg/ml in double-distilled water; Millipore) dishes (3 × 106 cells per 100-mm dish) or onto poly-d-lysine-coated coverslips in uncoated 100-mm dishes (3 × 106 cells per dish) to allow cells to settle and spread for overnight. NPCs were infected at a multiplicity of infection (MOI) of 3 in the next morning (based on the number of cells seeded), followed by incubation for 3 h to allow for virus absorption. The inoculum was removed, and infected cells were rinsed with PBS for twice and then replaced with culture medium consisting of 50% CM and 50% fresh GM. Then, 200-μl samples of culture supernatants were removed for virus titer determination at 24, 72, 96, and 120 h postinfection (hpi). Supernatant samples were adjusted to 1% DMSO and stored at −80°C. Supernatant samples were titered as described above. Means and standard deviations from at least two independent experiments are shown.
Immunofluorescence assay (IFA).
NPCs were grown on poly-d-lysine-coated coverslips and were either not infected, mock infected, or virus infected at an MOI of 3. Cells on coverslips were fixed with 3% formaldehyde, permeabilized with 1% Triton X-100, blocked with 30% fetal bovine serum in basic blocking solution (1% bovine serum albumin and 0.01% Tween 20 in PBS), and incubated with mouse monoclonal antibodies (MAbs) to GFAP (IgG2b; Fitzgerald Industries International, Inc.), Nestin (IgG1; Chemicon), or HCMV IE1 clone p63-27 (IgG2a) or with goat polyclonal antibodies to SOX2 or DCX (Santa Cruz Biotechnology). After a washing step, the coverslips were incubated, as appropriate, with the secondary antibodies Alexa Fluor 488-conjugated goat anti-mouse IgG1 and IgG2b (Molecular Probes), tetramethyl rhodamine isothiocyanate (TRITC)-conjugated anti-mouse IgG2a (Jackson ImmunoResearch Laboratories), or fluorescein-isothiocyanate-conjugated donkey anti-goat IgG (Jackson ImmunoResearch, Inc.). Nuclei were counterstained with Hoechst dye and coverslips were mounted with antifade mounting solution containing paraphenylene diamine. Images were obtained using a Nikon Eclipse 80i fluorescence microscope equipped with a Nikon DS-Ri1 camera and processed using the NIS-Elements F3.0 software. A total of 500 to 1,000 cells from five randomly selected fields of each coverslip were counted and at least three independent experiments were performed for each assay. Positive rates were calculated as the number of positive cells divided by total cell numbers.
Western blotting (WB).
NPCs were grown in poly-d-lysine-coated dishes and were either not infected, mock infected, or virus infected at an MOI of 3. The cells were digested with 1 ml of Accutase solution for ca. 3 to 5 min, pelleted by centrifugation, resuspended in ice-cold PBS, and counted. Cells were again pelleted by centrifugation, snap-frozen in liquid nitrogen, and stored at −80°C. Cell lysates were prepared as described elsewhere (55). The equivalent of 105 cells was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes (0.45 μm; Amersham Biosciences). HCMV proteins were detected using MAbs to IE1 clone p63-27 (IgG2a), gB clone 27-156 (IgG2b), or major capsid protein clone 28-4 (IgG 2a), or IE1/IE2 (CH16), pp65, or UL44 (IgG1; Virusys Corp.). NPC markers were detected by using MAbs to GFAP (IgG2b; Fitzgerald Industries International, Inc.), Nestin (IgG1; Chemicon), or Vim (IgM; Sigma) or by using goat polyclonal antibodies to SOX2 or DCX (IgG; Santa Cruz Biotechnology) or rabbit polyclonal antibody to Mus 1 (IgG; Abcam). HCMV receptors were detected by using rabbit polyclonal antibodies to epidermal growth factor receptor (EGFR), platelet-derived growth factor receptor α (PDGFRα), and integrin (INTG) β3 (IgG; Anbo). Secondary antibodies used were horseradish peroxidase-conjugated sheep anti-mouse IgG, donkey anti-rabbit IgG (Amersham Bioscience), or donkey anti-goat IgG (Proteintech Group). Two sets of independent infection experiments were performed and representative results are shown.
Live cell imaging.
Phase-contrast images (×300) of mock- and virus-infected NPCs were taken using a Nikon Eclipse TS100 inverted microscope equipped with a Nikon CoolPix P6000 camera. Representative images were collected from at least two independent experiments.
RT-PCR.
Primers used for reverse transcription-PCR (RT-PCR) are shown in Table 1. RNA was extracted from 3 × 106 NPCs using TRIzol Reagent (TaKaRa). DNA was removed by treatment with 10 U of DNase (TaKaRa), followed by ethanol precipitation. RNA (500 ng) was reverse transcribed with an RT Master Mix perfect Real Time kit (PrimeScript; TaKaRa) according to the manufacturer's instructions. Quantitative PCR (qPCR) was performed on a real-time thermocycler (Bio-Rad; Connect) with the CFX 96 software and using iTaq Universal SYBY-Green Supermix (Bio-Rad). Each 20-μl qPCR contained 2 μl of RT reaction and 10 μl of iTaq Universal SYBY-Green Supermix, a 0.25 μM concentration of forward (F) primer, and a 0.25 μM concentration of reverse (R) primer. Amplification was performed by denaturation at 95°C for 5 min, followed by 40 two-step cycles of 95°C for 10 s and 59°C for 10 s. Each reaction was performed in triplicate and GAPDH (glyceraldehyde-3-phosphate dehydrogenase) was analyzed as a control for each experiment. Two independent experiments were performed, and results are presented as the means ± the standard deviations. The data were compared by using the Student t test. A P value of <0.05 was considered statistically significant.
Table 1.
