

Abstract
Human cytomegalovirus (HCMV), a betaherpesvirus, can cause severe disease in immunosuppressed patients and following congenital infection. A vaccine that induces both humoral and cellular immunity may be required to prevent congenital infection. Dense bodies (DBs) are complex, noninfectious particles produced by HCMV-infected cells and may represent a vaccine option. As knowledge of the antigenicity and immunogenicity of DB is incomplete, we explored characterization methods and defined DB production methods, followed by systematic evaluation of neutralization and cell-mediated immune responses to the DB material in BALB/c mice. DBs purified from Towne-infected cultures treated with the viral terminase inhibitor 2-bromo-5,6-dichloro-1-beta-d-ribofuranosyl benzimidazole riboside (BDCRB) were characterized by nanoparticle tracking analysis (NTA), two-dimensional fluorescence difference gel electrophoresis (2D-DIGE), immunoblotting, quantitative enzyme-linked immunosorbent assay, and other methods. The humoral and cellular immune responses to DBs were compared to the immunogenicity of glycoprotein B (gB) administered with the adjuvant AddaVax (gB/AddaVax). DBs induced neutralizing antibodies that prevented viral infection of cultured fibroblasts and epithelial cells and robust cell-mediated immune responses to multiple viral proteins, including pp65, gB, and UL48. In contrast, gB/AddaVax failed to induce neutralizing antibodies that prevented infection of epithelial cells, highlighting a critical difference in the humoral responses induced by these vaccine candidates. Our data advance the potential for the DB vaccine approach, demonstrate important immunogenicity properties, and strongly support the further evaluation of DBs as a CMV vaccine candidate.
INTRODUCTION
The development of a vaccine to prevent disease associated with human cytomegalovirus (HCMV) infection remains a high priority (1, 2). Severe HCMV disease can occur following immune suppression or congenital infection. Congenital infection occurs at a frequency of 1% of all live births, of which 10% are symptomatic, indicating that the disease burden of congenital HCMV is a major public health concern. Humoral immune responses following infection in congenital and other disease settings have been reported (3–7), and these results support ongoing clinical evaluation of passive antibody approaches, such as those with hyperimmune globulins (CMV-HIGs), to prevent congenital disease (8–11). Promising outcomes of the passive antibody approach have spurred refined evaluation of glycoprotein epitopes that may be associated with the generation of highly potent neutralizing antibodies and continued evaluation of viral evasion strategies with the expectation that these studies will inform both prophylactic antibody treatments and vaccine approaches (12–17). Multiple HCMV glycoprotein complexes induce neutralizing antibodies, including glycoprotein B (gB), gH/gL/gO, gM/gN, and gH/gL/UL128/UL130/UL131A (12, 13, 18–20). In contrast to the broader roles for gB, gH/gL/gO, and gM/gN, the gH/gL/UL128/UL130/UL131A complex is more specialized but is considered to be required for viral entry into specific cell types, including epithelial and endothelial cells (21–23). A protective vaccine is expected to require neutralizing antibodies that prevent infection of epithelial and endothelial cells (24). In some animal models, the titers of neutralizing antibodies that prevented infection of epithelial and endothelial cells were increased by addition of the gH/gL/UL128/UL130/UL131A complex to a live virus vaccine (17). In other studies, gH/gL was sufficient to induce high-titer, broadly protective neutralizing antibodies (25). On the other hand, a vaccine that consisted solely of soluble gB protein formulated with adjuvant MF59 (gB/MF59) provided 50% efficacy in phase II clinical trials (26, 27), and this approach remains an important comparator for novel vaccine development. Overall, these studies suggest that the inclusion of multiple CMV antigens to expand the neutralizing antibody breadth may provide broader protection and increased efficacy.
The cellular immune response to HCMV has been shown to be protective in the transplant setting, but the role for cellular immunity in preventing congenital transmission is unclear. In transplant patients, adoptive transfer of HCMV-specific cytotoxic CD8+ T cells reduces HCMV disease and viremia (28, 29). The kinetics of CD4+ and CD8+ lymphoproliferative responses and the emergence of CD45RA+ revertant memory T cells have been evaluated in pregnant women (30, 31). These studies suggested that delayed CD4+ and possibly delayed CD8+ lymphoproliferative responses are associated with viral transmission to the fetus, while reversion to a CD45RA+ phenotype is associated with the control of viremia and vertical transmission. Overall, these observations suggest that a vaccine that induces a broad cellular immune response may reduce the viral load in the pregnant woman and be protective for the fetus. In general, cellular immune responses to HCMV are broad. One study documented the immunogenicity of 70% of the 213 open reading frames (ORFs) analyzed for CD4+ T cells, CD8+ T cells, or both (32). The most frequent responses by both T cell lineages are induced by viral proteins encoded by the ORFs UL55 (gB), UL83 (pp65), UL122, UL48, UL32, UL123 (IE1), UL99, and UL82. Inclusion of multiple antigens identified from this analysis may be appropriate to the goal of developing a vaccine to induce protective cellular immunity.
The breadth of the humoral and cellular responses to HCMV infection suggests that an effective vaccine will require a complex antigenic composition in order to induce a protective response. Live attenuated Towne virus was evaluated in phase I and II clinical trials as a potential vaccine expected to induce broad humoral and cellular responses (33–40). Towne was safe and reduced HCMV disease in renal transplant recipients; however, the immunity induced by Towne did not prevent infection of transplant recipients or, in a separate clinical trial, seronegative women with children excreting HCMV. In comparison to natural infection, recipients of the live attenuated Towne vaccine produced lower levels of neutralizing antibodies effective in preventing infection of epithelial cells (16). Specific antigenic differences between the attenuated virus and natural isolates and differences in the levels of antigen presented by the attenuated virus and potentially other factors may all have contributed to these differences. With the goal of improving the Towne vaccine, chimeric viruses were developed to transfer genes from the low-passage-number strain Toledo into the Towne vaccine strain, and these viruses were well tolerated in a small phase I trial (41, 42). Other live virus HCMV vaccine candidates may emerge from the efforts to restore the gH/gL/UL128/UL130/UL131A complex to an attenuated strain (17).
The dense body (DB) vaccine candidate offers many desirable qualities. Produced and released from HCMV-infected cells, the noninfectious DB particles are enveloped and incorporate multiple glycoproteins (43, 44). The DB tegument is also similar to that of viral particles, and overall, more than 20 viral glycoproteins and tegument proteins are common to both viral and DB particles (44). In addition, a DB vaccine would not require delivery of viral DNA or rely on replication, and both qualities would improve safety. Previous evaluations in mice have established that DBs purified by conventional methods induce neutralizing antibodies, preventing infection of fibroblasts in vitro, induce T cell responses specific for pp65, and are immunogenic without addition of adjuvant (45, 46). However, the specificity of the humoral immune response and the breadth of the cellular immune response to DBs have not been established. In addition, refined methods appropriate for evaluating the consistency of the antigenicity of DB particles have not been described. Here we report the immunogenicity of DBs in mice in comparison to that of gB/AddaVax, an approximation of the Chiron gB/MF59 (26, 27). Our results show that the DB antigen presentation induces more breadth in neutralizing antibody and cellular responses than gB/AddaVax, which makes the DB a promising CMV vaccine candidate.
MATERIALS AND METHODS
Viruses and cells.
MRC-5 fibroblast and ARPE-19 epithelial cells were obtained from the American Type Culture Collection (ATCC; Manassas, VA) and cultured as recommended. The origin and propagation of Towne virus and Toledo virus encoding green fluorescent protein (Toledo-GFP) have been described previously (41, 47). VR1814 was received as a gift from Lenore Pereira, University of California, San Francisco, CA, and was propagated by low-multiplicity infections of ARPE-19 epithelial cells. For all viruses, experimental stocks were prepared from infected cell supernatants by ultracentrifugation and resuspension of pellets in growth medium supplemented with SPG buffer (HyClone; Thermo Fisher Scientific, Waltham, MA) prior to storage at −80°C.
DB purification.
Culture supernatants were collected from Towne virus-infected MRC-5 cells and processed for recovery of noninfectious enveloped particles (NIEPs), virions, and DBs from glycerol-tartrate gradients as previously reported (48, 49). Protein concentrations were determined from comparisons to bovine serum albumin (BSA) standards using a micro-bicinchoninic acid (micro-BCA) kit (Thermo Fisher Scientific, Waltham, MA). For some experiments, 15 μM 2-bromo-5,6-dichloro-1-beta-d-ribofuranosyl benzimidazole riboside (BDCRB; obtained from the University of Michigan) was added beginning at 16 to 20 h postinfection for infections initiated at a multiplicity of infection (MOI) of 1.0 or at 6 days postinfection for infections initiated at an MOI of 0.01 to inhibit virion maturation. For either MOI, addition of BDCRB was followed by 4 days of culture prior to supernatant harvest.
NTA and transmission electron microscopy (TEM) analysis.
Nanoparticle tracking analysis (NTA) was performed with a NanoSight LM10 apparatus equipped with a sample chamber and a 640-nm red laser. Analyses were completed with NTA (version 2.1) software and included measurements acquired during 30-s tracking intervals with manual shutter and gain adjustments and acquisition in the extended dynamic range mode. The mean particle size and count corresponded to the arithmetic values calculated from particles analyzed across five repeated measurements. Electron microscopy was completed at Nanoimaging Services Inc. (La Jolla, CA). Samples, preserved in vitrified ice, were imaged using an FEI Tecnai T12 electron microscope at ×52,000 and ×21,000 magnifications. Percentages of DBs, virions, and NIEPs were determined from evaluations of 80 to 300 total particles per sample.
2D-DIGE, 2D picking gel, immunoblot analysis, and mass spectrometry.
