

Abstract
Nonstructural protein 4 (NSP4), encoded by rotavirus, exhibits various properties linked to viral pathogenesis, including enterotoxic activity. A recent study (O. V. Kavanagh et al., Vaccine 28:3106-3111, 2010) indicated that NSP4 also has adjuvant properties, suggesting a possible role in the innate immune response to rotavirus infection. We report here that NSP4 purified from the medium of rotavirus-infected Caco-2 cells triggers the secretion of proinflammatory cytokines from macrophage-like THP-1 cells and nitric oxide from murine RAW 264.7 cells. Secretion is accompanied by the stimulation of p38 and JNK mitogen-activated protein kinases (MAPKs) and nuclear factor NF-κB. NSP4 triggered the secretion of cytokines from murine macrophages derived from wild-type but not MyD88−/− or Toll-like receptor 2 (TLR2−/−) mice and induced secretion of interleukin-8 (IL-8) from human embryonic kidney cells transfected with TLR2 but not TLR4. Our studies identify NSP4 as a pathogen-associated molecular pattern (PAMP) encoded by rotavirus and provide a mechanism for the production of proinflammatory cytokines associated with the clinical symptoms of infection in humans and animals.
INTRODUCTION
Rotavirus is a nonenveloped, double-stranded RNA (dsRNA) virus responsible for acute gastroenteritis in the young of several animal species, including humans (1). The virus infects mature enterocytes present on intestinal villi. Transmission occurs via the fecal-oral route, although recent studies have confirmed extraintestinal spread and viremia in infected patients (2). Within the infected cell, six structural proteins (VP1 to VP4, VP6, and VP7) assemble into three concentric capsid layers that encase the segmented dsRNA genome (3, 4). Five nonstructural proteins (NSP1 to NSP5) are also synthesized in infected cells, and these play various roles in the replication of the viral genome, translation of viral mRNA, and virion morphogenesis (5).
NSP4 is a transmembrane glycoprotein with an essential role in the assembly of intermediate double-layered particles and their maturation to infectious virions within the infected cell. NSP4 is also actively secreted from virus-infected polarized epithelial cells in its full-length form after specific posttranslational modification (6). The range of cellular and physiological effects attributed to exogenous NSP4 includes the ability to act as a virus-encoded enterotoxin capable of inducing dose- and age-dependent diarrhea in mice and rats (7), potentiation of Cl− and water secretion from noninfected epithelia (8), disruption of transepithelial resistance (9), and direct inhibition of the Na+-d-glucose symporter transporter (10). Recombinant forms of NSP4 representing either the full-length protein or regions of the cytoplasmic domain have been demonstrated to cause the phospholipase C (PLC)-dependent elevation of intracellular Ca2+ ions (11) and secretion of 5-hydroxytryptamine (5-HT) from enteroendocrine cells (12).
Collectively, these studies indicate that NSP4 secreted from rotavirus-infected cells can interact with a range of cell types in vivo, initiate signaling pathways that affect the local physiology of the small intestine, and contribute to the clinical symptoms of rotavirus infection. Nevertheless, there remains a paucity of experimental data indicating how these apparently pleiotropic effects are regulated. For example, the nature and identity of plasma membrane receptors that activate intracellular signaling pathways in response to exogenous NSP4 are not well defined. Seo et al. reported that recombinant NSP4 can bind to the metal ion-dependent adhesion site (MIDAS) motif present on integrins α1β1 and α2β1 and activate intracellular signaling pathways that regulate cell spreading (13). Mutations in NSP4 that reduce integrin binding also correlated with an inability to cause diarrhea in mice. However, given the wide range of effects reported for NSP4, it is feasible that non-integrin receptors exist.
The majority of studies into the cellular effects of NSP4 have focused on primary enterocytes and cell lines of intestinal epithelial origin. We observed recently that NSP4, purified from the medium of rotavirus-infected cells, bound to a wide range of cells of distinct lineage, including immune cells (14). Furthermore, a recent paper has described the adjuvant-like property of NSP4 when coadministered with model antigens in mice, further suggesting a specific effect on immune cells (15). We have now extended the analysis of potential immunomodulatory functions possessed by NSP4 and report here the novel finding that NSP4 elicits intracellular signaling in macrophages, resulting in the secretion of inflammatory cytokines. Significantly, our study reveals that these effects are mediated through Toll-like receptor 2 (TLR2) and identify NSP4 as a novel agonist of a microbial pattern recognition receptor and a potential trigger of innate immunity to rotavirus infection in vivo.
MATERIALS AND METHODS
Cell culture.
MA104 and Caco-2 cells were cultured in Dulbecco's modified Eagle medium (DMEM) containing 10% fetal bovine serum (FBS), 1× nonessential amino acids, and 10 μg/ml ciprofloxacin. Cells were tested to be free of mycoplasma contamination using an e-Myco mycoplasma PCR detection kit (iNtRON Biotechinology). Human THP-1 cells and mouse RAW 264.7 cells were maintained in RPMI containing 10% FBS, 2 mM l-glutamine, penicillin-streptomycin (100 μg/ml), and 0.05 mM 2-mercaptomethanol. To differentiate monocytic THP-1 cells into macrophage-like cells, THP-1 cells were stimulated with 100 nM phorbol myristic acid (PMA) (Sigma) and seeded at a density of 2 × 105 per well in 96-well plates. Following a 72-h incubation at 37°C, medium was removed and the cells were washed twice with phosphate-buffered saline (PBS) and cultured in PMA-free medium for 5 days prior to use. Immortalized murine macrophages were a kind gift from Eicke Latz (University of Bonn, Germany) and were generated as previously described (16). Immortalized macrophages and RAW-ELAM cells were grown in DMEM supplemented with 10% FBS and l-glutamine. HEK293 cells transfected with plasmids encoding human TLR4, MD2, and CD14 were obtained from Eicke Latz (University of Bonn, Germany) and grown in DMEM plus 10% FBS without antibiotic selection for up to four passages prior to analysis of TLR activity.
Virus propagation and preparation of purified rotavirus TLPs.
Virus stocks for infection of Caco-2 cells were prepared in MA104 cells cultured in glass roller bottles (Schott Duran) in Medium 199 plus 10% FBS. Cells were infected with 0.5 PFU/cell of virus inoculum in 40 ml per bottle in serum-deficient Medium 199 containing 10% tryptose phosphate and 25 μg/ml trypsin-250. After overnight incubation, cells were pelleted by centrifugation and stored at −20°C. Pellets were thawed and virus liberated from cells by sonication on ice. Cell debris was removed by centrifugation at 200 × g for 10 min, and the supernatant was collected. Virus titers were measured by fluorescent focus assay in MA104 cells. Triple-layer particles (TLPs) were purified from cell pellets derived from 10 roller bottles using cesium chloride isopycnic centrifugation as described previously (17).
Purification of NSP4.
Bovine rotavirus (UK strain) was obtained from the late Ian Holmes, University of Melbourne, and propagated in MA104 cells as described previously (6). cDNA encoding the product of gene 10 (NSP4) was amplified following reverse transcription of viral mRNA and PCR amplification using the primers GGCTTTTAAAAGTTCTGTTCCGAGA (forward) and ATTAAGACCGTTCCTTCCATTAACG (reverse) and sequenced to confirm a 100% match to UK bovine rotavirus (GenBank accession number K03384.1). NSP4 was purified from the medium of 10-day postconfluent Caco-2 cells infected with bovine rotavirus (multiplicity of infection [MOI], 10) as previously described (14). Medium was removed after 36 h and centrifuged at 100,000 × g for 2 h to remove virions and cell debris. The centrifuged medium was concentrated ∼25-fold as concentrated medium (C.M) in an Amicon flow cell (molecular mass cutoff, 10 kDa). NSP4 was purified from the C.M by successive concanavalin A affinity and cation exchange (Mono S) chromatography in the absence of detergent and was determined to be >95% pure by SDS-PAGE and silver staining.
Depletion of NSP4 from infected cell medium.
Mouse monoclonal anti-NSP4 antibody (clone B4; a kind gift from Harry Greenberg), purified from hybridoma cell culture medium, was packed as a 1-ml column using an AminoLink plus immobilization kit (20394; Thermo Scientific). Concentrated medium from rotavirus-infected Caco-2 cells (1 ml) was applied to the NSP4 affinity column. Column flowthrough was collected as NSP4-depleted medium. The efficacy of NSP4 depletion was confirmed by Western blotting using anti-NSP4 antibody and silver staining.
Measurement of cytokines.
Either PMA-treated THP-1 cells or HEK293 cells were cultured in 96-well plates and stimulated with NSP4 or the agonist lipopolysaccharide (LPS) (from Salmonella enterica; Sigma) or Pam3Cys-Ser-(Lys)4 (Pam3CSK4; EMC Microcollections). Cell-free supernatants were collected and kept at −80°C until analysis. Tumor necrosis factor alpha (TNF-α), interleukin-6 (IL-6), and IL-8 (HEK293) were measured using the BD cytometric bead array flex set arrays (CBA flex; BD Bioscience). Data were acquired using a FACSAria II flow cytometer and analyzed using FCAP Array v1.0.1 software. Cytokines secreted from murine macrophages were measured by enzyme-linked immunoassay (ELISA). Macrophages were seeded at 2 × 104 cells per well in 96-well format 24 h prior to stimulation with NSP4 or TLR ligand Pam3CSK4 (20 ng/ml), poly(I·C) (20 μg/ml), or LPS (20 ng/ml) in triplicate. Culture supernatants were collected 24 h following stimulation and production of TNF-α and IL-6, then they were measured with DuoSet mouse TNF-α and IL-6 kits (R&D Systems) according to the manufacturer's instructions.
ELAM reporter assay.
RAW-ELAM cells were seeded at 4 × 104 cells per well 24 h prior to stimulation with the indicated ligands for 6 h. Cells were lysed with passive lysis buffer (Promega), and lysates were assayed for NF-κB-driven luciferase expression using luciferase assay reagent (Promega) and analyzed using FLUOstar Optima (BMG Technologies).
Immunoblot analysis.
Cells were incubated for 15 min at 4°C in lysis buffer (25 mM NaF, 1 mM sodium orthovanadate [Na3VO4], 50 mM Tris, pH 7.4, 100 mM KCl, 1 mM EDTA, 1 mM dithiothreitol [DTT], 10% glycerol, and 1% Triton X-100) and supplemented with a protease inhibitor cocktail (Complete EDTA-free; Roche, Indianapolis, IN). The lysates were centrifuged for 5 min at 13,000 rpm, and the supernatants were collected. Samples were diluted in Laemmli sample buffer, denatured by boiling for 5 min, subjected to 10% SDS-PAGE, and transferred to Immobilon NC membranes (Millipore, Billerica, MA). After treatment with blocking solution (5% bovine serum albumin [BSA], 0.05% Tween 20 in phosphate-buffered saline), the membranes were incubated with primary antibodies diluted in blocking solution overnight at 4°C. The membranes were rinsed with PBS-Tween 20, and bound antibodies were developed by incubation with a peroxidase-labeled secondary antibody and the chemiluminescence light analysis system (Invitrogen). Primary antibodies (Cell Signaling Technology [Danvers, MA]) used were phosphor-p38(Thr180/Tyr182) (9211), total p38 (9212), phosphor-SAPK/JNK(Thr183/Tyr185) (9251), phosphor-IKKα(Ser180)/β(ser181) (2681), and IKKβ (2684). Anti-α-tubulin (Sigma) was used as a loading control for JNK.
Confocal microscopy.