Primers used for RT-PCR
| Target | Orientationa | Sequence (5′–3′) | Product size (bp) |
|---|---|---|---|
| EGFR | F | CAAGTGTAAGAAGTGCGAAGG | 188 |
| R | GAGGAGGAGTATGTGTGAAGG | ||
| PDGFR | F | ACAACCACACTCAGACAGAAG | 134 |
| R | GGCAGAATCATCATCCTCCAC | ||
| ITGB3 | F | AACCTGCGGATTGGCTTC | 195 |
| R | CACACTCTGCTTCTTCACTTC | ||
| GFAP | F | ATCAACTCACCGCCAACAG | 194 |
| R | CGACTCAATCTTCCTCTCCAG | ||
| Sox2 | F | GCCGAGTGGAAACTTTTGTCG | 154 |
| R | GCAGCGTGTACTTATCCTTCTT | ||
| Nestin | F | CTGGAAGGTCGGCAGCAGC | 144 |
| R | GGGGTCCTGAAAGCTGAGGG | ||
| DCX | F | CCGCAATGGGGACCGCTACTTCAAG | 207 |
| R | CAGACATAGCTTTCCCCTTCCTCCAG | ||
| GAPDH | F | GAAGGTGAAGGTCGGAGTC | 225 |
| R | GAAGATGGTGATGGGATTTC |
F, forward; R, reverse.
RESULTS
Isolation and characterization of NPCs.
NPCs were initially cultivated from primary tissues in uncoated dishes as neurospheres or in fibronectin-coated dishes as monolayers (Fig. 1). NPCs were present on day 4 of culture in coated dishes, by day 8 formed colonies, and by day 14 colonies connected to form confluent monolayers (Fig. 2A). These cultures were designated passage 1 (P1). The neurospheres, subcultures on uncoated dishes, could revert to growth as a monolayer upon transfer to fibronectin-coated dishes (Fig. 2B). Monolayer NPCs could also convert to neurospheres when transferred to uncoated dishes (data not shown).
Fig 2.

Isolation and identification of NPCs. (A) Primary culture NPCs in fibronectin-coated dish showing NPC migration from dissociated brain tissue to form NPC colonies. (B) Subculture of NPCs. Transfer of NPCs from primary cultures to an uncoated dish results in formation of medium-size neurospheres (left); transfer of neurospheres to a fibronectin-coated dish (right) results in colonies on day 4 that coalesce by day 8. (C) Detection of NPC markers by IFA. NPCs growing on poly-d-lysine-coated coverslips were fixed, permeabilized, and then stained with antibodies to GFAP, SOX2, Nestin, or DCX and counterstained with Hoechst. Scale bar, 5 μm. (D) Detection of NPC markers by WB. Mock-infected (m) or virus-infected (v) NPCs were harvested at the indicated time points and assayed by WB with antibodies to the indicated marker proteins. Actin serves as a loading control.
NPC cultures were first evaluated by IFA for expression of NPC markers GFAP, SOX2, Nestin, DCX, Vim, and Mus 1 (40, 42, 53, 56, 57). NPC cultures were 100% positive for GFAP, SOX2, and Nestin and 40% positive for DCX (Fig. 2C). GFAP, SOX2, Nestin, DCX, Vim, and Mus1 were also detected by WB (Fig. 2D). NPCs derived from tissues at different gestational ages were also assayed by IFA and WB for expression of this same group of markers, but no notable differences as a function of gestational age were observed (data not shown).
Effects of viral infection on NPC marker expression.
To determine whether viral infection altered expression of NPC markers of the newly isolated NPCs, cultured NPCs were infected with HCMV strain Towne at an MOI of 3, and NPC markers were assayed by WB. Compared to mock-infected NPCs, virus-infected NPCs exhibited downregulation of GFAP, SOX2, Nestin, and DCX at 96 hpi (Fig. 2D). Vim levels were largely unaffected by infection, whereas Mus1 levels were increased at 24 hpi in infected cells and remained elevated through 120 hpi (Fig. 2D).
Early-passage NPCs are fully permissive for HCMV infection.
HCMV genes are temporally expressed in a series of phases of immediate-early (IE), early (E), and late (L). The IE genes are transcribed and translated immediately after virus entry. The IE proteins activate expression of E genes, which encode proteins that play key roles in viral replication and DNA synthesis. L proteins are mostly structural components and are expressed after initiation of viral-DNA replication. To investigate HCMV infection in early-passage NPCs, P3 NPC cultures established from early-gestation tissue were plated onto poly-d-lysine-coated dishes and infected with HCMV at an MOI of 3. CPE, viral protein expression, and progeny virus yield were examined (Fig. 3).
Fig 3.

Infection of early-passage (P3) NPCs. (A) NPCs at P3 from an early-gestation embryo were mock treated or infected with HCMV (MOI = 3), and representative live cell images were taken at the indicated times. Magnification, ×300. (B) Infected (v) or mock-treated (m) P3 NPC cultures were harvested at the indicated times (indicated above each lane in hpi) and analyzed by WB for the expression of viral proteins. Actin serves as a loading control. (C) Virus titers in the culture supernatants at different times postinfection were determined by plaque assay.
No obvious CPE was observed at 8 hpi, but CPE was evident at 12 hpi and progressed as increasingly rounded-up and clumped cells from 24 to 120 hpi (Fig. 3A). Expression of representative IE, E, and L viral proteins was examined by WB throughout the course of infection (Fig. 3B). IE1 was detectable at 4 hpi and peaked at 72 hpi. UL44, an E protein, was weakly detectable at 8 and 24 hpi, but levels increased substantially from 48 to 120 hpi. Low levels of pp65, an E/L protein, were observed at 4 to 24 hpi. This probably represents input pp65 carried by the infecting virus particles, whereas increases at later times reflect de novo pp65 synthesis. Major capsid protein (MCP) and glycoprotein B (gB) are L proteins. Both were first observed at 72 hpi, with levels increasing subsequently. The expressions of these viral genes were similar to that following infection in human fibroblasts (see Fig. S2 in the supplemental material).
These results suggest that P3 NPC cultures support HCMV infection and progression through the full cascade of viral gene expression. To determine whether this results in production of infectious viral progeny, titers of infectious virus in culture supernatants were determined at 24, 72, 96, and 120 hpi. The replication cycle of HCMV is 48 to 72 h, which is consistent with late viral protein expression (MCP and gB), as shown in Fig. 3B. Since the carryon virus at 24 hpi was <10 PFU/ml, only the virus titers at 72, 96, and 120 hpi are presented to show the infectious viral progeny. The results indicated that NPCs were productively infected and yielded increasing amounts of infectious virus, with titers as high as 3.3 × 105 PFU/ml at 120 hpi (Fig. 3C).
Gestational age of origin tissue does not impact HCMV replication in early-passage NPCs.