One milligram of pelleted DB particles was resuspended in 200 μl of lysis buffer (7 M urea, 2 M thiourea, 1% CHAPS {3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate}, 1% Triton X-100, 40 mM Tris, pH 8.8, 5 mM magnesium acetate, Complete mini-EDTA-free protease inhibitor cocktail [Roche Applied Sciences, Basel, Switzerland]). The lysates were sonicated and clarified by centrifugation at 4°C for 30 min at 13,000 × g and subsequently by a two-dimensional (2D) cleanup kit (GE Healthcare Biosciences, Piscataway, NJ) before protein quantitation by a 2D Quant kit (GE Healthcare Biosciences). For 2D fluorescence difference gel electrophoresis (2D-DIGE), 50 μg of proteins was labeled according to the manufacturer's protocol, a dye swap was performed to avoid bias in labeling differences, and proteins were separated by isoelectrofocusing on pH 4 to 7 strips, followed by SDS-PAGE on 10% acrylamide gels (Multiphor II system; GE Healthcare). Gel images obtained from a Typhoon FLA 9000 imager (GE Healthcare Biosciences) were analyzed by DeCyder 2D software (GE Healthcare Biosciences). For the picking gel, protein spots were stained with SYPRO Ruby (Invitrogen Life Technologies, Carlsbad, CA). For the 2D immunoblots, 50 μg of proteins was labeled with Cy3 before transfer to polyvinylidene difluoride membranes using a semidry system (NovaBlot kit; GE Healthcare Biosciences). Membranes were then incubated with serum or monoclonal antibodies and subsequently the appropriate horseradish peroxidase (HRP)-coupled secondary antibodies prior to detection of reactive protein spots by an ECL plus kit (GE Healthcare Biosciences). The spots of interest that also fit the criteria of perfect overlap (position and form) between the blot spot and reference gel were picked, destained, and digested in gel with sequencing-grade trypsin (Promega, Fitchburg, WI). The resulting peptides were extracted with acetonitrile and dried in a SpeedVac device (Thermo Fisher Scientific, Waltham, MA). Subsequent mass spectrometry identifications were performed by ProtTech Inc. (Phoenixville, PA) using a reverse-phase (RP) Agilent high-pressure liquid chromatography (HPLC) system coupled with an ion-trap mass spectrometer (LCQ DECA XP Plus; Thermo Fisher Scientific). The mass spectrometric data were searched against the data in the nonredundant protein database (NR Database, NCBI) with ProtTech's ProtQuest software suite.
Detection of residual viral DNA and MRC-5 GAPDH.
DB samples were treated with 10 units Benzonase (EMD Millipore, Billerica, MA) at 37°C for 2 h prior to quantitative PCRs (qPCRs) with primers 256F (CCGAGGTGGGTTACTACAACG) and 257R (GGAAGGGTAGAGGCTGGC) from the HCMV UL54 gene. The reaction mixtures included the sequence 5′-FAM-CCCCGTGGCCGTGTTCGACT-BHQ1-3′ (where FAM is 6-carboxyfluorescein and BHQ-1 is black hole quencher dye 1), and the viral DNA copy number was determined by comparison with the numbers on standard curves generated from CMV strain AD169 genomic DNA (Advanced Biotechnologies, Columbia, MD). Primers to detect MRC-5 GAPDH (glyceraldehyde-3-phosphate dehydrogenase) included 259F (CTCAAGATCATCAGCAATGCCTC) and 260R (GTCATGGATGACCTTGGCCAG). These reaction mixtures included the sequence 5′-HEX-TGCACCACCAACTGCTTAGCACC-BHQ2-3′. All qPCRs were completed using a Brilliant III Ultra-Fast qPCR master mix (Agilent Technologies, Santa Clara, CA).
Preparation of gB.
The Towne strain UL55 (gB) ORF was engineered to delete the transmembrane domain, amino acids 703 to 776, and cloned for secreted expression in CHO cells (50, 51). Purification by Ni-immobilized metal affinity chromatography (Ni-IMAC) and quaternized polyethyleneimine (HQ) chromatography resulted in a purity of 92 to 94%, as evaluated by GE ImageQuant analysis of Coomassie blue-stained protein gels and immunoblot analyses. The gB preparations also contained approximately 7 to 19 ng/mg of CHO cell protein (Gyros Immunoassay, Warren, NJ) and ∼1.2 ng/mg DNA (Invitrogen Quant-iT PicoGreen double-stranded DNA assay kit). Size exclusion chromatography (SEC-HPLC) and RP-HPLC analyses indicated a single major peak corresponding to the gB protein.
Microneutralization assays.
Serum samples were heat inactivated at 56°C for 30 to 45 min prior to serial dilutions in cell culture medium with or without 1% guinea pig complement (Lonza, Walkersville, MD). Virus was added, and the mixture was incubated at 37°C in 5% CO2 for 1 h prior to transfer to MRC-5 or ARPE-19 cells cultured in 96-well black-bottom Greiner culture plates. After overnight incubation at 37°C in 5% CO2, cells were fixed with 4% paraformaldehyde, permeabilized with 0.2% Triton X-100, and evaluated directly (Toledo-GFP) or, alternatively, were reacted with CH-160 (Virusys, Taneytown, MD) and goat anti-mouse IgG F(ab′)2 Alexa 488 (Invitrogen Life Technologies, Grand Island, NY) antibodies (Towne and VR1814). The numbers of infected cells were determined by fluorescence using an Isocyte reader (Dynamic Devices, Wilmington, DE), and 50% inhibitory concentration (IC50) values were determined using Prism software (GraphPad, San Diego, CA).
ELISA methods.
The quantities of cellular proteins in DB preparations were determined using commercially available quantitative enzyme-linked immunosorbent assay (ELISA) kits (MRC5 host cell proteins kit [Cyngus Technologies, Southport, NC]) according to the manufacturer's recommendations. The quantities of the gB and pp65 proteins in DB preparations were determined according to the following. Protein samples were disrupted with 8 M guanidine hydrochloride and sonication prior to dilution, and then ELISA plates were coated with serial dilutions of DBs (starting at 15 μg/well) or gB or pp65 protein standards (starting at 0.1 μg/well and 10 μg/well, respectively). Plates were blocked with Superblock (Thermo Fisher Scientific, Waltham, MA) before addition of detection antibodies (CH28 for gB and 3A12 for pp65) (Virusys, Taneytown, MD) and subsequent detection with goat anti-mouse IgG (H+L) HRP-coupled antibodies and reaction with 3,3′,5,5′-tetramethylbenzidine (TMB) substrate. Data on the absorbance at 450 nm were analyzed using the gB or pp65 protein standard dilution series to produce a standard curve and Softmax Pro software (Molecular Devices, Sunnyvale, CA) to calculate the 50% effective concentration (in μg/ml). To determine the quantities of IgG1, IgG2a, and total IgG antibodies binding gB and denatured gB, ELISA plates were coated with gB or denatured gB (at 100 ng/well). For these experiments, denatured gB was prepared by treatment with 8 M guanidine hydrochloride (Sigma, St. Louis, MO), followed by reduction and alkylation using a ReadyPrep kit (Bio-Rad, Hercules, CA) and dialysis into phosphate-buffered saline (PBS). Denaturation was verified by differential scanning fluorimetry on a C1000 thermal cycler equipped with a CFX96 detection system (Bio-Rad, Hercules, CA) and by ELISA binding of conformation-dependent antibody CH177. For determination of IgG1, IgG2a, and total IgG, plates were blocked with casein prior to addition of serial dilutions of experimental mouse serum or monoclonal antibody CH28 (total IgG and IgG1 positive control) or 21A4 (IgG2a positive control), acquired from Lenore Pereira (University of California, San Francisco, CA). Anti-IgG1 or anti-IgG2a antibodies (Jackson ImmunoResearch, West Grove, PA) or anti-IgG HRP-coupled antibodies (Thermo Fisher Scientific) were used for detection.
Depletion of antibody from serum.
CNBr-activated Sepharose beads (GE Healthcare, Little Chalfont, United Kingdom) were swollen in 1 mM HCl for 1 h and washed sequentially with an excess volume of 1 mM HCl and coupling buffer (100 mM NaHCO3, 500 mM NaCl, pH 8). The pelleted resin was resuspended in the presence of gB or denatured gB proteins in coupling buffer (at a ratio of 1 mg of protein per 0.1 g of resin) and incubated for 16 h at 4°C. Blocking buffer (200 mM glycine, pH 8) was added, and the mixture was incubated for an additional 2 h at room temperature. Next, the protein-coupled resin was washed sequentially with a large excess volume of coupling buffer and PBS, prior to addition of 200 μl of pooled mouse serum (diluted 1 in 2 in PBS) to 100 μl of pelleted beads. Following overnight incubation at 4°C on a rotator, the resin was removed by pelleting and the supernatant with gB antibody-depleted sera was collected and stored at 4°C.
Animals and study design.
All animal studies were approved and performed in accordance with MedImmune Institutional Animal Care and Use Committee policies. Female BALB/c mice (age, 7 to 8 weeks) from Charles River Laboratories (Hollister, CA) were housed under pathogen-free conditions in the animal facility at MedImmune. For injections, gB protein was premixed 1:1 (vol/vol) with AddaVax adjuvant (Invivogen, San Diego, CA) at 25°C by vortexing and incubation with shaking at 4°C for 60 min. At day 0, 13 or 14 and day 27 or 28, mice were anesthetized with isoflurane and injected intramuscularly with DBs in a 200-μl volume (100 μl per quadriceps) or with gB/AddaVax in a 100-μl volume (50 μl per quadriceps). A blood sample was collected prior to each vaccination and at 14 days after the last immunization from anesthetized mice via the retro-orbital sinus, and sera were stored at −20°C.
Isolation of cells from spleens and IFN-γ ELISPOT assay.
Following excision, spleens were processed to recover a single-cell suspension and then clarified with a cell strainer (BD Biosciences, Bedford, MA). Clarified cells were centrifuged (1,000 × g for 5 min at 4°C) and resuspended in 2 ml of ACK lysis buffer (Lonza, Walkersville, MD). Following 5 min incubation at room temperature, PBS plus 5% fetal bovine serum (FBS) was added to neutralize the ACK lysis buffer and the remaining cells were recovered by centrifugation. Resuspension of cell pellets, passage through a cell strainer, and a final centrifugation preceded resuspension in RPMI medium containing 10% FBS and determination of quantity and viability by a Vi-Cell viability analyzer (Beckman Coulter, Indianapolis, IN). From this preparation, the number of mouse splenocytes secreting gamma interferon (IFN-γ) was determined by enzyme-linked immunospot (ELISPOT) assay (BD Biosciences, San Diego, CA) according to the manufacturer's recommendations. For the in vitro stimulation, splenocytes from individual mice (5 × 105/well) were incubated with pp65 (Miltenyi, Auburn, CA) or gB (JPT, Berlin, Germany), overlapping peptides (15-mers overlapping by 11 amino acids), or a putative H2-Dd pp65 peptide (LGPISGHVL; Proimmune, Oxford, United Kingdom) at a concentration of 1 μg/ml per peptide. For assessment of cellular responses to more than 3 peptide pools, splenocytes of individual mice were pooled and aliquots were stimulated with pools of overlapping peptides representing 19 HCMV ORFs (JPT, Berlin, Germany) at a concentration of 1 μg/ml. Controls included splenocytes that were stimulated with phorbol myristate acetate (50 ng)-ionomycin (500 ng) or mock stimulated. Following 24 h of incubation in the presence of peptide at 37°C in a humidified incubator, the ELISPOT assay was completed and spots were counted by an ImmunoSpot ELISPOT assay reader (Cellular Technology Ltd., Cleveland, OH). For analysis, the spot counts in medium control wells were subtracted from the specific spot count after peptide stimulation, and the difference is reported as the number of spot-forming cells (SFC) per 1 × 106 splenocytes.
