

Abstract
We have shown that the circulating vaccine-derived polioviruses responsible for poliomyelitis outbreaks in Madagascar have recombinant genomes composed of sequences encoding capsid proteins derived from poliovaccine Sabin, mostly type 2 (PVS2), and sequences encoding nonstructural proteins derived from other human enteroviruses. Interestingly, almost all of these recombinant genomes encode a nonstructural 3A protein related to that of field coxsackievirus A17 (CV-A17) strains. Here, we investigated the repercussions of this exchange, by assessing the role of the 3A proteins of PVS2 and CV-A17 and their putative cellular partners in viral replication. We found that the Golgi protein acyl-coenzyme A binding domain-containing 3 (ACBD3), recently identified as an interactor for the 3A proteins of several picornaviruses, interacts with the 3A proteins of PVS2 and CV-A17 at viral RNA replication sites, in human neuroblastoma cells infected with either PVS2 or a PVS2 recombinant encoding a 3A protein from CV-A17 [PVS2-3A(CV-A17)]. The small interfering RNA-mediated downregulation of ACBD3 significantly increased the growth of both viruses, suggesting that ACBD3 slowed viral replication. This was confirmed with replicons. Furthermore, PVS2-3A(CV-A17) was more resistant to the replication-inhibiting effect of ACBD3 than the PVS2 strain, and the amino acid in position 12 of 3A was involved in modulating the sensitivity of viral replication to ACBD3. Overall, our results indicate that exchanges of nonstructural proteins can modify the relationships between enterovirus recombinants and cellular interactors and may thus be one of the factors favoring their emergence.
INTRODUCTION
Poliovirus (PV), a member of the genus Enterovirus (phylogenetic cluster C) from the Picornaviridae family, is the etiological agent of paralytic poliomyelitis (1). The World Health Organization program for the global eradication of poliomyelitis has been largely successful. However, this disease remains a public health issue, due partly to the rapid spread of PV in insufficiently immunized populations and the emergence of epidemic circulating Sabin vaccine-derived PV (cVDPV), which threatens to undermine the eradication program (2).
Enteroviruses are nonenveloped viruses with a single-stranded, positive-sense RNA genome. All viral proteins are encoded by a single large open reading frame (ORF). The resulting polyprotein is processed by viral proteases to yield mature viral proteins, including the capsid proteins (VP1 to VP4) and the nonstructural proteins required for viral replication (3). Most cVDPVs have mosaic genomes composed of mutated PV vaccine sequences encoding capsid proteins and some or all sequences encoding nonstructural proteins derived from other human enteroviruses of species C (HEV-C), such as coxsackievirus A (CV-A) (4). We have previously studied the cVDPVs responsible for two poliomyelitis outbreaks in Madagascar in 2002 and 2005 (5–7). These cVDPVs are all recombinant and most have mutated sequences encoding capsid proteins derived from the PV type 2 Sabin (PVS2) strain and some sequences in the region encoding the nonstructural proteins which are closely related to those of field CV-A17 isolates (5, 8).
PV infection, like all picornavirus infections, initiates a major remodeling of intracellular membranes, such that the cytoplasm of infected cells becomes filled with tightly associated vesicles (9, 10). Virus-induced vesicles are located in the perinuclear region of the cell and are mostly derived from the endoplasmic reticulum (ER), Golgi apparatus, and lysosomes of the host cell through the action of the nonstructural viral proteins 2BC and 3A (11–14). Enterovirus RNA replication occurs on the cytoplasmic surface of these membranous organelles, where all the nonstructural viral proteins required for RNA replication, including 3A and its precursor, 3AB, are located (15).
Analyses of enterovirus 3A proteins have led to the identification of several cellular partners of this protein. The 3A proteins of PV and of the related coxsackievirus B3 (CV-B3) can interact with the cellular Golgi brefeldin A-resistant guanine nucleotide exchange factor 1 (GBF1) (16, 17), which activates the GTPase ADP-ribosylation factor 1 (Arf1) (18, 19). Arf1 regulates the recruitment to membranes of coat protein complex I (COP-I), which is involved in protein transport between the ER and the Golgi apparatus (20, 21). By interacting with GBF1, 3A inhibits the cellular secretory pathway (13, 16, 17, 22). The 3A proteins of PV and CV-B3 also bind LIS1 (23), a component of a dynein motor complex required for Golgi apparatus integrity (24–26). The 3A-LIS1 interaction may therefore also contribute to changes in protein transport (23).
Another Arf1 effector, in addition to COP-I, is the phosphatidylinositol 4-kinase IIIβ (PI4KIIIβ), which catalyzes the production of phosphatidylinositol-4-phosphate (PI4P). The membrane-anchored 3A protein modulates GBF1/Arf1 activity, resulting in the preferential recruitment of PI4KIIIβ, rather than COP-I, to sites of viral RNA replication (27, 28). PI4KIIIβ recruitment leads to the enrichment of virus-induced membranous organelles in PI4P, which has been shown to facilitate viral RNA replication (28).
Aichi virus (AiV), another member of the Picornaviridae family, which belongs to the genus Kobuvirus and has been implicated in oyster-associated acute gastroenteritis (29), uses a different strategy to recruit PI4KIIIβ to the site of genome replication (30). AiV replication is dependent on the recruitment of PI4KIIIβ by a complex of 3A and the 60-kDa Golgi complex-associated protein (GCP60) also known as acyl-coenzyme A binding domain-containing 3 (ACBD3), a protein involved in the maintenance of Golgi apparatus structure (31). Thus, according to this model, the role of ACBD3 in the viral RNA replication machinery resembles that of GBF1 for enteroviruses (30). It has recently been shown that the 3A proteins of several other picornaviruses, including bovine kobuvirus, PV, CV-B3, and human rhinovirus 14, can be copurified with ACBD3 (32). In addition, a new host factor involved in PV replication, the valosin-containing protein (VCP/p97), has recently been shown to interact with the nonstructural viral proteins 3AB and 2BC (33).
Almost all of the recombinant genomes from PVS2-derived cVDPVs responsible for polio outbreaks in Madagascar (5, 8) encode a nonstructural 3A protein related to that of field coxsackievirus A17 (CV-A17) strains. Here, we investigated the repercussions of this exchange, by assessing the role of the 3A proteins of PVS2 and CV-A17 and their putative cellular partners in viral replication. We show that although the 3A proteins of PVS2 and CV-A17 interact with ACBD3, ACBD3 does not promote viral replication. Indeed, it actually seems to restrict this process. This inhibitory effect was more pronounced with PVS2 than with a recombinant PVS2 harboring a 3A protein from CV-A17 [PVS2-3A(CV-17)]. Furthermore, we have identified an amino acid in the 3A protein involved in modulating the sensitivity of viral replication to ACBD3.
MATERIALS AND METHODS
Cell lines, virus stocks, and viral infections.
Human neuroblastoma IMR5 cells, HeLa (MRL2 clone) cells, human embryonic kidney HEK-293T cells stably expressing the simian virus 40 large T antigen and HEK-293T-T7 cells stably expressing phage T7 polymerase were cultured in Dulbecco modified Eagle medium (DMEM; Gibco) supplemented with 10% (vol/vol) heat-inactivated fetal bovine serum (FBS; Gibco). Human HEp-2c cells were cultured in DMEM supplemented with 10% (vol/vol) newborn calf serum (Gibco). Cells were maintained at 37°C in a humidified atmosphere containing 95% air and 5% CO2.
The PVS2 strain was obtained from pT7S2 (generously provided by A. Macadam, National Institute for Biological Standards and Control (NIBSC), United Kingdom), a plasmid containing the full-length cDNA of the PVS2 genome downstream from the phage T7 RNA polymerase promoter (34). Stocks of PVS2 and PVS2 recombinants were generated on HEp-2c cells at 34°C and were stored at −80°C until use. In all of the experiments described here, subconfluent IMR5, HEK-293T, HeLa, or HEp-2c cells were inoculated with PVS2 or recombinant PVS2 at the multiplicity of infection (MOI) indicated, in DMEM supplemented with 10% FBS, as previously described (35). Time zero postinfection (p.i.) corresponds to the time at which virus inoculation was performed. Total virus yields (extracellular and intracellular) from infected cells were collected after freezing and thawing to release intracellular viruses. Virus titers were evaluated in HEp-2c cells by determining the number of 50% tissue culture infective dose units (TCID50) per ml, as previously described (36).
Antibodies and siRNAs.
Mouse anti-actin (A4700), rabbit anti-GST (G7781), rabbit anti-FLAG (F7425), and rabbit anti-ACBD3 (HPA015594) antibodies were obtained from Sigma-Aldrich. Donkey anti-rabbit Alexa Fluor 350 (A-10039), goat anti-mouse Alexa Fluor 488 (A-11029), donkey anti-mouse Alexa Fluor 546 (A-10036), and donkey anti-rabbit Alexa Fluor 546 (A-10040) antibodies were obtained from Invitrogen. Mouse anti-ACBD3 (sc-101277) antibody was obtained from Santa Cruz Biotechnology. Mouse anti-giantin (ab37266) and rabbit anti-PI4KIIIβ (ab109418) antibodies were obtained from Abcam. Horseradish peroxidase (HRP)-conjugated anti-mouse antibody (NA9310) and HRP-conjugated anti-rabbit (NA9340) antibodies were obtained from Amersham Biosciences. Mouse anti-dsRNA (MabJ2) antibody was purchased from English & Scientific Consulting. Mouse anti-GBF1 antibody (catalog no. 612116) was obtained from BD Biosciences. The small interfering RNAs, against ACBD3 (siRNA-ACBD3) used throughout the present study (ACBD3 ON-TARGETplus siRNA; catalog no. 64746) was purchased from Dharmacon. Another siRNA directed against ACBD3 (sc-78612) was obtained from Santa Cruz Biotechnology and was used in one experiment, as indicated in Results. A control siRNA (siRNA-ctrl; sc-37007) was also obtained from Santa Cruz Biotechnology.
ORF cloning and derived plasmids.
All ORFs were amplified by standard PCR methods (Ex Taq; TaKaRa) and inserted into the pDONR207 plasmid (Invitrogen) with an in vitro recombination-based cloning system (Gateway System; Invitrogen) as previously described (37). Plasmids were amplified by transforming Escherichia coli DH5α. All constructs were verified by nucleotide sequencing to ensure that no other mutations had occurred during the cloning process.
(i) ORFs and plasmids used in the yeast two-hybrid procedure.
ORFs encoding 3A(PVS2) and 3A(CV-A17) were obtained by the standard PCR method above, from pBR-S2 (38) and pBR-S2/CA17 (39), respectively, with the forward and reverse primers described in Table 1. For the recombination cloning of PCR products, the 5′ ends of the forward and reverse primers were fused to attB1.1 and attB2.1 recombination sequences, respectively, as previously described (40). The PCR products were transferred into pDONR207 by recombination, according to the manufacturer's instructions (BP cloning reaction; Invitrogen). The ORFs inserted into pDONR207 were then transferred, by recombination, into the Gateway-compatible Gal4-BD-fusion yeast expression vector pDEST32 (Invitrogen) according to the manufacturer's instructions (LR cloning reaction; Invitrogen).
Table 1.
Primer sequences used for engineering ORFs and truncated PVS2 3A plasmids
| Constructs | Sensea | Sequence (5′–3′)b | Genome positionc |
|---|---|---|---|
| ORF 3A(PVS2) | F | ggggacaactttgtacaaaaaagttggcatgGGACCACTGCAGTATAAAGATCTAAAA | 5110–5136* |
| R | ggggacaactttgtacaagaaagttggttaCTGGTGCCCAGCGAACAG | 5370–5353* | |
| ORF 3A(CV-A17) | F | ggggacaactttgtacaaaaaagttggcatgGGCCCCCTGCAGTACAAA | 5128–5145† |
| R | ggggacaactttgtacaagaaagttggttaCTGCTGTCCTGCAAAGAGC | 5388–5370† | |
| C-terminal truncated PVS2 3A | |||
| For the four plasmids below | F | taaccaactttcttgtacaaagttg | |
| p3A(PVS2)Δ30-87-GST | R | ATCAACTGCCTGGAGCAAATCGTTG | 5196–5172* |
| p3A(PVS2)Δ43-87-GST | R | CCAGCCTTTCTTTTCACAGTAATCT | 5235–5211* |
| p3A(PVS2)Δ59-87-GST | R | CCGGTTGATGTTCCTCTCTGTTTGA | 5283–5259* |
| p3A(PVS2)Δ80-87-GST | R | GTACATAACGTACACGACACCGGCT | 5349–5325* |
| N-terminal truncated PVS2 3A | |||
| For the five plasmids below | R | catgccaacttttttgtacaaactt | |
| p3A(PVS2)Δ1-18-GST | F | GAGTGTATCAACGATTTGCTCCAGG | 5164–5188* |
| p3A(PVS2)Δ1-29-GST | F | TCCCAGGAAGTGAGAGATTACTGTG | 5197–5221* |
| p3A(PVS2)Δ1-42-GST | F | ATTGTTAACATTACCAGTCAGGTTC | 5236–5260* |
| p3A(PVS2)Δ1-58-GST | F | GCGATGACTATCCTACAAGCAGTAAC | 5284–5309* |
| p3A(PVS2)Δ1-80-GST | F | AAGCTGTTCGCTGGGCACCAgtaacc | 5350–5370* |
F, forward; R, reverse.
Sequences complementary to those of plasmid vectors are indicated in lowercase.
*, According to PVS2 numbering; †, according to CV-A17 67591 numbering.
(ii) p3A(PVS2)-GST, p3A(CV-A17)-GST, and pACBD3-GST plasmids.
Plasmids encoding glutathione S-transferase (GST)-tagged 3A from PVS2 [p3A(PVS2)-GST] and from CV-A17 [p3A(CV-A17)-GST] were generated by transferring the ORFs encoding 3A(PVS2) and 3A(CV-A17), respectively, from pDONR207 into pDEST27 Gateway-compatible GST-fusion expression vectors (Invitrogen) by recombination, according to the manufacturer's instructions (LR cloning reaction; Invitrogen). For the generation of pACBD3-GST plasmids, the ORF encoding ACBD3 in pENTR221-ACBD3 (Invitrogen) was transferred to pDEST27 by recombination.
(iii) p3A(PVS2-V12I)-GST plasmid.
The plasmid encoding a GST-tagged 3A(V12I) [p3A(PVS2-V12I)-GST], consisting of the 3A(PVS2) sequence harboring a GTT→ATC substitution in the 3A-encoding sequence, leading to the replacement of the Val residue in position 12 with an Ile residue, was generated by site-directed mutagenesis, with the QuikChange mutagenesis kit (Stratagene), according to the manufacturer's instructions. We used p3A(PVS2)-GST as the template. The forward and reverse primers used are described in Table 2.
Table 2.
Primers sequences used to generate modified PVS2 genomes
| Primera | Template | Sequence (5′–3′)b | Genome positionc |
|---|---|---|---|
| Fragment A | pBR-ΔLS2 | ||
| pBR322-3568F* | GTTCGCCAGTTAATAGTTTG | ||
| S2.063.c | CTTAACATCTATTTTTAGATCTTTATAtcccttgtcatcgtcatccttgtaatcaccCTGCAGTGGTCCCTGGAATAGAGC | 5121–5098* | |
| Fragment B | pS2Δ5′ | ||
| S2.064.d | GCTCTATTCCAGGGACCACTGCAGgattacaaggatgacgatgacaagTATAAAGATCTAAAAATAGATGTTAAG | 5122–5148* | |
| pBR322-91R* | GATTTCATACACGGTGCC | ||
| Fragment C | pBR-ΔLS2 | ||
| pBR322-3568F* | GTTCGCCAGTTAATAGTTTG | ||
| S2.060.c | GGGGGTGTAGTCTTTATGTCAATCTTTAGGTCTTTGTACTGCAGGGGGCCCTGGAATAGAGCTTCCATGCAATT | 5109–5086* | |
| Fragment D | pBR-ΔLCA17 | ||
| 67591.01.d | AGAGAAACAGAAGATCAAACATTGGTAATTGCATGGAAGCTCTATTCCAGGGCCCCCTGCAGTACAAAGACCTAA | 5128–5152† | |
| 67591.02.c | CTGATAGTGGGTACATTGGGTCGTTTATTTGGCAAACCAGTGTATGCACCCTGCTGTCCTGCAAAGAGCTTGTA | 5388–5365† | |
| Fragment E | pS2Δ5′ | ||
| S2.059.d | CTGTTGCTGGAGTAGTGTATGTGATGTACAAGCTCTTTGCAGGACAGCAGGGTGCATACACTGGTTTGCCAAAT | 5371–5394* | |
| pBR322-91R* | GATTTCATACACGGTGCC | ||
| Fragment F | pBR-ΔLS2 | ||
| pBR322-3568F* | GTTCGCCAGTTAATAGTTTG | ||
| S2.066.c | TCTTTGTAtcccttgtcatcgtcatccttgtaatcaccCTGCAGGGGGCCCTGGAATAGAGCTTCCATGCAATT | 5109–5086* | |
| Fragment G | pBR-ΔLCA17 | ||
| 67591.04.d | CATTGGTAATTGCATGGAAGCTCTATTCCAGGGCCCCCTGCAGggtgattacaaggatgacgatgacaagggaTACAAAGACCTAAAGATTGACATAAAG | 5140–5166† | |
| 67591.02.c | CTGATAGTGGGTACATTGGGTCGTTTATTTGGCAAACCAGTGTATGCACCCTGCTGTCCTGCAAAGAGCTTGTA | 5388–5365† | |
| 5 T7 extension | Fused fragments | ||
| S2.017.d | GGGTAATACGACTCACTATAGGTTAAAACAGCTCTGGGGTTGTACC | 1–24* | |
| S3.002.c | GGCGCGCCGTTTAAACTTTTTTTTTTTTTTTTTTTTTTTTCCTCCGAATTAAAGAAAAATTTAC | 7417-poly(A)* | |
| 3A V12I mutagenesis | p3A(PVS2)-GST and pT7-Luc-PVS2ΔP1 | ||
| V12I.F | GAGTGTATCAACGATTTGCTCCAGG | 5164–5188* | |
| V12I.R | CGGAGGGGGACTGGTCTTGATATCTATTTTTAGATC | 5163–5128* |
*, From Bessaud & Delpeyroux (41).
In the sequences, 5′ additional overlapping extensions are indicated in italics, the FLAG-tag-encoding sequence is shown in lowercase, the T7 promoter is underlined, and mutated nucleotides are indicated in boldface.
*, According to PVS2 numbering; †, according to CV-A17 67591 numbering.
(iv) pACBD3-3×FLAG plasmid.
The plasmid encoding the triple-flag-epitope sequence (3×FLAG)-tagged ACBD3 (pACBD3-3×FLAG) was generated by transferring the ORF encoding ACBD3 from pENTR221-ACBD3 (Invitrogen) to pCI-neo Gateway-compatible 3×FLAG fusion expression vectors (Promega) by recombination, according to the manufacturer's instructions (LR cloning reaction; Invitrogen).
(v) C-terminally and N-terminally truncated 3A(PVS2)-GST plasmids.
ORFs encoding 3A(PVS2) proteins with C- and N-terminal truncations were generated from the 3A(PVS2) ORF inserted into pDONR207, by PCR with a QuikChange Lightning site-directed mutagenesis kit (Stratagene), according to the manufacturer's instructions, with the forward and reverse primers described in Table 1. The truncated PVS2 3A ORFs inserted into pDONR207 were then transferred, by recombination, into the pDEST27 Gateway-compatible GST-fusion expression vector (Invitrogen), according to the manufacturer's instructions (LR cloning reaction; Invitrogen).
Yeast two-hybrid screen.
The yeast two-hybrid procedure has been described elsewhere (40). Briefly, full-length 3A proteins from PVS2 or CV-A17 were inserted into the pDEST32 vector for expression as a fusion protein with the Gal4 DNA-binding domain (Gal4-BD). These plasmids were used to transform the AH109 yeast strain (Clontech), and Gal4-BD-fused PVS2 or CV-A17 was used as bait in a mating strategy for the screening of a human spleen cDNA library. The human spleen cDNA library was inserted into the pPC86 vector (Invitrogen) for expression as fusions with the Gal4 activation domain (Gal4-AD) and was maintained in the Y187 strain of yeast (Clontech). Transformed AH109 and Y187 yeast cells were mixed together for mating, and diploids were plated on a selective medium lacking histidine to select for interaction-dependent transactivation of the HIS3 reporter gene. Gal4-AD-cDNA from (His+) colonies was amplified by PCR and sequenced to identify the host proteins interacting with the 3A proteins of PVS2 or CV-A17. Mating-based screening was carried out to obtain 8.4-fold coverage of the complexity of the cDNA library.
Construction of PVS2-3A-FLAG, PVS2-3A(CV-A17), and PVS2-3A(CV-A17)-FLAG viruses.
The fusion PCR-based procedure previously described (41) was used to produce in vitro the three modified PVS2 genomes (Fig. 1A). The PVS2-3A-FLAG genome consists of the PVS2 genome with a 30-nucleotide (nt) insertion after nt 5122, encoding a FLAG epitope (GDYKDDDDKG) inserted after residue 4 of the 3A protein. The FLAG epitope was flanked by two Gly residues to increase its accessibility. The PVS2-3A(CV-A17) genome consists of the PVS2 genome but with the sequence encoding the 3A protein replaced with that encoding the 3A protein of CV-A17. The PVS2-3A(CV-A17)-FLAG genome consists of the PVS2 genome but with the 3A-encoding sequence replaced with that of 3A(CV-A17), with the FLAG-epitope insertion (described above) after residue 4.
Fig 1.

Schematic diagram of the organization of PVS2 and PVS2-derived recombinant genomes engineered by fusion PCR. (A) For each genome, overlapping PCR fragments (labeled A to G) were generated with the primers described in Table 2. Genome fragments derived from PVS2 and CV-A17 are shown in white and gray, respectively. The FLAG sequence shown in black, encoding a FLAG epitope flanked by two glycine residues (GDYKDDDDKG), was inserted after nt 5122, corresponding to residue 4 of 3A(PVS2) or 3A(CV-A17). (B) FLAG epitope insertion did not affect viral growth: one-step growth curves comparing PVS2 with PVS2-3A(FLAG) (left panel) and PVS2-3A(CV-A17) with PVS2-3A(CV-A17)-FLAG (right panel), in IMR5 cells infected at an MOI of 10 TCID50/cell. Each point represents the mean total virus titer for three independent experiments; error bars indicate the standard deviations.
The fusion PCR (41) allowed the synthesis of modified full-length genomes placed under the control of the T7 promoter, by the fusion of overlapping PCR fragments (Fig. 1A) generated with forward and reverse primers containing the 5′ extension described in Table 2. We ensured that the final recombinant cDNA was not contaminated with full-length PVS2 or CV-A17 cDNAs by performing PCR on three plasmids containing PVS2 and CV-A17 partial sequences (Table 2): pBR-ΔLS2 contained a defective PVS2 cDNA with a 1,205-nt deletion (nt 6251 to 7455) encompassing part of the 3D-encoding sequence and the whole 3′-noncoding region, pS2Δ5′ contained a defective PVS2 cDNA with a 188-nt deletion (nt 291 to 478) within the 5′-noncoding region, and pBR-ΔLCA17 contained a defective CV-A17 cDNA genome carrying a 1,734-nt deletion (nt 33 to 1766) encompassing part of the 5′-noncoding region and VP2 encoding sequences.
Finally, the recombinant PVS2 were recovered, as previously described (41), by transfecting HEK-293T-T7 cells (kindly provided by Pierre Charneau, Institut Pasteur, Paris, France), which constitutively express the T7 polymerase, with the modified full-length PVS2 genomes. Transfected cells were incubated at 34°C until a cytopathogenic effect was observed. The supernatants containing the viruses were collected and clarified by centrifugation. The genomic modifications were checked by sequencing with the BigDye Terminator v3.1 kit (Applied Biosystems) on an ABI Prism 3140 automated sequencer (Applied Biosystems). Virus stocks were generated after two additional passages on HEp-2c cells and were stored at −80°C until use. The stability of FLAG insertion in the PVS2-3A-FLAG and PVS2-3A(CV-A17)-FLAG recovered from the second passage was confirmed by immunoblotting whole-cell extracts of infected cells with an anti-FLAG antibody (data not shown). We confirmed that FLAG epitope insertion had no effect on viral growth, by comparison with the parental PVS2 and PVS2-3A(CV-A17), respectively (Fig. 1B).
Luciferase-encoding replicons and luciferase assays. (i) Luc-PVS2, Luc-PVS2-3A(CV-A17), and Luc-PVS2-3A(V12I).
The three luciferase-encoding replicons Luc-PVS2, Luc-PVS2-3A(CV-A17), and Luc-PVS2-3A(V12I) were generated by in vitro transcription from pT7-Luc-PVS2ΔP1, pT7-Luc-PVS2ΔP1-3A(CV-A17), and pT7-Luc-PVS2ΔP1-3A(V12I), respectively, constructed as described below. In vitro transcription was performed after the linearization of these plasmids by EcoRI digestion, with a RiboMAX Large-Scale T7 RNA production system (Promega), according to the manufacturer's instructions.
The pT7-Luc-PVS2ΔP1 plasmid was constructed from pT7S2, in which the major part of the P1-encoding region (nt 751 to 3336) was replaced by the firefly luciferase gene. Briefly, pT7S2 was digested at positions 11097 and 3341 with AscI and PsiI, respectively, to delete both the 5′ NC and the P1-encoding region. A 5′ NC sequence, flanked by the AscI and PsiI restriction sites at its 5′ and 3′ ends, respectively, was amplified by PCR from a full-length pT7S2 with the forward and reverse primers 5′-GTTCGCCAGTTAATAGTTTG-3′ and 5′-CCATTTATAATGTAGTATTGTTGTTTTATCC-3′, respectively, and was inserted into pT7S2 previously digested with AscI and PsiI to give pT7-PVS2ΔP1, with Quick Ligase (New England BioLabs). Finally, the firefly luciferase-encoding sequence was amplified by PCR from pGL4.13 (Promega) with the forward and reverse primers 5′-ATGGAAGATGCCAAAAACATTAAG-3′ and 5′-TACACGGCGATCTTGCCGCCC-3′, respectively. It was introduced into the PsiI site of pT7-PVS2ΔP1 at position 750, according to the Quick Ligase protocol (New England BioLabs), to generate pT7-Luc-PVS2ΔP1.
The pT7-Luc-PVS2ΔP1-3A(CV-A17) plasmid was generated from pT7-Luc-PVS2ΔP1, in which the sequence of the 3A of PVS2 was replaced with that of CV-A17. An intermediate p12AAQ52P-S2Luc3ACA17-pMA-T plasmid was generated with GeneArt (Life Technologies). p12AAQ52P-S2Luc3ACA17-pMA-T carried a SnabI-NcoI (nt 4453 to 5462) fragment from the PVS2 sequence, in which the 3A sequence was replaced with that of 3A(CV-A17). The SnabI-NcoI fragment was inserted, with Quick Ligase (New England BioLabs), into pT7-Luc-PVS2ΔP1 previously digested with SnabI and NcoI, to generate pT7-Luc-PVS2ΔP1-3A(CV-A17).
The pT7-Luc-PVS2ΔP1-3A(V12I) plasmid consists of a pT7-Luc-PVS2ΔP1 plasmid with a GTT→ATC substitution at nt 5143 to 5145 (according to PVS2 sequence numbering) was introduced to generate a Val→Ile substitution at amino acid 12 of 3A(PVS2). Nucleotide substitutions were generated by site-directed mutagenesis with the V12I.F and V12I.R primers (Table 2) and the QuikChange mutagenesis kit (Stratagene) according to the manufacturer's instructions.
(ii) Luciferase activity.
Luciferase activity in cells transfected with luciferase-encoding replicons was measured with a Bright-Glo luciferase reporter gene assay kit according to the manufacturer's instructions (Fluoprobes) and a Tecan Infinite 200 reader.
Plasmids, replicons, and siRNA transfection.
HEK-293T cells in six-well or 24-well plates were transfected with plasmids (600 or 200 ng of plasmid/well, respectively) in the presence of the JetPRIME reagent according to the manufacturer's instructions (Polyplus transfection).
Luciferase-encoding replicons were subjected to in vitro transcription and the products were used to transfect HEK-293T cells in 96-well dishes (250 ng of replicon/well) in the presence of Lipofectamine 2000 (Invitrogen), according to the manufacturer's instructions. IMR5, HEK-293T, HeLa, or HEp-2c cells were transfected with siRNA (10 nM) in the presence of Lipofectamine RNAiMAX (Invitrogen), according to the manufacturer's instructions. When siRNA transfection was carried out before viral infection or plasmid transfection, the siRNA-transfected cells were cultured for 36 h before being infected with virus or transfected with plasmids.
Pulldown assay.
HEK-293T cells were plated in six-well culture dishes at a density of 106 cells per well in DMEM supplemented with 10% (vol/vol) FBS and then cultured for 24 h. Cells were transfected with 600 ng of plasmids encoding GST-tagged proteins or GST alone, in the presence of JetPRIME reagent (Polyplus transfection), according to the manufacturer's instructions. Two days after transfection, cells were either mock infected or infected by incubation with PVS2-3A-FLAG or PVS2-3A(CV-A17)-FLAG for 16 h. The cells were then harvested, pelleted, washed once in phosphate-buffered saline (PBS), and resuspended in ice-cold lysis buffer (0.5% NP-40, 20 mM Tris-HCl [pH 8], 120 mM NaCl, and 1 mM EDTA) supplemented with Complete protease inhibitor cocktail (Roche) and then incubated for 20 min at 4°C with gentle shaking. The suspension was then clarified by centrifugation at 14,000 × g for 10 min. For pulldown analysis, cell lysates were incubated for 2 h at 4°C, with gentle shaking, with 25 μl of glutathione-Sepharose beads (Amersham Biosciences) for the purification of GST-tagged proteins. Beads were then washed three times in ice-cold lysis buffer, and proteins were recovered by boiling in denaturing loading buffer (Invitrogen). Purified complexes and protein extracts were then analyzed by Western blotting.
Whole-cell extracts.
Cells were collected, washed with PBS, and resuspended in lysis buffer (20 mM Tris [pH 7.5], 135 mM NaCl, 2 mM EDTA, 1% NP-40, 1% sodium deoxycholate, and 0.1% sodium dodecyl sulfate) supplemented with a protease inhibitor cocktail (Roche). The extracts were then incubated on ice for 20 min, and the lysates were clarified by centrifugation for 10 min at 1,200 × g.
Western blot analysis.
Protein concentrations were determined with a bicinchoninic acid protein assay kit (Pierce). Samples containing equal amounts of protein were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (10 to 20% Tricine gels; Novex), as previously described (35). The proteins were transferred to nitrocellulose membranes (Amersham Biosciences). Nonspecific sites were blocked with nonfat milk, as previously described (35), and the membranes were incubated for 2 h at room temperature with the primary antibody. Membranes then were washed in 0.1% Tween 20 in PBS (PBST; pH 7.4) and treated with the appropriate HRP-conjugated secondary antibody for 2 h at room temperature. The immunoblots were washed in PBST, and proteins were detected with an enhanced chemiluminescence detection kit (Amersham Biosciences) and a G-box (SynGene). Anti-actin antibody was used to control for equal protein loading.
Assessment of the surface expression of TNFR1.
Aliquots of 4 × 105 HEp2-c cells were harvested, pelleted, and incubated in blocking buffer (2% bovine serum albumin in PBS) with a primary mouse anti-human tumor necrosis factor receptor 1 (TNFR1) antibody (R&D Systems) for 1 h at 4°C with gentle shaking. The cells were then rinsed and incubated with a secondary goat anti-mouse Alexa Fluor 488-conjugated antibody (Invitrogen) for 1 h at 4°C. Staining with antibodies was followed by incubation with 1 μg of propidium iodide (PI; Sigma)/ml. PI-positive cells, corresponding to cells with the disrupted plasma membranes, were excluded from the analysis. The percentage of TNFR1-positive cells was determined by flow cytometry. We analyzed at least 10,000 cells per sample. Fluorescence was measured with a FACScan machine (Becton Dickinson). The data were analyzed with CellQuest software (Becton Dickinson).
Immunofluorescence staining.
IMR5 cells grown on polylysine-coated (10 μg/ml) slides were fixed by incubation for 15 min at 4°C in paraformaldehyde (4%) and were permeabilized by treatment with 0.2% Triton X-100 in PBS for 5 min. The cells were incubated for 1 h in blocking buffer (2% bovine serum albumin in PBS) and then for 2 h at room temperature with the primary antibodies. The cells were washed three times, for 5 min each time, in blocking buffer and then incubated for 2 h with the appropriate Alexa Fluor-conjugated secondary antibodies at room temperature. The slides were then washed three times in PBS and mounted in ProLong Gold antifade reagent (P-36931; Invitrogen) containing DAPI (4′,6′-diamidino-2-phenylindole) for nuclear staining. When indicated, staining of nuclei was performed by the incubation of slides with 1 μM TO-PRO-3 iodide (T3605; Invitrogen) for 15 min. The slides were then washed three times in PBS and mounted in ProLong Gold antifade reagent (P-36930; Invitrogen). Images were acquired with a Zeiss Axioplan 2 microscope using the Zeiss ApoTome system and Zeiss Axiovision 4.4 software.
Statistical analysis.
Data are expressed as means ± the standard deviations for three independent experiments. A Student t test was used to compare experimental conditions and controls. A P value of <0.05 was considered significant.
Pearson correlation coefficient calculation.
We assessed the degree of colocalization of two components statistically by calculating Pearson correlation coefficients for image intensity in the red and green or blue channels, with the JACoP tool, using Costes' automatic threshold (42). Pearson coefficient values of ≥0.5 are considered to indicate a significant positive correlation.
RESULTS
ACBD3 is a cellular partner interacting with the 3A proteins of PVS2 and CV-A17.
We used a standard yeast two-hybrid screening procedure to identify cellular proteins binding to the 3A proteins from PVS2 and CV-A17. Each 3A protein was used as bait for the screening of a human spleen cDNA library. Positive yeast colonies were selected and found to express either ACBD3 (PVS2, 32 colonies; CV-A17, 14 colonies) or the cyclic AMP-responsive element-binding protein 3 (CREB3) (PVS2, 5 colonies; CV-A17, 4 colonies). Since ACBD3 is associated with the Golgi apparatus and plays a role in the replication of some picornaviruses (30–32), we decided to focus on this cellular partner to study possible differences in viral replication as a function of its relationships with the 3A proteins from PVS2 and CV-A17. The functional role of 3A interacting with CREB3 is under investigation.
We first confirmed the interaction between 3A from PVS2 or CV-A17 and endogenous ACBD3. HEK-293T cells were transfected with expression vectors encoding GST alone or GST fused to the 3A protein from PVS2 [p3A(PVS2)-GST] or CV-A17 [p3A(CV-A17)-GST]. The 3A-GST proteins were then purified with glutathione-Sepharose beads, and samples were analyzed for the presence of ACBD3. Cellular ACBD3 was copurified with 3A-GST from PVS2 or CV-A17 but not with GST alone (Fig. 2A). These results confirm the interaction between 3A from PVS2 or CV-A17 and ACBD3.
Fig 2.
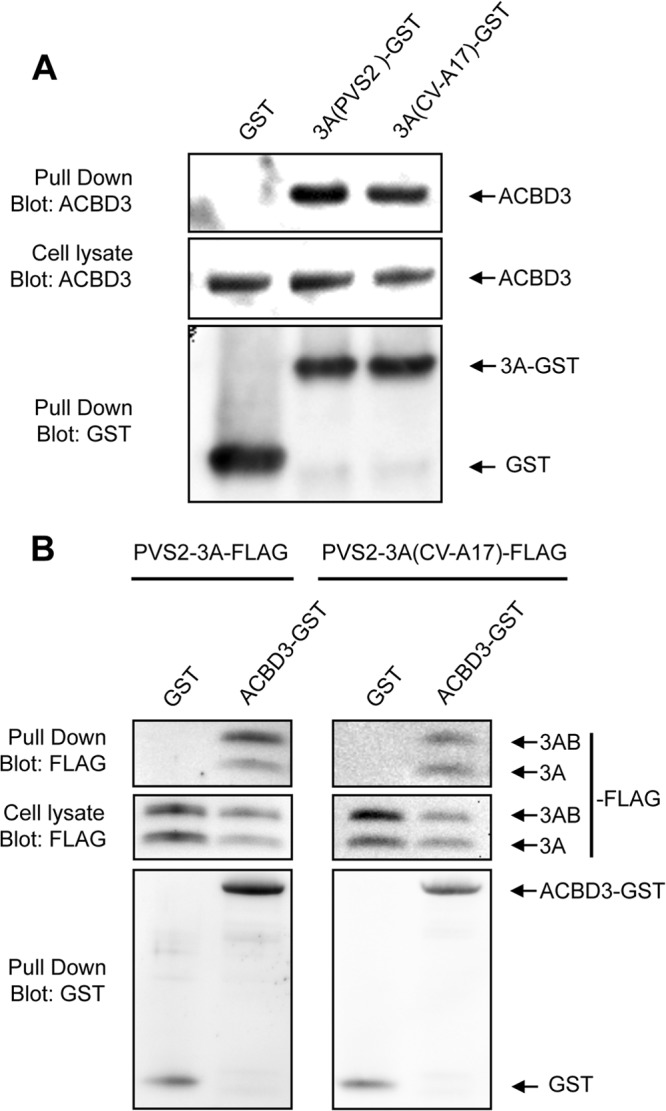
ACBD3 interacts with the 3A proteins of both PVS2 and CV-A17. (A) GST pulldown assay on lysates from HEK-293T cells transfected with expression vectors encoding 3A(PVS2)-GST, 3A(CV-A17)-GST, or GST alone. Two days posttransfection, the cells were collected and subjected to GST pulldown analysis. Endogenous ACBD3 and GST were detected by Western blotting with antibodies directed against ACBD3 and GST, respectively. (B) GST pulldown assay on lysates from HEK-293T cells infected with PVS2-3A-FLAG (left) or PVS2-3A(CV-A17)-FLAG (right). Before infection, HEK-293T cells were transfected with expression vectors encoding GST or ACBD3-GST. Cell lysates were collected at 16 h p.i. and subjected to GST pulldown analysis. FLAG and GST were detected by Western blotting with antibodies directed against FLAG and GST, respectively.
Localization of 3A and ACBD3 following viral infection.
We investigated the distribution of 3A (from PVS2 and CV-A17) and ACBD3 during viral infection, by constructing a PVS2 recombinant synthesizing a 3A protein tagged with a 10-amino-acid FLAG epitope, inserted into its N-terminal region after residue 4 (PVS2-3A-FLAG), and a PVS2 recombinant in which the 3A protein was replaced with the 3A protein from CV-A17, with a FLAG epitope in the same position [PVS2-3A(CV-A17)-FLAG]. The N-terminal region of 3A has been shown to tolerate small insertions, which do not prevent the generation of viruses (43–45). We first confirmed that FLAG epitope insertion had no effect on viral growth compared to the parental PVS2 and PVS2-3A(CV-A17) (Fig. 1B). We also investigated whether the inserted tag had an effect on 3A binding to ACBD3. HEK-293T cells were transfected with expression vectors encoding GST alone or fused to the ACBD3 protein (ACBD3-GST) and then infected with PVS2-3A-FLAG or PVS2-3A(CV-A17)-FLAG, at an MOI of 50 TCID50/cell, for 16 h. GST pulldown assays were then carried out (Fig. 2B). The 3A-FLAG proteins from PVS2 and CV-A17 and their corresponding tagged 3AB precursors were pulled down with ACBD3-GST, but not with GST alone. Thus, the insertion of a tag in the N-terminal region of the 3A protein had no effect on 3A binding to ACBD3, for either PVS2 or PVS2-3A(CV-A17).
We then investigated, by immunofluorescence microscopy, whether the 3A protein colocalized with ACBD3 in PV-infected neuroblastoma IMR5 cells, a cell model more relevant than HEK-293T cells for PV infection. IMR5 cells were mock infected or infected at an MOI of 50 TCID50/cell with PVS2-3A-FLAG, and then tagged 3A and 3AB proteins and endogenous ACBD3 proteins were immunolabeled at the time points indicated, with anti-FLAG and anti-ACBD3 antibodies, respectively (Fig. 3A and B). As expected, in PVS2-3A-FLAG-infected cells, 3A/3AB proteins labeling, which was clearly detected from 4 h p.i. (Fig. 3B), formed foci concentrated in the perinuclear region, as previously described in PV-infected cells (46). This pattern, typical of the remodeling of host intracellular membranes, has been shown to correspond to viral RNA replication organelles (46). Endogenous ACBD3 labeling in mock-infected cells was observed in a region immediately adjacent to the nucleus, corresponding to the Golgi apparatus, as indicated by the labeling of a Golgi marker, the endogenous giantin protein, with an anti-giantin antibody (Fig. 3A and B). We assessed the colocalization of 3A/3AB and ACBD3 after PVS2-3A-FLAG infection at the indicated time points by calculating Pearson correlation coefficients. 3AB/3A proteins colocalized with ACBD3 in the perinuclear foci at 4, 6, and 8 h p.i. (Fig. 3B), whereas total cellular ACBD3 levels remained unchanged, as determined by Western blotting with the anti-ACBD3 antibody (data not shown).
Fig 3.

PV 3A colocalizes with ACBD3 throughout the viral cycle, at sites of viral RNA replication, in IMR5 cells. (A) Subcellular localization of ACBD3 in uninfected cells. Cells were subjected to immunofluorescence staining with antibodies directed against giantin (green; left panel) and ACBD3 (red; middle panel), respectively. The merged image (right panel) is an overlay of the ACBD3 and the giantin images with the nuclei stained with DAPI (white). (B) Colocalization of FLAG-tagged 3A and ACBD3 proteins during PVS2-3A-FLAG infection. Cells were mock infected or infected, and FLAG-tagged 3A/3AB and endogenous ACBD3 proteins were subjected to immunofluorescence staining at the indicated times p.i. with antibodies directed against FLAG (blue; left panel) and ACBD3 (red; middle panel), respectively. The merged image (right panel) is an overlay of the 3A/3AB and ACBD3 images, with the nuclei stained with TO-PRO-3 (white). (C) Colocalization of ACBD3 with dsRNA during PVS2-3A-FLAG infection. Cells were mock infected or infected and endogenous ACBD3 protein and dsRNA were subjected to immunofluorescence staining (at the indicated times p.i.) with antibodies directed against ACBD3 (red; left panel) and dsRNA (green; middle panel), respectively. The merged image (right panel) is an overlay of the ACBD3 and the dsRNA images, with the nuclei stained with DAPI (white). (D) Colocalization of FLAG-tagged 3A/3AB(CV-A17) and ACBD3 proteins during PVS2-3A(CV-A17)-FLAG infection. At 6 h p.i., FLAG-tagged 3A/3AB(CV-A17) and endogenous ACBD3 proteins were subjected to immunofluorescence staining with antibodies directed against FLAG (blue; left panel) and ACBD3 (red; middle panel), respectively. The merged image (right panel) is an overlay of the 3A/3AB(CV-A17) and the ACBD3 images, with the nuclei stained with TO-PRO-3 (white). (E) Colocalization of ACBD3 with dsRNA during PVS2-3A(CV-A17)-FLAG infection. At 6 h p.i., endogenous ACBD3 protein and dsRNA were subjected to immunofluorescence staining with antibodies directed against ACBD3 (red; left panel) and dsRNA (green; middle panel), respectively. The merged image (right panel) is an overlay of the ACBD3 and the dsRNA images, with the nuclei stained with DAPI (white). Pearson correlation coefficients (r) were calculated to assess the degree of colocalization of ACBD3 with giantin proteins (A), 3A/3AB (B and D) or dsRNA (C and E). *, Pearson correlation coefficient values of ≥0.5 are considered to indicate a significant positive correlation.
For confirmation of the localization of ACBD3 at viral RNA replication organelles, we investigated the distributions of ACBD3 and viral RNA replication in PVS2-infected IMR5 cells by immunofluorescence microscopy. Replicating viral RNA was detected by staining double-stranded replicative intermediates and replicative forms with an anti-dsRNA antibody (47). ACBD3 and replicating viral RNA had similar distributions in the perinuclear region of cells at 4 h p.i. and were colocalized at 6 and 8 h p.i., as confirmed by the Pearson correlation coefficients (Fig. 3C). ACBD3 was therefore present at the sites of viral RNA replication. Similar results were obtained with IMR5 cells infected with PVS2-3A(CV-A17)-FLAG, as illustrated in Fig. 3D and E for the 6-h p.i. time point. Thus, the 3A proteins of both PVS2 and CV-A17 seem to localize with ACBD3 at viral RNA replication organelles.
We then investigated whether ACBD3 plays a role in the subcellular distribution of the 3A/3AB proteins during viral infection by using immunofluorescence microscopy in ACBD3-silenced cells. IMR5 cells were transfected with siRNA-ACBD3 or siRNA-ctrl and then mock infected or infected with PVS2-3A-FLAG or PVS2-3A(CV-A17)-FLAG. Endogenous ACBD3 and tagged 3A and 3AB proteins were immunolabeled at 6 h p.i. with anti-ACBD3 and anti-FLAG, antibodies, respectively. In both siRNA-ctrl and siRNA-ACBD3-transfected cells, 3A/3AB proteins labeling formed foci concentrated in the perinuclear region, as previously observed at this time point (Fig. 4). Thus, ACBD3 does not seem to be involved in the subcellular distribution of the 3A/3AB proteins during infection with PVS2 or PVS2-3A(CV-A17).
Fig 4.
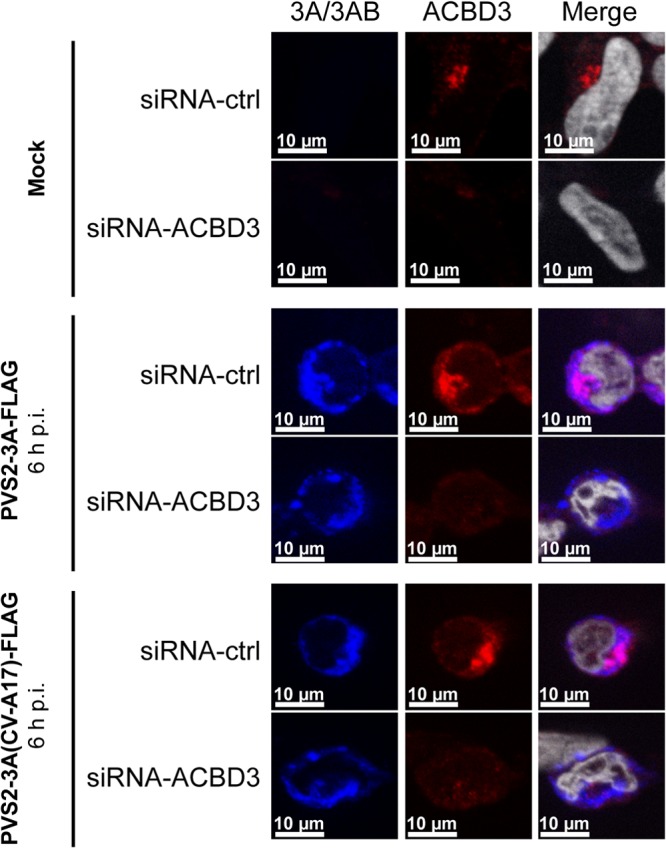
Subcellular localization of FLAG-tagged 3A/3AB proteins in siRNA-ctrl- or siRNA-ACBD3-transfected IMR5 cells infected with PVS2-3A-FLAG or PVS2-3A(CV-A17)-FLAG. Cells were transfected with siRNA-ctrl or siRNA-ACBD3. Two days posttransfection, the cells were either mock infected (upper panel) or infected with PVS2-3A-FLAG (middle panel) or PVS2-3A(CV-A17)-FLAG (bottom panel) at an MOI of 50 TCID50/cell. FLAG-tagged 3A/3AB and endogenous ACBD3 proteins were subjected to immunofluorescence staining at 6 h p.i., with antibodies directed against FLAG (blue) and ACBD3 (red), respectively. The merged image is an overlay of the 3A/3AB and ACBD3 images, in which the nuclei are stained with TO-PRO-3 (white).
ACBD3 is not involved in inhibition of the protein secretory pathway in infected cells but delays the growth of PVS2 and PVS2-3A(CV-A17).
The PV 3A protein itself disrupts ER-to-Golgi protein transport (48). We investigated the possible involvement of an interaction between ACBD3 and 3A in this process. Protein transport was investigated by analyzing the presentation of TNF receptor I (TNFRI) at the cell surface, as previously described (23, 49). After ACBD3 downregulation with siRNA, no change was observed in the inhibition of protein transport in cells infected with PVS2 or PVS2-3A(CV-A17), suggesting that ACBD3 does not play a role in this inhibition (data not shown).
We then investigated the role of ACBD3 in viral growth by evaluating the effect of ACBD3 silencing on viral yield in IMR5 cells infected with PVS2 or PVS2-3A(CV-A17). The efficacy of siRNA-ACBD3-mediated silencing of ACBD3 expression in IMR5 cells was checked by Western blotting with a specific antibody: as expected, ACBD3 levels were significantly lower in cells transfected with siRNA-ACBD3 than in cells transfected with siRNA-ctrl (Fig. 5A).
Fig 5.

ACBD3 silencing enhances PVS2 growth in IMR5, HEK-293T and HeLa cells. (A) Silencing of cellular ACBD3. IMR5 (left panel), HEK-293T (middle panel), or HeLa (right panel) cells were transfected with 10 nM control siRNA (siRNA-ctrl) or siRNA-ACBD3. Two days after transfection with siRNA, cell lysates were collected and ACBD3 was detected by Western blotting with an anti-ACBD3 antibody. Actin was used as a protein-loading control. (B) Effect of ACBD3 silencing on PVS2 and PVS2-3A(CV-A17) growth in IMR5 cells infected at an MOI of 10 TCID50/cell. Cells were transfected with siRNA-ctrl or siRNA-ACBD3. Two days posttransfection, the cells were infected with PVS2 (left) or PVS2-3A(CV-A17) (right). (C) Effect of ACBD3 silencing on PVS2 growth in HEK-293T or HeLa cells. Cells were transfected with siRNA-ctrl or siRNA-ACBD3. Two days posttransfection, HEK-293T cells (left panel) or HeLa cells (right panel) were infected with PVS2 at an MOI of 10 TCID50/cell. (D) Effect of ACBD3 silencing on PVS2 and PVS2-3A(CV-A17) growth in IMR5 cells infected at an MOI of 0.1 TCID50/cell. Cells were transfected with siRNA-ctrl or siRNA-ACBD3. Two days posttransfection, cells were infected with PVS2 (left) or PVS2-3A(CV-A17) (right). (E and F) Effect of ACBD3 overexpression on PVS2 and PVS2-3A(CV-A17) growth in HEK-293T cells. Cells were transfected with 200 ng per well of a vector encoding ACBD3-3×FLAG or 3×FLAG alone. Two days after transfection: (E) cell lysates were collected and ACBD3 was detected by Western blotting with an anti-ACBD3 antibody (actin was used as a protein-loading control) or (F) cells were infected with PVS2 (left panel) or PVS2-3A(CV-A17) (right panel) at an MOI of 10 TCID50/cell. Each point represents the mean total virus titer for three independent experiments; error bars indicate standard deviations. *, P < 0.05 in Student t tests comparing ACBD3-overexpressing cells with mock-transfected cells.
Surprisingly, after PVS2 infection (MOI of 10 TCID50/cell) viral growth rates were significantly higher in ACBD3-silenced cells than in siRNA-ctrl-transfected cells, at the 4- and 6-h p.i. time points (Fig. 5B, left panel). These results suggest that ACBD3 delays PVS2 growth. The inhibitory effect of ACBD3 on PVS2 growth was checked with another siRNA-ACBD3 (Santa Cruz) (data not shown). We also investigated whether ACBD3 delayed PVS2 growth in two other cell lines, HEK-293T and HeLa cells. The efficacy of siRNA-ACBD3-mediated silencing of ACBD3 expression in these cells was checked as described above (Fig. 5A). The inhibitory effect of ACBD3 on PVS2 growth was confirmed at 4 h p.i. in both cell lines (Fig. 5C).
We then investigated whether ACBD3 also inhibited viral growth of the PVS2 expressing the 3A protein of CV-A17, PVS2-3A(CV-A17) (Fig. 5B, right panel). In cells expressing endogenous ACBD3 (siRNA-ctrl-transfected cells), PVS2-3A(CV-A17) grew more rapidly than PVS2 at 4 h p.i. (Fig. 5B, left panel), suggesting that the 3A protein of CV-A17 provides some advantage in terms of viral growth. Furthermore, in ACBD3-silenced cells, the growth rate of PVS2-3A(CV-A17) was slightly higher than that in siRNA-ctrl-transfected cells at the 4-h p.i. time point, but no difference was observed at later time points (Fig. 5B, right panel). Thus, the inhibition of viral growth by endogenous ACBD3 seems to be weaker for PVS2-3A(CV-A17) than for PVS2, with the difference in viral growth abolished by the silencing of endogenous ACBD3 expression. These results suggest that viral growth depends on the nonstructural 3A protein expressed, which determines the sensitivity of viral growth to the inhibitory effect of ACBD3. The differential effects of endogenous ACBD3 on the growth of PVS2 and PVS2-3A(CV-A17) were also observed in cells infected at a lower MOI (0.1 TCID50/cell) (Fig. 5D).
We then investigated whether the inhibition of viral growth by ACBD3 was enhanced in cells overexpressing ACBD3. We transfected HEK-293T cells (which are highly transfectable) with 200 ng/well of an expression vector encoding a triple peptide FLAG (3×FLAG) alone (pCI-neo-3×FLAG) or fused to ACBD3 (pACBD3-3×FLAG). The overexpression of ACBD3 in HEK-293T cells was checked by Western blotting with an anti-ACBD3 antibody (Fig. 5E). As expected, after infection with PVS2 or PVS2-3A(CV-A17), viral growth rates were significantly lower at 4 and 6 h p.i. in ACBD3-overexpressing cells than in pCI-neo-3×FLAG-transfected cells (Fig. 5F). These results support the conclusion that ACBD3 inhibits the growth of PVS2 and PVS2-3A(CV-A17).
The amino acid at position 12 in the 3A protein can modulate the sensitivity of PVS2 replication to ACBD3.
The 3A proteins of PVS2 and CV-A17 differ by only three amino acids, at positions 12 (Val→Ile), 15 (Ser→Thr) and 86 (His→Gln) (Fig. 6). The amino acid difference at position 12 is the only one that results in the acquisition in the recombinant PVS2-3A(CV-A17) of a residue (Ile) present in the other Sabin strains (types 1 and 3) and reference wild-type PVs 1, 2, and 3 (50–53). Furthermore, most of the PVS2 cVDPVs responsible for poliomyelitis outbreaks in Madagascar and cocirculating field CV-A17 strains had an Ile residue in this position of their 3A proteins (5, 8). We therefore investigated whether the inhibitory effect of ACBD3 on viral growth involved the replication step in our model and whether this effect could be modulated by the amino acid in position 12 of the 3A protein. We constructed luciferase-encoding replicons, Luc-PVS2 and Luc-PVS2-3A(CV-A17), in which the PVS2 capsid genes were replaced with the luciferase gene in the full-length PVS2 and PVS2-3A(CV-A17) genomes, respectively. We also constructed a Luc-PVS2 replicon in which the Val residue in position 12 of the 3A protein was replaced with an Ile residue, Luc-PVS2-3A(V12I). To transfect replicons, HEK-293T cells were used, because these cells are particularly permissive to RNA transfection. HEK-293T cells treated with siRNA targeting ACBD3 or with the control siRNA were transfected with each of the three luciferase-expressing replicons (Fig. 7A). In cells transfected with siRNA-ctrl, the replication levels of Luc-PVS2-3A(CV-A17) and Luc-PVS2-3A(V12I) were similar and higher than that of Luc-PVS2. In ACBD3-silenced cells, the three replicons had similar replication levels, higher than those in mock-silenced cells. Similar experiments were performed in IMR5 and HeLa cells (Fig. 7B and C). As expected, the level of viral replication was low in these cells, but confirmed the results obtained in HEK-293T cells.
Fig 6.

Amino acid sequence alignment of 3A proteins from enteroviruses. The sequences of 3A proteins from poliovirus vaccine strains (Sabin 1, 2, and 3), wild-type poliovirus (Mahoney, Lansing, and Leon strains), PVS2-derived cVDPVs responsible for poliomyelitis outbreaks in Madagascar in 2002 (MAD004 and MAD0029 strains), and a representative cocirculating field CV-A17 strain, shown in an alignment generated with CLC Main Workbench software v.6.0.1. The GenBank accession numbers for each sequence are shown in brackets.
Fig 7.

Effect of ACBD3 silencing on Luc-PVS2, Luc-PVS2-3A(CV-A17) and Luc-PVS2-3A(V12I) replication in HEK-293T (A), IMR5 (B), and HeLa (C) cells. Cells were transfected with siRNA-ctrl (continuous lines) or siRNA-ACBD3 (broken lines). Two days after siRNA transfection, cells were transfected with Luc-PVS2, Luc-PVS2-3A(CV-A17), or Luc-PVS2-3A(V12I) RNA, as indicated. The luciferase activity (RLU) was measured at the indicated time posttransfection (p.t.) with luciferase-encoding replicons. The data shown are the means from three independent experiments. Error bars indicate standard deviations.
Thus, ACBD3 inhibited the replication of both Luc-PVS2 and Luc-PVS2-3A(CV-A17). This inhibitory effect was less pronounced with the Luc-PVS2 replicon containing the 3A sequence from CV-A17 than with that containing the 3A sequence from PVS2. Our results also suggest that the Ile residue in position 12 of the 3A(CV-A17) protein is associated with a lower sensitivity of PVS2-3A(CV-A17) replication to ACBD3.
Interestingly, the residue in position 12 of the 3A sequence was previously found to be located in the N-terminal region of 3A interacting with another cellular factor important for PV replication, GBF1 (54). We investigated whether the potential advantage of the PVS2-3A(CV-A17) over PVS2 resulted from a more favorable interaction of GBF1 with 3A(CV-A17) than with 3A(PVS2). HEK-293T cells were transfected with expression vectors encoding GST alone or GST fused to the 3A protein from PVS2, CV-A17, or a 3A(PVS2) protein in which the Val residue in position 12 was replaced with an Ile residue [p3A(PVS2-V12I)-GST]. GBF1 was copurified similarly with 3A-GST from PVS2 and CV-A17 and with 3A(PVS2-V12I) (Fig. 8). Thus, the Ile residue in position 12 of 3A from CV-A17 does not seem to modify the interaction between 3A and GBF1. These results suggest that this amino acid can modulate the sensitivity of PVS2 replication to ACBD3 without altering the binding of 3A to GBF1.
Fig 8.
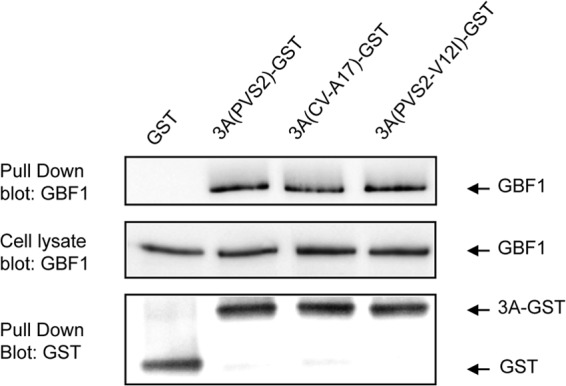
GST pulldown assay on lysates from HEK-293T cells transfected with expression vectors encoding 3A(PVS2)-GST, 3A(CV-A17)-GST, 3A(PVS2-V12I)-GST, or GST alone. Two days posttransfection, the cells were collected and subjected to GST pulldown analysis. Endogenous GBF1 and GST were detected by Western blotting with antibodies directed against GBF1 and GST, respectively (the order of the samples on the gel has been modified in this figure).
ACBD3 does not seem to be involved in the increase in PI4KIIIβ recruitment to replication sites in PVS2- and PVS2-3A(CV-A17)-infected cells.
In the AiV model, a complex of 3A and ACBD3 is involved in the recruitment of PI4KIIIβ to membranous organelles to promote genome replication (30). We therefore investigated, by immunofluorescence microscopy, whether ACBD3 downregulated PV replication in our model by altering the recruitment of PI4KIIIβ to the virus-induced foci concentrated in the perinuclear region shown in Fig. 3B.
IMR5 cells were transfected with siRNA-ACBD3 or siRNA-ctrl and then mock infected or infected with PVS2 or PVS2-3A(CA-17). Endogenous ACBD3 and PI4KIIIβ proteins were immunolabeled at the time points indicated, with anti-ACBD3 and anti-PI4KIIIβ antibodies, respectively. In mock-infected cells transfected with siRNA-ctrl, the endogenous ACBD3 and PI4KIIIβ proteins colocalized in a region corresponding to the Golgi apparatus (Fig. 9). In siRNA-ctrl-transfected cells infected with PVS2 or PVS2-3A(CA-17), PI4KIIIβ colocalized with ACBD3 at 6 h p.i. (Fig. 9), in foci concentrated in the perinuclear region corresponding to the replication sites previously described (Fig. 3B). This colocalization was confirmed by calculating the Pearson correlation coefficient. The silencing of ACBD3 in mock-infected cells or cells infected with PVS2 or PVS2-3A(CV-A17) had no effect on the subcellular distribution of PI4KIIIβ. Thus, the downregulation of PV replication by ACBD3 does not seem to involve a change in the recruitment of PI4KIIIβ to replication sites in our model.
Fig 9.

Subcellular localization of PI4KIIIβ in siRNA-ctrl or siRNA-ACBD3-transfected IMR5 cells infected with PVS2 or PVS2-3A(CV-A17). Cells were transfected with siRNA-ctrl or siRNA-ACBD3. Two days after transfection, cells were mock infected (upper panel) or infected with PVS2 (middle panel) or PVS2-3A(CV-A17) (bottom panel) at an MOI of 50 TCID50/cell. Six hours after infection, endogenous ACBD3 and PI4KIIIβ proteins were subjected to immunofluorescence staining with antibodies directed against ACBD3 (red) and PI4KIIIβ (green), respectively. The merge image is an overlay of the ACBD3 and the PI4KIIIβ images, in which the nuclei are stained with DAPI (white). Pearson correlation coefficients (r) were calculated to assess the degree of colocalization of ACBD3 with PI4KIIIβ. *, Pearson correlation coefficient values of ≥0.5 are considered to indicate a significant positive correlation.
The residue in position 12 of 3A maps outside of the region required for ACBD3 binding.
We investigated whether the amino acid in position 12 of 3A was located in the portion of 3A mediating binding to ACBD3, by generating a series of 3A proteins with N- and C-terminal truncations (3A Δ), named according to the amino acids deleted (Fig. 10A). These truncated 3A proteins were fused to GST, and their ability to interact with endogenous ACBD3 was assessed in GST pulldown assays, as described above. GST alone and full-length 3A fused to GST were used as negative and positive controls, respectively. The deleted proteins 3A(Δ1-18) and 3A(Δ59-87) interacted with ACBD3, whereas 3A proteins with more extensive N- or C-terminal deletions did not (Fig. 10B). These results suggest that residues 19 to 58, corresponding to the unfolded region and part of the alpha helix of the structured region of the 3A protein, include the region required for ACBD3 binding. This result is consistent with the findings reported by Greninger et al. (55) while the present study was under review.
Fig 10.

Mapping of the ACBD3 binding site on 3A(PVS2). (A) Schematic representation of the 3A(PVS2) proteins with N-terminal and C-terminal truncations. Gray bars represent each of the truncated 3A(PVS2) proteins, with the deleted amino acid positions indicated. Truncated 3A(PVS2) proteins were named according to the amino acid sequences deleted. The complete amino acid sequence of PVS2 3A is shown (dark bar) with the Val residue in position 12 indicated with a star. The conserved hydrophobic domain and structured region are indicated. The ACBD3-3A binding data are summarized on the right. The dotted lines indicate the 3A protein region containing the amino acids required for ACBD3 binding. (B) Analysis of interaction between ACBD3 and C-terminally (left panel) or N-terminally (right panel) truncated 3A(PVS2) proteins in GST pulldown assays. HEK-293T cells were transfected with expression vectors encoding truncated 3A(PVS2) proteins fused to GST or with GST alone. Two days posttransfection, the cell lysates were subjected to GST pulldown assays. Endogenous ACBD3 and GST proteins were detected by Western blotting with antibodies directed against ACBD3 and GST, respectively. (C) GST pulldown assay on lysates from HEK-293T cells transfected with expression vectors encoding 3A(PVS2)-GST, 3A(CV-A17)-GST, 3A(PVS2-V12I)-GST, or GST alone. Two days posttransfection, the cells were collected and subjected to GST pulldown analysis. Endogenous ACBD3 and GST were detected by Western blotting with antibodies directed against ACBD3 and GST, respectively.
The residue in position 12 is located close to the region required for ACBD3 binding. We therefore assessed whether the V12I substitution in 3A affected the interaction with ACBD3 in a GST pulldown assay on HEK-293T cells transfected with expression vectors encoding GST alone or GST fused to 3A(PVS2), 3A(CV-A17), or 3A(PVS2-V12I). ACBD3 copurified similarly with all three forms of 3A-GST (Fig. 10C). Thus, the residue in position 12 associated with the sensitivity to ACBD3 of PVS2 replication seems to be located outside the region required for ACBD3 binding, and the V12I substitution does not seem to affect the interaction with ACBD3.
DISCUSSION
In most of the genomes of the PVS2-derived cVDPVs responsible for polio outbreaks in Madagascar, the region encoding nonstructural protein 3A is related to that of field CV-A17 strains (5, 8). We investigated the repercussions of this exchange by assessing the role of the 3A proteins of PVS2 and CV-A17 and their putative cellular partners in viral replication. We showed that the 3A proteins of both PVS2 and CV-A17 interacted with a Golgi protein, ACBD3. These findings are consistent with reports, published while the present study was in progress, showing that ACBD3 binds 3A from AiV and other picornaviruses, including PV, human rhinovirus 14, coxsackieviruses B2, B3, and B5, and bovine kobuvirus (30, 32). The 3A proteins of some other picornaviruses (cardioviruses and enterovirus 71) do not appear to associate with ACBD3 (32).
In neuroblastoma IMR5 cells infected with either PVS2 or a recombinant PVS2 encoding a 3A protein from CV-A17, we showed that ACBD3 colocalized with the 3A protein and with replicating viral RNAs. The 3A protein also inhibits cellular protein transport in PV-infected cells (48). We therefore investigated the possible role of an interaction between ACBD3 and 3A in this process. The downregulation of ACBD3 levels with siRNA did not seem to affect the inhibition of protein transport in cells infected with PVS2 or PVS2-3A(CV-A17), suggesting that ACBD3 is not involved in this inhibition (data not shown). However, ACBD3 downregulation led to a significant increase in viral growth for PVS2 and PVS2-3A(CV-A17) at early time points after infection. Similar results were obtained with replicons derived from PVS2 and PVS2-3A(CV-A17), in ACBD3-silenced cells. Our results, therefore, strongly suggest that ACBD3 delayed viral growth at the level of replication in our model. Furthermore, this inhibitory effect of ACBD3 on viral replication does not seem to depend on the cell model because we obtained similar results in several cell lines.
Interestingly, ACBD3 inhibited viral replication more strongly with PVS2 than with PVS2-3A(CV-A17). Furthermore, the amino acid in position 12 of 3A appeared to be involved in modulating the sensitivity of viral replication to ACBD3, although it does not seem to be located in the region required for ACBD3 binding (55; the present study). Thus, our data suggest that the residue in position 12 of 3A plays no role in binding to ACBD3, but it could influence the functional outcome of this interaction. Indeed, this residue is located in the N-terminal region of 3A interacting with GBF1, another cellular factor important for replication (54). However, replacement of the Val residue in position 12 of the 3A from PVS2 with an Ile residue, as in the 3A from CV-A17, did not modify the binding of 3A to GBF1. Thus, the potential advantage of PVS2-3A(CV-A17) over PVS2 does not seem to result from a more favorable interaction of GBF1 with 3A(CV-A17) than with 3A(PVS2).
In the AiV model, Sasaki et al. (30) showed that ACBD3 interacts with several nonstructural viral proteins, including 3A, and recruits PI4KIIIβ to sites of viral replication. Using AiV replicons, these authors showed that the silencing of ACBD3 inhibits AiV RNA replication, indicating that ACBD3 plays an important role in viral replication. Greninger et al. (32) obtained similar results but, surprisingly, they also found that ACBD3 silencing prevented the replication of PV replicons. Finally, in the CV-B3 model, viral replication seems to be stimulated, rather than inhibited, upon ACDB3 knockdown (Frank van Kuppeveld, personal communication). All of these results, together with our own, suggest that the role of ACBD3 in viral replication may depend on the viral species and strain considered.
It has recently been shown that the ACBD3-binding region of AiV 3A involves multiple portions of this protein, whereas it appears to be well delimited in the poliovirus 3A protein (55). Thus, differences between the ACBD3-binding sites of 3A may reflect differences in the role of ACBD3 in replication between picornaviruses.
Other cellular factors, such as ACBD3-interacting factors, may also play a role in the differential effects of this cellular protein on picornavirus replication. Indeed, during the revision of this article, Greninger et al. (55) identified the putative Rab33 GTPase-activating proteins TBC1D22A and TBC1D22B as new ACBD3-interacting factors. They also showed that TBC1D22A competes directly with PI4KIIIβ for binding to the same site on ACBD3. Furthermore, poliovirus and AiV 3A proteins may differentially modulate the interaction of PI4KIIIβ and TBC1D22A/B with ACBD3 (55). In the present study, we found that the downregulation of viral replication by ACBD3 in PVS2-infected cells, or, to a lesser extent, in PVS2-3A(CV-A17)-infected cells, did not seem to be due to a failure of PI4KIIIβ recruitment to the perinuclear region corresponding to the replication sites of infected cells. Van der Schaar et al. (56) recently described 3A CV-B3 mutants that can bypass host factor PI4KIIIβ. Thus, other, as-yet-unidentified alternative pathways may be involved in the downregulation of viral replication by ACBD3 in our model.
Another ACBD3-interacting factor, the metal-dependent protein phosphatase 1L (PPM1L), was also recently identified (57). PPM1L was shown to be involved in the regulation of ceramide trafficking at ER-Golgi membrane contact sites (58). It would be interesting to assess whether inhibition of viral replication by ACBD3 in our model is related to the regulation of ceramide trafficking.
As outlined above, the effects of ACBD3 on viral replication may differ according to picornavirus strains. A single cellular protein may thus help the virus by promoting its replication or, conversely, defend the cell against the virus by delaying its replication. As mentioned above, a recently identified host factor involved in PV replication, VCP/p97, has been shown to interact with the nonstructural viral proteins 3AB and 2BC (33). Interestingly, as for ACBD3, the effect of VCP/p97 downregulation on viral replication differs between picornaviruses, with strong suppression for PV, an apparent lack of effect for CV-B3, and enhancement for AiV. The authors of that study suggested that alternative or opposite pathways might be involved in the replication of these viruses or that a difference in host factor levels might be required for the optimal replication of these viruses.
In conclusion, we have shown that a recombinant PVS2 encoding a 3A protein from CV-A17 is more resistant to the inhibitory effect of ACBD3 than the parental vaccine PVS2 strain. The mechanism by which ACBD3 attenuates these viruses remains to be clarified, but our results suggest that exchanges of nonstructural proteins can modify the relationships between enterovirus recombinants and cellular interactors and may thus be one of the factors favoring their emergence.
ACKNOWLEDGMENTS
We thank Florence Colbère-Garapin for invaluable support throughout this study and for fruitful discussions. We also thank Pierre Charneau, who provided the HEK-293T-T7 cells, and Emmanuelle Perret and Anne Danckaert (Plate-Forme d'Imagerie Dynamique, Institut Pasteur, Paris, France) for assistance with fluorescence microscopy and the Plateforme de Génotypage des Pathogènes et Santé Publique (Institut Pasteur, Paris, France) for sequencing. We thank Santos Susin and Victor Yuste (Institut Pasteur, Paris, France) for providing IMR5 cells.
This study was supported by grants from the Institut Pasteur (Transverse Research Program PTR 276), the Agence Nationale de la Recherche (ANR-09-MIEN-019), and the Fondation pour la Recherche Médicale (DMI20091117313). F.T. and C.B. were supported by grants from the Ministère de l'Enseignement Supérieur et de la Recherche. S.J. received funding from the French Délégation Générale pour l'Armement and the Centre National pour la Recherche Scientifique. M.C. was supported by a stipend from the Pasteur-Paris University International Ph.D. program and by the Institut Carnot Pasteur Maladies Infectieuses.
Footnotes
Published ahead of print 8 August 2013
REFERENCES
- 1.Pallansch M, Roos R. 2007. Enteroviruses: polioviruses, coxsackieviruses, echoviruses, and newer enteroviruses, p 839–893. In Knipe DM, Howley PM. (ed), Fields virology, 4th ed, vol 1 Lippincott/Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Kew OM, Sutter RW, de Gourville EM, Dowdle WR, Pallansch MA. 2005. Vaccine-derived polioviruses and the endgame strategy for global polio eradication. Annu. Rev. Microbiol. 59:587–635. [DOI] [PubMed] [Google Scholar]
- 3.Racaniello VR. 2007. Picornaviridae: the viruses and their replication, p 795–838. In Knipe DM, Howley PM. (ed), Fields virology, 4th ed, vol 1 Lippincott/Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 4.Combelas N, Holmblat B, Joffret ML, Colbere-Garapin F, Delpeyroux F. 2012. Recombination between poliovirus and coxsackie A viruses of species C: a model of viral genetic plasticity and emergence. Viruses 3:1460–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rakoto-Andrianarivelo M, Guillot S, Iber J, Balanant J, Blondel B, Riquet F, Martin J, Kew O, Randriamanalina B, Razafinimpiasa L, Rousset D, Delpeyroux F. 2007. Co-circulation and evolution of polioviruses and species C enteroviruses in a district of Madagascar. PLoS Pathog. 3:e191. 10.1371/journal.ppat.0030191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rakoto-Andrianarivelo M, Gumede N, Jegouic S, Balanant J, Andriamamonjy SN, Rabemanantsoa S, Birmingham M, Randriamanalina B, Nkolomoni L, Venter M, Schoub BD, Delpeyroux F, Reynes JM. 2008. Reemergence of recombinant vaccine-derived poliovirus outbreak in Madagascar. J. Infect. Dis. 197:1427–1435. [DOI] [PubMed] [Google Scholar]
- 7.Rousset D, Rakoto-Andrianarivelo M, Razafindratsimandresy R, Randriamanalina B, Guillot S, Balanant J, Mauclere P, Delpeyroux F. 2003. Recombinant vaccine-derived poliovirus in Madagascar. Emerg. Infect. Dis. 9:885–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Joffret ML, Jegouic S, Bessaud M, Balanant J, Tran C, Caro V, Holmblat B, Razafindratsimandresy R, Reynes JM, Rakoto-Andrianarivelo M, Delpeyroux F. 2012. Common and diverse features of cocirculating type 2 and 3 recombinant vaccine-derived polioviruses isolated from patients with poliomyelitis and healthy children. J. Infect. Dis. 205:1363–1373. [DOI] [PubMed] [Google Scholar]
- 9.Bienz K, Egger D, Pfister T, Troxler M. 1992. Structural and functional characterization of the poliovirus replication complex. J. Virol. 66:2740–2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bienz K, Egger D, Pfister T. 1994. Characteristics of the poliovirus replication complex. Arch. Virol. 1994(Suppl 9):147–157. [DOI] [PubMed] [Google Scholar]
- 11.Cho MW, Teterina N, Egger D, Bienz K, Ehrenfeld E. 1994. Membrane rearrangement and vesicle induction by recombinant poliovirus 2C and 2BC in human cells. Virology 202:129–145. [DOI] [PubMed] [Google Scholar]
- 12.Suhy DA, Giddings TH, Jr, Kirkegaard K. 2000. Remodeling the endoplasmic reticulum by poliovirus infection and by individual viral proteins: an autophagy-like origin for virus-induced vesicles. J. Virol. 74:8953–8965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Belov GA, Feng Q, Nikovics K, Jackson CL, Ehrenfeld E. 2008. A critical role of a cellular membrane traffic protein in poliovirus RNA replication. PLoS Pathog. 4:e1000216. 10.1371/journal.ppat.1000216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Egger D, Teterina N, Ehrenfeld E, Bienz K. 2000. Formation of the poliovirus replication complex requires coupled viral translation, vesicle production, and viral RNA synthesis. J. Virol. 74:6570–6580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kirkegaard K, Semler BL. 2010. Genome replication II: the process, p 127–140. In Ehrenfeld E, Domingo E, Ross RP. (ed), The picornaviruses. ASM Press, Washington, DC. [Google Scholar]
- 16.Wessels E, Duijsings D, Lanke KH, van Dooren SH, Jackson CL, Melchers WJ, van Kuppeveld FJ. 2006. Effects of picornavirus 3A proteins on protein transport and GBF1-dependent COP-I recruitment. J. Virol. 80:11852–11860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wessels E, Duijsings D, Niu TK, Neumann S, Oorschot VM, de Lange F, Lanke KH, Klumperman J, Henke A, Jackson CL, Melchers WJ, van Kuppeveld FJ. 2006. A viral protein that blocks Arf1-mediated COP-I assembly by inhibiting the guanine nucleotide exchange factor GBF1. Dev. Cell 11:191–201. [DOI] [PubMed] [Google Scholar]
- 18.Bui QT, Golinelli-Cohen MP, Jackson CL. 2009. Large Arf1 guanine nucleotide exchange factors: evolution, domain structure, and roles in membrane trafficking and human disease. Mol. Genet. Genomics 282:329–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Casanova JE. 2007. Regulation of Arf activation: the Sec7 family of guanine nucleotide exchange factors. Traffic 8:1476–1485. [DOI] [PubMed] [Google Scholar]
- 20.Kawamoto K, Yoshida Y, Tamaki H, Torii S, Shinotsuka C, Yamashina S, Nakayama K. 2002. GBF1, a guanine nucleotide exchange factor for ADP-ribosylation factors, is localized to the cis-Golgi and involved in membrane association of the COPI coat. Traffic 3:483–495. [DOI] [PubMed] [Google Scholar]
- 21.Zhao X, Claude A, Chun J, Shields DJ, Presley JF, Melancon P. 2006. GBF1, a cis-Golgi and VTCs-localized ARF-GEF, is implicated in ER-to-Golgi protein traffic. J. Cell Sci. 119:3743–3753. [DOI] [PubMed] [Google Scholar]
- 22.Lanke KH, van der Schaar HM, Belov GA, Feng Q, Duijsings D, Jackson CL, Ehrenfeld E, van Kuppeveld FJ. 2009. GBF1, a guanine nucleotide exchange factor for Arf, is crucial for coxsackievirus B3 RNA replication. J. Virol. 83:11940–11949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kondratova AA, Neznanov N, Kondratov RV, Gudkov AV. 2005. Poliovirus protein 3A binds and inactivates LIS1, causing block of membrane protein trafficking and deregulation of cell division. Cell Cycle 4:1403–1410. [DOI] [PubMed] [Google Scholar]
- 24.Faulkner NE, Dujardin DL, Tai CY, Vaughan KT, O'Connell CB, Wang Y, Vallee RB. 2000. A role for the lissencephaly gene LIS1 in mitosis and cytoplasmic dynein function. Nat. Cell Biol. 2:784–791. [DOI] [PubMed] [Google Scholar]
- 25.Reiner O, Carrozzo R, Shen Y, Wehnert M, Faustinella F, Dobyns WB, Caskey CT, Ledbetter DH. 1993. Isolation of a Miller-Dieker lissencephaly gene containing G protein beta-subunit-like repeats. Nature 364:717–721. [DOI] [PubMed] [Google Scholar]
- 26.Smith DS, Niethammer M, Ayala R, Zhou Y, Gambello MJ, Wynshaw-Boris A, Tsai LH. 2000. Regulation of cytoplasmic dynein behaviour and microtubule organization by mammalian Lis1. Nat. Cell Biol. 2:767–775. [DOI] [PubMed] [Google Scholar]
- 27.Delang L, Paeshuyse J, Neyts J. 2012. The role of phosphatidylinositol 4-kinases and phosphatidylinositol 4-phosphate during viral replication. Biochem. Pharmacol. 84:1400–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hsu NY, Ilnytska O, Belov G, Santiana M, Chen YH, Takvorian PM, Pau C, van der Schaar H, Kaushik-Basu N, Balla T, Cameron CE, Ehrenfeld E, van Kuppeveld FJ, Altan-Bonnet N. 2010. Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell 141:799–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yamashita T, Sakae K, Tsuzuki H, Suzuki Y, Ishikawa N, Takeda N, Miyamura T, Yamazaki S. 1998. Complete nucleotide sequence and genetic organization of Aichi virus, a distinct member of the Picornaviridae associated with acute gastroenteritis in humans. J. Virol. 72:8408–8412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sasaki J, Ishikawa K, Arita M, Taniguchi K. 2012. ACBD3-mediated recruitment of PI4KB to picornavirus RNA replication sites. EMBO J. 31:754–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sohda M, Misumi Y, Yamamoto A, Yano A, Nakamura N, Ikehara Y. 2001. Identification and characterization of a novel Golgi protein, GCP60, that interacts with the integral membrane protein giantin. J. Biol. Chem. 276:45298–45306. [DOI] [PubMed] [Google Scholar]
- 32.Greninger AL, Knudsen GM, Betegon M, Burlingame AL, Derisi JL. 2012. The 3A protein from multiple picornaviruses utilizes the Golgi adaptor protein ACBD3 to recruit PI4KIIIβ. J. Virol. 86:3605–3616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arita M, Wakita T, Shimizu H. 2012. Valosin-containing protein (VCP/p97) is required for poliovirus replication and is involved in cellular protein secretion pathway in poliovirus infection. J. Virol. 86:5541–5553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pollard SR, Dunn G, Cammack N, Minor PD, Almond JW. 1989. Nucleotide sequence of a neurovirulent variant of the type 2 oral poliovirus vaccine. J. Virol. 63:4949–4951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Autret A, Martin-Latil S, Mousson L, Wirotius A, Petit F, Arnoult D, Colbere-Garapin F, Estaquier J, Blondel B. 2007. Poliovirus induces Bax-dependent cell death mediated by c-Jun NH2-terminal kinase. J. Virol. 81:7504–7516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reed LJ, Muench M. 1938. A simple method for estimating fifty percent endpoints. Am. J. Hyg. (Lond.) 1938:493–497. [Google Scholar]
- 37.Pellet J, Tafforeau L, Lucas-Hourani M, Navratil V, Meyniel L, Achaz G, Guironnet-Paquet A, Aublin-Gex A, Caignard G, Cassonnet P, Chaboud A, Chantier T, Deloire A, Demeret C, Le Breton M, Neveu G, Jacotot L, Vaglio P, Delmotte S, Gautier C, Combet C, Deleage G, Favre M, Tangy F, Jacob Y, Andre P, Lotteau V, Rabourdin-Combe C, Vidalain PO. 2010. ViralORFeome: an integrated database to generate a versatile collection of viral ORFs. Nucleic Acids Res. 38:D371–D378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Riquet FB, Blanchard C, Jegouic S, Balanant J, Guillot S, Vibet MA, Rakoto-Andrianarivelo M, Delpeyroux F. 2008. Impact of exogenous sequences on the characteristics of an epidemic type 2 recombinant vaccine-derived poliovirus. J. Virol. 82:8927–8932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jegouic S, Joffret ML, Blanchard C, Riquet FB, Perret C, Pelletier I, Colbere-Garapin F, Rakoto-Andrianarivelo M, Delpeyroux F. 2009. Recombination between polioviruses and co-circulating coxsackie A viruses: role in the emergence of pathogenic vaccine-derived polioviruses. PLoS Pathog. 5:e1000412. 10.1371/journal.ppat.1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Caignard G, Guerbois M, Labernardiere JL, Jacob Y, Jones LM, Wild F, Tangy F, Vidalain PO. 2007. Measles virus V protein blocks Jak1-mediated phosphorylation of STAT1 to escape IFN-alpha/beta signaling. Virology 368:351–362. [DOI] [PubMed] [Google Scholar]
- 41.Bessaud M, Delpeyroux F. 2012. Development of a simple and rapid protocol for the production of customized intertypic recombinant polioviruses. J. Virol. Methods 186:104–108. [DOI] [PubMed] [Google Scholar]
- 42.Bolte S, Cordelieres FP. 2006. A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 224:213–232. [DOI] [PubMed] [Google Scholar]
- 43.Strauss DM, Glustrom LW, Wuttke DS. 2003. Towards an understanding of the poliovirus replication complex: the solution structure of the soluble domain of the poliovirus 3A protein. J. Mol. Biol. 330:225–234. [DOI] [PubMed] [Google Scholar]
- 44.Teterina NL, Pinto Y, Weaver JD, Jensen KS, Ehrenfeld E. 2011. Analysis of poliovirus protein 3A interactions with viral and cellular proteins in infected cells. J. Virol. 85:4284–4296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Teterina NL, Lauber C, Jensen KS, Levenson EA, Gorbalenya AE, Ehrenfeld E. 2011. Identification of tolerated insertion sites in poliovirus nonstructural proteins. Virology 409:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bienz K, Egger D, Rasser Y, Bossart W. 1983. Intracellular distribution of poliovirus proteins and the induction of virus-specific cytoplasmic structures. Virology 131:39–48. [DOI] [PubMed] [Google Scholar]
- 47.Jurgeit A, Moese S, Roulin P, Dorsch A, Lotzerich M, Lee WM, Greber UF. 2010. An RNA replication-center assay for high content image-based quantifications of human rhinovirus and coxsackievirus infections. Virol. J. 7:264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Doedens JR, Kirkegaard K. 1995. Inhibition of cellular protein secretion by poliovirus proteins 2B and 3A. EMBO J. 14:894–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Neznanov N, Kondratova A, Chumakov KM, Angres B, Zhumabayeva B, Agol VI, Gudkov AV. 2001. Poliovirus protein 3A inhibits tumor necrosis factor (TNF)-induced apoptosis by eliminating the TNF receptor from the cell surface. J. Virol. 75:10409–10420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kitamura N, Semler BL, Rothberg PG, Larsen GR, Adler CJ, Dorner AJ, Emini EA, Hanecak R, Lee JJ, van der Werf S, Anderson CW, Wimmer E. 1981. Primary structure, gene organization and polypeptide expression of poliovirus RNA. Nature 291:547–553. [DOI] [PubMed] [Google Scholar]
- 51.La Monica N, Meriam C, Racaniello VR. 1986. Mapping of sequences required for mouse neurovirulence of poliovirus type 2 Lansing. J. Virol. 57:515–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stanway G, Hughes PJ, Mountford RC, Reeve P, Minor PD, Schild GC, Almond JW. 1984. Comparison of the complete nucleotide sequences of the genomes of the neurovirulent poliovirus P3/Leon/37 and its attenuated Sabin vaccine derivative P3/Leon 12a1b. Proc. Natl. Acad. Sci. U. S. A. 81:1539–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Toyoda H, Kohara M, Kataoka Y, Suganuma T, Omata T, Imura N, Nomoto A. 1984. Complete nucleotide sequences of all three poliovirus serotype genomes. Implication for genetic relationship, gene function and antigenic determinants. J. Mol. Biol. 174:561–585. [DOI] [PubMed] [Google Scholar]
- 54.Wessels E, Duijsings D, Lanke KH, Melchers WJ, Jackson CL, van Kuppeveld FJ. 2007. Molecular determinants of the interaction between coxsackievirus protein 3A and guanine nucleotide exchange factor GBF1. J. Virol. 81:5238–5245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Greninger AL, Knudsen GM, Betegon M, Burlingame AL, DeRisi JL. 2013. ACBD3 interaction with TBC1 domain 22 protein is differentially affected by enteroviral and kobuviral 3A protein binding. mBio 4:e00098-00013. 10.1128/mBio.00098-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.van der Schaar HM, van der Linden L, Lanke KH, Strating JR, Purstinger G, de Vries E, de Haan CA, Neyts J, van Kuppeveld FJ. 2012. Coxsackievirus mutants that can bypass host factor PI4KIIIβ and the need for high levels of PI4P lipids for replication. Cell Res. 22:1576–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shinoda Y, Fujita K, Saito S, Matsui H, Kanto Y, Nagaura Y, Fukunaga K, Tamura S, Kobayashi T. 2012. Acyl-CoA binding domain containing 3 (ACBD3) recruits the protein phosphatase PPM1L to ER-Golgi membrane contact sites. FEBS Lett. 586:3024–3029. [DOI] [PubMed] [Google Scholar]
- 58.Saito S, Matsui H, Kawano M, Kumagai K, Tomishige N, Hanada K, Echigo S, Tamura S, Kobayashi T. 2008. Protein phosphatase 2Cεis an endoplasmic reticulum integral membrane protein that dephosphorylates the ceramide transport protein CERT to enhance its association with organelle membranes. J. Biol. Chem. 283:6584–6593. [DOI] [PubMed] [Google Scholar]