As noted above, the gestational stage of tissues from which NPC cultures were established had no notable impact on NPC marker expression. To determine whether gestational stage has any effect on HCMV replication in NPCs, the viral infection studies described above were conducted using early-passage (P3 to P9) NPC cultures established from early (n = 3, NPC-E1, -E2, and -E3)-, mid (n = 5, NPC-M1 to -M5)-, and late (n = 4, NPC-L1 to -L4)-gestational-stage cases.
Early-passage NPCs had a similar morphological appearance irrespective of the gestational ages of origin tissue (examples of uninfected P3 cultures from early-, mid-, and late-gestation tissues are shown in Fig. 4). The progression of viral CPE was also similar (Fig. 4), and no notable differences in viral protein expression or infectious virus yield in culture supernatants were observed (data not shown).
Fig 4.

Comparison of cell morphology and HCMV-induced CPE between early-passage NPCs (P3) from tissues at different gestational ages. P3 NPCs from early (A), mid (B), or late (C) gestational embryos were mock treated or infected with HCMV (MOI = 3), and representative live cell images were taken at the indicated times. Magnification, ×300.
Effect of passage on HCMV infection in NPCs.
To investigate the effect of passage of NPC on HCMV replication, infections were compared between early-passage NPCs at P3 and P6 from either the same or different sources of NPCs from early, mid, and late gestations. Similar results were obtained from P3 and P6 cultures from the same or different donor tissue. For example, a comparison of the results from P3 and P6 NPC cultures from the same donor is shown in Fig. 5. No obvious difference in CPE development was observed (Fig. 5A). Slight elevations in IE2 levels (1.2- to 2.6-fold) were detected in P6 versus P3 cultures, but the expression of IE1, UL44, and gB levels after infection were unaffected by the limited passage number (Fig. 5B). Differences in virus titers (Fig. 5C) were also small (<3.2-fold) and not significant.
Fig 5.

Comparison of HCMV infection in early-passage NPCs derived from different mid gestation cases. NPC-M1 (P6) and NPC-M2 (P3) cultures, obtained from midgestation tissues, were mock treated or infected with HCMV (MOI = 3). (A) Representative live cell images were taken at the indicated times. Magnification, ×300. (B) Mock-treated (m) or infected (M1/M2) cultures were harvested at the indicated times (indicated above each lanes in hpi) and analyzed by WB for the expression of viral proteins. IE2 (86 kDa) migrates more slowly than IE1 (72 kDa). Actin serves as loading control. (C) Virus titers in the culture supernatants at different times postinfection were determined by plaque assay. The ratios of titers from infected M1 and M2 cultures at each time point were plotted (left panel). The numbers above each bar indicate the fold differences (right panel).
In contrast, it was consistently observed that regardless of gestational age, the morphologies of NPCs changed between early passages (up to P9) and extended passages (P11 to P20), with extended-passage NPCs having shorter cell processes and becoming thicker and more aggregated on poly-d-lysine-coated plates (Fig. 6A). However, NPCs were still 100% positive for GFAP, SOX2, and Nestin and 35% positive for DCX, values which are similar to those noted above. There were no significant changes in NPC marker positive rates, as assessed by IFA (data not shown).
Fig 6.

Comparison of HCMV infection in early-passage versus extended-passage NPCs. P3 and P11 NPCs cultures derived from the same tissue were mock treated or infected with HCMV (MOI = 3). (A) Representative live cell images were taken at the indicated times. Magnification, ×300. (B) Mock treated (m) or infected (P3/P11) cultures were harvested at the hpi indicated (above each lane) and analyzed by WB for expression of viral proteins. IE2 (86 kDa) migrates more slowly than IE1 (72 kDa). The signal strength for gB at 72 hpi (dashed box) was quantitated by densitometry. Actin served as a loading control. (C) Virus titers in the culture supernatants at different times postinfection were determined by plaque assay, and the ratios of titers from infected P11 and P3 cultures at each time point were plotted (left panel). The numbers above each bar indicate the fold differences (right panel). *, P < 0.05.
Differences in HCMV infection were further investigated between early-passage and extended-passage NPCs from the same sources. NPC cultures derived from two different donor tissues with early, mid, and late gestational ages were used to assay the HCMV infections, including CPE, viral gene expression, and release of infectious progeny. However, again, no significant differences were detected with respect to gestational age of origin tissues. Therefore, the results from one representative comparison with midgestational age between early- and extended-passage NPCs are shown (Fig. 6).
CPE appeared to develop faster in P11 versus P3 cultures (Fig. 6A), although this may be due partly to differences in cell morphology/distribution at the time of infection. There were minor differences in IE2 and no differences in UL44 expression between P3 and P11 cultures, but gB levels were significantly higher (2.4-fold) in P11 cultures at 72 hpi (Fig. 6B). Virus titers for P11 versus P3 cultures were 6.4-, 7.8-, and 5.5-fold higher at 72 hpi (P = 0.047), 96 hpi (P = 0.048), and 120 hpi (P = 0.016), respectively (Fig. 6C). The differences in virus yield were consistently observed in early- and later-passage NPCs derived from tissue of different gestational age (data not shown).
Extended passage of NPCs results in increased GFAP expression.
In myeloid progenitor lineages permissivity for HCMV replication is dependent on cell differentiation (18, 19, 31). Although changes in expression of NPC markers between early and extended passages were not evident by IFA, the fact that striking morphological changes were observed suggests that differentiation may be occurring, but they are not reflected by profound changes in marker positive rates. In particular, since all (i.e., 100%) NPCs were GFAP positive, it was impossible to evaluate the change in GFAP protein expression by IFA without fluorescence signal quantification. As an alternative approach for the detection of subtle changes in differentiation specific gene expression, we used the more sensitive and quantitative methods of both RT-PCR and WB (Fig. 7). GFAP mRNA was significantly elevated (3.17-fold, P < 0.05) in extended-passage versus early-passage NPCs, while SOX2 and Nestin levels were unchanged, and DCX was significantly decreased (3.7-fold, P < 0.05) (Fig. 7A). The GFAP protein level was dramatically increased in extended-passage versus early-passage NPCs, but SOX2, Nestin, and DCX levels were not obviously changed (Fig. 7B). The expression levels of Vim and Mus1were not changed (data not shown).
Fig 7.