RESULTS
Production of DBs composed of multiple glycoproteins and tegument proteins inducing humoral and cellular immune responses during natural infection.
For production of DBs composed of multiple glycoproteins and tegument proteins inducing humoral and cellular immune responses during natural infection, we employed a conventional purification method for DBs that utilizes negative-viscosity, positive-density glycerol-tartrate gradients (48, 49). To rapidly assess the consistency in particle size distribution, we utilized nanoparticle tracking analysis (NTA). NTA is well suited for characterization of particles 30 to 1,000 nm in size and analyses of polydisperse materials in liquid suspension (52). In this method, laser light scattering and a charge-coupled device camera are used to track the movements of individual particles, and subsequently, size is determined from diffusion rates. For DB analysis, we compared the NTA sizing results from fractions recovered from the gradients (Fig. 1). As reported previously (48, 49), two sharply defined bands of noninfectious enveloped particles (NIEPs) and virions and a broad band of DBs were observed by illumination of the gradient (Fig. 1A and data not shown). For evaluation of NTA, fraction 1 was selected to include NIEPs and virions, while fractions 2 to 4 partitioned the DB band. The mean particle size determined by NTA differed for each of these fractions (Fig. 1B), consistent with the expected correlation between distance along the gradient and DB size (Fig. 1A). Thus, the particle mean size was the smallest in fraction 2 and the largest in fraction 4 and ranged from ≤200 nm to ≥500 nm, consistent with the expectation that the DB size distribution is ∼250 to 600 nm (53). By NTA, fraction 1 resolved into two overlapping peaks, and the average particle size for fraction 1 was 230 nm (weighted average), as expected from published measurements of intact virions and NIEPs (53). Overall, this evaluation indicated that NTA was appropriate for rapid determination of DB size. To obtain materials for composition analyses, we recovered two fractions, labeled dense bodies and virions in Fig. 1A, and completed the remaining gradient centrifugation steps as reported previously (48, 49). NTA and TEM evaluations (Fig. 2) indicated that 96% of the particles in the DB fraction lacked capsids and were of an average size of 306 nm (Table 1). In the virion fraction, 75 to 80% of the particles included capsids, and the average particle size was 180 nm by NTA and 180 to 215 nm by TEM.
Fig 1.
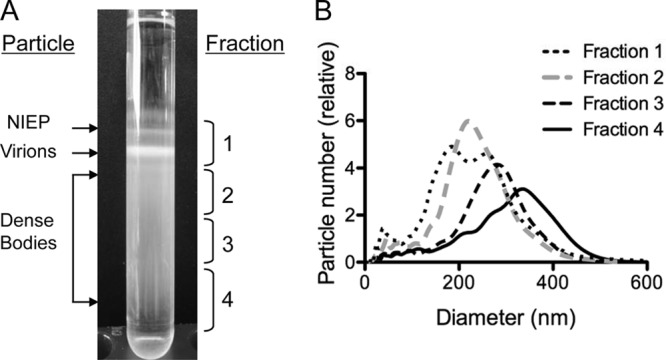
Fractions and particle sizes from glycerol-tartrate gradients monitored by NTA. (A) Image of a representative gradient, with brackets on the right indicating the relative positions of fractions 1 to 4 recovered for evaluation of particle size, as determined by NTA. Indicated on the left are the relative positions of DB and virion fractions selected for imaging and composition analyses. (B) Composite image of NTA results from five independent particle size and number evaluations for each of the fractions 1 to 4 recovered from glycerol-tartrate gradients. The particle counts graphed correspond to the arithmetic mean values calculated from five repeated measurements.
Fig 2.
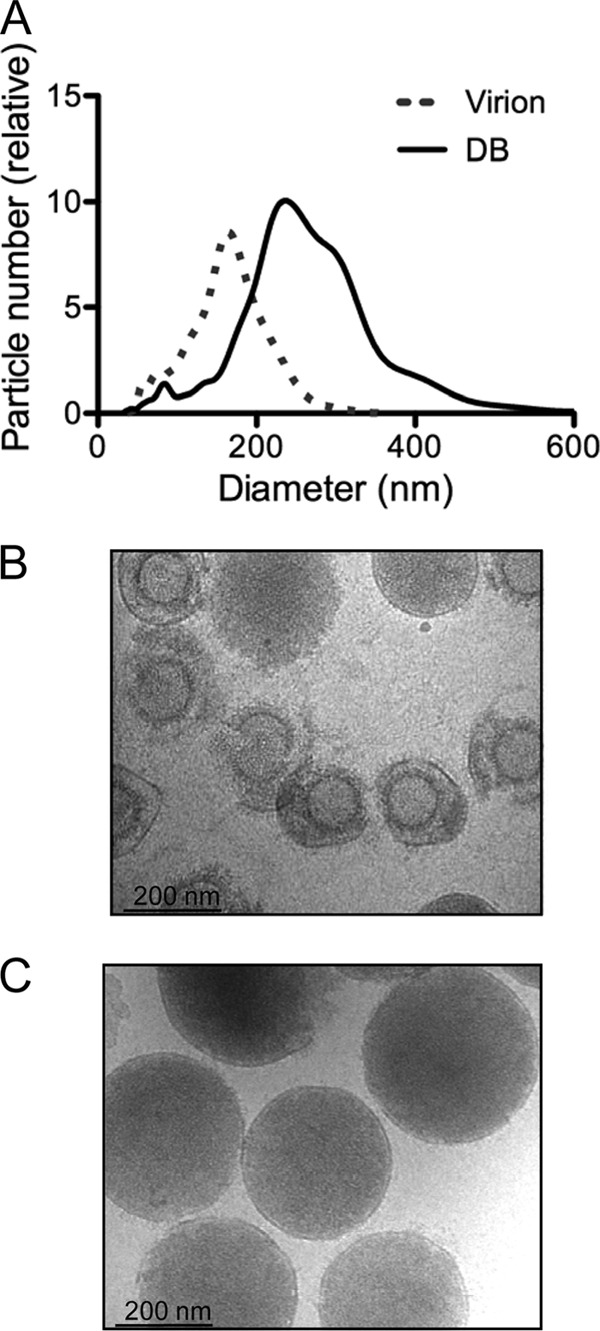
DB and virion preparations for immunoblot and 2D-DIGE analyses of composition. (A) Composite image of NTA results produced from five independent assessments of virion and DB fractions recovered from glycerol-tartrate gradients, as indicated in the legend to Fig. 1A. Graphed particle counts were determined as indicated in the legend to Fig. 1B. (B and C) Representative TEM images acquired from the virion (B) or DB (C) fractions recovered from glycerol-tartrate gradients, as indicated in the legend to Fig. 1A.
Table 1.
Physical properties of virion and DB fractions recovered from glycerol-tartrate gradients
| Gradient fraction and preparation no. | % DBs | % capsid-containing particles | Avg size (nm) by: |
|
|---|---|---|---|---|
| TEM | NTAa | |||
| Virions | ||||
| 1 | 25 | 75 | 180–215 | 180 |
| 2 | 20 | 80 | ||
| DBs | ||||
| 1 | 96 | 4 | 250–344 | 306 |
| 2 | 99 | 1 | ||
z-average particle size.
In previous studies of DB antigenicity, in-gel comparisons of DB lysates and BSA standards have been standard (45, 46, 54). Here, we first determined protein quantity by BCA and then compared DBs with BSA by gel analysis (Fig. 3A). As expected (46), DB lysates revealed a complex composition of >10 protein bands visible by silver staining (Fig. 3A) or Coomassie blue staining (not shown), reflective of the inclusion of multiple tegument proteins and glycoproteins (44). Relative to the findings for BSA, DB titration revealed the expected pp65 concentration of ∼60% (44) (Fig. 3A). This analysis indicated that the BCA methodology was appropriate for DB protein determinations, and BCA quantitation was applied for the remaining studies.
Fig 3.

DB fractions evaluated with specific monoclonal antibodies or Cytogam. (A) Image of silver-stained DBs or BSA separated by denaturing gel electrophoresis, with the arrowhead indicating the expected migration of pp65 in DBs. (B) Immunoblot analysis of DBs reacted with pp65-specific antibodies. Arrowhead, expected migration of pp65; MM, molecular mass marker. (C) Immunoblot analysis of DBs reacted with gB-specific antibodies. Arrow, expected migration of the mature 55-kDa gB. (D) 2D immunoblot analysis of DBs reacted with pp65 antibodies. (E) 2D immunoblot analysis of DBs reacted with gB antibodies. (F) 2D gel and immunoblot analysis of DBs reacted with Cytogam. Regions of the 2D gel selected for mass spectrometry are indicated by rectangles 1 to 6. Viral peptides identified within the region marked by rectangles 1 to 6: 1, gB (UL55); 2, tegument protein pp71 (UL82); 3, UL51; 4, pp65 (UL83); 5, tegument protein (UL88); 6, myristylated tegument protein (UL99). (G) 2D gel and immunoblot analysis of virions reacted with Cytogam. Rectangles indicating gel regions selected for mass spectrometry and identification are as described in the legend to panel F.