RAW 264.7 cells seeded on glass coverslips were treated with NSP4 or medium only at 37°C for 30 min and then washed extensively in PBS. Nuclei were stained with Hoechst 33342 immediately prior to fixation of the cells with 4% paraformaldehyde and permeabilization in acetone-methanol (1:1, vol/vol) at −20°C for 15 min. The permeabilized cells were blocked in 10% bovine serum albumin (BSA) and incubated with a rabbit polyclonal antibody against p65 (622601; BioLegend). Cells were washed with PBS and incubated for 30 min with Alexa Fluor 594-conjugated anti-rabbit immunoglobulin G (10 μg/ml; Invitrogen, Carlsbad, CA). Coverslips washed in PBS were mounted on microscope slides with ProLong Gold antifade reagent (Invitrogen). Images were obtained with an Andor Revolution confocal microscope at ×1,000 magnification.
RESULTS
NSP4 induces secretion of inflammatory cytokines and nitric oxide from human and murine macrophage-like cell lines.
Previous studies of the pathophysiological role of NSP4 in rotavirus infection have focused on its ability to induce signaling in cell lines of intestinal epithelial origin. Rotavirus infection is known to increase intestinal permeability, and infection of extraintestinal tissues has been reported (2). Thus, it is feasible that NSP4 could activate signaling pathways in cells other than intestinal epithelia. Consistent with this notion, a recent paper has described adjuvant properties of NSP4 in mice (15). Based on these observations, we hypothesized that NSP4 stimulates innate immunity, potentially via the activation of microbial pattern recognition receptors (PRRs). To address this, we measured the ability of purified NSP4 to stimulate the secretion of cytokines from macrophages, critical cellular mediators of innate immune responses. While primary human tissue macrophages cannot be readily expanded ex vivo, monocyte-derived cell lines can be used to model macrophage function following chemical induction of differentiation. Treatment of the monocytic cell line THP-1 with PMA activates protein kinase C and induces differentiation toward a macrophage phenotype (18). Therefore, we assessed the ability of NSP4 to elicit the secretion of cytokines from PMA-treated THP-1 (pTHP-1) cells as a model of tissue macrophages.
NSP4 induced a dose-dependent increase in the secretion of TNF-α and IL-6 from pTHP-1 cells (Fig. 1A and B). As little as 0.1 μg/ml NSP4 caused a significant elevation of TNF-α above the background level, whereas a 10-fold greater concentration was required to elicit comparable quantities of IL-6. To confirm that cytokine secretion was not due to a contamination with rotavirus, triple-layered particles (TLPs) purified by CsCl gradient centrifugation were added as a control. Notably, pTHP-1 cells were not susceptible to rotavirus infection, and no expression of NSP4 was detected in the cells by immunofluorescence microscopy, even using an inoculum equivalent to an MOI of 100 calculated in MA104 cells (data not shown). TLPs failed to elicit secretion of significant quantities of either cytokine. Trypsinization of virus prior to addition was not performed to avoid potential proteolysis of cell surface receptors, but the infectious status of the virus was confirmed by titration of the infectivity on MA104 cells.
Fig 1.

NSP4 induces secretion of proinflammatory cytokines from THP-1 cells. (A and B) Dose-dependent secretion of TNF-α and IL-6 from pTHP-1 cells. PMA-treated THP-1 cells (2 × 105 cells/well) were incubated with increasing concentrations of purified NSP4 (0.1, 1.0, and 10 μg/ml) or CsCl-purified TLPs (MOI of 1, 10, and 100). The amount of cytokine secreted was compared to that of untreated pTHP-1 cells (UT), cells incubated with medium from uninfected Caco-2 cells (M), or cells treated with control agonists LPS (0.1 μg/ml) and Pam3CSK4 (1 μg/ml) for 24 h. (C) Secretion of TNF-α from pTHP-1 cells in response to 10×-concentrated medium (C.M) from rotavirus-infected cells after ultracentrifugation to remove virus particles and the same medium following immunodepletion of NSP4. Western blot analysis (inset) confirms removal of NSP4 from the concentrated medium. Purified NSP4 (1 μg/ml) was used as a positive control. Cytokine concentrations in the medium were determined by cytometric bead array assay. Data are the means ± SEM of duplicate samples. (D) RAW 264.7 cells were stimulated with increasing concentrations of purified NSP4 (0.1, 1.0, and 10 μg/ml) or identical amounts of CsCl-purified TLPs. Pam3CSK4 (10 μg/ml), LPS (1 μg/ml), and medium from mock-infected Caco-2 cells were used as controls. After 24 h of stimulation, cell-free medium was collected and assayed for nitrite using the Griess assay. Data are means ± SEM from triplicate samples.
Therefore, NSP4 secreted from rotavirus-infected cells can trigger the secretion of inflammatory cytokines from macrophage-like cells in the absence of virions or viral nucleic acid, suggesting that NSP4 is a trigger of innate immunity in rotavirus-infected animals. To confirm that NSP4 specifically was responsible for the secretion of TNF-α from pTHP-1 cells, we examined the ability of conditioned medium from virus-infected cells to elicit cytokine production before and after immune depletion of NSP4 (Fig. 1C). Medium was ultracentrifuged and concentrated 10-fold by ultrafiltration, and NSP4 was removed by repeated passage over immobilized anti-NSP4 monoclonal antibody. Removal of NSP4 was confirmed by Western blotting (Fig. 1C, inset). The NSP4-depleted medium did not cause TNF production from pTHP-1 cells even when a 3-fold excess of protein in the immune-depleted sample was used. Purified NSP4 also caused the secretion of nitric oxide from the murine macrophage cell line RAW 264.7. Nitrite concentrations in the medium were measured in response to treatment with NSP4 and other proinflammatory stimuli as a proxy marker of gaseous NO (Fig. 1D).
NSP4 activates NF-κB and the MAPKs p38 and JNK.
Upon recognition of viruses, diverse PRRs activate intracellular signaling pathways that culminate in the release of NF-κB from its endogenous inhibitor IκB, whereupon it translocates to the nucleus to initiate transcription of various proinflammatory genes. In addition, the expression of various inflammatory cytokines is regulated by chromatin remodeling induced by mitogen-activated protein kinase (MAPK) signaling (19). Three distinctly regulated MAPKs in mammals are activated by various TLR ligands: extracellular signal-regulated kinase 1/2 (ERK1/2), p38 proteins (p38α/β/γ/δ), and c-Jun N-terminal kinases (JNKs) (20). We investigated the potential of NSP4 to activate both NF-κB and two members of the MAPK family activated by TLRs, p38 and JNK. NSP4 induced the phosphorylation of IKKα and β subunits, p38, and JNK within 15 min (Fig. 2). NSP4 also caused the translocation of the p65 subunit of NF-κB from the cytoplasm to the nucleus of RAW 264.7 cells following the phosphorylation and degradation of the inhibitory subunit (Fig. 3).
Fig 2.
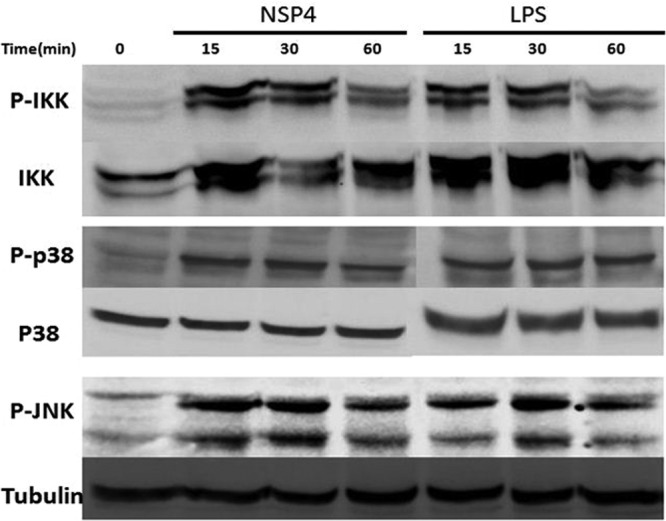
NSP4 activates IKK, p38, and JNK in PMA-treated THP-1 cells. Lysates (15 μg protein/lane) of cells treated with NSP4 (10 μg/ml) or LPS (0.1 μg/ml) for the periods indicated were blotted after SDS-PAGE and probed with antibodies to phosphorylated forms of the indicated proteins. The same membrane was then stripped and reblotted to measure amounts of total IKKβ p38 or α-tubulin in each sample.
Fig 3.
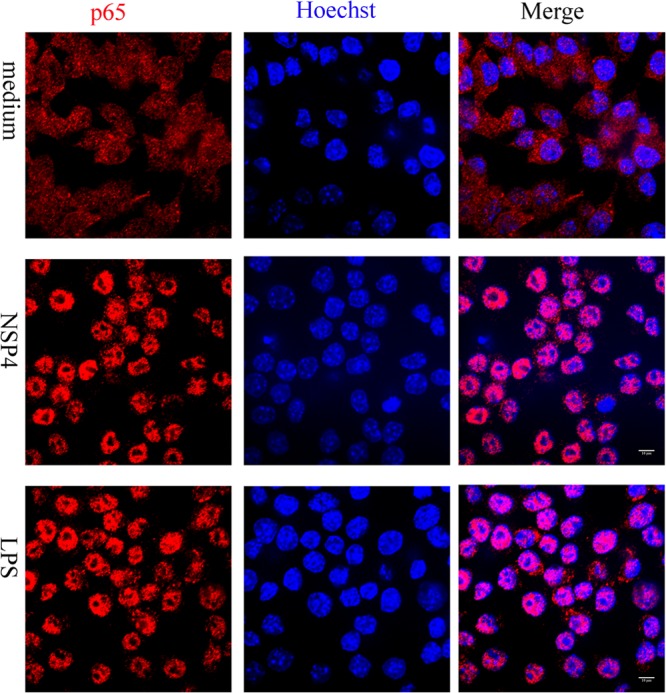
NSP4 stimulates nuclear translocation of NF-κB. RAW 264.7 cells seeded on glass coverslips were incubated with normal medium or medium supplemented with either NSP4 (1 μg/ml) or LPS (100 ng/ml) on ice for 10 min, after which the temperature was raised to 37°C for 30 min. Images were obtained by confocal microscopy after labeling with anti-p65 (red) antibody and Hoechst (blue) as described in Materials and Methods. Nuclear translocation of p65 is revealed by purple color in the merged image. Scale bar, 10 μm.
Induction of cytokine secretion by NSP4 is dependent on MyD88 and TLR2.
The previous experiments suggest that NSP4 interacts with a TLR family member on the surface of immune cells, resulting in secretion of cytokines. Most TLRs initiate intracellular signaling in response to ligand binding by recruitment of the adaptor molecule MyD88 to the cytoplasmic domain of the receptor. TLR3, on the other hand, utilizes TRIF as an alternative adaptor while TLR4 can use either MyD88 or TRIF (21). We first established that NSP4 was able to activate NF-κB in mouse macrophages at nanogram concentrations, consistent with our findings in human cells (Fig. 4A). To investigate if NSP4-induced inflammation is mediated by the TLR signaling pathway, we examined the secretion of cytokines from immortalized macrophages derived from wild-type mice or mice lacking both MyD88 and TRIF (MyD88/TRIF−/−), which would ablate TLR-mediated responses. MyD88/TRIF−/− macrophages displayed ablated TNF-α production following both NSP4 and LPS stimulation, consistent with a role for TLRs in NSP4 immune recognition (Fig. 4B). Furthermore, mice doubly deficient in MyD88 and MyD88-adaptor-like (Mal) (MyD88/Mal−/−) displayed inhibited IL-6 and TNF-α secretion (Fig. 4C and D, respectively) following NSP4, Pam3Cys, or LPS, but not poly I·C, stimulation, consistent with a critical role for MyD88 and Mal in mediating NSP4 proinflammatory responses. Mal, also known as TIRAP, is necessary to recruit Myd88 to TLR2 and TLR4 (11). Importantly, TRIF was not required to mediate NSP4-mediated inflammation.
Fig 4.

NSP4 induces proinflammatory cytokine production via TLR2. (A) RAW 264.7 cells stably expressing the NF-κB-dependent promoter for ELAM linked to luciferase were seeded at 4 × 104 cells 24 h prior to stimulation with NSP4 or LPS where indicated. Cells were cultured for 6 h and lysed, and luciferase activity was assessed to determine NF-κB activation. NS, nonstimulated control cells. (B to F) Wild-type or knockout immortalized macrophages were seeded at 2 × 104 cells 24 h prior to stimulation with the ligand NSP4 (10 to 0.5 μg/ml), Pam3Cys (20 ng/ml), poly(I·C) (20 μg/ml), or LPS (20 ng/ml). Cultured supernatants were collected 24 h after ligand addition and assessed for IL-6 and TNF-α protein expression by ELISA. All data are presented as the means ± standard errors of the means from triplicate samples.
To determine which TLR family member is recognized by NSP4, macrophages were generated from mice lacking either TLR2 or TLR4, and the secretion of TNF-α and IL-6 from these cells was measured in response to NSP4 or TLR ligand stimulation. Secretion of IL-6 and TNF-α from macrophages derived from TLR2−/− mice was severely inhibited in response to both NSP4 and Pam3CSK4 (Fig. 4B and C). Conversely, macrophages from wild type, TLR3−/−, and TLR4−/− mice showed a dose-dependent secretion of cytokines in response to NSP4, although the absolute amount of IL-6 and TNF-α secreted by knockout cells was slightly decreased compared to that of wild-type cells. Taken together, these results suggest that NSP4 induces proinflammatory responses via TLR2.
NSP4 triggers secretion of IL-8 in HEK293 cells transfected with TLR2.
To confirm that NSP4 can activate intracellular signaling through TLR2, we transfected HEK293 cells with a plasmid expressing TLR2. Untransfected cells failed to secrete IL-8 when incubated with the agonists shown in Fig. 5. Upon transient expression of TLR2, the cells secreted IL-8 in response to NSP4 and Pam3CSK4 but not TLPs. To further confirm that TLR4 was not responsible for NSP4 activity, 293 cells transfected with a combination of TLR4, MD2, and CD14 were used in the same assay. Only LPS elicited secretion of IL-8 in these cells that did not respond to the viral glycoprotein. These results further confirm our findings with murine macrophages and support the conclusion that NSP4 induces NF-κB activation and downstream cytokine secretion in human cells via TLR2.
Fig 5.
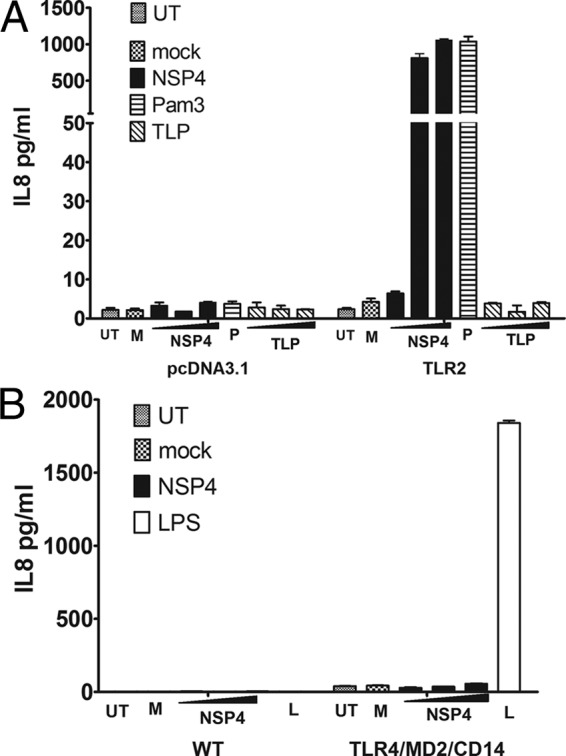
NSP4 activates NF-κB via TLR2 but not TLR4. (A) HEK 293T cells (3 × 104 per well) were transfected with either pcDNA3.1 or pTLR2. Twenty-four h later, the cells were incubated with 0.1, 1.0, or 10 μg/ml NSP4 or CsCl-purified TLPs, Pam3CSK4 (P; 10 μg/ml), or medium from noninfected Caco-2 cells (M), or they were left untreated (UT). After a further 16 h, the medium was assayed for IL-8 by cytometric bead array assay. (B) The same concentrations of NSP4 did not trigger IL-8 secretion from HEK293 cells stably transfected with TLR4/MD2/CD14.
DISCUSSION
Recognition of viruses by the innate immune system is mediated by a broad range of microbial pattern recognition receptors (PRRs). Viral nucleic acids represent the major trigger of innate immunity through activation of cytoplasmic receptors, including RIG-I, MDA5, AIM2, and IFI16, and/or members of the TLR family located on endosomal membranes that recognize either dsRNA (TLR3) or single-stranded RNA species (TLR7 and TLR8) (reviewed in reference 22). These receptors collectively enable nucleic acids from highly diverse viral species to be distinguished from host DNA/RNA and trigger activation of antiviral effectors, principally type I interferon (IFN).
The innate immune response appears to play a significant role in controlling rotavirus replication and the resolution of rotavirus disease, principally through the induction of an IFN response. Rotavirus infection of murine intestinal epithelial cells can induce type I (IFN-β) and type III (IFN-λ) interferons, and mice lacking receptors for either cytokine exhibit higher levels of viral replication than wild-type mice (23). In response, the virus has evolved mechanisms to antagonize the effect of IFN-α/β through the activity of NSP1 that mediates the degradation of at least two interferon regulatory factors within infected cells (24–26). Rotaviruses trigger the host innate immune response through MDA-5- and RIG-I-dependent signaling mechanisms and their common adaptor protein, MAVS/IPS-1 (27, 28). Interestingly, these studies revealed that TLR3/TRIF was dispensable for the induction of the early innate transcriptional response in rotavirus-infected cells. In contrast, a recent study suggests that age-dependent TLR3 expression in intestinal epithelium contributes to rotavirus susceptibility in infants (29). Further studies into innate immune control of rotavirus replication have highlighted the critical role of plasmacytoid dendritic cells (pDCs) in the production of type I IFN in response to rotavirus structural proteins and dsRNA (30).
Unlike the nucleic acid-sensing family of cytosolic and endosomal receptors, the subfamily of TLR2 receptors was originally thought to activate only NF-κB- and AP-1-regulated genes, including the inflammatory cytokines TNF-α and IL-6, but not the activation of IFN regulatory factors necessary for the production of type I IFN (31). The simplicity of this view has been challenged by the observation that viral (but not bacterial) ligands of TLR2 can elicit production of type I IFN in a subset of inflammatory monocytes present in mice. Only a highly specialized subset of cells, defined by the markers Ly6Chi CD11b+ CD11c− B220−, was able to secrete IFN-β in response to vaccinia virus in a TLR2-dependent manner (32). Whether an analogous population of cells is present in humans is unknown. pTHP-1 and RAW 247.6 cells did not produce type I IFN in response to TLR2 ligands in our hands (data not shown). However, the possibility remains that in vivo, a viral TLR2 ligand like NSP4 contributes to an antiviral innate immune response through production of type I IFN by specialized cells and augments the (likely greater) amounts produced by pDCs that are activated by viral nucleic acids.
Conversely, the activation of TLR2 by NSP4 may play an important role in viral pathogenesis and reflect a viral manipulation of host innate immunity for improved transmission and spread following the production of inflammatory mediators in the gut. Elevated levels of inflammatory cytokines correlate directly with the severity and frequency of diarrheal episodes during the acute phase of rotavirus infection in naturally infected children (33, 34) and are also observed in experimentally infected gnotobiotic pigs (35, 36). Therefore, the observations reported here are consistent with markers of systemic inflammation observed during rotavirus infection in vivo. Additional studies on children with rotavirus diarrhea suggest that multiple TLRs, including TLR2, regulate the production of inflammatory cytokines during the acute phase of infection (37). Viral activation of TLR2 through secreted NSP4 could also account for other symptoms of rotavirus infection. A link between inflammatory cytokine production and diarrhea is suggested by studies that reveal the effect of TNF-α on intestinal ion secretion in isolated porcine intestinal epithelia and cultured human HT-29 cells (38, 39). The production of nitric oxide by macrophages exposed to NSP4, as reported earlier (40), could also be triggered through TLR2, possibly augmented by secreted IFN-γ, providing a mechanistic link to the induction of diarrhea.
TLR2 recognizes lipoproteins ubiquitous to all bacteria, but the receptor recognizes a remarkable range of molecules, including diacyl and triacylglycerol moieties, proteins, and polysaccharides (41). Despite the apparent lack of the canonical ligand motif, an increasing number of viruses and viral proteins have been found to activate TLR2 signaling, and several studies point to the importance of TLR2-mediated responses in regulating viral infectivity and pathogenesis in vivo (42–45). Notably, TLR2 is critical in the control of vaccinia virus infection by the innate immune system despite the presence of additional receptors capable of sensing DNA viruses (46). Envelope glycoproteins have emerged as candidates for viral recognition by TLR2, prompting speculation that conserved features of glycoprotein structure are sufficient to trigger innate defense mechanisms, synchronizing innate immunity with attachment and entry, the earliest steps in the virus replication cycle. This idea is plausible, because a lack of amino acid sequence conservation among envelope glycoproteins from distinct virus families could be offset by the presence of conserved structural features, for example, peptide conformations common to multiple viral glycoproteins involved in membrane fusion (47). An exception to this paradigm is hepatitis C virus nonstructural protein (NS3), a serine protease and helicase that stimulates TLR2-mediated signaling in macrophages (48).
The results presented here now extend the range of known viral TLR2 agonists to rotavirus NSP4. We recently reported the purification of NSP4 secreted from polarized Caco-2 cells infected with rotavirus. Unusually, the hydrophobic N terminus, including the region proposed to act as the transmembrane domain, remains present in the secreted protein. NSP4 was secreted as large oligomers in a complex with noncovalently associated lipid. SDS-PAGE analysis indicated that a minor proportion of the protein is present as a shorter 26-kDa form, likely as a result of proteolysis of the flexible C-terminal tail. It is interesting to speculate whether the oligomeric status, associated lipid component, or another form of posttranslational modification is responsible for TLR2 ligand activity of the protein. However, this activity was not due to contaminating virions, as TLPs purified by CsCl gradient centrifugation were unable to stimulate production of inflammatory cytokines in THP-1 cells or IL-8 in TLR2-transfected 293 cells.
Finally, NSP4 has been reported to exhibit mucosal adjuvant-like properties when coadministered with model antigens in mice (15). Although the mechanism by which NSP4 enhanced the antibody response to coadministered antigens was not defined, the induction of immunomodulatory cytokines through an interaction with TLR2 represents a plausible mechanism by which secreted NSP4 might influence both innate and adaptive immunity to rotavirus infection. Further studies into the role of TLR2 in immunity to rotavirus employing TLR knockout mice may be valuable in resolving the role of this and other pattern recognition receptors in controlling the replication and shedding of rotavirus in vivo.
Footnotes
Published ahead of print 7 August 2013
REFERENCES
- 1.Greenberg HB, Estes MK. 2009. Rotaviruses: from pathogenesis to vaccination. Gastroenterology 136:1939–1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blutt SE, Conner ME. 2007. Rotavirus: to the gut and beyond! Curr. Opin. Gastroenterol. 23:39–43. [DOI] [PubMed] [Google Scholar]
- 3.Chen JZ, Settembre EC, Aoki ST, Zhang X, Bellamy AR, Dormitzer PR, Harrison SC, Grigorieff N. 2009. Molecular interactions in rotavirus assembly and uncoating seen by high-resolution cryo-EM. Proc. Natl. Acad. Sci. U. S. A. 106:10644–10648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Trask SD, Ogden KM, Patton JT. 2012. Interactions among capsid proteins orchestrate rotavirus particle functions. Curr. Opin. Virol. 2:373–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hu L, Crawford SE, Hyser JM, Estes MK, Prasad BV. 2012. Rotavirus non-structural proteins: structure and function. Curr. Opin. Virol. 2:380–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bugarcic A, Taylor JA. 2006. Rotavirus nonstructural glycoprotein NSP4 is secreted from the apical surfaces of polarized epithelial cells. J. Virol. 80:12343–12349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ball JM, Tian P, Zeng CQ, Morris AP, Estes MK. 1996. Age-dependent diarrhea induced by a rotaviral nonstructural glycoprotein. Science 272:101–104. [DOI] [PubMed] [Google Scholar]
- 8.Morris AP, Scott JK, Ball JM, Zeng CQ, O'Neal WK, Estes MK. 1999. NSP4 elicits age-dependent diarrhea and Ca(2+) mediated I(-) influx into intestinal crypts of CF mice. Am. J. Physiol. 277:G431–G444. [DOI] [PubMed] [Google Scholar]
- 9.Tafazoli F, Zeng CQ, Estes MK, Magnusson KE, Svensson L. 2001. NSP4 enterotoxin of rotavirus induces paracellular leakage in polarized epithelial cells. J. Virol. 75:1540–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Halaihel N, Lievin V, Ball JM, Estes MK, Alvarado F, Vasseur M. 2000. Direct inhibitory effect of rotavirus NSP4(114–135) peptide on the Na(+)-D-glucose symporter of rabbit intestinal brush border membrane. J. Virol. 74:9464–9470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fitzgerald KA, Palsson-McDermott EM, Bowie AG, Jefferies CA, Mansell AS, Brady G, Brint E, Dunne A, Gray P, Harte MT, McMurray D, Smith DE, Sims JE, Bird TA, O'Neill LA. 2001. Mal (MyD88-adapter-like) is required for Toll-like receptor-4 signal transduction. Nature 413:78–83. [DOI] [PubMed] [Google Scholar]
- 12.Hagbom M, Istrate C, Engblom D, Karlsson T, Rodriguez-Diaz J, Buesa J, Taylor JA, Loitto VM, Magnusson KE, Ahlman H, Lundgren O, Svensson L. 2011. Rotavirus stimulates release of serotonin (5-HT) from human enterochromaffin cells and activates brain structures involved in nausea and vomiting. PLoS Pathog. 7:e1002115. 10.1371/journal.ppat.1002115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seo NS, Zeng CQ, Hyser JM, Utama B, Crawford SE, Kim KJ, Hook M, Estes MK. 2008. Inaugural article: integrins alpha1beta1 and alpha2beta1 are receptors for the rotavirus enterotoxin. Proc. Natl. Acad. Sci. U. S. A. 105:8811–8818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Didsbury A, Wang C, Verdon D, Sewell MA, MacIntosh J, Taylor JA. 2011. Rotavirus NSP4 is secreted from infected cells as an oligomeric lipoprotein and binds to glycosaminoglycans on the surface of non-infected cells. Virol. J. 20:551–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kavanagh OV, Ajami NJ, Cheng E, Ciarlet M, Guerrero RA, Zeng CQ, Crawford SE, Estes MK. 2010. Rotavirus enterotoxin NSP4 has mucosal adjuvant properties. Vaccine 28:3106–3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, Fitzgerald KA, Latz E. 2008. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 9:847–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Greig SL, Berriman JA, O'Brien JA, Taylor JA, Bellamy AR, Yeager MJ, Mitra AK. 2006. Structural determinants of rotavirus subgroup specificity mapped by cryo-electron microscopy. J. Mol. Biol. 356:209–221. [DOI] [PubMed] [Google Scholar]
- 18.Daigneault M, Preston JA, Marriott HM, Whyte MK, Dockrell DH. 2010. The identification of markers of macrophage differentiation in PMA-stimulated THP-1 cells and monocyte-derived macrophages. PLoS One 5:e8668. 10.1371/journal.pone.0008668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saccani S, Pantano S, Natoli G. 2002. p38-Dependent marking of inflammatory genes for increased NF-kappa B recruitment. Nat. Immunol. 3:69–75. [DOI] [PubMed] [Google Scholar]
- 20.Chang L, Karin M. 2001. Mammalian MAP kinase signalling cascades. Nature 410:37–40. [DOI] [PubMed] [Google Scholar]
- 21.Fitzgerald KA, Palsson-McDermott EM, Bowie AG, Jefferies CA, Mansell AS, Brady G, Brint E, Dunne A, Gray P, Harte MT, McMurray D, Smith DE, Sims JE, Bird TA, O'Neill LA. 2001. Mal (MyD88-adapter-like) is required for Toll-like receptor-4 signal transduction. Nature 413:78–83. [DOI] [PubMed] [Google Scholar]
- 22.O'Neill LA, Bowie AG. 2010. Sensing and signaling in antiviral innate immunity. Curr. Biol. 20:R328–R333. [DOI] [PubMed] [Google Scholar]
- 23.Pott J, Mahlakõiv T, Mordstein M, Duerr CU, Michiels T, Stockinger S, Staeheli P, Hornef MW. 2011. IFN-λ determines the intestinal epithelial antiviral host defense. Proc. Natl. Acad. Sci. U. S. A. 108:7944–7949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barro M, Patton JT. 2005. Rotavirus nonstructural protein 1 subverts innate immune response by inducing degradation of IFN regulatory factor 3. Proc. Natl. Acad. Sci. U. S. A. 102:4114–4119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barro M, Patton JT. 2007. Rotavirus NSP1 inhibits expression of type I interferon by antagonizing the function of interferon regulatory factors IRF3, IRF5, and IRF7. J. Virol. 81:4473–4481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Graff JW, Ettayebi K, Hardy ME. 2009. Rotavirus NSP1 inhibits NFkappaB activation by inducing proteasome-dependent degradation of beta-TrCP: a novel mechanism of IFN antagonism. PLoS Pathog. 5:e100028. 10.1371/journal.ppat.1000280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Broquet AH, Hirata Y, McAllister CS, Kagnoff MF. 2011. RIG-I/MDA5/MAVS are required to signal a protective IFN response in rotavirus-infected intestinal epithelium. J. Immunol. 186:1618–1626. [DOI] [PubMed] [Google Scholar]
- 28.Sen A, Prijssers AJ, Dermody TS, García-Sastre A, Greenberg HB. 2011. The early interferon response to rotavirus is regulated by PKR and depends on MAVS/IPS-1, RIG-I, MDA-5, and IRF3. J. Virol. 85:3717–3732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pott J, Stockinger S, Torow N, Smoczek A, Lindner C, McInerney G, Bäckhed F, Baumann U, Pabst O, Bleich A, Hornef MW. 2012. Age-dependent TLR3 expression of the intestinal epithelium contributes to rotavirus susceptibility. PLoS Pathog. 8:e1002670. 10.1371/journal.ppat.1002670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Deal EM, Jaimes MC, Crawford SE, Estes MK, Greenberg HB. 2010. Rotavirus structural proteins and dsRNA are required for the human primary plasmacytoid dendritic cell IFNalpha response. PLoS Pathog. 6:e1000931. 10.1371/journal.ppat.1000931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Toshchakov V, Jones BW, Perera PY, Thomas K, Cody MJ, Zhang S, Williams BR, Major J, Hamilton TA, Fenton MJ, Vogel SN. 2002. TLR4, but not TLR2, mediates IFN-beta-induced STAT1alpha/beta-dependent gene expression in macrophages. Nat. Immunol. 3:392–398. [DOI] [PubMed] [Google Scholar]
- 32.Barbalat R, Lau L, Locksley RM, Barton GM. 2009. Toll-like receptor 2 on inflammatory monocytes induces type I interferon in response to viral but not bacterial ligands. Nat. Immunol. 10:1200–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jiang B, Snipes-Magaldi L, Dennehy P, Keyserling H, Holman RC, Bresee J, Gentsch J, Glass RI. 2003. Cytokines as mediators for or effectors against rotavirus disease in children. Clin. Diagn. Lab. Immunol. 10:995–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang Y, Dennehy PH, Keyserling HL, Tang K, Gentsch JR, Glass RI, Jiang B. 2007. Rotavirus infection alters peripheral T-cell homeostasis in children with acute diarrhea. J. Virol. 81:3904–3912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Azevedo MS, Yuan L, Pouly S, Gonzales AM, Jeong KI, Nguyen TV, Saif LJ. 2006. Cytokine responses in gnotobiotic pigs after infection with virulent or attenuated human rotavirus. J. Virol. 80:372–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gonzalez AM, Azevedo MS, Jung K, Vlasova A, Zhang W, Saif LJ. 2010. Innate immune responses to human rotavirus in the neonatal gnotobiotic piglet disease model. Immunology 131:242–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu J, Yang Y, Sun J, Ding Y, Su L, Shao C, Jiang B. 2006. Expression of Toll-like receptors and their association with cytokine responses in peripheral blood mononuclear cells of children with acute rotavirus diarrhoea. Clin. Exp. Immunol. 144:376–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kandil HM, Berschneider HM, Argenzio RA. 1994. Tumour necrosis factor alpha changes porcine intestinal ion transport through a paracrine mechanism involving prostaglandins. Gut 35:934–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oprins JC, Meijer HP, Groot JA. 2000. TNF-alpha potentiates the ion secretion induced by muscarinic receptor activation in HT29cl.19A cells. Am. J. Physiol. Cell Physiol. 278:C463–C472. [DOI] [PubMed] [Google Scholar]
- 40.Borghan MA, Mori Y, El-Mahmoudy AB, Ito N, Sugiyama M, Takewaki T, Minamoto N. 2007. Induction of nitric oxide synthase by rotavirus enterotoxin NSP4: implication for rotavirus pathogenicity. J. Gen. Virol. 88:2064–2072. [DOI] [PubMed] [Google Scholar]
- 41.Kang JY, Nan X, Jin MS, Youn SJ, Ryu YH, Mah S, Han SH, Lee H, Paik SG, Lee JO. 2009. Recognition of lipopeptide patterns by Toll-like receptor 2-Toll-like receptor 6 heterodimer. Immunity 31:873–884. [DOI] [PubMed] [Google Scholar]
- 42.Bieback K, Lien E, Klagge IM, Avota E, Schneider-Schaulies J, Duprex WP, Wagner H, Kirschning CJ, Ter Meulen V, Schneider-Schaulies S. 2002. Hemagglutinin protein of wild-type measles virus activates toll-like receptor 2 signaling. J. Virol. 76:8729–8736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Boehme KW, Guerrero M, Compton T. 2006. Human cytomegalovirus envelope glycoproteins B and H are necessary for TLR2 activation in permissive cells. J. Immunol. 177:7094–7102. [DOI] [PubMed] [Google Scholar]
- 44.Compton T, Kurt-Jones EA, Boehme KW, Belko J, Latz E, Golenbock DT, Finberg RW. 2003. Human cytomegalovirus activates inflammatory cytokine responses via CD14 and Toll-like receptor 2. J. Virol. 77:4588–4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kurt-Jones EA, Chan M, Zhou S, Wang J, Reed G, Bronson R, Arnold MM, Knipe DM, Finberg RW. 2004. Herpes simplex virus 1 interaction with Toll-like receptor 2 contributes to lethal encephalitis. Proc. Natl. Acad. Sci. U. S. A. 101:1315–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhu J, Martinez J, Huang X, Yang Y. 2007. Innate immunity against vaccinia virus is mediated by TLR2 and requires TLR-independent production of IFN-beta. Blood 109:619–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Harrison SC. 2008. Viral membrane fusion. Nat. Struct. Mol. Biol. 15:690–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kang JY, Nan X, Jin MS, Youn SJ, Ryu YH, Mah S, Han SH, Lee H, Paik SG, Lee JO. 2009. Recognition of lipopeptide patterns by Toll-like receptor 2-Toll-like receptor 6 heterodimer. Immunity 31:873–884. [DOI] [PubMed] [Google Scholar]