NPC marker expression in early- and later-passage NPCs. Early-passage (P3) and later-passage (P11) NPCs from the same tissues were cultured and harvested, and RNA was extracted from uninfected samples. NPC markers expressions were determined. Representative samples of the GFAP, SOX2, Nestin, and DCX mRNA levels were determined by quantitative RT-PCR (A), and the protein levels were determined by WB (B). *, P < 0.05.
Entry efficiency in extended-passage NPCs was enhanced.
The efficiency with which HCMV enters cells in culture can be measured by the frequency of cells that stain positive for IE1 antigen at 24 to 48 hpi. To determine whether entry efficiency differed between the early- and extended-passage NPCs, cultures were infected at an MOI of 0.5 or 3 and then fixed and stained for IE1 antigen at 24 and 48 hpi. The results revealed that virus entry into early-passage NPCs was less efficient than into extended-passage NPCs (Fig. 8). At an MOI of 0.5, higher-passage cultures exhibited increased infection rates (from 20.8 to 29% at 24 hpi and from 31.9 to 41.6% at 48 hpi), but these differences are not statistically significant. At an MOI of 3, however, higher-passage cultures exhibited statistically significant (P ≤ 0.001) increases in infection rates from 43.9 to 65.5% at 24 hpi and from 48.7 to 83.4% at 48 hpi (Fig. 8A).
Fig 8.

Viral entry and HCMV receptor expression in early- and later-passage NPCs. (A) Early-passage (P3) and later-passage (P11) NPCs grown on coverslips were infected with HCMV at an MOI of 0.5 or 3 and then stained for IE1 antigen expression with Hoechst at 24 and 48 hpi. Representative images (left panel) are shown, and the magnification is ×300 (scale bar, 20 μm). IE1-positive cells were counted, and the percentages of total cells that were IE1 positive were calculated (right panel). *, P < 0.05. (B) Expression levels of HCMV receptors. Uninfected NPCs were analyzed for EGFR, PDGFRα (PDGFR), or INTG αVβ3 (ITGB3) mRNA levels by quantitative RT-PCR (left panel) or protein levels by WB (right panel). *, P < 0.05; **, P < 0.01.
To determine whether increased entry efficiency corresponded to increased HCMV receptor expression, the mRNA and protein levels of putative HCMV receptors EGFR, PDGFRα, and INTG αVβ3 (58–60) were determined by RT-PCR and WB (Fig. 8B). The mRNA levels of INTG β3 were significantly higher (P < 0.01), those of EGFR were significantly (P < 0.05) lower, and those of PDGFRα were unchanged in extended-passage versus early-passage NPCs. Correspondingly, both of the protein levels of PDGFRα and INTG αVβ3 were present at higher levels, and EGFR was slightly lower in extended-passage versus early-passage uninfected NPCs. The PDGFRα protein level was higher, whereas mRNA did not appear higher in extended passages. These protein levels, however, did not completely correspond to the RT-PCR results; the HCMV receptor levels were probably higher in extended-passage versus early-passage NPCs.
DISCUSSION
cCMV infection is the most common infectious causes of neurological developmental disorders in infants and children. The long-term outcome of cCMV infection is highly variable, ranging from severe infants exhibiting significant neurological disabilities to asymptomatic infants who achieve normal development milestones. It has been proposed that the outcome is more severe when congenital infection is acquired in early gestation, especially during the first trimester (4).
The SVZ of the fetal brain where NPCs are the predominant cell type is a susceptible site of infection and is affected frequently by cCMV infection (2, 22, 23, 31, 37, 38). Several studies have demonstrated that human NPCs are permissive for HCMV infection. However, these previous studies of HCMV infection in NPCs has only been performed in NPCs established from the same gestational age tissue at early passages (40, 42, 43, 45, 46, 49). Variation in HCMV infection in NPCs either from different gestational age brain tissue or at different passages, especially at extended passages after long-term culture, has not yet been characterized.
To determine whether the NPCs from early gestational age were more permissive for HCMV infection, stem cell characteristics (e.g., differentiation status) of NPCs, the corresponding biological parameters of NPCs, as well as virological parameters of HCMV infection in NPCs established from different cases at different gestational ages, and at early and later passages were thoroughly investigated. Our study is the first to consider these questions and results are informative. They demonstrate that NPCs are fully permissive for HCMV infection and HCMV infection among NPCs from early-, mid-, or late-gestational tissues are indistinguishable. Furthermore, our results have defined differences (summarized in Fig. 9) in HCMV infections between early-passage (P3 to 9) and later-passage (P11 to 20) NPCs.
Fig 9.
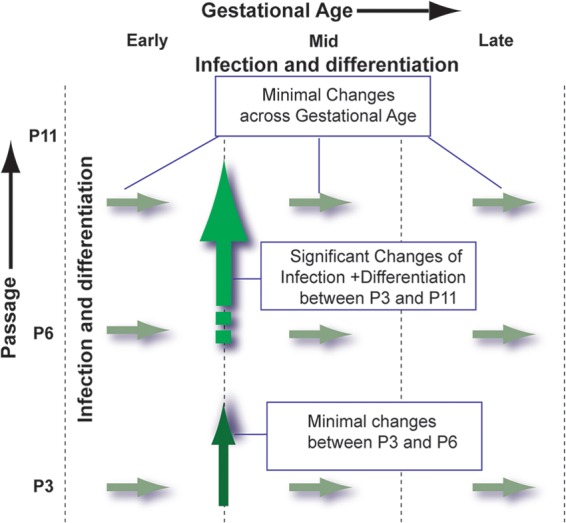
Schematic illustrating changes in HCMV infection and NPC characteristics with gestational ages and passages. Infections, cell morphology, and NPC marker expression were identical between NPCs at the same passage and were unaffected by gestational age of source tissues. Differences in morphology, viral receptor expression, entry efficiency, viral gene expression, and production of viral progeny were observed between early- and later-passage NPCs.
In this work, NPCs were isolated from fetal brain tissue and the homogeneity of NPC cultures was confirmed by staining for NPC-specific markers. All NPCs were positive for GFAP, SOX2, Nestin, Vim, and Mus1, and 40% of NPCs were DCX positive. These markers did not show notable differences in expression among NPCs that were derived from tissue from all gestational ages when assayed by either IFA or WB (data not shown). We noted that GFAP, SOX2, Nestin and DCX were downregulated by HCMV infection (Fig. 2), which are consistent with previously reported results (42). In addition, cell morphology was identical among early-passage NPCs that were derived from different gestational ages; all NPCs were fully permissive for HCMV infection, and no obvious differences were observed among NPCs originating from tissues of different gestational ages (Fig. 3, Fig. 4). Notably, there were no differences in NPC markers expression, including the differentiation status and cell morphology among early-passage cells, suggesting that the NPCs were indistinguishable irrespective of the gestational ages at which they were established (summarized in Fig. 9).