For protein composition, we initially determined DB lysate reactivity with pp65-specific (Fig. 3B) or gB-specific (Fig. 3C) monoclonal antibodies by conventional immunoblotting. Each of these antibodies bound multiple proteins that differed by apparent molecular mass, suggesting that DB evaluation would benefit from techniques providing an increased resolution for complex protein samples, such as 2D gel electrophoresis and immunoblotting (2D-IB) (Fig. 3D and E). When resolved on a 2D gel, immunoblotting with the same specific monoclonal antibodies indicated that the DB preparations included (i) pp65-related proteins that differed by isoelectric point (pI) and molecular mass and (ii) gB-related proteins of a single molecular mass but different pIs. Identification by mass spectrometry confirmed that the proteins bound by the monoclonal antibodies were pp65 and gB. In combination, these analyses also indicated that pp65-related proteins that migrated at a higher molecular mass than predicted were likely aggregates, while those that migrated at a lower molecular mass were likely peptide fragments resulting from degradation. The immature form of gB is 130 kDa, while proteolytic processing produces a mature, 55-kDa gB protein resident to extracellular virions and derived from the carboxyl-terminal region of the larger protein (50, 51, 55, 56). The epitope bound by the specific monoclonal antibody used here maps to the carboxyl-terminal region (57), but only the 55-kDa gB was identified by 2D-IB analysis. To evaluate the potential presence of the 130-kDa gB as well as to identify additional viral proteins, we analyzed DB and virion lysates with human CMV immune globulin (Cytogam) (Fig. 3F and G), which contains antibodies to a much broader range of naturally occurring CMV epitopes. As anticipated for CMV-seropositive, pooled human sera (58), Cytogam bound multiple proteins (Fig. 3F and G). Of the six viral proteins identified by mass spectrometry, three tegument proteins (pp65 [UL83], pp71 [UL82], and UL99) and glycoprotein gB (UL55) are known to be immunogenic for CD4+ and CD8+ T cells in naturally seropositive individuals (32), suggesting that broad cell-mediated immunity would be expected using DBs as a vaccine. In addition, Cytogam bound both the mature and uncleaved gB protein forms. Overall, this evaluation identified additional proteins that could easily be monitored by immunoblotting and would be expected to induce cell-mediated immune responses; however, with the exception of gB, the analyses did not reveal additional glycoprotein targets of the humoral response.
To evaluate DB components more broadly and incorporate quantitative assessments of variance, we monitored DB preparations by 2D-DIGE and principal component analysis (PCA) (Fig. 4A and B). In other applications, this approach has been rigorously tested both within and between laboratories for reproducibility of results (59). Here, two different preparations of virus and DB fractions were recovered from glycerol-tartrate gradients and compared by 2D-DIGE. For the replicates, materials that had been labeled with Cy3 in the first evaluation were instead labeled with Cy5 to reduce any potential biases in protein labeling. In comparison with one-dimensional gels and conventional stains (Fig. 3A), 2D-DIGE resolved far greater numbers of proteins (Fig. 4A). In addition, the comparisons of labeled DB and virion lysates revealed protein distribution patterns unique to each material. This was anticipated and consistent with the published results of protein analyses of the abundance of these closely related particles purified by different methods (44). When PCA was applied to the results, virion preparations clustered distinctly from DB preparations, confirming the unique compositions of each. Comparable variances indicated the consistency of the composition between different preparations of DBs and virions. Next, from comparisons of DB and virion lysates, 22 spots on 2D gels (Fig. 4C) were selected for protein identification by mass spectrometry (Table 2). The results indicated that among the most abundant or differentially abundant proteins were 9 virus-encoded proteins and 13 host cell-encoded proteins. In the virus-encoded protein category, pp65 was the only protein of elevated abundance in DB fractions relative to virion fractions. In contrast, five virus-encoded proteins were more abundant in virion fractions. Of these, three (gB, UL51, and pp71) remained easily identified by the reactivity of DBs with Cytogam (Fig. 3D). Two proteins enriched in the virion fractions (UL85 and UL86) are capsid proteins and therefore not expected to be structural components of DBs, suggesting that residual virus may contribute to the presence of capsid protein in our DB fractions. Importantly, none of the host cell proteins were more abundant in DB fractions than in virion fractions (Table 2), consistent with previous reports (44). Several viral and host cell proteins were equally abundant in virion fractions and DB fractions. Included among these was gL. Identification of gL in the glycerol-tartrate gradient fraction is consistent with the findings for DB particles purified by Nycodenz gradients and evaluated with different methods (44). Although we did not identify gH or other glycoproteins, more extensive proteomic evaluations have reported gH and other glycoproteins resident to both virions and DBs (44) that may have been excluded from identification by our selection criteria. Overall, these analyses demonstrated the utility of 2D-DIGE for evaluation of DB preparations and indicated that the composition could be tracked by sensitive and quantitative methods from preparation to preparation.
Fig 4.
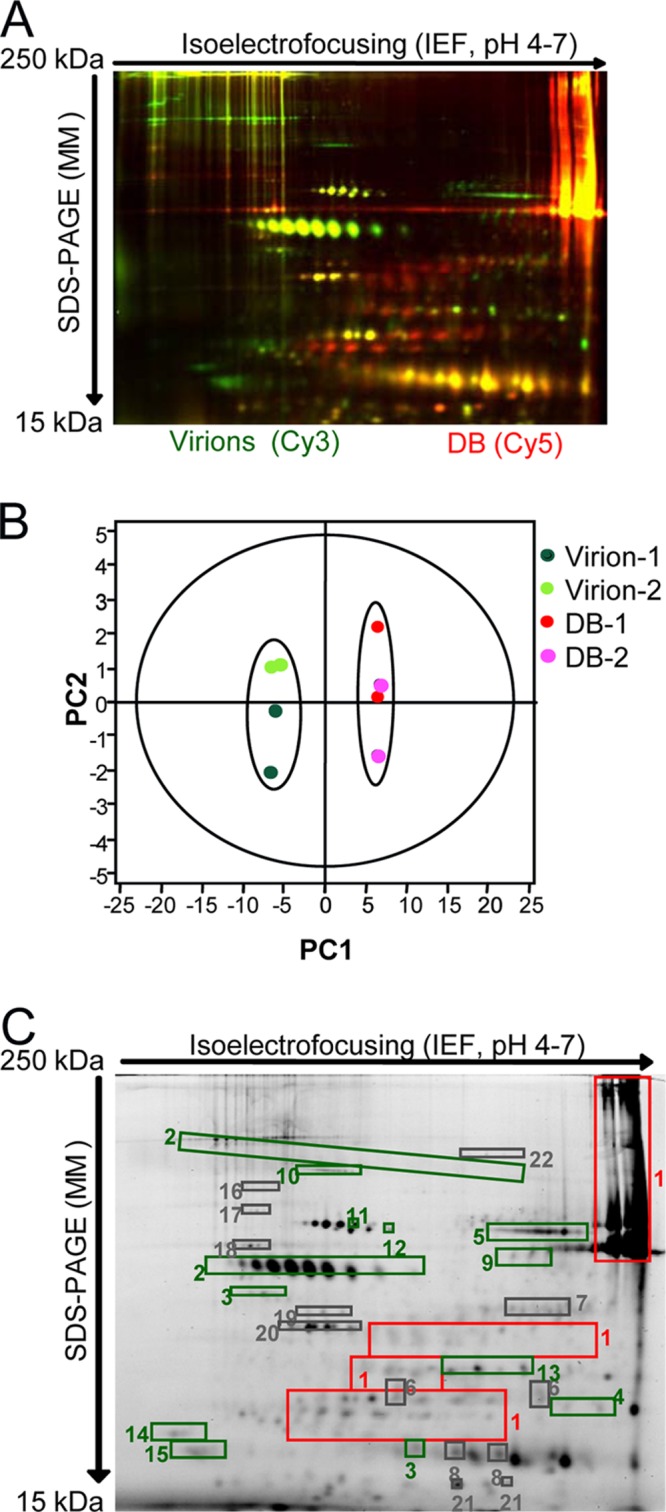
2D-DIGE of DB and virion fractions recovered from glycerol-tartrate gradients. (A) Image from 2D-DIGE analysis of virion lysates labeled with Cy3 (green) in comparison to DB lysates labeled with Cy5 (red). (B) PCA of purification and DIGE replicates to evaluate the consistency in differences between virion (light and dark green) and DB (pink and red) fractions. Virion-1 and DB-1, purification replicate for preparation 1; Virion-2 and DB-2 purification replicate for preparation 2. 2D-DIGE replicates are indicated by symbol color changes (pink versus red and light green versus dark green). (C) Abundant and differentially abundant proteins identified by 2D-DIGE and selected for identification by mass spectrometry. DB/virion fold change differences determined by 2D-DIGE are indicated by the color of the rectangles placed over the gel regions selected for mass spectrometry. For fold changes of <1, the negative of the inverse is reported. Red rectangles, fold change of >2; green rectangles fold change of <−2; gray rectangles, fold change with an absolute value of <2.
Table 2.
Identities of 22 proteins from DB and virion fractions determined by 2D gel electrophoresis and mass spectrometry of isolated peptides
| Protein and spot no. | Name | gi no. | Peptide | Molecular size (kDa) | Fold change (DBs/virus)a |
|---|---|---|---|---|---|
| Viral proteins | |||||
| 1 | 65-kDa phosphoprotein (pp65) | 130714 | 33 | 62.2 | 3 |
| 2 | Envelope gB | 138192 | 16 | 102.8 | −4 |
| 3 | UL51 | 222354501 | 10 | 40.4 | −2.5 |
| 4 | Capsid triplex subunit 2 (UL85) | 52138258 | 3 | 34.9 | −2 |
| 5 | Tegument protein pp71 (UL82) | 254770942 | 49 | 62.2 | −6 |
| 6 | Envelope gL (UL115) | 52139280 | 4 | 31.4 | <2 |
| 7 | Tegument protein (UL88) | 222354518 | 5 | 48.2 | <2 |
| 8 | Myristylated tegument protein (UL99) | 52139271 | 2 | 21.2 | <2 |
| 9 | Major capsid protein (UL86) | 222354516 | 10 | 155 | −4.5 |
| MRC-5 proteins | |||||
| 10 | Protein bicaudal D homolog 2 isoform 1 | 1479166 | 21 | 97.2 | −8.5 |
| 11 | Heat shock cognate 71-kDa protein isoform 1 | 5729877 | 20 | 71 | −2.5 |
| 12 | Heat shock 70-kDa protein 1A/1B | 167466173 | 12 | 70.9 | −2.5 |
| 13 | Serine/threonine-protein phosphatase PP1-alpha catalytic subunit isoform 1 | 4506003 | 6 | 37.9 | −2.5 |
| 14 | 14-3-3 protein epsilon | 5803225 | 9 | 29.3 | −4 |
| 15 | 14-3-3 protein zeta/delta | 4507953 | 8 | 27.9 | −5 |
| 16 | 90-kDa heat shock protein | 306891 | 7 | 83.5 | <2 |
| 17 | 78-kDa glucose-regulated protein precursor | 16507237 | 14 | 72.4 | <2 |
| 18 | Phosphatase 2A regulatory subunit | 189428 | 6 | 65.9 | <2 |
| 19 | Eukaryotic initiation factor 4A-I isoform 1 | 4503529 | 8 | 46.3 | <2 |
| 20 | Actin, cytoplasmic 1 | 4501885 | 7 | 42 | <2 |
| 21 | Growth factor receptor-bound protein 2 isoform 1 | 4504111 | 3 | 25.3 | <2 |
| 22 | Vinculin | 24657579 | 2 | 117.2 | <2 |
The fold change represents the average of the ratios of multiple spots corresponding to the same protein. A fold change of <1 is reported as the negative of the inverse. A fold change with an absolute value of <2 was not considered to be a significant difference.
Glycerol-tartrate gradients provide enriched DB fractions with levels of virion contamination of 1 to 4%. To further reduce viral contamination, we prepared materials from cultures treated with the viral terminase inhibitor 2-bromo-5,6-dichloro-1-beta-d-ribofuranosyl benzimidazole riboside (BDCRB) that prevents DNA encapsidation and, as a result, virion maturation (60, 61). Previous studies have shown that BDCRB does not interfere with DB maturation (62). Thus, this approach aimed to increase the purity of the DB preparation by reducing the quantity of virions released into the culture supernatant. For immunogenicity studies, multiple lots of DBs were prepared from cultures treated with BDCRB, and their compositions were evaluated by NTA, 2D-DIGE, gB, pp65, and MRC-5 protein ELISAs, qPCRs, and virus titration (Table 3 and data not shown). Culture and preparative methods were varied to produce DB lots with different quantities of viral DNA-positive particles. All DB preparations included <1 ng/μg MRC-5 cell proteins. Overall, the results of these studies showed that cultures treated with BDCRB at a high MOI produced DBs of >99.9% purity, as determined from the viral DNA genome copy and particle numbers.
Table 3.
Physical properties of DB fractions prepared from cultures treated with BDCRB
| DB preparation | MOI for production | Storage temp (°C) | No. of particles/μga | Residual amt of virus (PFU/μg) | No. of viral DNA-positive particles/μg | z-average particle size (nm)a | Avg particle size (nm)a | pp65/gB ratiob | Host cell protein concn (ng/μg)b |
|---|---|---|---|---|---|---|---|---|---|
| DB-3 | 1 | −80 | 2.5 × 108 | 3.0 × 102 | 2.4 × 105 | 304 | 245 | 41 | 0.22 |
| DB-4 | 0.01 | −80 | 2.8 × 108 | 1.28 × 103 | 4.45 × 106 | 305 | 244 | 45 | 0.35 |
| DB-5 | 1 | 4 | NDc | ND | 2.5 × 105 | ND | 246 | ND | 0.4 |
Determined by NTA.
Determined by ELISA.
ND, not determined.
DBs induce high-titer complement-independent neutralizing antibodies that prevent Toledo infection of fibroblasts.
For humoral and cellular immune response comparisons of the subunit CMV vaccine gB/Addavax and DBs, doses of 10 μg gB and 100 μg DBs were used in all studies. Dose selection was based on preliminary titration studies of gB in the presence of AddaVax (Fig. 5A) and an independent but similar titration study for DBs (data not shown) that yielded results expected from previous reports (46). The 10-μg gB and 100-μg DB doses selected for direct comparison induced consistent gB-specific antibody titers or neutralizing antibody titers that did not increase with higher doses (Fig. 5A and data not shown). In the direct comparison following immunizations of BALB/c mice, both complement-dependent and complement-independent neutralizing activity was measured in vitro using Toledo-GFP (47) as the target virus. The gB/Addavax-inoculated mice generated high-titer neutralizing antibody that was poorly detected in the absence of complement (Fig. 5B) but readily detected in the presence of complement (Fig. 5C). These data indicate that gB/AddaVax induced high-titer, largely complement-dependent neutralizing antibodies in mice. In contrast, the DB-immunized groups had high-titer neutralizing antibodies in the presence or absence of complement (Fig. 5B and C). Overall, Towne-derived DBs induced highly consistent neutralizing antibody titers across multiple preparation methods and animal experiments.
Fig 5.
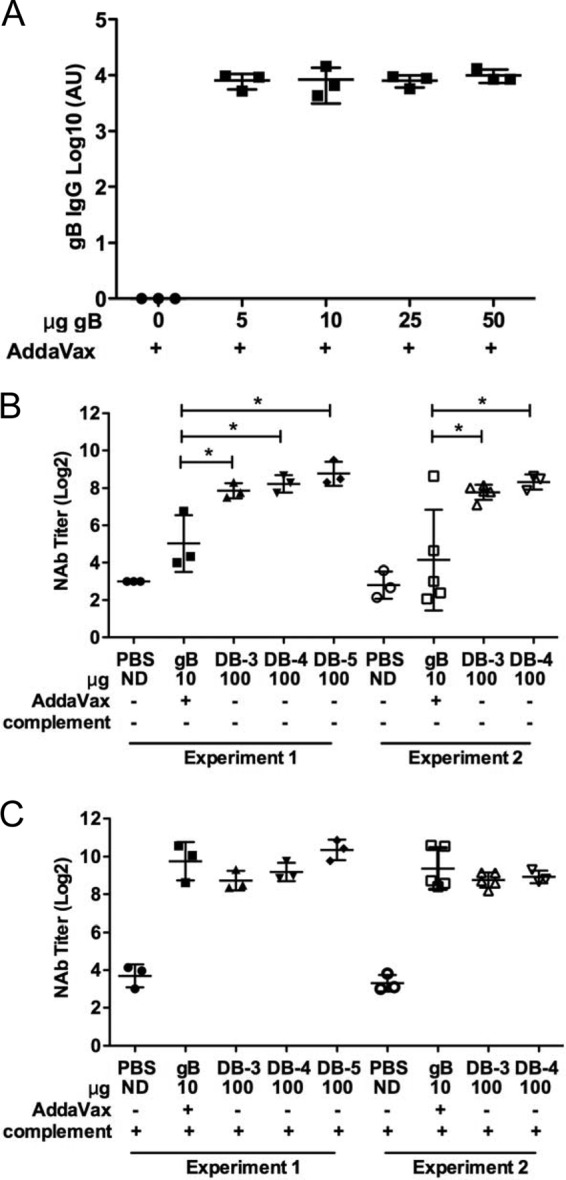
DBs induce high-titer broadly protective complement-independent neutralizing antibodies. (A) gB-specific total IgG titers induced by gB/Addavax following three inoculations in BALB/c mice. AU, absorbance units. (B and C) Comparisons of neutralizing antibody (NAb) titers induced following vaccination with three different DB preparations (DB-3, DB-4, and DB-5) in two different animal experiments (closed or open symbols). Neutralizing antibody titers (IC50s) in mouse serum collected after three inoculations were determined in vitro from neutralization of Toledo-GFP infection of MRC-5 fibroblast cells. Neutralization assays whose results are reported in panels B and C were completed at the same time. For panel B, heat-inactivated serum samples were evaluated without addition of complement. *, P < 0.05. For DB-3, DB-4, and DB-5 in comparison to PBS, all P values were <0.0001, except for DB-2 experiment 2 (P = 0.0003). For panel C, 1% guinea pig complement was added to the heat-inactivated serum samples during the neutralization assay. For experiments 1 and 2, P values in comparison to the results obtained with PBS were <0.0009 for gB/AddaVax, DB-3, DB-4, and DB-5. ND, not determined.
DBs but not gB/AddaVax induce neutralizing antibody titers preventing VR1814 infection of fibroblasts and epithelial cells.
Towne-derived DB particles include gB and other glycoproteins (Table 2; Fig. 3 and 4) that may also induce neutralizing antibodies. The potential for gB to induce neutralizing antibodies in mice that prevent infection of fibroblasts and epithelial cells was addressed by neutralization of VR1814, a low-passage-number isolate that retains endothelial/epithelial cell tropism (63). Sera of mice immunized with gB/AddaVax neutralized VR1814 infection of MRC-5 cells (Fig. 6A). However, titers varied over 17-fold within the experimental group when evaluated in the absence of complement and were not significantly different from those for the controls. In the presence of complement, increased titers were observed, and the mean titer compared to that for the controls was significantly increased (∼18-fold). Despite the variability, these data indicate that gB-specific neutralizing antibodies prevent VR1814 infection of MRC-5 fibroblasts. In contrast, the same sera failed to prevent neutralization of VR1814 infection of epithelial cells (Fig. 6B). In comparison, in sera of DB-immunized mice, high levels of neutralizing antibodies that prevented infection of VR1814 on both MRC-5 or ARPE-19 cells were detected. Furthermore, robust titers were observed in the presence or absence of complement. These neutralizing antibody levels were significantly different from those for the gB/Addavax-immunized and placebo-treated control mice (Fig. 6A and B) and suggest that differences in the gB protein present on DBs (such as both cleaved and uncleaved gB protein [Fig. 3B]) or the presence of additional glycoproteins (Fig. 4; Table 2) may contribute to the broader activity of induced antibodies. Overall, these results demonstrate that the neutralizing responses elicited by Towne-derived DBs were highly effective at preventing infection of either cell type.
Fig 6.

DBs induce complement-independent neutralizing antibody titers that prevent infection of ARPE-19 epithelial cells. Neutralizing antibody titers (IC50s) in mouse sera collected after three inoculations were determined by in vitro neutralization of VR1814 infection. Neutralization assays whose results are reported in panels A and B were completed at the same time. (A) Neutralizing antibody titers were determined from infections of MRC-5 fibroblast cells following incubation of heat-inactivated mouse serum and VR1814 virus in the presence or absence of 1% guinea pig complement, as indicated. *, P < 0.05; **, P < 0.01. P values were <0.0001 for DB-3 and DB-4 in comparison to PBS in the presence or absence of added complement. (B) Neutralizing antibody titers were determined from infections of ARPE-19 epithelial cells, as reported for panel A. ***, P < 0.0005.
DBs induce gB-specific antibodies that prevent infection of epithelial cells.
Linear and conformation-dependent epitopes comprise the targets of gB-specific neutralizing antibodies in immunized mouse and human serum (16, 58–60). To determine the role of gB-specific antibodies in the observed differences in neutralizing antibody responses induced by DBs and gB/AddaVax, we first determined gB-specific antibody titers by ELISA (Fig. 7). Relative to immunization with gB/AddaVax, sera from mice immunized with DBs had lower titers of gB-specific total IgG antibodies (Fig. 7A and B), corresponding to lower IgG1 (Fig. 7C and D) but similar IgG2a (Fig. 7E and F) antibody titers. The gB-specific antibodies from gB/Addavax-immunized mice bound intact and denatured gB. The DB-induced IgG2a antibodies also bound intact and denatured gB, but in most animals, DBs induced IgG1 antibodies that did not bind denatured gB. Thus, antibody isotype differences and conformation-dependent binding were two qualities that distinguished the humoral responses to DBs and gB/AddaVax. Next, neutralization titers were determined before and after depletion of gB- and denatured gB-binding antibodies in assays of fibroblast and epithelial cell infection (Fig. 8 and data not shown). Multiple depletions with intact gB reduced the ELISA titer of sera from gB/AddaVax-immunized mice by only 10-fold. Multiple depletions with denatured gB reduced the ELISA titer of sera from gB/AddaVax-immunized mice by 100-fold (data not shown). However, neither depletion method reduced the neutralization of Towne virus infection of MRC-5 cells by more than 35% (data not shown), consistent with the presence of high-titer gB-specific antibodies in the sera of gB/AddaVax-immunized mice (Fig. 7). In contrast, depletions of Cytogam reduced gB- and denatured gB-binding antibody titers to levels that were undetectable in the ELISAs (Fig. 8A). Here, the depletion reduced the neutralization of Towne virus infection of MRC-5 cells by 85% (Fig. 8B). In combination, these results are consistent with an expectation of low levels of gB-specific antibodies (12) that nonetheless can contribute to neutralization of HCMV. As a consequence, we used Cytogam as control for the evaluation of sera from DB-immunized mice. Following depletion of antibodies from the DB-immunized mouse serum, gB-binding antibodies were reduced by ∼100-fold, while denatured gB-binding antibodies were undetectable by ELISA (Fig. 8A). Importantly, depletion reduced the neutralization of Towne virus infection of MRC-5 cells by 65%, indicating that DB induced gB-specific neutralizing antibodies (Fig. 8B). Neutralization of VR1814 infection of MRC-5 cells was also reduced by 75% following depletion of Cytogam, but depletion had a minimal impact on sera from mice immunized with DB (Fig. 8C). In contrast, neutralizing titers determined by VR1814 infection of ARPE-19 epithelial cells were reduced by 50 to 60% following depletion of denatured gB-binding antibodies from Cytogam or sera from DB-immunized mice (Fig. 8D). These results suggest a role for a linear epitope in induction of gB-specific antibodies that neutralize VR1814 infection of epithelial cells. In combination with binding studies, the results suggest that the IgG2a antibodies induced by DB likely contribute to preventing infection of epithelial cells. Overall, these results support the conclusion that DBs induced gB-specific neutralizing antibodies that prevent infection of fibroblast and epithelial cells.
Fig 7.

DBs induce lower gB-specific total IgG and IgG1 than gB/AddaVax. ELISA titers of total IgG (A and B) or IgG1 (C and D) and IgG2a (E and F) isotypes in mouse sera obtained following inoculations with gB/AddaVax or DBs are shown. The protein used to determine binding by ELISA was gB for the titers reported in panels A, C, and E and denatured gB for the titers reported in panels B, D, and F.
Fig 8.

DB-induced gB-specific antibodies contribute to neutralization of VR1814 infection of epithelial cells. (A) Total IgG ELISA titer results before and after depletion of pooled sera from mice immunized with DBs with gB or denatured gB from Cytogam. The gB or denatured gB protein used to deplete sera was subsequently used to determine binding in ELISA, as indicated in the depletion and detection lines beneath the graph. (B) Neutralizing antibody titers (IC50s) determined by Towne infection of MRC-5 fibroblasts following depletion of gB-binding or denatured gB-binding antibodies and graphed as the percentage of the neutralization titer in nondepleted sera. (C) Neutralizing antibody titers determined from neutralization of VR1814 virus infection of MRC-5 fibroblast cells. Sera were depleted with gB or denatured gB, and results are graphed as reported for panel B. (D) Neutralizing antibody titers determined from neutralization of VR1814 virus infection of ARPE-19 epithelial cells. Sera were depleted with gB or denatured gB, and results are graphed as reported for panel B.
Towne vaccine virus-derived DBs induce a broad T cell response.
Previous reports of DB immunogenicity have focused on T cell responses specific for the predominant component, pp65 (45, 46, 54), yet the complexity of the DB composition may promote cellular responses to multiple viral proteins. Here, cellular immunity was first evaluated by confirming the pp65-specific response. Splenic lymphocytes from mice immunized with DBs were evaluated by IFN-γ ELISPOT assay with pools of overlapping peptides that span the pp65 protein (Fig. 9A). Splenic lymphocytes collected 2 weeks after the third immunization showed robust pp65-specific T cell responses with a mean number of spot-forming cells (SFC) per 106 splenocytes of 489 ± 117. For further analysis, a second sample of the same splenocytes was evaluated with a putative immunodominant major histocompatibility complex (MHC) class I H2-Dd pp65-specific epitope (LGPISGHVL) in the IFN-γ ELISPOT assay (64) (Fig. 9A). The mean number of SFC per 106 splenocytes after in vitro stimulation with the H2-Dd epitope was 437 ± 110, suggesting that after DB immunization of BALB/c mice the majority of pp65-specific T cells were directed against this epitope and were CD8+ T cells. Magnetic bead depletion studies confirmed this result (data not shown). Next, the induction of gB-specific memory T cells by immunization with DB and gB/AddaVax was evaluated (Fig. 9B). Mean responses were 94 ± 32 SFC per 106 splenocytes for gB/AddaVax-immunized mice and 23 ± 15 SFC per 106 splenocytes for DB-immunized mice. Overall, these results indicate that T cells are induced by at least two components of DBs.
Fig 9.

DBs induce a broad T cell response. IFN-γ ELISPOT assay results from splenocytes of mice analyzed after immunization with DB (DB-4) or gB/AddaVax, as indicated. (A) Responses to a pp65-specific peptide pool and to a putative immunodominant MHC class I H2-Dd pp65 T cell epitope. Responses were determined following three immunizations and are graphed as the number of SFC per 106 splenocytes. ***, P ≤0.0006. (B) Responses to a gB-specific peptide pool determined and graphed as reported for panel A. Solid symbols, results from experiment 1; open symbols, results from experiment 2. **, P = 0.004; ***, P = 0.0002; ****, P < 0.0001. (C) Responses determined in triplicate by IFN-γ ELISPOT assay with peptides spanning 17 HCMV ORFs. Black bars, proteins expected to be components of DBs (44); white bars, proteins not reported as DB components. (D) IFN-γ responses to IE1-, pp65-, or gB-specific peptide pools in splenocytes recovered from mice following two immunizations with UV-treated DBs or PBS. Responses were determined and results are graphed as reported for panel A. ***, P = 0.0004; ****, P < 0.0001; ns, not significant.
To further evaluate the breadth of the T cell response induced by DBs, splenic lymphocyte responses to 17 different HCMV ORF-specific peptide pools were determined by IFN-γ ELISPOT assay (Fig. 9C). By combination with analyses of pp65 and gB, these ORF peptide pools provide a profile of the response to dominant T cell targets identified following natural HCMV infection of human hosts (32). By proteomic methods, 12/17 of the viral proteins represented in the ORF peptide pool are suggested to be DB structural components (44). Here, splenocytes from five DB-immunized mice were combined and aliquots were stimulated with the overlapping peptide pools (Fig. 9C). The highest responses were detected after UL48 (114 SFC per 106 splenocytes), IE1 (48 SFC), and pp71 (UL82) (32 SFC) peptide stimulation. Low-level responses to IE-2, UL32, UL94, US24, and US32 peptide pools were detectable. The IE1- and IE2-specific T cell responses were unexpected, as these viral proteins are not structural components of either DBs or virions and HCMV is a species-restricted virus. However, despite species restriction, immediate early (IE) protein expression can follow infection of mouse cells (65), and the DB fractions did include residual viral DNA and infectivity (Table 3). To determine the role of infectivity, DBs were exposed to UV prior to immunization and the IFN-γ responses of stimulated lymphocytes were reevaluated (Fig. 9D). These results indicated that the pp65- and gB-specific responses were significantly higher in T cells recovered from DB-immunized mice than in T cells from controls. In contrast, responses to IE1 peptides were not significantly different from those of the controls. Overall, these results indicate that the cellular response to DB is more complex than that to gB/AddaVax, consistent with expectations from analyses of protein composition.
DISCUSSION
The studies presented here have evaluated the humoral and cellular immune responses of mice to gB/AddaVax and the HCMV vaccine candidate DBs. We relied on the natural adjuvant properties of DBs but added AddaVax to gB because it is a squalene-based oil-in-water nanoemulsion like MF59, the proprietary adjuvant used with gB in previous clinical trials (26, 27). We report that DBs, but not gB/AddaVax, induced antibodies preventing infection of both fibroblasts and epithelial cells. From assays of fibroblast infection, we also report that the animal-to-animal variability in neutralizing antibody titers observed following gB/AddaVax immunization was reduced following DB immunization. The observed variability in neutralizing antibody titers determined with viral strain Toledo and sera from mice immunized with gB/AddaVax correlated with complement-dependent antibodies, while the complement-independent antibodies present after immunization with DBs were also more consistently induced. In contrast, the addition of complement to neutralization assays that employed VR1814 had much less of an impact on the observed variability in titers. Primary sequence alignments (not shown) indicated that the gB ORFs encoded by these viruses have few differences within antigenic domains, but we have not addressed the specific contribution of these differences to neutralization. Likewise, differences in complement evasion by these viruses may contribute but were not evaluated. Regardless of the potential contributions from these factors, our studies suggest that DBs elicit a more consistent humoral response in mice than gB/AddaVax.
As DBs, but not gB/AddaVax, induced antibodies that prevented VR1814 infection of epithelial cells in vitro, we evaluated the contribution of gB-specific antibodies to the neutralizing activity induced by DB immunization. By depleting gB-specific antibodies, we determined that the gB-specific neutralizing antibodies induced by DBs did contribute to preventing infection of both fibroblasts and epithelial cells. Moreover, by evaluating the role of antibodies that bound denatured gB, we uncovered a role for antibodies that bound linear epitopes in the neutralization of VR1814 infection of epithelial cells. Evidence for the generation by DB immunization of gB-specific antibodies that neutralize epithelial cell infection and the apparent failure of gB/AddaVax immunization to induce antibodies that neutralize epithelial cell infection may implicate the multiple forms of gB present on DBs. In addition, the combination of other glycoproteins in DBs may also prove to contribute or even dominate the neutralizing antibody responses that prevent infection. Towne fails to produce the gH/gL/UL128/UL130/UL131A complex that induces high-titer antibodies (12, 17) due to a frameshift mutation in UL130 (66), but gH/gL or other glycoproteins may also contribute to the neutralizing antibodies induced by DBs that prevent VR1814 infection of fibroblasts and epithelial cells. In this regard, others have reported that antibodies to gH are induced in mice immunized with DBs but did not determine whether the antibodies were effective at neutralization (45). We have confirmed the inclusion of gL in the DBs used for immunizations in this study, and other studies have reported that gH/gL is sufficient to induce antibodies that neutralize infection of epithelial cells (25). Although additional effort will be required to determine the contributions of the multiple glycoproteins included in DBs to induction of neutralizing antibodies, differences in isotype, binding to denatured gB, and the combination of viruses used in our neutralization assays have revealed important differences in the humoral responses induced by gB/AddaVax and DBs.
Our analyses of cellular immune responses to DB and gB/AddaVax showed that DBs elicited a broad T cell response in mice, including responses to pp65, gB, and UL48, which are also among the most frequently recognized viral proteins in the human host (32). In comparison to gB/AddaVax, DBs induced slightly fewer gB-specific T cells under the conditions that we employed. However, the overall increase in the breadth of the T cell responses may be an advantage for a CMV vaccine. As anticipated from the composition of DBs, the majority of the T cell responses in mice were pp65 specific. Here, the total pp65-specific T cells were primarily directed against an MHC class I H2-Dd T cell epitope originally predicted in silico (67). We also found that the detection of IE1-specific responses is apparently sensitive to low levels of residual virus and evaluated UV treatment as a method to interfere with this response. Importantly, UV treatment did not prevent the induction of neutralizing antibody responses by DBs. Overall, our analyses of the cellular responses to DBs demonstrate the potential of these complex particles to elicit a broad response in a biological model of immunogenicity. Mice are not permissive to HCMV, rendering direct comparisons to the live, attenuated Towne virus unapproachable. However, in combination with the described properties of the humoral immune responses, our studies suggest that DBs provide qualities highly desired for an HCMV vaccine that are not provided by gB/AddaVax.
We have reported multiple methodologies to characterize the quality and protein composition of DBs purified by glycerol-tartrate gradients (45, 46, 54). The results from the combined approaches suggest that DB consistency can be evaluated by quantitative measurements and statistical methods. The evaluation of NTA indicated that the methodology is appropriate for the analysis of DB particle size distribution. The use of 2D-DIGE provided a thorough evaluation of composition that had a sensitivity that far exceeded the sensitivity of conventional staining approaches due to increased resolution and the potential for direct comparisons with a reference material, here demonstrated by comparison to virion lysates. Using measurements of particle numbers in combination with rapid infectivity and genome copy number assessments to evaluate quantities, we confirmed that BDCRB does not prevent DB maturation. Importantly, BDCRB did increase the purity of DBs, and we achieved preparations of >99.9% purity with regard to residual virus. We explored the impact of UV inactivation on DB immunogenicity after finding the cellular response to IE1 to be a sensitive indicator of residual virus. The results indicated that the DB immunogen induced broad cellular and humoral responses with or without inactivation of residual virus, suggesting that the combination of BDCRB and virus inactivation presents an approachable method for DB vaccine production. In combination, these studies have provided refined methods to produce and characterize DBs.
As an HCMV vaccine candidate, DBs offer multiple qualities important for safety and protective immunity. DB immunogenicity does not require replication or the introduction of viral DNA. The production methodology presented here incorporated a small-molecule inhibitor, BDCRB, to reduce residual virus. Our studies did not evaluate the levels of BDCRB removed by glycerol-tartrate gradient fractionation, but scalable DB production and purification processes would likely achieve this goal and, as well, remove residual viral DNA. Further, the natural adjuvant properties, the complexity of the DB protein composition, and the potential for genetic modification of the producing virus all combine to make these particles of interest for development of a vaccine to prevent HCMV congenital infection.
ACKNOWLEDGMENTS
We are grateful for the contributions of Althaf Hussain, Floro Cataniag, Joseph Horwitz, Murali Bilikallahalli, Joy Barnitz, Roderick Tang, and all the members of the MedImmune Tissue Culture and Animal Care Facilities. We also thank Edward Mocarski for suggestions regarding BDCRB, John Drach for the generous gift of BDCRB, and George Kemble for insightful comments made during preparation of the manuscript.
All of us were affiliated with MedImmune at the time that the work was performed.
Footnotes
Published ahead of print 7 August 2013
REFERENCES
- 1.Arvin AM, Fast P, Myers M, Plotkin S, Rabinovich R. 2004. Vaccine development to prevent cytomegalovirus disease: report from the National Vaccine Advisory Committee. Clin. Infect. Dis. 39:233–239. [DOI] [PubMed] [Google Scholar]
- 2.Plotkin SA. 2004. Congenital cytomegalovirus infection and its prevention. Clin. Infect. Dis. 38:1038–1039. [DOI] [PubMed] [Google Scholar]
- 3.Boppana SB, Rivera LB, Fowler KB, Mach M, Britt WJ. 2001. Intrauterine transmission of cytomegalovirus to infants of women with preconceptional immunity. N. Engl. J. Med. 344:1366–1371. [DOI] [PubMed] [Google Scholar]
- 4.Ishibashi K, Tokumoto T, Tanabe K, Shirakawa H, Hashimoto K, Kushida N, Yanagida T, Inoue N, Yamaguchi O, Toma H, Suzutani T. 2007. Association of the outcome of renal transplantation with antibody response to cytomegalovirus strain-specific glycoprotein H epitopes. Clin. Infect. Dis. 45:60–67. [DOI] [PubMed] [Google Scholar]
- 5.Ross SA, Arora N, Novak Z, Fowler KB, Britt WJ, Boppana SB. 2010. Cytomegalovirus reinfections in healthy seroimmune women. J. Infect. Dis. 201:386–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gerna G, Sarasini A, Patrone M, Percivalle E, Fiorina L, Campanini G, Gallina A, Baldanti F, Revello MG. 2008. Human cytomegalovirus serum neutralizing antibodies block virus infection of endothelial/epithelial cells, but not fibroblasts, early during primary infection. J. Gen. Virol. 89:853–865. [DOI] [PubMed] [Google Scholar]
- 7.Fowler KB, Stagno S, Pass RF, Britt WJ, Boll TJ, Alford CA. 1992. The outcome of congenital cytomegalovirus infection in relation to maternal antibody status. N. Engl. J. Med. 326:663–667. [DOI] [PubMed] [Google Scholar]
- 8.Nigro G, Adler SP, La Torre R, Best AM. 2005. Passive immunization during pregnancy for congenital cytomegalovirus infection. N. Engl. J. Med. 353:1350–1362. [DOI] [PubMed] [Google Scholar]
- 9.Maidji E, Nigro G, Tabata T, McDonagh S, Nozawa N, Shiboski S, Muci S, Anceschi MM, Aziz N, Adler SP, Pereira L. 2010. Antibody treatment promotes compensation for human cytomegalovirus-induced pathogenesis and a hypoxia-like condition in placentas with congenital infection. Am. J. Pathol. 177:1298–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Adler SP, Nigro G. 2009. Findings and conclusions from CMV hyperimmune globulin treatment trials. J. Clin. Virol. 46(Suppl 4):S54–S57. [DOI] [PubMed] [Google Scholar]
- 11.Polilli E, Parruti G, D'Arcangelo F, Tracanna E, Clerico L, Savini V, D'Antonio F, Rosati M, Manzoli L, D'Antonio D, Nigro G. 2012. Preliminary evaluation of the safety and efficacy of standard intravenous immunoglobulins in pregnant women with primary cytomegalovirus infection. Clin. Vaccine Immunol. 19:1991–1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fouts AE, Chan P, Stephan JP, Vandlen R, Feierbach B. 2012. Antibodies against the gH/gL/UL128/UL130/UL131 complex comprise the majority of the anti-cytomegalovirus (anti-CMV) neutralizing antibody response in CMV hyperimmune globulin. J. Virol. 86:7444–7447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Potzsch S, Spindler N, Wiegers AK, Fisch T, Rucker P, Sticht H, Grieb N, Baroti T, Weisel F, Stamminger T, Martin-Parras L, Mach M, Winkler TH. 2011. B cell repertoire analysis identifies new antigenic domains on glycoprotein B of human cytomegalovirus which are target of neutralizing antibodies. PLoS Pathog. 7:e1002172. 10.1371/journal.ppat.1002172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kropff B, Burkhardt C, Schott J, Nentwich J, Fisch T, Britt W, Mach M. 2012. Glycoprotein N of human cytomegalovirus protects the virus from neutralizing antibodies. PLoS Pathog. 8:e1002999. 10.1371/journal.ppat.1002999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Axelsson F, Adler SP, Lamarre A, Ohlin M. 2007. Humoral immunity targeting site I of antigenic domain 2 of glycoprotein B upon immunization with different cytomegalovirus candidate vaccines. Vaccine 26:41–46. [DOI] [PubMed] [Google Scholar]
- 16.Cui X, Meza BP, Adler SP, McVoy MA. 2008. Cytomegalovirus vaccines fail to induce epithelial entry neutralizing antibodies comparable to natural infection. Vaccine 26:5760–5766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fu TM, Wang D, Freed DC, Tang A, Li F, He X, Cole S, Dubey S, Finnefrock AC, ter Meulen J, Shiver JW, Casimiro DR. 2012. Restoration of viral epithelial tropism improves immunogenicity in rabbits and rhesus macaques for a whole virion vaccine of human cytomegalovirus. Vaccine 30:7469–7474. [DOI] [PubMed] [Google Scholar]
- 18.Britt WJ, Mach M. 1996. Human cytomegalovirus glycoproteins. Intervirology 39:401–412. [DOI] [PubMed] [Google Scholar]
- 19.Urban M, Klein M, Britt WJ, Hassfurther E, Mach M. 1996. Glycoprotein H of human cytomegalovirus is a major antigen for the neutralizing humoral immune response. J. Gen. Virol. 77(Pt 7):1537–1547. [DOI] [PubMed] [Google Scholar]
- 20.Shimamura M, Mach M, Britt WJ. 2006. Human cytomegalovirus infection elicits a glycoprotein M (gM)/gN-specific virus-neutralizing antibody response. J. Virol. 80:4591–4600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Revello MG, Gerna G. 2010. Human cytomegalovirus tropism for endothelial/epithelial cells: scientific background and clinical implications. Rev. Med. Virol. 20:136–155. [DOI] [PubMed] [Google Scholar]
- 22.Ryckman BJ, Jarvis MA, Drummond DD, Nelson JA, Johnson DC. 2006. Human cytomegalovirus entry into epithelial and endothelial cells depends on genes UL128 to UL150 and occurs by endocytosis and low-pH fusion. J. Virol. 80:710–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ryckman BJ, Rainish BL, Chase MC, Borton JA, Nelson JA, Jarvis MA, Johnson DC. 2008. Characterization of the human cytomegalovirus gH/gL/UL128-131 complex that mediates entry into epithelial and endothelial cells. J. Virol. 82:60–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sung H, Schleiss MR. 2010. Update on the current status of cytomegalovirus vaccines. Expert Rev. Vaccines 9:1303–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Loomis RJ, Lilja AE, Monroe J, Balabanis KA, Brito LA, Palladino G, Franti M, Mandl CW, Barnett SW, Mason PW. 2013. Vectored co-delivery of human cytomegalovirus gH and gL proteins elicits potent complement-independent neutralizing antibodies. Vaccine 31:919–926. [DOI] [PubMed] [Google Scholar]
- 26.Pass RF. 2009. Development and evidence for efficacy of CMV glycoprotein B vaccine with MF59 adjuvant. J. Clin. Virol. 46(Suppl 4):S73–S76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pass RF, Zhang C, Evans A, Simpson T, Andrews W, Huang ML, Corey L, Hill J, Davis E, Flanigan C, Cloud G. 2009. Vaccine prevention of maternal cytomegalovirus infection. N. Engl. J. Med. 360:1191–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Walter EA, Greenberg PD, Gilbert MJ, Finch RJ, Watanabe KS, Thomas ED, Riddell SR. 1995. Reconstitution of cellular immunity against cytomegalovirus in recipients of allogeneic bone marrow by transfer of T-cell clones from the donor. N. Engl. J. Med. 333:1038–1044. [DOI] [PubMed] [Google Scholar]
- 29.Einsele H, Roosnek E, Rufer N, Sinzger C, Riegler S, Loffler J, Grigoleit U, Moris A, Rammensee HG, Kanz L, Kleihauer A, Frank F, Jahn G, Hebart H. 2002. Infusion of cytomegalovirus (CMV)-specific T cells for the treatment of CMV infection not responding to antiviral chemotherapy. Blood 99:3916–3922. [DOI] [PubMed] [Google Scholar]
- 30.Lilleri D, Fornara C, Furione M, Zavattoni M, Revello MG, Gerna G. 2007. Development of human cytomegalovirus-specific T cell immunity during primary infection of pregnant women and its correlation with virus transmission to the fetus. J. Infect. Dis. 195:1062–1070. [DOI] [PubMed] [Google Scholar]
- 31.Lilleri D, Fornara C, Revello MG, Gerna G. 2008. Human cytomegalovirus-specific memory CD8+ and CD4+ T cell differentiation after primary infection. J. Infect. Dis. 198:536–543. [DOI] [PubMed] [Google Scholar]
- 32.Sylwester AW, Mitchell BL, Edgar JB, Taormina C, Pelte C, Ruchti F, Sleath PR, Grabstein KH, Hosken NA, Kern F, Nelson JA, Picker LJ. 2005. Broadly targeted human cytomegalovirus-specific CD4+ and CD8+ T cells dominate the memory compartments of exposed subjects. J. Exp. Med. 202:673–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Plotkin SA, Farquhar J, Horberger E. 1976. Clinical trials of immunization with the Towne 125 strain of human cytomegalovirus. J. Infect. Dis. 134:470–475. [DOI] [PubMed] [Google Scholar]
- 34.Plotkin SA, Huang ES. 1985. Cytomegalovirus vaccine virus (Towne strain) does not induce latency. J. Infect. Dis. 152:395–397. [DOI] [PubMed] [Google Scholar]
- 35.Plotkin SA, Smiley ML, Friedman HM, Starr SE, Fleisher GR, Wlodaver C, Dafoe DC, Friedman AD, Grossman RA, Barker CF. 1984. Towne-vaccine-induced prevention of cytomegalovirus disease after renal transplants. Lancet i:528–530. [DOI] [PubMed] [Google Scholar]
- 36.Plotkin SA, Smiley ML, Friedman HM, Starr SE, Fleisher GR, Wlodaver C, Dafoe DC, Friedman AD, Grossman RA, Barker CF. 1984. Prevention of cytomegalovirus disease by Towne strain live attenuated vaccine. Birth Defects Orig. Artic. Ser. 20:271–287. [PubMed] [Google Scholar]
- 37.Plotkin SA, Starr SE, Friedman HM, Brayman K, Harris S, Jackson S, Tustin NB, Grossman R, Dafoe D, Barker C. 1991. Effect of Towne live virus vaccine on cytomegalovirus disease after renal transplant. A controlled trial. Ann. Intern. Med. 114:525–531. [DOI] [PubMed] [Google Scholar]
- 38.Plotkin SA, Starr SE, Friedman HM, Gonczol E, Weibel RE. 1989. Protective effects of Towne cytomegalovirus vaccine against low-passage cytomegalovirus administered as a challenge. J. Infect. Dis. 159:860–865. [DOI] [PubMed] [Google Scholar]
- 39.Plotkin SA, Higgins R, Kurtz JB, Morris PJ, Campbell DA, Jr, Shope TC, Spector SA, Dankner WM. 1994. Multicenter trial of Towne strain attenuated virus vaccine in seronegative renal transplant recipients. Transplantation 58:1176–1178. [PubMed] [Google Scholar]
- 40.Adler SP, Starr SE, Plotkin SA, Hempfling SH, Buis J, Manning ML, Best AM. 1995. Immunity induced by primary human cytomegalovirus infection protects against secondary infection among women of childbearing age. J. Infect. Dis. 171:26–32. [DOI] [PubMed] [Google Scholar]
- 41.Kemble G, Duke G, Winter R, Spaete R. 1996. Defined large-scale alterations of the human cytomegalovirus genome constructed by cotransfection of overlapping cosmids. J. Virol. 70:2044–2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Heineman TC, Schleiss M, Bernstein DI, Spaete RR, Yan L, Duke G, Prichard M, Wang Z, Yan Q, Sharp MA, Klein N, Arvin AM, Kemble G. 2006. A phase 1 study of 4 live, recombinant human cytomegalovirus Towne/Toledo chimeric vaccines. J. Infect. Dis. 193:1350–1360. [DOI] [PubMed] [Google Scholar]
- 43.Stannard LM, Rider JR, Farrar GH. 1989. Morphology and distribution of gp52 on extracellular human cytomegalovirus (HCMV) supports biochemical evidence that it represents the HCMV glycoprotein B. J. Gen. Virol. 70(Pt 6):1553–1560. [DOI] [PubMed] [Google Scholar]
- 44.Varnum SM, Streblow DN, Monroe ME, Smith P, Auberry KJ, Pasa-Tolic L, Wang D, Camp DG, II, Rodland K, Wiley S, Britt W, Shenk T, Smith RD, Nelson JA. 2004. Identification of proteins in human cytomegalovirus (HCMV) particles: the HCMV proteome. J. Virol. 78:10960–10966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pepperl S, Munster J, Mach M, Harris JR, Plachter B. 2000. Dense bodies of human cytomegalovirus induce both humoral and cellular immune responses in the absence of viral gene expression. J. Virol. 74:6132–6146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pepperl-Klindworth S, Frankenberg N, Plachter B. 2002. Development of novel vaccine strategies against human cytomegalovirus infection based on subviral particles. J. Clin. Virol. 25(Suppl 2):S75–S85. [DOI] [PubMed] [Google Scholar]
- 47.Wang Z, Mo C, Kemble G, Duke G. 2004. Development of an efficient fluorescence-based microneutralization assay using recombinant human cytomegalovirus strains expressing green fluorescent protein. J. Virol. Methods 120:207–215. [DOI] [PubMed] [Google Scholar]
- 48.Irmiere A, Gibson W. 1983. Isolation and characterization of a noninfectious virion-like particle released from cells infected with human strains of cytomegalovirus. Virology 130:118–133. [DOI] [PubMed] [Google Scholar]
- 49.Talbot P, Almeida JD. 1977. Human cytomegalovirus: purification of enveloped virions and dense bodies. J. Gen. Virol. 36:345–349. [DOI] [PubMed] [Google Scholar]
- 50.Spaete RR, Saxena A, Scott PI, Song GJ, Probert WS, Britt WJ, Gibson W, Rasmussen L, Pachl C. 1990. Sequence requirements for proteolytic processing of glycoprotein B of human cytomegalovirus strain Towne. J. Virol. 64:2922–2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Spaete RR, Thayer RM, Probert WS, Masiarz FR, Chamberlain SH, Rasmussen L, Merigan TC, Pachl C. 1988. Human cytomegalovirus strain Towne glycoprotein B is processed by proteolytic cleavage. Virology 167:207–225. [DOI] [PubMed] [Google Scholar]
- 52.Filipe V, Hawe A, Jiskoot W. 2010. Critical evaluation of nanoparticle tracking analysis (NTA) by NanoSight for the measurement of nanoparticles and protein aggregates. Pharm. Res. 27:796–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gibson W. 2008. Structure and formation of the cytomegalovirus virion. Curr. Top. Microbiol. Immunol. 325:187–204. [DOI] [PubMed] [Google Scholar]
- 54.Pepperl-Klindworth S, Frankenberg N, Riegler S, Plachter B. 2003. Protein delivery by subviral particles of human cytomegalovirus. Gene Ther. 10:278–284. [DOI] [PubMed] [Google Scholar]
- 55.Law KM, Wilton-Smith P, Farrar GH. 1985. A murine monoclonal antibody recognising a single glycoprotein within a human cytomegalovirus virion envelope glycoprotein complex. J. Med. Virol. 17:255–266. [DOI] [PubMed] [Google Scholar]
- 56.Rasmussen L, Mullenax J, Nelson R, Merigan TC. 1985. Viral polypeptides detected by a complement-dependent neutralizing murine monoclonal antibody to human cytomegalovirus. J. Virol. 55:274–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Basgoz N, Qadri I, Navarro D, Sears A, Lennette E, Youngblom J, Pereira L. 1992. The amino terminus of human cytomegalovirus glycoprotein B contains epitopes that vary among strains. J. Gen. Virol. 73(Pt 4):983–988. [DOI] [PubMed] [Google Scholar]
- 58.Mocarski ES, Kemble GW, Lyle JM, Greaves RF. 1996. A deletion mutant in the human cytomegalovirus gene encoding IE1(491aa) is replication defective due to a failure in autoregulation. Proc. Natl. Acad. Sci. U. S. A. 93:11321–11326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bech-Serra JJ, Borthwick A, Colome N, Albar JP, Wells M, Sanchez del Pino M, Canals F. 2009. A multi-laboratory study assessing reproducibility of a 2D-DIGE differential proteomic experiment. J. Biomol. Tech. 20:293–296. [PMC free article] [PubMed] [Google Scholar]
- 60.Underwood MR, Harvey RJ, