

Abstract
The interleukin-6 (IL-6) receptor, which exists as membrane-bound and soluble forms, plays critical roles in the immune response. The soluble IL-6 receptor (sIL6R) has been identified as a potential therapeutic target for preventing coronary heart disease. However, little is known about the role of this receptor during viral infection. In this study, we show that sIL6R, but not IL-6, is induced by viral infection via the cyclooxygenase-2 pathway. Interestingly, sIL6R, but not IL-6, exhibited extensive antiviral activity against DNA and RNA viruses, including hepatitis B virus, influenza virus, human enterovirus 71, and vesicular stomatitis virus. No synergistic effects on antiviral action were observed by combining sIL6R and IL-6. Furthermore, sIL6R mediated antiviral action via the p28 pathway and induced alpha interferon (IFN-α) by promoting the nuclear translocation of IFN regulatory factor 3 (IRF3) and NF-κB, which led to the activation of downstream IFN effectors, including 2′,5′-oligoadenylate synthetase (OAS), double-stranded RNA-dependent protein kinase (PKR), and myxovirus resistance protein (Mx). Thus, our results demonstrate that sIL6R, but not IL-6, plays an important role in the host antiviral response.
INTRODUCTION
Interleukin 6 (IL-6), an inflammatory cytokine produced mainly by T cells, macrophages, and adipocytes, promotes inflammatory responses via two types of receptors. The membrane-bound IL-6 receptor (IL6R) is expressed predominantly by hepatocytes, neutrophils, monocytes/macrophages, and some lymphocytes (1, 2). The circulating soluble form of the IL6R (sIL6R), which can be detected in various bodily fluids and is secreted by monocytes, hepatocytes, and endothelial cells (3), is generated by two independent mechanisms, namely, limited proteolysis of the membrane-bound protein and translation from an alternatively spliced mRNA (4). Two distinct isoforms of sIL6R have been identified. The first is shed from the cell surface via proteolytic cleavage of the membrane-bound IL6R (PC-sIL6R) (5, 6), whereas the second is the product of differential mRNA splicing (DS-sIL6R) (7, 8). Peters et al. (2) have shown that TAPI, a specific inhibitor of the mammalian shedding metalloproteinases, inhibits IL6R shedding (8). The two modes of IL-6 activation are presented as either classical IL-6 activation via membrane-bound IL6R (classical IL-6 signaling) or sIL6R-mediated cell signaling (IL-6 trans-signaling). In both cases, responses are elicited through engagement with the membrane-bound gp130 receptor subunit. Classical IL-6 signaling is unaffected by soluble gp130 (sgp130) but preferentially binds the IL-6/sIL6R complex to antagonize IL-6 trans-signaling (9).
Human ciliary neurotrophic factor (CNTF) is a neurotrophic cytokine that exerts a neuroprotective effect in multiple sclerosis and amyotrophic lateral sclerosis. Although CNTF and its receptor are expressed mostly in the nervous system, an extracellular portion of its receptor has been shown to be homologous with the IL6R (10). CNTF can use both the membrane-bound and the soluble form of human IL6R as a substitute for its cognate α-receptor (11). IL-27 consists of the cytokine subunit p28 and the nonsignaling α-receptor EBI3. Liu et al. (12) have shown that IL-27 activates STAT1/STAT2 and STAT3 signaling and double-stranded RNA (dsRNA)-dependent protein kinase (PKR), indicating that alpha interferon (IFN-α) contributes in part to IL-27-mediated antiviral function. Crabé et al. (13) have shown that p28 forms a complex with the IL6R and induces STAT1 and STAT3 signal transduction in IL-27-responsive cells. p28 has been shown to act additionally via the nonsignaling membrane-bound IL-6 receptor as an agonistic cytokine but also as a gp130 β-receptor antagonist, leading to inhibition of IL-6 signaling (14).
Cyclooxygenase (COX) is the rate-limiting enzyme in the biosynthesis of prostaglandins and thromboxanes from arachidonic acid. Two COX isoforms have been discovered: COX-1 and COX-2. COX-1 is constitutively expressed in almost all human tissues, and COX-2 is induced by inflammatory stimuli, resulting in increased prostanoid synthesis in inflamed tissues. Research has demonstrated that COX-2 expression is also stimulated by viral proteins, such as influenza A virus (IAV) nonstructural protein 1 (NS1) (15), Epstein-Barr virus latent membrane protein 1 (16), the hepatitis C virus core and NS5A proteins (17), and hepatitis B virus (HBV) HBx (18). Moreover, COX-2 is overexpressed in liver cirrhosis, contributing to the overproduction of prostaglandins, which are major effectors of the inflammation and hyperdynamic circulation associated with hepatocellular carcinoma development in cirrhosis (19).
Type I IFNs, primarily IFN-α/β, produced by virus-infected cells induce the expression of more than 400 interferon-stimulated genes (ISGs), whose products cooperate to induce an antiviral state (20). ISG15, the Mx proteins, the 2′,5′-oligoadenylate synthetase (OAS)-directed RNase L pathway, and PKR show differing levels of responsiveness to type I IFNs. In humans, these cytokines comprise 13 IFN-α subtypes and engage the ubiquitously expressed IFN-α receptor (IFNAR) complex, which is composed of IFNAR1 and IFNAR2 (21, 22). The production of type I IFNs is controlled by the transcription factor NF-κB, which has served as a standard for inducible transcription factors for more than 20 years. The NF-κB family consists of five members, p50, p52, p65 (RelA), c-Rel, and RelB, encoded by NF-κB1, NF-κB2, RELA, REL, and RELB, respectively. Each member possesses an N-terminal Rel homology domain, which is responsible for DNA binding and homo- and heterodimerization (23).
Numerous published studies have focused on the association between IL-6 and viral infection. In accordance with the role of IL-6 as a proinflammatory cytokine, increased levels in plasma have been correlated with disease severity (24, 25). IL-6 production in follicular B cells in the draining lymph node is a necessary early event during the antiviral response that is sufficient to induce critical cytokines, including IL-21 (26). There are several type of cells do not express the membrane-bound IL6R, so the IL-6/sIL6R complex was used as a model to stimulate cells and activate IL-6 trans-signaling responses. The IL-6/sIL6R complex is critically involved in the maintenance of a disease state, by promoting the transition from acute to chronic inflammation (9). However, little is known about the role of the IL6R during viral infection. Nevertheless, recent research has indicated that the IL6R could play a crucial role in preventing coronary heart disease (27). Membrane-bound IL6R also serves as a target of miR-124 in the microRNA feedback-inflammatory loop, during which hepatocyte nuclear factor 4α (HNF4α) initiates hepatocellular transformation (1, 28). Limited expression of the membrane-bound IL6R has hindered further research on its biological function; however, the sIL6R could play an important role in the systemic inflammatory response by circulating among different bodily fluids. In this study, we show that expression of the sIL6R, but not IL-6, induced by viral infection may be regulated by COX-2. Furthermore, unlike the membrane-bound receptor, the soluble form elicits extensive antiviral activity in response to infection by DNA and RNA viruses via activation of the type I IFN pathway. Our results uncover a distinct role for the sIL6R in the host cellular response to viral infection, thus providing a potential candidate and strategy for the development of novel antiviral therapeutics.
MATERIALS AND METHODS
Clinical samples.
Serum and throat swab samples were collected from 17 healthy individuals and 17 IAV-infected patients admitted to the Hubei Provincial Center for Disease Control and Prevention. Serum and peripheral blood samples were obtained from 22 healthy individuals with no history of liver disease and 22 HBV-infected patients admitted to Renmin Hospital (Wuhan University). The study was conducted according to the principles of the Declaration of Helsinki and was approved by the Institutional Review Board of the College of Life Science, Wuhan University, in accordance with the guidelines for the protection of human subjects. Written informed consent was obtained from each participant.
Cell culture and virus.
A549 cells were cultured in F12K medium containing 10% fetal bovine serum (FBS). Huh7, Huh7.5.1, MDCK, and Vero cells were cultured in Dulbecco's modified Eagle's medium (DMEM) containing 10% FBS. RD cells were cultured in RPMI 1640 medium containing 10% FBS. The influenza virus A/Hong Kong/498/97 (H3N2) strain used in this study was provided by the China Center for Type Culture Collection. A recombinant vesicular stomatitis virus carrying the enhanced green fluorescent protein gene (VSV-eGFP) was provided by Mingzhou Chen of Wuhan University. The human enterovirus 71 (EV71) strain used in this study is from Xiangyang (GenBank accession number JN230523.1).
Plasmids, siRNA, antibodies, chemical reagents, and inhibitors.
The COX-2 expression plasmid pCMV-COX-2 has been described previously (29). pHBV-1.3 (adw) was generated from the HBV genome as described previously (30). All constructs was confirmed by DNA sequencing. All small interfering RNAs (siRNAs) and irrelevant control siRNAs (si-nc) were purchased from GenePharma with the following sequences: si-COX-2, 5′-GGACUUAUUGGGUAAUGUUATT-3′; si-p28, 5′-UCCCUUGCUCCUGGUUCAATT-3′; si-CNTF, 5′-GCUGAUGGGAUGCCUATUUAT-3′; si-gp130, 5′-GCACUUGCAACAUUCUUTT-3′.
Antibodies against OAS, PKR, IFN regulatory factor 3 (IRF3), RelA, NF-κB1, IFNAR1, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies against sIL6R (eBioscience, San Diego, CA, USA), lamin A (Epitomics), and COX-2 (Cayman Chemical, Ann Arbor, MI, USA) were also used. Neutralizing antibodies (NAb) against sIL6R and IL-6 were purchased from R&D Systems, while neutralizing antibodies against human IFN-α (Hu-IFN-α) and Hu-IFN-β were purchased from PBL Interferon Source.
Recombinant human sIL6R (rhsIL6R) protein was purchased from R&D Systems, and recombinant human IL-6 (rhIL-6) protein was purchased from eBioscience. Prostaglandin E2 (PGE2) and NS398 were purchased from Sigma-Aldrich (St. Louis, MO, USA). TAPI was purchased from Santa Cruz Biotechnology.
Isolation of PBMCs.
Peripheral blood mononuclear cells (PBMCs) were obtained by density centrifugation of blood samples diluted 1:1 in pyrogen-free saline over Histopaque (Hao Yang Biotech) as described previously (31). Cells were washed twice in saline and were cultured in RPMI 1640 medium.
Isolation and viral infection of human AT II cells.
Human alveolar type II (AT II) cells were isolated from deidentified human lungs that were not suitable for transplantation and were donated for medical research. We purchased AT II cells from Wuhan PriCells Biotechnology & Medicine Co., Ltd. Primary cultured differentiated human AT II cells were inoculated with influenza A viruses for 1 h at a multiplicity of infection (MOI) of 1 and were harvested at 24 hpi for quantitative reverse transcription-PCR (qRT-PCR) analysis.
MTT method.
Methylthiazolyldiphenyl-tetrazolium bromide (5 mg/ml) was dissolved in phosphate-buffered saline (PBS) (0.01 M; pH 7.4) and was stirred with a constant-temperature magnetic stirrer for 30 min. Then it was passed through a 0.22-μm microfiltration membrane to remove bacteria and was kept no longer than 2 weeks. A549 cells were seeded in a 96-well culture plate at a density of 104/cm2, and then 200 μl DMEM containing 10% FBS was added to each well. After the cells had been cultured for 24 h, either they were left untreated or NS398 (20 μM, 40 μM, or 80 μM) and/or dimethyl sulfoxide (DMSO) (0.25%, 0.05%, or 0.1%) was added to the medium. After 3 days, the cell culture plate was taken out for the MTT assay. We added 20 μl MTT reagents to each well and then cultured the cells for 4 h. Afterwards, the medium was removed, and 150 μl DMSO was added to each well to dissolve formazan. The 96-well culture plate was agitated for 10 min on a shaker. Finally, a well with DMSO but without cells was used to adjust the zero value, and the optical density (OD) value of each well was determined at 490 nm.
qRT-PCR analysis.
qRT-PCR analysis was performed to determine relative mRNA levels. Total RNA was isolated with TRIzol (Invitrogen, Carlsbad, CA, USA). Cellular RNA samples were reverse transcribed with random primers. qRT-PCR was performed using a LightCycler 480 system (Roche, Indianapolis, IN, USA) with the following primers: for GAPDH, 5′-AAGGCTGTGGGCAAGG-3′ (sense) and 5′-TGGAGGAGTGGGTGTCG-3′ (antisense); for sIL6R, 5′-GCGACAAGCCTCCCAGGTTC-3′ (sense) and 5′-GTGCCACCCAGCCAGCTATC-3′ (antisense); for COX-2, 5′-TGCATTCTTTGCCCAGCACT-3′ (sense) and 5′-AAAGGCGCAGTTTACGCTGT-3′ (antisense); for IL-6, 5′-GGTACATCCTCGACGGCATCTCA-3′ (sense) and 5′-TGCACAGCTCTGGCTTGTTCCTC-3′ (antisense); for IFN-α, 5′-TTTCTCCTGCCTGAAGGACAG-3′ (sense) and 5′-GCTCATGATTTCTGCTCTGACA-3′ (antisense); for IFN-β, 5′-AAAGAAGCAGCAATTTTCAGC-3′ (sense) and 5′-CCTTGGCCTTCAGGTAATGCA-3′ (antisense); for Mx, 5′-GCCGGCTGTGGATATGCTA-3′ (sense) and 5′-TTTATCGAAACATCTGTGAAAGCAA-3′ (antisense); for OAS, 5′-AGAAGGCAGCTCACGAAACC-3′ (sense) and 5′-CCACCACCCAAGTTTCCTGTA-3′ (antisense); for PKR, 5′-AGAGTAACCGTTGGTGACATAACCT-3′ (sense) and 5′-GCAGCCTCTGCAGCTCTATGTT-3′ (antisense); for IFNAR1, 5′-AAAATGGCAATGATAGG-3′ (sense) and 5′-CAGGCTATGCACCCTCCTTCC-3′ (antisense); for VP-1, 5′-CCCTTTAGTGGTTAGGATTT-3′ (sense) and 5′-CACCAGTTGGTTTAATGGAG-3′ (antisense).
Transfection and luciferase reporter assays.
Cells were plated at a density of 4 × 105 per 24-well or 6-well plate, depending on the experiment, and were grown to 80% confluence prior to transfection. Cells were transfected with Lipofectamine 200 (Invitrogen) for 24 h, serum starved for an additional 24 h, and then harvested. A Renilla luciferase reporter assay system (Promega, Madison, WI, USA) was used to measure the luciferase activity of each sample 48 h after transfection. Renilla luciferase activities were determined as internal controls for transfection efficiency.
Western blotting, nuclear extraction, and enzyme-linked immunosorbent assays (ELISA).
Whole-cell lysates were prepared by lysing cells in PBS containing 0.01% Triton X-100, 0.01% EDTA, and 10% protease inhibitor mixture (Roche). To separate and collect the cytosolic and nuclear protein fractions, cells were washed with ice-cold PBS and were collected by centrifugation. The resulting pellets were resuspended in hypotonic buffer (10 mM HEPES [pH 7.9], 10 mM KCl, 0.5 mM dithiothreitol [DTT], 10% protease mixture inhibitor) for 15 min on ice and were vortexed for 10 s. Nuclei were pelleted by centrifugation at 13,000 × g for 1 min, and the pellets and cytosolic protein-containing supernatants were collected. The protein concentration of each sample was determined using a Bradford assay kit (Bio-Rad, Hercules, CA, USA). A 100-μg aliquot of each sample was subjected to 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and was transferred to a nitrocellulose membrane (Amersham). Blots were blocked with nonfat dry milk prior to incubation with primary and secondary antibodies. Bands were detected using the SuperSignal chemiluminescent reagent (Pierce, Rockford, IL).
A standard ELISA kit was used to quantify hepatitis B virus early antigen (HBeAg) (Shanghai Kehua Biotechnology, Shanghai, China). To determine the amounts of sIL6R and IL-6 secreted into culture supernatants, a human sIL6R Quantikine ELISA kit and a human IL-6 Valukine ELISA kit were used (R&D Systems).
Quantitation of HBV DNA replicative intermediates by qRT-PCR.
HBV DNA replicative intermediates, which are found in HBV core particles within transfected cells, were analyzed by qRT-PCR. Cells were lysed and centrifuged, and then magnesium chloride was added to the supernatant. DNA that was not protected by HBV core protein was digested with DNase I, while cell lysates were treated with proteinase K. DNA was extracted with phenol-chloroform. Core-associated HBV DNA was recovered by ethanol precipitation and was quantified by qRT-PCR using HBV primers 5′-ATCCTGCTGCTATGCCTCATCTT-3′ (sense) and 5′-ACAGTGGGGAAAGCCCTACGAA-3′ (antisense) and HBV probe 5′-TGGCTAGTTTACTAGTGCAATTTTG-3′. PCR was performed, and the results analyzed, using a LightCycler 480 system (Roche).
Measurement of influenza virus replication.
A549 cells were infected with influenza virus A/Hong Kong/498/97 (H3N2) as described previously (32) at an MOI of 1. Viral titers were measured at various time points postinfection by a hemagglutination assay in U-shaped plates, as described previously (33). Relative levels of IAV nucleoprotein (NP)-specific viral RNA (NP-vRNA), NP-cRNA, and NP-mRNA were detected with reverse transcription primers and qRT-PCR test primers. The following primers were used for reverse transcription: for NP-vRNA, 5′-CTCACCGAGTGACATCAACATCATG-3′; for NP-cRNA, 5′-AGTAGAAACAAGGGTATTTTTCTTTAATTGTCAT-3′; and for NP-mRNA, oligo(dT). The following primers were used for qRT-PCR: for NP, 5′-ATCAGACCGAACGAGAATCCAGC-3′ (sense) and 5′-GGAGGCCCTCTGTTGATTAGTGT-3′ (antisense); for γ-actin, 5′-TCTGTCAGGGTTGGAAAGTC-3′ (sense) and 5′-AAATGCAAACCGCTTCCAAC-3′ (antisense) (29).
Statistical analysis.
All experiments were performed in duplicate or quadruplicate. Statistical analysis was performed using the two-tailed Student t test for comparison between two groups. A P value of <0.05 was considered statistically significant.
RESULTS
Influenza A virus-induced sIL6R expression is downregulated following COX-2 inhibition.
sIL6R expression was assessed by qRT-PCR in A549 cells infected with influenza virus A/Hong Kong/498/97 (H3N2) at an MOI of 1 at various time points of infection. IAV infection induced sIL6R expression in a time-dependent manner: mRNA levels were upregulated at least 5-fold over those in mock-infected cells beginning at 4 h postinfection (hpi) and showed a >25-fold increase at 24 hpi. We also examined sIL6R expression in freshly isolated IAV-infected PBMCs at the time points indicated in Fig. 1A. Results similar to those obtained with A549 cells (Fig. 1A) and primary human alveolar type II cells (Fig. 1B) were observed. These data suggest that significant expression of sIL6R was induced by IAV infection.
Fig 1.
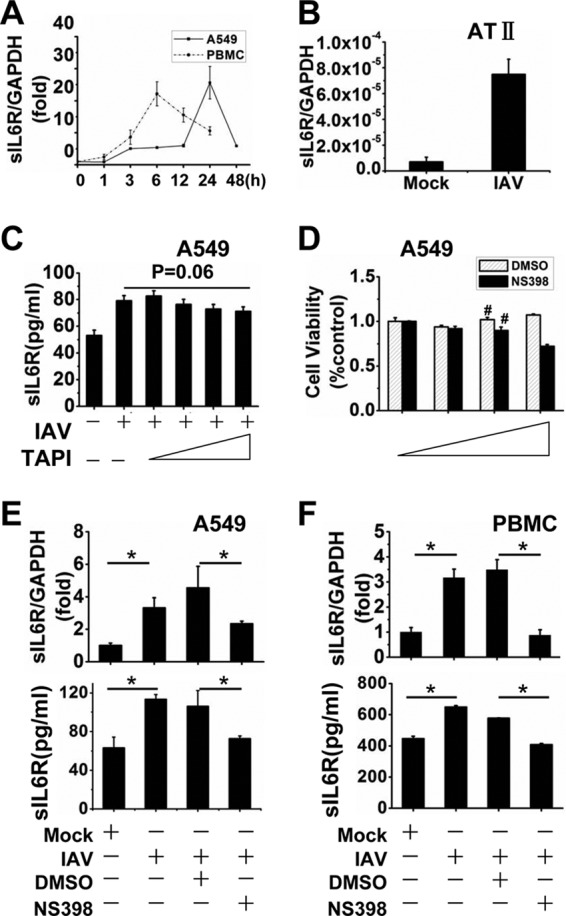
IAV induces sIL6R expression, and NS398 downregulates sIL6R. (A) A549 cells and freshly isolated PBMCs were infected with IAV A/Hong Kong/498/97 (H3N2) at an MOI of 1, and the cells were harvested at the indicated time points. sIL6R mRNA levels were analyzed by qRT-PCR. (B) AT II cells were inoculated with IAV for 1 h at an MOI of 1 and were harvested at 24 hpi. sIL6R mRNA levels were measured by qRT-PCR. (C) A549 cells were incubated with (5 nM, 10 nM, or 20 nM) or without TAPI for 8 h and were then infected with IAV (MOI, 1) for 4 h. The levels of sIL6R protein were measured by ELISA. (D) A549 cells were incubated with (20 μM, 40 μM, or 80 μM) or without NS398 or with (0.25%, 0.05%, or 0.1%) or without DMSO for 24 h. Cell viability was measured by an MTT assay. Number signs (#) indicate the concentrations of NS398 (40 μM) and DMSO (0.05%) selected for use in this study. (E and F) A549 cells (E) and freshly isolated PBMCs (F) were incubated with NS398 (40 μM) or with dimethyl sulfoxide as a control for 8 h and were then infected with IAV (MOI, 1) or heat-inactivated IAV (mock) for 4 h. Cell lysates and culture supernatants were then prepared and collected. The levels of sIL6R mRNA (top graphs) and secreted sIL6R protein (bottom graphs) were determined by qRT-PCR and ELISA, respectively. *, P < 0.05. All graphs represent means ± standard deviations for 3 experiments.
To clarify the mechanism of sIL6R production, we used a shedding inhibitor to block proteolytic cleavage of the membrane-bound IL6R. Hydroxamic acid-based metalloprotease inhibitors, such as TAPI, are known to prevent the shedding of various cell surface proteins (34–36), including the IL-6R (6, 37). Surprisingly, IAV-induced release of the sIL6R in A549 cells was not blocked by TAPI (Fig. 1C). This indicated that the IAV-induced soluble IL6R is a truncated protein produced by differential splicing of the IL6R mRNA (DS-sIL6R) and is not shed from the cell surface by proteolytic cleavage of the membrane-bound IL6R (PC-sIL6R).
In this study, NS398 was used as the selective COX-2 inhibitor in the following experiments, and DMSO was used as the solvent of NS398. To study the cytotoxic effect, either NS398 (20 μM, 40 μM, or 80 μM), DMSO (0.25%, 0.05%, or 0.1%), or neither was added to the culture medium of A549 cells, and cell viability was determined by MTT assays (38). No significant effect on cell viability was observed in the presence of 40 μM NS398 or 0.05% DMSO (Fig. 1D). Our previous protein chip results suggested that sIL6R was one of the cellular factors whose expression was affected by COX-2 (data not shown). To investigate the role of COX-2 in the induction of sIL6R expression, A549 cells were treated with the selective COX-2 inhibitor NS398 (40 μM) prior to infection with IAV (MOI, 1) for 4 h. We found that NS398 treatment suppressed sIL6R mRNA and protein levels significantly relative to those for the dimethyl sulfoxide control (Fig. 1E). To ensure that this was not a cell-specific event, human PBMCs were treated and examined similarly. Not surprisingly, NS398 treatment decreased both sIL6R mRNA and protein levels in PBMCs (Fig. 1F), suggesting that COX-2 plays a role in IAV-mediated sIL6R expression.
sIL6R expression is regulated by the COX-2 pathway.
We further investigated the role of COX-2 in sIL6R expression by overexpressing the enzyme in A549 cells with the pCMV-COX-2 plasmid. Cells were transfected with the COX-2-expressing vector or a control vector for 24 h before the examination of sIL6R mRNA levels and at various concentrations for 48 h before the examination of sIL6R protein levels by ELISA. Our data demonstrate that COX-2-mediated induction of sIL6R expression is dose dependent (Fig. 2A and B). Analysis of sIL6R protein levels in A549 cells transfected with a fixed amount of plasmid pCMV-COX-2 for varying times indicated that COX-2 increased sIL6R expression in a time-dependent manner (Fig. 2C). This regulation of sIL6R expression by COX-2 was further confirmed using an RNA interference (RNAi) approach with COX-2-specific siRNA (si-COX-2). A549 cells were transfected with si-COX-2 or a nonsense control siRNA (si-nc) for 4 h and were then infected with IAV (MOI, 1) for 24 h, followed by quantitation of sIL6R mRNA and protein by qRT-PCR and ELISA, respectively. COX-2 knockdown inhibited sIL6R expression at both the mRNA and protein levels (Fig. 2D). Prostaglandin E2, the main metabolite of COX-2, also induced sIL6R mRNA expression in a dose- and time-dependent manner in PBMCs (Fig. 2E and F). Taken together, these results suggest that the COX-2 pathway regulates sIL6R expression in IAV-infected cells.
Fig 2.
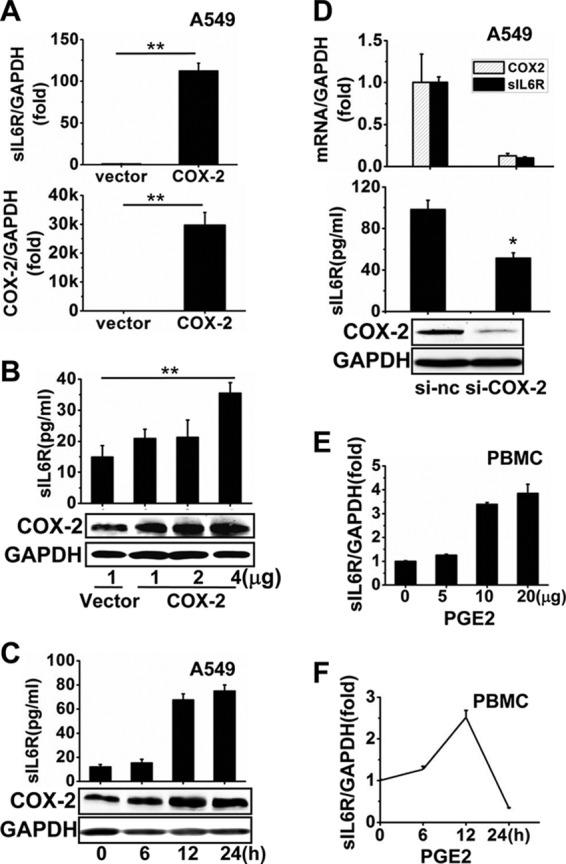
Determination of the effect of COX-2 on the regulation of sIL6R expression. (A) A549 cells were transfected with pCMV-COX-2 or a vector control for 24 h. Levels of COX-2 and sIL6R mRNAs were determined by qRT-PCR. **, P < 0.01. (B) A549 cells were transfected with pCMV-COX-2 or a vector control at different concentrations, as indicated, for 48 h. Levels of sIL6R and COX-2 proteins were determined by ELISA and Western blotting. **, P < 0.01. (C) A549 cells were transfected with pCMV-COX-2 for the indicated times. Levels of secreted sIL6R protein and COX-2 protein were determined by ELISA and Western blotting, respectively. (D) A549 cells were transfected with COX-2-specific siRNA or nonsense control siRNA for 4 h before infection with IAV (MOI, 1) for 24 h. Levels of COX-2 and sIL6R mRNAs, sIL6R protein, and COX-2 protein were determined by qRT-PCR, ELISA, and Western blotting, respectively. *, P < 0.05. (E and F) Freshly isolated PBMCs were treated with PGE2 at different concentrations (E) or with 5 μg PGE2 for different periods (F) as indicated. Levels of sIL6R mRNA were measured by qRT-PCR. All graphs represent means ± standard deviations for 3 experiments.
COX-2-mediated regulation of sIL6R expression is independent of IL-6.
Our results thus far showed that IAV infection led to increased sIL6R expression via COX-2. To test whether COX-2 affects IL-6 expression simultaneously, A549 cells were transfected with the pCMV-COX-2 plasmid at various concentrations for 48 h, and then IL-6 mRNA and protein levels were assessed. Our data demonstrate that COX-2 overexpression had no effect on IL-6 mRNA or protein levels, suggesting that this enzyme is likely not involved in regulating IL-6 expression in these cells (Fig. 3A). Furthermore, to explore whether IL-6 expression influences sIL6R expression, we examined sIL6R mRNA and protein levels in A549 cells treated with varying concentrations of rhIL-6 protein for 24 h (Fig. 3B). In addition, we measured sIL6R levels in A549 cells treated with 40 ng/ml rhIL-6 protein for varying times (Fig. 3C). The results from both of these experiments demonstrate that IL-6 did not affect sIL6R expression at the mRNA or protein level. Thus, COX-2-induced sIL6R expression may be independent of IL-6, which itself is unable to stimulate the expression of sIL6R.
Fig 3.
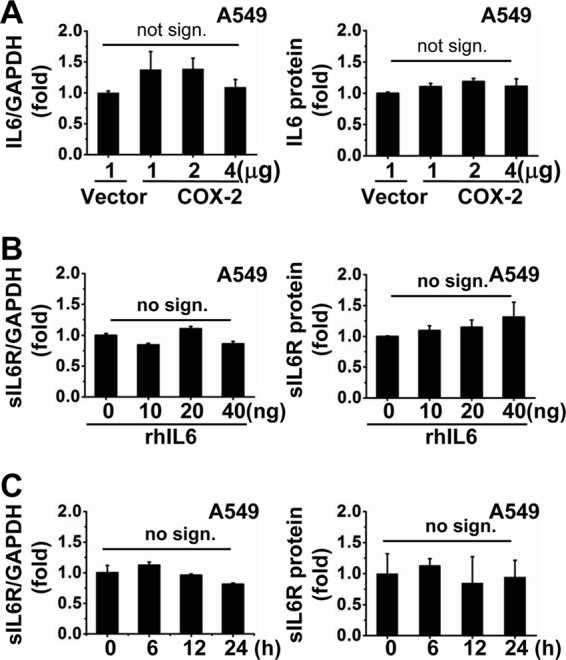
COX-2-regulated sIL6R expression is independent of IL-6. (A) A549 cells were transfected with pCMV-COX-2 at the indicated concentrations for 48 h. Levels of IL-6 mRNA and protein were determined by qRT-PCR and ELISA, respectively. (B and C) A549 cells were treated with recombinant human IL-6 protein (rhIL-6) at the indicated concentrations (B) or for the indicated times (C). Levels of sIL6R mRNA and protein were measured by qRT-PCR and ELISA, respectively. All graphs represent means ± standard deviations for 3 experiments. no sign., no significant difference.
sIL6R but not IL-6 elicits extensive antiviral activity.
Because IAV induced sIL6R production, we investigated whether the sIL6R plays a role in IAV replication. We constructed sIL6R and IL-6 overexpression plasmids and purchased recombinant human sIL6R and IL-6 proteins. Much larger quantities of the sIL6R and IL-6 were detected in the supernatants of transfected cells than in control supernatants (Fig. 4A and B). The phosphorylation of STAT3 was detected by Western blotting to confirm the biological activities of both the rhIL-6 and rhsIL6R proteins (Fig. 4C). MDCK cells were either left untreated or treated with rhsIL6R protein (40 ng/ml) for 6 h. The cells were infected with IAV (MOI, 1), and viral NP gene expression levels, including vRNA, cRNA and mRNA, were measured at 3 hpi. Treatment with rhsIL6R led to significant decreases in the expression of all three types of viral RNA examined (Fig. 4D).
Fig 4.
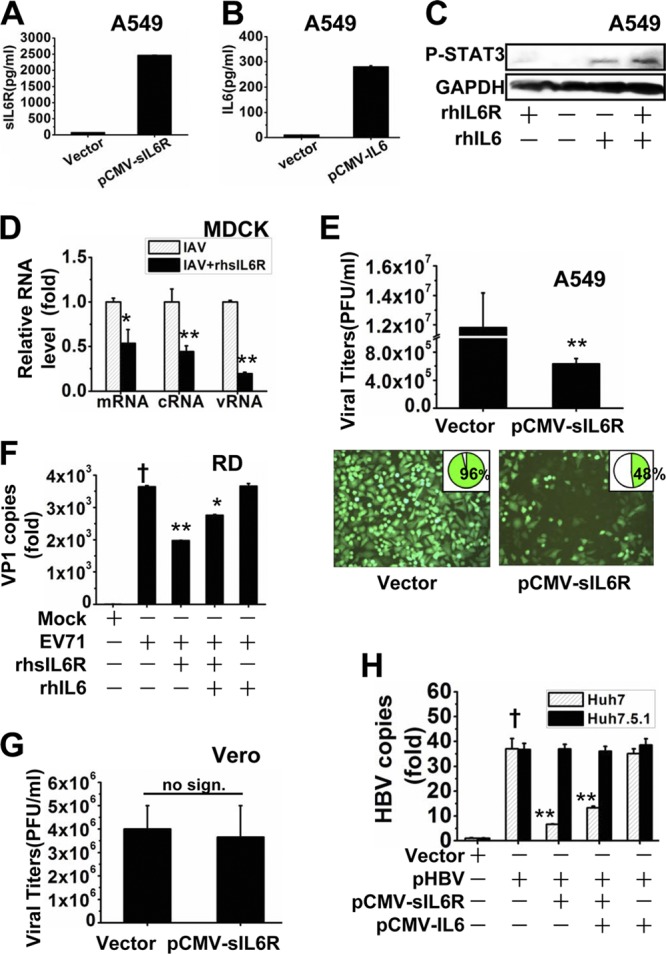
Antiviral activity of sIL6R. (A) A549 cells were transfected with pCMV-sIL6R for 48 h. The levels of sIL6R protein were measured by ELISA. (B) A549 cells were transfected with pCMV-IL-6 for 48 h. The levels of IL-6 protein were measured by ELISA. (C) A549 cells were treated with recombinant human sIL6R (rhsIL6R) protein (40 ng/ml), rhIL-6 protein (40 ng/ml), or both rhsIL6R (20 ng/ml) and rhIL-6 (20 ng/ml) and were cultured for 24 h. Levels of STAT3 phosphorylation (P-STAT3) were measured by Western blotting. (D) MDCK cells were either left untreated or treated with rhsIL6R (40 ng/ml) for 6 h, infected with IAV (MOI, 1), and then harvested at 3 hpi. Relative levels of NP-specific mRNA, cRNA, and vRNA were measured by qRT-PCR. Asterisks indicate significant differences (*, P < 0.05; **, P < 0.01) from IAV-infected cells without rhsIL6R. (E) A549 cells were transfected with pCMV-sIL6R or a vector control for 4 h and were then infected with VSV-eGFP (MOI, 1) for 6 h. (Bottom) VSV-eGFP replication was measured by flow cytometry for eGFP expression (data shown in insets) and was visualized by fluorescence microscopy. (Top) Supernatants were harvested at 24 hpi and were analyzed for VSV production, estimated on Vero cells by using a standard plaque assay. (F) RD cells were treated with rhsIL6R (40 ng/ml) or rhIL-6 (40 ng/ml) for 6 h, infected with EV71 (MOI, 1) or inactivated EV71 (mock), and then cultured for 12 h. Supernatants were harvested, and the number of EV71 copies was determined by qRT-PCR. The dagger indicates a significant difference (P < 0.01) from cells infected with inactivated EV71. Asterisks indicate significant differences (*, P < 0.05; **, P < 0.01) from untreated EV71-infected cells. (G) Vero cells were transfected with pCMV-sIL6R or a vector control for 8 h and were then infected with VSV (MOI, 1). Supernatants were harvested at 24 hpi and were analyzed for VSV production using a standard plaque assay. no sign., no significant difference. (H) Huh7 cells and Huh7.5.1 cells were cotransfected with pHBV and either pCMV-sIL6R, pCMV-IL-6, or both and were cultured for 48 h. HBV capsid-associated DNA levels were assessed by qRT-PCR. The dagger indicates a significant difference (P < 0.01) from control vector-transfected cells. Asterisks indicate significant differences (**, P < 0.01) from untreated pHBV-transfected cells. All graphs represent means ± standard deviations for 3 experiments.
To test whether sIL6R promotes a universal antiviral function, we assessed the effects of sIL6R on the production of the recombinant virus VSV-eGFP in A549 cells. Viral titers were significantly lower in cells overexpressing sIL6R than in control cells (Fig. 4E, top). VSV-eGFP replication was determined by measuring eGFP expression by flow cytometry and was visualized by fluorescence microscopy (Fig. 4E, bottom). sIL6R-treated cells exhibited a lower cytopathic effect than control cells at 24 hpi. Moreover, the number of infected cells decreased from 96% to 48% following sIL6R overexpression (Fig. 4E, bottom, insets).
Since IL-6 also functions by binding its membrane-bound receptor, we next examined whether IL-6 and the sIL6R have a synergistic effect on viral infection. RD cells were treated with either rhsIL6R protein (40 ng/ml), rhIL-6 protein (40 ng/ml), or both rhsIL6R (20 ng/ml) and rhIL-6 (20 ng/ml) for 6 h. The cells were then infected with either EV71 (MOI, 1) or inactivated EV71, which served as a control. Supernatants were harvested at 12 hpi, and the level of EV71 mRNA was measured. Treatment with rhsIL6R effectively reduced the level of viral VP1 mRNA (Fig. 4F). Interestingly, rhsIL6R combined with rhIL-6 also inhibited EV71 replication, but no synergistic effect on viral infection was observed. Not surprisingly, IL-6 alone had no effect on EV71 replication. To test whether the antiviral function of sIL6R depends on the presence of interferon (IFN), Vero cells (a cell line lacking functional type I IFN genes [39, 40]) were chosen for investigation of the antiviral function of sIL6R against VSV. No antiviral activity was observed in Vero cells (Fig. 4G). We then experimented with Huh7.5.1 cells, a cell line derived from Huh7 cells with a single point mutation in the dsRNA sensor retinoic acid-inducible gene I (RIG-I) (41–43), which have much lower type I IFN levels than Huh7 cells. HBV-1.3, a 1.3-fold-long version of the HBV genome (subtype ayw or adw) that retains the ability to produce mature HBV virions, was cotransfected into Huh7 cells or Huh7.5.1 cells along with pCMV-sIL6R, pCMV-IL-6, or both. We found that sIL6R significantly suppressed HBV DNA replication in Huh7 cells but not in Huh7.5.1 cells. In addition, cotransfection of sIL6R with IL-6 also suppressed HBV replication, but not as effectively as sIL6R alone, in Huh7 cells. IL-6 alone had no effect on HBV DNA replication either in Huh7 cells or in Huh7.5.1 cells (Fig. 4H). Taken together, our data indicate that sIL6R but not IL-6 exerts a universal antiviral function in response to infection by different viruses and that the antiviral function depends on the presence of type I IFN.
sIL6R induces type I IFN production in A549 cells.
Type I IFNs exhibit universal antiviral activity against different viral infections, so they are among the most common antiviral agents used to treat chronic viral infections. Since our results demonstrated that sIL6R can inhibit viral replication and that its antiviral activity depends on the presence of IFN, we proceeded to investigate whether the sIL6R affects the antiviral activity of type I IFN by using VSV, a pathogen that is extremely sensitive to the action of type I IFN (44). To this end, we examined the effect of sIL6R on VSV infection. A549 cells were transfected with pCMV-sIL6R and were then incubated with neutralizing antibodies for 6 h before infection with VSV (MOI, 1) for 12 h. As expected, VSV titers decreased significantly after sIL6R expression. This suppression was reversed by treatment with neutralizing antibodies against the sIL6R (sIL6R-NAb), IFN-α (IFN-α-NAb), or IFN-β (IFN-β-NAb) but not by treatment with NAb against IL-6 (IL-6-NAb) (Fig. 5A). qRT-PCR of A549 cells and PBMCs incubated with rhsIL6R for 24 h showed that IFN-α expression increased significantly in the presence of rhsIL6R in both cell types (Fig. 5B and C). Meanwhile, the level of IFN-β mRNA increased dramatically in A549 cells but only slightly in PBMCs.
Fig 5.
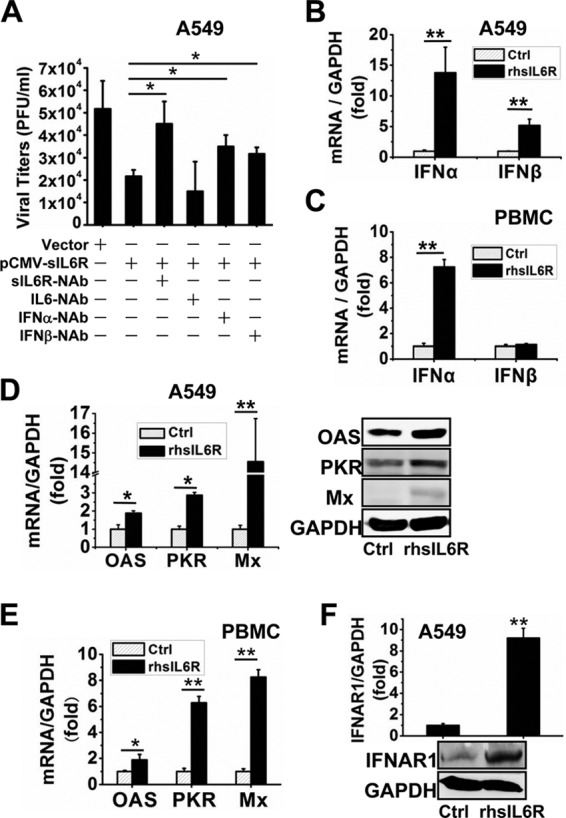
Type I IFN is induced by sIL6R. (A) A549 cells were transfected with pCMV-sIL6R or a vector control for 6 h and were then infected with VSV (MOI, 1) for 12 h. The cells were then incubated with neutralizing antibodies against the sIL6R (sIL6R-NAb), IL-6 (IL-6-NAb), IFN-α (IFN-α-NAb), or IFN-β (IFN-β-NAb) (each at 2 μg/ml) for 12 h. Supernatants were harvested and were analyzed for VSV production, estimated on Vero cells using a standard plaque assay. *, P < 0.05. (B and C) A549 cells (B) and freshly isolated PBMCs (C) were treated with rhsIL6R protein (40 ng) or a control (Ctrl) and were cultured for 24 h. Levels of IFN-α and IFN-β mRNAs were measured by qRT-PCR. **, P < 0.01. (D to F) A549 cells and freshly isolated PBMCs were treated with rhsIL6R protein (40 ng) or a control and were cultured for 24 h. (D) Levels of OAS, PKR, and Mx mRNA and protein in A549 cells were determined by qRT-PCR and Western blotting, respectively. (E) Levels of OAS, PKR, and Mx mRNA in PBMCs were measured by qRT-PCR. (F) Levels of IFNAR1 mRNA and protein in A549 cells were determined by qRT-PCR and Western blotting, respectively. *, P < 0.05; **, P < 0.01. All graphs represent means ± standard deviations for 3 experiments.
We next assessed whether sIL6R induced IFN-α downstream effectors. A549 cells and PBMCs were treated with rhsIL6R, and qRT-PCR and Western blotting were performed to determine the levels of OAS, PKR, and Mx induced by IFN-α. As expected, sIL6R increased the mRNA and protein levels of each IFN-α effector examined (Fig. 5D and E). sIL6R also increased the mRNA levels of the IFN-α receptor IFNAR1 in A549 cells (Fig. 5F). Collectively, our results demonstrate that sIL6R induces type I IFN expression and activates IFN downstream effectors.
sIL6R-mediated antiviral function via p28.
We confirmed that sIL6R acts independently of IL-6. Since it has been reported that IL6R can also bind to CNTF (11), p28 (14), and gp130, here we further investigated whether these three cellular factors play a role in sIL6R-mediated antiviral function.
First, we examined the expression levels of the three cellular factors in IAV-infected cells, including A549 cells, alveolar type II (AT II) epithelial cells, and PMBCs (Fig. 6A to C). AT II cells are one of the primary targets for influenza A virus pneumonia, and differentiated human AT II cells support productive IAV infection (45). Cells were infected with IAV for 24 h, and the mRNA levels of the three cellular factors were measured by qRT-PCR. The results showed that p28 levels were significantly increased in all three types of cells. CNTF levels were significantly increased in A549 and AT II cells but not in PBMCs. gp130 levels were increased in AT II cells and PBMCs but not in A549 cells. Meanwhile, PBMCs were either left untreated or treated with rhsIL6R protein (40 ng/ml) and were cultured for 24 h before measurement of the mRNA levels of the three factors (Fig. 6D). The results showed that none of them are affected by rhsIL6R.
Fig 6.
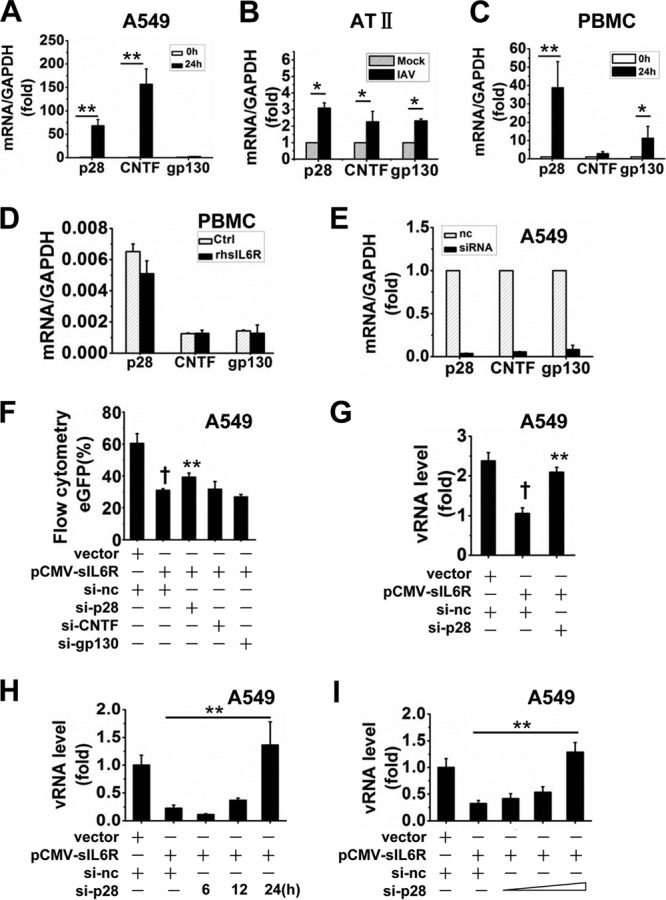
p28 contributes to sIL6R-mediated antiviral function. (A to C) A549 cells (A), AT II cells (B), and PBMCs (C) were infected with IAV (MOI, 1) for 24 h. Levels of p28, CNTF, and gp130 mRNA were analyzed by qRT-PCR. *, P < 0.05; **, P < 0.01. (D) PBMCs were either left untreated or treated with rhsIL6R protein (40 ng) and were cultured for 24 h. Levels of p28, CNTF, and gp130 were measured by qRT-PCR. (E) A549 cells were transfected with individual specific siRNAs against p28, CNTF, and gp130 (si-p28, si-CNTF, and si-gp130) for 24 h, and mRNA levels of p28, CNTF, and gp130 were analyzed by qRT-PCR. A nonsense siRNA (si-nc) was used as a control. (F) A549 cells were cotransfected with each siRNA (si-nc, si-p28, si-CNTF, and si-gp130) and pCMV-sIL6R for 6 h and were then infected with VSV-eGFP (MOI, 1) for 4 h. VSV-eGFP replication was measured by flow cytometry for eGFP expression. A dagger indicates a significant difference (P < 0.01) from control vector-transfected cells. Asterisks indicate significant differences (P < 0.01) from cells cotransfected with si-nc and pCMV-sIL6R. (G) A549 cells were cotransfected with si-p28 and pCMV-sIL6R for 8 h and were then infected with IAV (MOI, 1) for 4 h. Relative levels of NP-specific vRNA were measured by qRT-PCR. (H) A549 cells were cotransfected with si-p28 and pCMV-sIL6R for 8 h and were then infected with IAV (MOI, 1) for the indicated times. Relative levels of NP-specific vRNA were measured by qRT-PCR. (I) A549 cells were cotransfected with pCMV-sIL6R and si-p28 at different concentrations, as indicated, for 8 h and were then infected with IAV (MOI, 1) for 12 h. Relative levels of NP-specific vRNA were measured by qRT-PCR. All graphs represent means ± standard deviations for 3 experiments.
We next investigated whether the sIL6R exerts its antiviral function via any of the three factors. Specific siRNA (si-p28, si-CNTF, or si-gp130) or nonsense control siRNA was synthesized for the following antiviral assays, and their silencing efficacies were confirmed at the RNA level (Fig. 6E). Each specific siRNA was cotransfected into A549 cells along with pCMV-sIL6R, and the cells were then infected with VSV-eGFP for 4 h before flow cytometry analysis (Fig. 6F). The results showed that eGFP expression was reduced after sIL6R overexpression and that this reduction could be partly reversed after silencing of the p28 by si-p28. No similar results were observed with si-CNTF or si-gp130. In a further experiment, si-p28 and pCMV-sIL6R were cotransfected into A549 cells, which were then infected with IAV (MOI, 1). The results showed that IAV replication was reduced after overexpression of sIL6R and that this reduction was reversed by si-p28 as well (Fig. 6G). The silencing of p28 by si-p28 could reduce the antiviral effect of sIL6R in a time- and dose-dependent manner (Fig. 6H and I). Taking these findings together, we conclude that sIL6R mediated antiviral activity, at least in part, through the p28 pathway.
Effects of sIL6R on the nuclear translocation of IRF3 and NF-κB.
IRF3 and NF-κB are required for the induction of type I IFNs (46). To investigate the mechanism of sIL6R-induced IFN-α production, the cellular localization of these transcription factors was assessed in A549 cells transfected with pCMV-sIL6R for different times (0, 24, and 48 h). Cytosolic and nuclear fractions were prepared from sIL6R-transfected cells, and Western blot analysis was performed. Our data revealed that the levels of IRF3, RelA, and NF-κB1 protein within the nucleus increased, while their cytosolic levels decreased, over time in the sIL6R-transfected cells (Fig. 7A). Similar results were obtained with immunofluorescence assays 48 h after transfection (Fig. 7B to D). These results are consistent with the increased production of type I IFNs observed.
Fig 7.
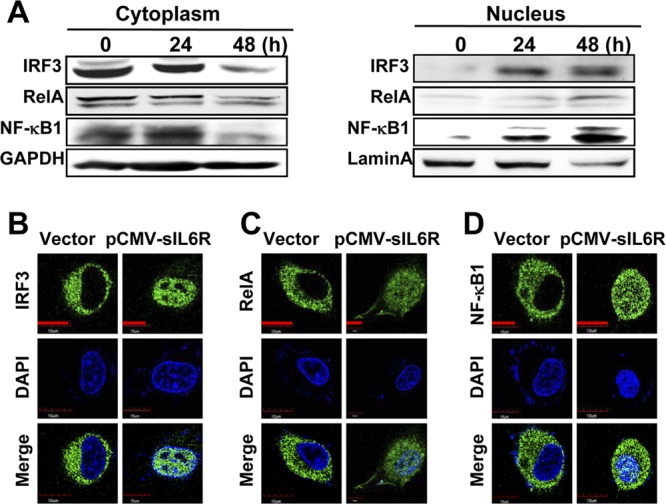
Effects of sIL6R on IRF3 and NF-κB translocation. (A) A549 cells were transfected with pCMV-sIL6R or a control vector. Then cytosolic and nuclear extracts were prepared at the indicated time points and were subjected to Western blot analysis. GAPDH and lamin A were used as markers for the cytosolic and nuclear fractions, respectively. Levels of IRF3, RelA, and NF-κB1 proteins were detected by Western blotting. (B to D) A549 cells were transfected with pCMV-sIL6R or a control vector for 48 h. After fixation, the cells were immunostained with antibodies against IRF3 (B), RelA (C), and NF-κB1 (D). Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) (blue). Bars, 10 μm.
sIL6R expression is elevated in patients infected with influenza virus or HBV.
To determine the clinical relevance of our findings, we measured sIL6R levels in clinical samples. Throat swab samples were collected from 17 healthy individuals and 17 patients infected with seasonal IAV. In addition, PBMCs were obtained from 22 healthy individuals and 22 HBV patients. sIL6R mRNA levels from throat swab samples were approximately 3-fold higher in IAV patients than in healthy individuals (Table 1). For the 22 HBV patients, sIL6R mRNA levels in PBMCs were greater than those for healthy individuals (Table 2). Because IAV and sIL6R expression have been linked on the basis of cell culture experiments, we analyzed the relationship between these factors in clinical samples. Indeed, viral NP mRNA levels correlated positively with sIL6R mRNA levels in throat swabs (R = 0.61) (Fig. 8A), further supporting an association between the induction of sIL6R expression and viral infection.
Table 1.
Summary of sIL6R mRNA levels in throat swab samples from healthy individuals and influenza A virus-infected patients
| Characteristica | Value for: |
|
|---|---|---|
| Healthy individuals (n = 17)b | Patients (n = 17) | |
| Age (yr) | 40.6 ± 11 | 39.2 ± 13.5 |
| Gender (no. male/female) | 6/11 | 9/8 |
| No. (%) of individuals | ||
| Asian race or ethnicity | 17 (100) | 17 (100) |
| Sample collection between 11/8/2011 and 1/19/2012 | 17 (100) | 17 (100) |
| Residence in Hubei province, China | 17 (100) | 17 (100) |
| HA antigen positive | NA | 17 (100) |
| Anti-HA antibody positive | NA | 17 (100) |
| No. with viral genotype A (H3/H1) | NA | 17 (13/4) |
| sIL6R/GAPDH mRNA level (fold) | 2.9 ± 2.7 | 8.4 ± 6.3 |
HA, hemagglutinin.
NA, not applicable.
Table 2.
Summary of sIL6R levels in PBMCs from healthy individuals and HBV-infected patients
| Characteristica | Value for: |
|
|---|---|---|
| Healthy individuals (n = 22)b | Patients (n = 22) | |
| Age (yr) | 39.6 ± 10.0 | 44.1 ± 16.0 |
| Gender (no. male/female) | 9/13 | 11/11 |
| No. (%) of individuals | ||
| Asian race or ethnicity | 22 (100) | 22 (100) |
| Sample collection between 3/14/2012 and 5/11/2012 | 22 (100) | 22 (100) |
| Residence in Hubei province, China | 22 (100) | 22 (100) |
| No. positive/negative for HBsAg | 0/22 | 22/0 |
| No. with HBV genotype b/c | NA | 4/18 |
| ALT concn (U/liter) | 14.0 ± 7.4 | 42.6 ± 54.5 |
| No. of HBV DNA copies/ml | <500 | 4,852 ± 3,780 |
| sIL6R/GAPDH mRNA level (fold) | 5.2 ± 5.0 | 10.0 ± 6.5 |
ALT, alanine aminotransferase.
NA, not applicable.
Fig 8.
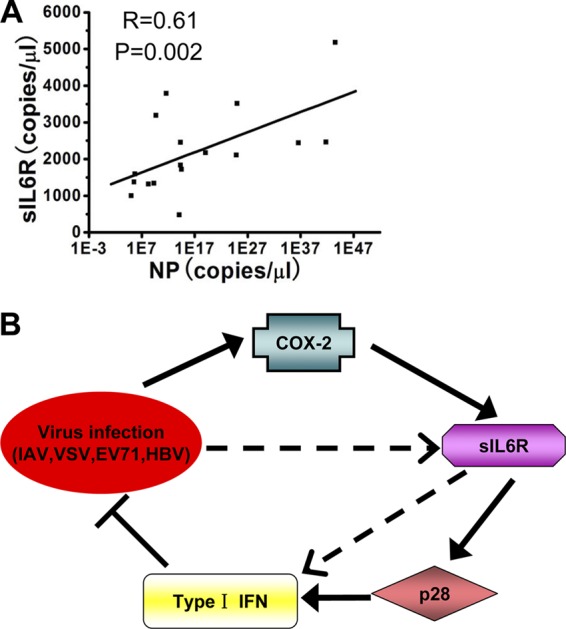
(A) The relative sIL6R and viral NP levels in throat swab samples were subjected to correlation analysis (n = 23). (B) Hypothetical model for COX-2-mediated sIL6R expression. Solid arrows represent signaling pathways identified in this study. Broken arrows indicate potential signaling pathways. Viruses (IAV, VSV, EV71, and HBV) induce sIL6R expression through the COX-2 pathway. The enhanced sIL6R activates type I IFN expression through the p28 pathway, leading to the activation of downstream effectors and the inhibition of viral replication. In addition, there are other potential signaling pathways that may regulate sIL6R-mediated antiviral function.
DISCUSSION
In this study, we investigated the role and underlying mechanism of the sIL6R in the immune response to viral infection. Recently, two papers reported the IL6R as a target for preventing coronary heart disease on the basis of a Mendelian randomization analysis (27, 47). These reports inspired us to investigate whether the sIL6R plays a novel role in diseases independently of IL-6. PBMCs are involved mainly in immune responses and play a major role in regulating host defense mechanisms against microbial infections (48). As such, these cells are widely used to research the immediate immune responses to viral and other microbial infections (12, 31, 48). In fact, our previous study demonstrated that PBMCs infected by influenza virus serve as a clean and efficient model for studying the antiviral immune response (12).
Here we identified cellular signaling pathways that contribute to sIL6R expression during IAV infection, as well as the antiviral mechanism of the sIL6R. Our data demonstrate that IAV infection induces sIL6R expression in three different types of cells. IAV-induced sIL6R was mostly in the DS form, not in the PC form. This increase in expression was suppressed by the COX-2 inhibitor NS398 in both A549 cells and PBMCs, confirming the involvement of COX-2 in regulating sIL6R expression. Interestingly, COX-2 overexpression did not affect IL-6 expression in A549 cells. Moreover, IL-6 had no effect on sIL6R mRNA or protein levels. As we know, the IL6R was named the IL-6 receptor because IL-6 first binds to the IL6R and the complex associated with gp130, thereby promoting IL-6/IL6R/gp130 dimerization and the subsequent initiation of intracellular signaling (9, 49, 50). However, there was no evidence to prove the correlation of expression between the IL6R and IL-6. Our results indicated that IL-6 had no effect on sIL6R expression. Taking the data together, we demonstrated that sIL6R was induced by COX-2 independently of IL-6, suggesting a novel pathway of regulating sIL6R expression.
To understand the biological function of sIL6R during IAV infection, we examined the effect of treatment with rhsIL6R protein on IAV replication. Our data showed that the levels of IAV RNAs were reduced significantly by rhsIL6R protein. Titers of the recombinant virus VSV-eGFP were suppressed by a sIL6R plasmid. The results also indicated that sIL6R, but not IL-6, suppressed EV71 and HBV replication. In fact, no synergistic effects on antiviral activity were observed when cells were treated with both sIL6R and IL-6. The fact that antiviral action was eliminated in type I IFN-deficient cells (Vero and Huh7.5.1 cells) suggests that the antiviral function of sIL6R is dependent on the expression of type I IFN.
VSV is a pathogen that is extremely sensitive to the action of type I IFNs (51), and our results demonstrate sIL6R-mediated repression of VSV replication. Thus, we investigated the effects of neutralizing antibodies against IFN-α, IFN-β, sIL6R, and IL-6 on the sIL6R-mediated suppression of VSV titers. In A549 cells, the abilities of neutralizing antibodies against IFN-α and IFN-β to reverse the sIL6R-induced inhibition of VSV further confirmed the activation of sIL6R-mediated antiviral effects by type I IFNs. As expected, IL-6 had no effect on sIL6R-mediated antiviral activity. Treatment with rhsIL6R protein showed that both IFN-α and IFN-β were induced by the sIL6R in A549 cells, while only IFN-α levels were significantly increased in PBMCs. This discrepancy may be due to inherent differences in these cell types (52). We also confirmed the expression of genes encoding type I IFN downstream effectors, including OAS, PKR, Mx, and IFNAR1 (22), in sIL6R-treated A549 cells. Moreover, increases in OAS, PKR, and Mx mRNA levels were observed in rhsIL6R-treated PBMCs. Thus, the sIL6R can enhance the production of type I IFNs and their downstream effectors.
To find out whether other cellular proteins bind to the sIL6R and exert antiviral functions together, we detected the expression of three factors that have been reported to be correlated with sIL6R: p28, CNTF, and gp130. Only p28 expression was significantly increased in all three types of IAV-infected cells. Meanwhile, VSV-eGFP infection was suppressed after sIL6R overexpression, and this suppression could be partly reversed after the silencing of p28 by specific siRNA. Further, the NP-vRNA level was reduced after sIL6R overexpression and then was restored after p28 silencing. All these results demonstrate that p28 plays an important role in sIL6R-mediated antiviral function. In agreement with a previous study (12) showing that IL-27 (p28) could active IFN-α downstream effectors, our results demonstrated that p28 contributes, at least in part, to sIL6R-mediated antiviral function. But other potential pathways may also regulate sIL6R-mediated antiviral action. sIL6R may also associate directly with IFN pathway-related factors and influence IFN expression. Further study is needed to address this hypothesis.
The production of type I IFN is controlled primarily at the level of transcription, in which IRF3 and NF-κB play pivotal roles (53). Inactive IRF3 resides predominantly in the cytoplasm but translocates to the nucleus upon phosphorylation induced by viruses recognized by pattern recognition receptors (54). NF-κB proteins are present in the cytoplasm in association with the inhibitory IκB proteins (55). Upon viral infection, phosphorylated IκB kinase (IKK) complexes phosphorylate IκBα, which is subsequently ubiquitinated and degraded via the proteasome pathway (56, 57). NF-κB is then released and translocates to the nucleus. Both activated IRF3 and NF-κB bind specific promoter elements of type I IFN genes to activate gene expression. Our data show that in A549 cells, the sIL6R activates the nuclear translocation of IRF3 and NF-κB, further supporting the activation of type I IFN production by the sIL6R during the antiviral response.
Our findings were confirmed by throat swab samples from patients infected with IAV and from healthy individuals (58), as well as by blood samples from HBV patients and healthy individuals. We found that sIL6R levels were significantly higher in IAV and HBV patients than in healthy individuals. Heinz et al. (59) showed that sIL6R concentrations increased significantly in patients who responded to IFN-α therapy by virus elimination and that, conversely, sIL6R concentrations in the sera of nonresponding patients did not change. Comparison of serum IL-6 concentrations uncovered no differences between responders and nonresponders. These findings suggested a possible role of the sIL6R in the elimination of chronic HBV infection. There are some conflicting findings about IL-6 function during hepatitis B virus infection. One study reported that IL-6 affected HBV at the level of transcription and downregulated the expression of HNF1 and HNF4, two transcription factors essential for HBV promoter activity (60). However, Galun et al. (61) showed that IL-6 supports HBV infection in hepatocytes, and Ohno et al. (62) indicated that IL-6 increases the HBV replication rate in infected hepatocytes, since IL-6-signaling affects HBV enhancer 1 in the HBV genome. This contradiction may be explained by considering that, on the one hand, IL-6 does not interact directly with HBV (59), while, on the other hand, IL-6 may have different functions in different phases of virus infection.
Based on these results, we propose a hypothetical model for the role of the sIL6R in the virus-triggered induction of type I IFNs during the antiviral immune response (Fig. 8B). In this model, viral infection induces sIL6R expression through the COX-2 pathway. This enhanced sIL6R activates type I IFN expression through the p28 pathway, leading to the activation of downstream effectors and the inhibition of viral replication. Although more studies are needed to better understand the complex regulatory mechanisms of sIL6R during viral replication and antiviral responses, this study demonstrates the distinct role of the sIL6R during viral infection, indicating the potential clinical use of the sIL6R for antiviral therapy.
ACKNOWLEDGMENTS
We thank Mingzhou Chen, Wuhan University, for providing VSV-eGFP. We also thank the Hubei Provincial Center for Disease Control and Prevention (Hubei CDC) and Renmin Hospital (Wuhan University) for generous assistance in collecting samples from healthy individuals, patients seropositive for influenza A virus antigen, and HBV patients.
This work was supported by research grants from the Major State Basic Research Development Program of China (grants 2013CB911102 and 2009CB522506), the National Natural Science Foundation of China (81271821), and the National Mega Project on Major Infectious Diseases Prevention (2012ZX10004503-004). The funding agencies had no role in the study design, data collection or analysis, the decision to publish, or the preparation of the manuscript.
Footnotes
Published ahead of print 14 August 2013
REFERENCES
- 1.Hatziapostolou M, Polytarchou C, Aggelidou E, Drakaki A, Poultsides GA, Jaeger SA, Ogata H, Karin M, Struhl K, Hadzopoulou-Cladaras M, Iliopoulos D. 2011. An HNF4α-miRNA inflammatory feedback circuit regulates hepatocellular oncogenesis. Cell 147:1233–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Peters BM, Jacobs S, Ehlers M, Vollmer P, Müllberg J, Wolf E, Brem G, Meyer zum Büschenfelde KH, Rose-John S. 1996. The function of the soluble interleukin 6 (IL-6) receptor in vivo: sensitization of human soluble IL-6 receptor transgenic mice towards IL-6 and prolongation of the plasma half-life of IL-6. J. Exp. Med. 183:1399–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nilsson MB. 2005. Interleukin-6, secreted by human ovarian carcinoma cells, is a potent proangiogenic cytokine. Cancer Res. 65:10794–10800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Matthews V, Schuster B, Schutze S, Bussmeyer I, Ludwig A, Hundhausen C, Sadowski T, Saftig P, Hartmann D, Kallen KJ, Rose-John S. 2003. Cellular cholesterol depletion triggers shedding of the human interleukin-6 receptor by ADAM10 and ADAM17 (TACE). J. Biol. Chem. 278:38829–38839. [DOI] [PubMed] [Google Scholar]
- 5.Müllberg J, Schooltink H, Stoyan T, Günther M, Graeve L, Buse G, Mackiewicz A, Heinrich PC, Rose-John S. 1993. The soluble interleukin-6 receptor is generated by shedding. Eur. J. Immunol. 23:473–480. [DOI] [PubMed] [Google Scholar]
- 6.Müllberg J, Durie FH, Otten-Evans C, Alderson MR, Rose-John S, Cosman D, Black RA, Mohler KM. 1995. A metalloprotease inhibitor blocks shedding of the IL-6 receptor and the p60 TNF receptor. J. Immunol. 155:5198–5205. [PubMed] [Google Scholar]
- 7.Horiuchi S, Koyanagi Y, Zhou Y, Miyamoto H, Tanaka Y, Waki M, Matsumoto A, Yamamoto M, Yamamoto N. 1994. Soluble interleukin-6 receptors released from T cell or granulocyte/macrophage cell lines and human peripheral blood mononuclear cells are generated through an alternative splicing mechanism. Eur. J. Immunol. 24:1945–1948. [DOI] [PubMed] [Google Scholar]
- 8.Jones SA, Novick D, Horiuchi S, Yamanoto N, Szalai AJ, Fuller GM. 1999. C-reactive protein: a physiological activator of interleukin 6 receptor shedding. J. Exp. Med. 189:599–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rose-John S, Scheller J, Elson G, Jones SA. 2006. Interleukin-6 biology is coordinated by membrane-bound and soluble receptors: role in inflammation and cancer. J. Leukoc. Biol. 80:227–236. [DOI] [PubMed] [Google Scholar]
- 10.Kishimoto T, Akira S, Narazaki M, Taga T. 1995. Interleukin-6 family of cytokines and gp130. Blood 86:1243–1254. [PubMed] [Google Scholar]
- 11.Schuster B. 2003. Signaling of human ciliary neurotrophic factor (CNTF) revisited. The interleukin-6 receptor can serve as an α-receptor for CNTF. J. Biol. Chem. 278:9528–9535. [DOI] [PubMed] [Google Scholar]
- 12.Liu L, Cao Z, Chen J, Li R, Cao Y, Zhu C, Wu K, Wu J, Liu F, Zhu Y. 2012. Influenza A virus induces interleukin-27 through cyclooxygenase-2 and protein kinase A signaling. J. Biol. Chem. 287:11899–11910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crabé S, Guay-Giroux A, Tormo AJ, Duluc D, Lissilaa R, Guilhot F, Mavoungou-Bigouagou U, Lefouili F, Cognet I, Ferlin W, Elson G, Jeannin P, Gauchat JF. 2009. The IL-27 p28 subunit binds cytokine-like factor 1 to form a cytokine regulating NK and T cell activities requiring IL-6R for signaling. J. Immunol. 183:7692–7702. [DOI] [PubMed] [Google Scholar]
- 14.Garbers C, Spudy B, Aparicio-Siegmund S, Waetzig GH, Sommer J, Holscher C, Rose-John S, Grotzinger J, Lorenzen I, Scheller J. 2013. An interleukin-6 receptor-dependent molecular switch mediates signal transduction of the IL-27 cytokine subunit p28 (IL-30) via a gp130 protein receptor homodimer. J. Biol. Chem. 288:4346–4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li W, Yang F, Liu Y, Gong R, Liu L, Feng Y, Hu P, Sun W, Hao Q, Kang L, Wu J, Zhu Y. 2009. Negative feedback regulation of IL-32 production by iNOS activation in response to dsRNA or influenza virus infection. Eur. J. Immunol. 39:1019–1024. [DOI] [PubMed] [Google Scholar]
- 16.Murono S, Inoue H, Tanabe T, Joab I, Yoshizaki T, Furukawa M, Pagano JS. 2001. Induction of cyclooxygenase-2 by Epstein-Barr virus latent membrane protein 1 is involved in vascular endothelial growth factor production in nasopharyngeal carcinoma cells. Proc. Natl. Acad. Sci. U. S. A. 98:6905–6910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nuñez O. 2004. Increased intrahepatic cyclooxygenase 2, matrix metalloproteinase 2, and matrix metalloproteinase 9 expression is associated with progressive liver disease in chronic hepatitis C virus infection: role of viral core and NS5A proteins. Gut 53:1665–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lara-Pezzi E, Gomez-Gaviro MV, Galvez BG, Mira E, Iniguez MA, Fresno M, Martinez C, Arroyo AG, Lopez-Cabrera M. 2002. The hepatitis B virus X protein promotes tumor cell invasion by inducing membrane-type matrix metalloproteinase-1 and cyclooxygenase-2 expression. J. Clin. Invest. 110:1831–1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mohammed NA, El-Aleem SA, El-Hafiz HA, McMahon RFT. 2004. Distribution of constitutive (COX-1) and inducible (COX-2) cyclooxygenase in postviral human liver cirrhosis: a possible role for COX-2 in the pathogenesis of liver cirrhosis. J. Clin. Pathol. 57:350–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhao L, Jha BK, Wu A, Elliott R, Ziebuhr J, Gorbalenya AE, Silverman RH, Weiss SR. 2012. Antagonism of the interferon-induced OAS-RNase L pathway by murine coronavirus ns2 protein is required for virus replication and liver pathology. Cell Host Microbe 11:607–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sadler AJ, Williams BRG. 2008. Interferon-inducible antiviral effectors. Nat. Rev. Immunol. 8:559–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Q, Gong R, Qu J, Zhou Y, Liu W, Chen M, Liu Y, Zhu Y, Wu J. 2012. Activation of the Ras/Raf/MEK pathway facilitates hepatitis C virus replication via attenuation of the interferon-JAK-STAT pathway. J. Virol. 86:1544–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hayden MS, Ghosh S. 2008. Shared principles in NF-κB signaling. Cell 132:344–362. [DOI] [PubMed] [Google Scholar]
- 24.Arankalle VA, Lole KS, Arya RP, Tripathy AS, Ramdasi AY, Chadha MS, Sangle SA, Kadam DB. 2010. Role of host immune response and viral load in the differential outcome of pandemic H1N1 (2009) influenza virus infection in Indian patients. PLoS One 5:e13099. 10.1371/journal.pone.0013099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ylöstalo JH, Bartosh TJ, Coble K, Prockop DJ. 2012. Human mesenchymal stem/stromal cells cultured as spheroids are self-activated to produce prostaglandin E2 that directs stimulated macrophages into an anti-inflammatory phenotype. Stem Cells 30:2283–2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Karnowski A, Chevrier S, Belz GT, Mount A, Emslie D, D'Costa K, Tarlinton DM, Kallies A, Corcoran LM. 2012. B and T cells collaborate in antiviral responses via IL-6, IL-21, and transcriptional activator and coactivator, Oct2 and OBF-1. J. Exp. Med. 209:2049–2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Interleukin-6 Receptor Mendelian Randomisation Analysis (IL6R MR) Consortium, Hingorani AD, Casas JP. 2012. The interleukin-6 receptor as a target for prevention of coronary heart disease: a Mendelian randomisation analysis. Lancet 379:1214–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cairo S, Buendia MA. 2012. How transient becomes stable: an epigenetic switch linking liver inflammation and tumorigenesis. J. Hepatol. 57:910–912. [DOI] [PubMed] [Google Scholar]
- 29.Li W, Liu Y, Mukhtar MM, Gong R, Pan Y, Rasool ST, Gao Y, Kang L, Hao Q, Peng G, Chen Y, Chen X, Wu J, Zhu Y. 2008. Activation of interleukin-32 pro-inflammatory pathway in response to influenza A virus infection. PLoS One 3:e1985. 10.1371/journal.pone.0001985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yue X, Wang H, Zhao F, Liu S, Wu J, Ren W, Zhu Y. 2012. Hepatitis B virus-induced calreticulin protein is involved in IFN resistance. J. Immunol. 189:279–286. [DOI] [PubMed] [Google Scholar]
- 31.Netea MG, Azam T, Lewis EC, Joosten LAB, Wang M, Langenberg D, Meng X, Chan ED, Yoon D-Y, Ottenhoff T, Kim S-H, Dinarello CA. 2006. Mycobacterium tuberculosis induces interleukin-32 production through a caspase-1/IL-18/interferon-gamma-dependent mechanism. PLoS Med. 3:e277. 10.1371/journal.pmed.0030277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zou F, Liu Y, Liu L, Wu K, Wei W, Zhu Y, Wu J. 2007. Retinoic acid activates human inducible nitric oxide synthase gene through binding of RARα/RXRα heterodimer to a novel retinoic acid response element in the promoter. Biochem. Biophys. Res. Commun. 355:494–500. [DOI] [PubMed] [Google Scholar]
- 33.Mukhtar MM, Li S, Li W, Wan T, Mu Y, Wei W, Kang L, Rasool ST, Xiao Y, Zhu Y, Wu J. 2009. Single-chain intracellular antibodies inhibit influenza virus replication by disrupting interaction of proteins involved in viral replication and transcription. Int. J. Biochem. Cell Biol. 41:554–560. [DOI] [PubMed] [Google Scholar]
- 34.Arribas J, Coodly L, Vollmer P, Kishimoto TK, Rose-John S, Massagué J. 1996. Diverse cell surface protein ectodomains are shed by a system sensitive to metalloprotease inhibitors. J. Biol. Chem. 271:11376–11382. [DOI] [PubMed] [Google Scholar]
- 35.Bennett TA, Lynam EB, Sklar LA, Rogelj S. 1996. Hydroxamate-based metalloprotease inhibitor blocks shedding of l-selectin adhesion molecule from leukocytes: functional consequences for neutrophil aggregation. J. Immunol. 156:3093–3097. [PubMed] [Google Scholar]
- 36.Mohler KM, Sleath PR, Fitzner JN, Cerretti DP, Alderson M, Kerwar SS, Torrance DS, Otten-Evans C, Greenstreet T, Weerawarna K. 1994. Protection against a lethal dose of endotoxin by an inhibitor of tumour necrosis factor processing. Nature 370:218–220. [DOI] [PubMed] [Google Scholar]
- 37.Jones SA, Horiuchi S, Novick D, Yamamoto N, Fuller GM. 1998. Shedding of the soluble IL-6 receptor is triggered by Ca2+ mobilization, while basal release is predominantly the product of differential mRNA splicing in THP-1 cells. Eur. J. Immunol. 28:3514–3522. [DOI] [PubMed] [Google Scholar]
- 38.Yu Y, Gong R, Mu Y, Chen Y, Zhu C, Sun Z, Chen M, Liu Y, Zhu Y, Wu J. 2011. Hepatitis B virus induces a novel inflammation network involving three inflammatory factors, IL-29, IL-8, and cyclooxygenase-2. J. Immunol. 187:4844–4860. [DOI] [PubMed] [Google Scholar]
- 39.Pauli E-K, Schmolke M, Wolff T, Viemann D, Roth J, Bode JG, Ludwig S. 2008. Influenza A virus inhibits type I IFN signaling via NF-κB-dependent induction of SOCS-3 expression. PLoS Pathog. 4:e1000196. 10.1371/journal.ppat.1000196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Prescott J, Hall P, Acuna-Retamar M, Ye C, Wathelet MG, Ebihara H, Feldmann H, Hjelle B. 2010. New World hantaviruses activate IFNλ production in type I IFN-deficient Vero E6 cells. PLoS One 5:e11159. 10.1371/journal.pone.0011159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Blight KJ, McKeating JA, Rice CM. 2002. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J. Virol. 76:13001–13014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sumpter R, Loo YM, Foy E, Li K, Yoneyama M, Fujita T, Lemon SM, Gale M. 2005. Regulating intracellular antiviral defense and permissiveness to hepatitis C virus RNA replication through a cellular RNA helicase, RIG-I. J. Virol. 79:2689–2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhong J. 2005. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. U. S. A. 102:9294–9299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li Q, Tainsky MA. 2011. Epigenetic silencing of IRF7 and/or IRF5 in lung cancer cells leads to increased sensitivity to oncolytic viruses. PLoS One 6:e28683. 10.1371/journal.pone.0028683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang J, Oberley-Deegan R, Wang S, Nikrad M, Funk CJ, Hartshorn KL, Mason RJ. 2009. Differentiated human alveolar type II cells secrete antiviral IL-29 (IFN-λ1) in response to influenza A infection. J. Immunol. 182:1296–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hiscott J, Grandvaux N, Sharma S, Tenoever BR, Servant MJ, Lin RT. 2003. Convergence of the NF-κB and interferon signaling pathways in the regulation of antiviral defense and apoptosis. Ann. N. Y. Acad. Sci. 1010:237–248. [DOI] [PubMed] [Google Scholar]
- 47.IL6R Genetics Consortium Emerging Risk Factors Collaboration, Sarwar N, Butterworth AS, Freitag DF, Gregson J, Willeit P, Gorman DN, Gao P, Salaheen D, Rendon A, Nelson CP, Braund PS, Hall AS, Chasman DI, Tybjærg-Hansen A, Chambers JC, Benjamin EJ, Franks PW, Clarke R, Wilde AA, Trip MD, Steri M, Witteman JC, Qi L, van der Schoot CE, de Faire U, Erdmann J, Stringham HM, Koenig W, Rader DJ, Melzer D, Reich D, Psaty BM, Kleber ME, Panagiotakos DB, Willeit J, Wennberg P, Woodward M, Adamovic S, Rimm EB, Meade TW, Gillum RF, Shaffer JA, Hofman A, Onat A, Sundström J, Wassertheil-Smoller S, Mellström D, Gallacher J, Cushman M, et al. 2012. Interleukin-6 receptor pathways in coronary heart disease: a collaborative meta-analysis of 82 studies. Lancet 379:1205–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ronni T, Sareneva T, Pirhonen J, Julkunen I. 1995. Activation of IFN-α, IFN-γ, MxA, and IFN regulatory factor 1 genes in influenza A virus-infected human peripheral blood mononuclear cells. J. Immunol. 154:2764–2774. [PubMed] [Google Scholar]
- 49.Rose-John S. 2001. Coordination of interleukin-6 biology by membrane bound and soluble receptors. Adv. Exp. Med. Biol. 495:145–151. [DOI] [PubMed] [Google Scholar]
- 50.Taga T. 1997. The signal transducer gp130 is shared by interleukin-6 family of haematopoietic and neurotrophic cytokines. Ann. Med. 29:63–72. [DOI] [PubMed] [Google Scholar]
- 51.Liu S, Hao Q, Peng N, Yue X, Wang Y, Chen Y, Wu J, Zhu Y. 2012. Major vault protein: a virus-induced host factor against viral replication through the induction of type-I interferon. Hepatology 56:57–66. [DOI] [PubMed] [Google Scholar]
- 52.Hata N, Sato M, Takaoka A, Asagiri M, Tanaka N, Taniguchi T. 2001. Constitutive IFN-α/β signal for efficient IFN-α/β gene induction by virus. Biochem. Biophys. Res. Commun. 285:518–525. [DOI] [PubMed] [Google Scholar]
- 53.Sato M, Suemori H, Hata N, Asagiri M, Ogasawara K, Nakao K, Nakaya T, Katsuki M, Noguchi S, Tanaka N, Taniguchi T. 2000. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-α/β gene induction. Immunity 13:539–548. [DOI] [PubMed] [Google Scholar]
- 54.Honda K, Takaoka A, Taniguchi T. 2006. Type I interferon gene induction by the interferon regulatory factor family of transcription factors. Immunity 25:349–360. [DOI] [PubMed] [Google Scholar]
- 55.Li Q, Verma IM. 2002. NF-κB regulation in the immune system. Nat. Rev. Immunol. 2:725–734. [DOI] [PubMed] [Google Scholar]
- 56.Mercurio FH. 1997. IKK-1 and IKK-2: cytokine-activated IκB kinases essential for NF-κB activation. Science 278:860. [DOI] [PubMed] [Google Scholar]
- 57.Zandi E, Rothwarf DM, Delhase M, Hayakawa M, Karin M. 1997. The IκB kinase complex (IKK) contains two kinase subunits, IKKα and IKKβ, necessary for IκB phosphorylation and NFκB activation. Cell 91:243–252. [DOI] [PubMed] [Google Scholar]
- 58.Yin J, Liu S, Zhu Y. 2013. An overview of the highly pathogenic H5N1 influenza virus. Virol. Sin. 28:3–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Heinz D, Peters M, Prange R, Gerken G, Rose-John S. 2001. Possible role of human interleukin-6 and soluble interleukin-6 receptor in hepatitis B virus infection. J. Viral Hepat. 8:186–193. [DOI] [PubMed] [Google Scholar]
- 60.Hösel M, Quasdorff M, Wiegmann K, Webb D, Zedler U, Broxtermann M, Tedjokusumo R, Esser K, Arzberger S, Kirschning CJ, Langenkamp A, Falk C, Büning H, Rose-John S, Protzer U. 2009. Not interferon, but interleukin-6 controls early gene expression in hepatitis B virus infection. Hepatology 50:1773–1782. [DOI] [PubMed] [Google Scholar]
- 61.Galun E, Nahor O, Eid A, Jurim O, Rose-John S, Blum HE, Nussbaum O, Ilan E, Daudi N, Shouval D, Reisner Y, Dagan S. 2000. Human interleukin-6 facilitates hepatitis B virus infection in vitro and in vivo. Virology 270:299–309. [DOI] [PubMed] [Google Scholar]
- 62.Ohno H, Kaneko S, Kobayashi K, Murakami S. 1997. Human hepatitis B virus enhancer 1 is responsive to human interleukin-6. J. Med. Virol. 52:413–418. [DOI] [PubMed] [Google Scholar]