Cell morphologies were also indistinguishable among later-passage NPCs, but obviously different from the early-passage NPCs (Fig. 3 to 6). The features and changes of the cell morphology with extended-passage NPCs imply that NPC characteristics change during extended passages, a finding consistent with earlier results (51, 61, 62), and correlate with changes in NPC marker expression levels (Fig. 7) and HCMV infection changes (Fig. 6, Fig. 8).
Following HCMV infection of these NPCs, CPE was observed, a full range of viral proteins were expressed and high titers of infectious virus were produced, all of which confirmed that NPCs were fully permissive for HCMV infection (Fig. 3 to 6 and 8). These results are consistent with results described previously (40, 42, 43, 45, 63, 64). No notable differences in infection were observed among the same-passage NPCs originating from different gestational ages (Fig. 4). These results indicate that gestational age of the source tissue has no effect on HCMV infection, i.e., contrary to our initial expectation, that NPCs originating from early-gestational-age tissue are more permissive. However, at an earlier gestational age, NPCs are the key players in fetal brain development and give rise to neurons and glia. The fetal brain possesses a greater abundance of NPCs at earlier gestational ages, thus, it can be argued that the neurological outcome following HCMV infection will be worse in early gestation.
Differences in HCMV infection were observed within early-passage NPCs (P6 versus P3), but not statistically significant (Fig. 5). In contrast, differences in HCMV infection between early (P3 to P9)- and later (P11 to P20)-passage NPCs were significant (Fig. 6 and 8). Two aspects could contribute to the HCMV infection differences. One is virus, and the other is NPCs. The same HCMV virus stock was used for all of the experiments, so the difference caused from virus could be excluded. As for NPCs, cell morphology was notably changed, suggesting that NPCs had differentiated. The levels of GFAP mRNA and protein were elevated in NPCs between extended passage and early passage, whereas the levels of DCX mRNAs were reduced (although the DCX protein levels were unchanged; Fig. 7), indicating NPCs had possibly differentiated toward astrocytes. Taken together, these changes in cell biology and HCMV infection suggest that extended-passage NPCs are at least partially differentiated, which correlates with the idea that HCMV infection is highly dependent on cell differentiation. These results are consistent with the previous reports that have shown that the extent of permissiveness of ePSCs and myeloid progenitor cells for HCMV infection are highly dependent on cell differentiation status (31, 32, 35, 65, 66). In addition, our most recent study has shown that human ePSCs become susceptible to HCMV infection during differentiation into spinal cord and forebrain neural stem/progenitor cells and the extent of susceptibility is associated with the degree of differentiation (data not shown). Similar results have been observed previously (24–26, 32, 67, 68). The significant difference in virus infection correlates with obvious cell morphology change and difference in NPC marker expression. Concomitant with differentiation, virus entry efficiency was significantly increased upon infection with an (high) MOI of 3. Virus entry is a receptor-dependent event, and higher receptor levels explain the mechanism for higher entry efficiency in later-passage NPCs. Our studies of the expression of putative receptors for HCMV do not point to an obvious explanation for the increase in the entry of HCMV into later-passage NPCs and may suggest that these molecules are not representative of receptors key for entry into NPCs. All of the evidence supports that later-passage NPCs are differentiated and are more permissive for HCMV infection, thus it is possible that receptor expression is also linked to the state of differentiation of these cells. Later-passage NPCs being differentiated and more permissive for HCMV infection suggests that the infection in the fetal brain disrupts the NPCs and might result in further neural damage crossing the SVZ to area beyond SVZ, such as the cortex plate, during infection at later times. Immature astrocytes and neurons could be involved and cause greater loss of glia and neurons. Similar results have been reported by using mouse model (37, 66).
All NPCs from different cases at the early, mid, and late gestational ages are fully permissive for HCMV infection in vitro. Similar HCMV infections (in terms of CPE development, viral gene expression, and virus shedding) were found in similar-passage NPCs across the early, mid, and late gestational ages. These results suggest that naturally there are no significant differences in HCMV infection among all NPCs. In the early gestational period, there are greater abundance of HCMV targets (i.e., NPCs), presenting a greater potential for more severe damage since the brain is not well developed. In addition, later-passage NPCs, being differentiated and more permissive, provide a chance for further and deeper neural damage. Other factors, including maternal immunity, the maturity of the fetal brain-blood barrier, fetal immunity, and different virus strains, may be additional reasons for the different outcomes (69, 70). Identification of the major receptor among EGFR, PDGFR, and INTG β3 utilized by HCMV, as well as of the major cellular factor(s) that causes the significant difference in the initiation of HCMV infection, needs to be further studied.
In summary, several groups have proven that NPCs are fully permissive for CMV infection, whereas the susceptibility of NPCs from different gestational ages and at different passages to HCMV infection has not been studied. Our work demonstrated that NPCs isolated from all of the source tissues were similar according to several criteria and that there was no difference in the permissiveness to HCMV infection irrespective of the gestational ages of the donor tissue. Furthermore, it was found that initiation of differentiation occurred in later in vitro-passaged NPCs, at least partially, toward astrocytes and that these NPCs exhibited higher permissiveness to infection than did earlier-passage cells.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by the National Program on Key Basic Research Project (973 programs 2011CB504804 and 2012CB519003), the National Natural Science Foundation of China (81071350, 81271850, 31170155, and 31000090), Scientific Innovation Project of Chinese Academy of Sciences (XDB02050500 and XDA0104107), and a Seed Grant from the University of Idaho (YDP-764) to M.-H.L.
We appreciate the critical reading by Michael McVoy and Sean M. Amberg. We thank Rong-Ge Yang for kindly providing the qPCR device before our own equipment was set up.
Footnotes
Published ahead of print 31 July 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.01120-13.
REFERENCES
- 1.Ross DS, Dollard SC, Victor M, Sumartojo E, Cannon MJ. 2006. The epidemiology and prevention of congenital cytomegalovirus infection and disease: activities of the Centers for Disease Control and Prevention Workgroup. J. Womens Health (Larchmt.) 15:224–229. [DOI] [PubMed] [Google Scholar]
- 2.Boppana SB, Ross SA, Shimamura M, Palmer AL, Ahmed A, Michaels MG, Sanchez PJ, Bernstein DI, Tolan RW, Jr, Novak Z, Chowdhury N, Britt WJ, Fowler KB. 2011. Saliva polymerase-chain-reaction assay for cytomegalovirus screening in newborns. N. Engl. J. Med. 364:2111–2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Misono S, Sie KC, Weiss NS, Huang ML, Boeckh M, Norton SJ, Yueh B. 2011. Congenital cytomegalovirus infection in pediatric hearing loss. Arch. Otolaryngol. Head Neck Surg. 137:47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stagno S, Pass RF, Cloud G, Britt WJ, Henderson RE, Walton PD, Veren DA, Page F, Alford CA. 1986. Primary cytomegalovirus infection in pregnancy: incidence, transmission to fetus, and clinical outcome. JAMA 256:1904–1908. [PubMed] [Google Scholar]
- 5.Dar L, Pati SK, Patro AR, Deorari AK, Rai S, Kant S, Broor S, Fowler KB, Britt WJ, Boppana SB. 2008. Congenital cytomegalovirus infection in a highly seropositive semi-urban population in India. Pediatr. Infect. Dis. J. 27:841–843. [DOI] [PubMed] [Google Scholar]
- 6.Wen L, Wu S, Lu S. 1996. The epidemiological study on human cytomegalovirus infection of pregnant women and the maternal-fetal transmission in three Chinese metropolis. Zhonghua Fu Chan Ke Za Zhi 31:714–717. (In Chinese.) [PubMed] [Google Scholar]
- 7.Zhong XY, Ma TY. 1993. A clinical study of cytomegalovirus infections during pregnancy. J. Tongji Med. Univ. 13:60–64. [DOI] [PubMed] [Google Scholar]
- 8.Bale JF., Jr 1984. Human cytomegalovirus infection and disorders of the nervous system. Arch. Neurol. 41:310–320. [DOI] [PubMed] [Google Scholar]
- 9.Becroft DM. 1981. Prenatal cytomegalovirus infection: epidemiology, pathology, and pathogenesis. Perspect. Pediatr. Pathol. 6:203–241. [PubMed] [Google Scholar]
- 10.Conboy TJ, Pass RF, Stagno S, Britt WJ, Alford CA, McFarland CE, Boll TJ. 1986. Intellectual development in school-aged children with asymptomatic congenital cytomegalovirus infection. Pediatrics 77:801–806. [PubMed] [Google Scholar]
- 11.Grosse SD, Ross DS, Dollard SC. 2008. Congenital cytomegalovirus (CMV) infection as a cause of permanent bilateral hearing loss: a quantitative assessment. J. Clin. Virol. 41:57–62. [DOI] [PubMed] [Google Scholar]
- 12.Pass RF, Stagno S, Myers GJ, Alford CA. 1980. Outcome of symptomatic congenital cytomegalovirus infection: results of long-term longitudinal follow-up. Pediatrics 66:758–762. [PubMed] [Google Scholar]
- 13.Revello MG, Zavattoni M, Furione M, Fabbri E, Gerna G. 2006. Preconceptional primary human cytomegalovirus infection and risk of congenital infection. J. Infect. Dis. 193:783–787. [DOI] [PubMed] [Google Scholar]
- 14.Schachtele SJ, Mutnal MB, Schleiss MR, Lokensgard JR. 2011. Cytomegalovirus-induced sensorineural hearing loss with persistent cochlear inflammation in neonatal mice. J. Neurovirol. 17:201–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Engman ML, Lewensohn-Fuchs I, Mosskin M, Malm G. Congenital cytomegalovirus infection: the impact of cerebral cortical malformations. Acta Paediatr. 99:1344–1349. [DOI] [PubMed] [Google Scholar]
- 16.Sweeten TL, Posey DJ, McDougle CJ. 2004. Brief report: autistic disorder in three children with cytomegalovirus infection. J. Autism Dev. Disord. 34:583–586. [DOI] [PubMed] [Google Scholar]
- 17.Boppana SB, Fowler KB, Vaid Y, Hedlund G, Stagno S, Britt WJ, Pass RF. 1997. Neuroradiographic findings in the newborn period and long-term outcome in children with symptomatic congenital cytomegalovirus infection. Pediatrics 99:409–414. [DOI] [PubMed] [Google Scholar]
- 18.Boppana SB, Pass RF, Britt WJ, Stagno S, Alford CA. 1992. Symptomatic congenital cytomegalovirus infection: neonatal morbidity and mortality. Pediatr. Infect. Dis. J. 11:93–99. [DOI] [PubMed] [Google Scholar]
- 19.Dahle AJ, Fowler KB, Wright JD, Boppana SB, Britt WJ, Pass RF. 2000. Longitudinal investigation of hearing disorders in children with congenital cytomegalovirus. J. Am. Acad. Audiol. 11:283–290. [PubMed] [Google Scholar]
- 20.Karltorp E, Hellstrom S, Lewensohn-Fuchs I, Carlsson-Hansen E, Carlsson PI, Engman ML. Congenital cytomegalovirus infection: a common cause of hearing loss of unknown aetiology. Acta Paediatr. 101:e357–e362. [DOI] [PubMed] [Google Scholar]
- 21.McMullan BJ, Palasanthiran P, Jones CA, Hall BM, Robertson PW, Howard J, Rawlinson WD. 2011. Congenital cytomegalovirus. Time to diagnosis, management, and clinical sequelae in Australia: opportunities for earlier identification. Med. J. Aust. 194:625–629. [DOI] [PubMed] [Google Scholar]
- 22.Barkovich AJ, Lindan CE. 1994. Congenital cytomegalovirus infection of the brain: imaging analysis and embryologic considerations. Am. J. Neuroradiol. 15:703–715. [PMC free article] [PubMed] [Google Scholar]
- 23.Perlman JM, Argyle C. 1992. Lethal cytomegalovirus infection in preterm infants: clinical, radiological, and neuropathological findings. Ann. Neurol. 31:64–68. [DOI] [PubMed] [Google Scholar]
- 24.Kashiwai A, Kawamura N, Kadota C, Tsutsui Y. 1992. Susceptibility of mouse embryo to murine cytomegalovirus infection in early and mid-gestation stages. Arch. Virol. 127:37–48. [DOI] [PubMed] [Google Scholar]
- 25.Tebourbi L, Testart J, Cerutti I, Moussu JP, Loeuillet A, Courtot AM. 2002. Failure to infect embryos after virus injection in mouse zygotes. Hum. Reprod. 17:760–764. 10.1093/humrep/17.3.760. [DOI] [PubMed] [Google Scholar]
- 26.Tsutsui Y, Naruse I. 1987. Murine cytomegalovirus infection of cultured mouse embryos. Am. J. Pathol. 127:262–270. [PMC free article] [PubMed] [Google Scholar]
- 27.Tsutsui Y, Kashiwai A, Kawamura N, Kadota C. 1993. Microphthalmia and cerebral atrophy induced in mouse embryos by infection with murine cytomegalovirus in midgestation. Am. J. Pathol. 143:804–813. [PMC free article] [PubMed] [Google Scholar]
- 28.Melnick M, Mocarski ES, Abichaker G, Huang J, Jaskoll T. 2006. Cytomegalovirus-induced embryopathology: mouse submandibular salivary gland epithelial-mesenchymal ontogeny as a model. BMC Dev. Biol. 6:42. 10.1186/1471-213X-6-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shimamura M, Murphy-Ullrich JE, Britt WJ. Human cytomegalovirus induces TGF-β1 activation in renal tubular epithelial cells after epithelial-to-mesenchymal transition. PLoS Pathog. 6:e1001170. 10.1371/journal.ppat.1001170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tsutsui Y, Kashiwai A, Kawamura N, Kadota C, Nagahama M. 1991. Postnatal porencephaly induced in mouse by murine cytomegalovirus. Acta Neuropathol. 82:435–441. [DOI] [PubMed] [Google Scholar]
- 31.Kawasaki H, Kosugi I, Arai Y, Iwashita T, Tsutsui Y. 2011. Mouse embryonic stem cells inhibit murine cytomegalovirus infection through a multi-step process. PLoS One 6:e17492. 10.1371/journal.pone.0017492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matsukage S, Kosugi I, Kawasaski H, Miura K, Kitani H, Tsutsui Y. 2006. Mouse embryonic stem cells are not susceptible to cytomegalovirus but acquire susceptibility during differentiation. Birth Defects Res. A Clin. Mol. Teratol. 76:115–125. [DOI] [PubMed] [Google Scholar]
- 33.Kosugi I, Kawasaki H, Arai Y, Tsutsui Y. 2002. Innate immune responses to cytomegalovirus infection in the developing mouse brain and their evasion by virus-infected neurons. Am. J. Pathol. 161:919–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li RY, Baba S, Kosugi I, Arai Y, Kawasaki H, Shinmura Y, Sakakibara SI, Okano H, Tsutsui Y. 2001. Activation of murine cytomegalovirus immediate-early promoter in cerebral ventricular zone and glial progenitor cells in transgenic mice. Glia 35:41–52. [DOI] [PubMed] [Google Scholar]
- 35.Shinmura Y, Aiba-Masago S, Kosugi I, Li RY, Baba S, Tsutsui Y. 1997. Differential expression of the immediate-early and early antigens in neuronal and glial cells of developing mouse brains infected with murine cytomegalovirus. Am. J. Pathol. 151:1331–1340. [PMC free article] [PubMed] [Google Scholar]
- 36.Tsutsui Y, Kawasaki H, Kosugi I. 2002. Reactivation of latent cytomegalovirus infection in mouse brain cells detected after transfer to brain slice cultures. J. Virol. 76:7247–7254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tsutsui Y, Kosugi I, Kawasaki H. 2005. Neuropathogenesis in cytomegalovirus infection: indication of the mechanisms using mouse models. Rev. Med. Virol. 15:327–345. [DOI] [PubMed] [Google Scholar]
- 38.Chang WL, Tarantal AF, Zhou SS, Borowsky AD, Barry PA. 2002. A recombinant rhesus cytomegalovirus expressing enhanced green fluorescent protein retains the wild-type phenotype and pathogenicity in fetal macaques. J. Virol. 76:9493–9504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kawasaki H, Kosugi I, Arai Y, Tsutsui Y. 2002. The amount of immature glial cells in organotypic brain slices determines the susceptibility to murine cytomegalovirus infection. Lab. Invest. 82:1347–1358. [DOI] [PubMed] [Google Scholar]
- 40.Cheeran MC, Hu S, Ni HT, Sheng W, Palmquist JM, Peterson PK, Lokensgard JR. 2005. Neural precursor cell susceptibility to human cytomegalovirus diverges along glial or neuronal differentiation pathways. J. Neurosci. Res. 82:839–850. [DOI] [PubMed] [Google Scholar]
- 41.Lokensgard JR, Cheeran MC, Gekker G, Hu S, Chao CC, Peterson PK. 1999. Human cytomegalovirus replication and modulation of apoptosis in astrocytes. J. Hum. Virol. 2:91–101. [PubMed] [Google Scholar]
- 42.Luo MH, Hannemann H, Kulkarni AS, Schwartz PH, O'Dowd JM, Fortunato EA. 2010. Human cytomegalovirus infection causes premature and abnormal differentiation of human neural progenitor cells. J. Virol. 84:3528–3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Luo MH, Schwartz PH, Fortunato EA. 2008. Neonatal neural progenitor cells and their neuronal and glial cell derivatives are fully permissive for human cytomegalovirus infection. J. Virol. 82:9994–10007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McCarthy M, Auger D, Whittemore SR. 2000. Human cytomegalovirus causes productive infection and neuronal injury in differentiating fetal human central nervous system neuroepithelial precursor cells. J. Hum. Virol. 3:215–228. [PubMed] [Google Scholar]
- 45.Odeberg J, Wolmer N, Falci S, Westgren M, Seiger A, Soderberg-Naucler C. 2006. Human cytomegalovirus inhibits neuronal differentiation and induces apoptosis in human neural precursor cells. J. Virol. 80:8929–8939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Odeberg J, Wolmer N, Falci S, Westgren M, Sundtrom E, Seiger A, Soderberg-Naucler C. 2007. Late human cytomegalovirus (HCMV) proteins inhibit differentiation of human neural precursor cells into astrocytes. J. Neurosci. Res. 85:583–593. [DOI] [PubMed] [Google Scholar]
- 47.Lecointe D, Hery C, Janabi N, Dussaix E, Tardieu M. 1999. Differences in kinetics of human cytomegalovirus cell-free viral release after in vitro infection of human microglial cells, astrocytes and monocyte-derived macrophages. J. Neurovirol. 5:308–313. [DOI] [PubMed] [Google Scholar]
- 48.Luo MH, Fortunato EA. 2007. Long-term infection and shedding of human cytomegalovirus in T98G glioblastoma cells. J. Virol. 81:10424–10436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McCarthy M, Resnick L, Taub F, Stewart RV, Dix RD. 1991. Infection of human neural cell aggregate cultures with a clinical isolate of cytomegalovirus. J. Neuropathol. Exp. Neurol. 50:441–450. [DOI] [PubMed] [Google Scholar]
- 50.Sun T, Wang XJ, Xie SS, Zhang DL, Wang XP, Li BQ, Ma W, Xin H. A comparison of proliferative capacity and passaging potential between neural stem and progenitor cells in adherent and neurosphere cultures. Int. J. Dev. Neurosci. 29:723–731. [DOI] [PubMed] [Google Scholar]
- 51.Zietlow R, Precious SV, Kelly CM, Dunnett SB, Rosser AE. Long-term expansion of human foetal neural progenitors leads to reduced graft viability in the neonatal rat brain. Exp. Neurol. 235:563–573. [DOI] [PubMed] [Google Scholar]
- 52.Cobbs CS, Harkins L, Samanta M, Gillespie GY, Bharara S, King PH, Nabors LB, Cobbs CG, Britt WJ. 2002. Human cytomegalovirus infection and expression in human malignant glioma. Cancer Res. 62:3347–3350. [PubMed] [Google Scholar]
- 53.Schwartz PH, Bryant PJ, Fuja TJ, Su H, O'Dowd DK, Klassen H. 2003. Isolation and characterization of neural progenitor cells from post-mortem human cortex. J. Neurosci. Res. 74:838–851. [DOI] [PubMed] [Google Scholar]
- 54.Tamashiro JC, Hock LJ, Spector DH. 1982. Construction of a cloned library of the EcoRI fragments from the human cytomegalovirus genome (strain AD169). J. Virol. 42:547–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Luo MH, Rosenke K, Czornak K, Fortunato EA. 2007. Human cytomegalovirus disrupts both ataxia telangiectasia mutated protein (ATM)- and ATM-Rad3-related kinase-mediated DNA damage responses during lytic infection. J. Virol. 81:1934–1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kosugi I, Shinmura Y, Kawasaki H, Arai Y, Li RY, Baba S, Tsutsui Y. 2000. Cytomegalovirus infection of the central nervous system stem cells from mouse embryo: a model for developmental brain disorders induced by cytomegalovirus. Lab. Invest. 80:1373–1383. [DOI] [PubMed] [Google Scholar]
- 57.Suh H, Consiglio A, Ray J, Sawai T, D'Amour KA, Gage FH. 2007. In vivo fate analysis reveals the multipotent and self-renewal capacities of Sox2+ neural stem cells in the adult hippocampus. Cell Stem Cell 1:515–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Soroceanu L, Akhavan A, Cobbs CS. 2008. Platelet-derived growth factor-alpha receptor activation is required for human cytomegalovirus infection. Nature 455:391–395. [DOI] [PubMed] [Google Scholar]
- 59.Wang X, Huang DY, Huong SM, Huang ES. 2005. Integrin alphavbeta3 is a coreceptor for human cytomegalovirus. Nat. Med. 11:515–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang X, Huong SM, Chiu ML, Raab-Traub N, Huang ES. 2003. Epidermal growth factor receptor is a cellular receptor for human cytomegalovirus. Nature 424:456–461. [DOI] [PubMed] [Google Scholar]
- 61.Koehler KR, Tropel P, Theile JW, Kondo T, Cummins TR, Viville S, Hashino E. Extended passaging increases the efficiency of neural differentiation from induced pluripotent stem cells. BMC Neurosci. 12:82. 10.1186/1471-2202-12-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zaim M, Karaman S, Cetin G, Isik S. Donor age and long-term culture affect differentiation and proliferation of human bone marrow mesenchymal stem cells. Ann. Hematol. 91:1175–1186. [DOI] [PubMed] [Google Scholar]
- 63.Casavant NC, Luo MH, Rosenke K, Winegardner T, Zurawska A, Fortunato EA. 2006. Potential role for p53 in the permissive life cycle of human cytomegalovirus. J. Virol. 80:8390–8401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tsutsui Y, Nogami-Satake T. 1990. Differential expression of the major immediate-early gene of human cytomegalovirus. J. Gen. Virol. 71(Pt 1):115–124. [DOI] [PubMed] [Google Scholar]
- 65.Soderberg-Naucler C, Fish KN, Nelson JA. 1997. Interferon-gamma and tumor necrosis factor-alpha specifically induce formation of cytomegalovirus-permissive monocyte-derived macrophages that are refractory to the antiviral activity of these cytokines. J. Clin. Invest. 100:3154–3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tsutsui Y. 2009. Effects of cytomegalovirus infection on embryogenesis and brain development. Congenit. Anom. (Kyoto) 49:47–55. [DOI] [PubMed] [Google Scholar]
- 67.Poland SD, Bambrick LL, Dekaban GA, Rice GP. 1994. The extent of human cytomegalovirus replication in primary neurons is dependent on host cell differentiation. J. Infect. Dis. 170:1267–1271. [DOI] [PubMed] [Google Scholar]
- 68.Poland SD, Costello P, Dekaban GA, Rice GP. 1990. Cytomegalovirus in the brain: in vitro infection of human brain-derived cells. J. Infect. Dis. 162:1252–1262. [DOI] [PubMed] [Google Scholar]
- 69.Boppana SB, Miller J, Britt WJ. 1996. Transplacentally acquired antiviral antibodies and outcome in congenital human cytomegalovirus infection. Viral Immunol. 9:211–218. [DOI] [PubMed] [Google Scholar]
- 70.Schleiss MR. 2008. Cytomegalovirus vaccine development. Curr. Top. Microbiol. Immunol. 325:361–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.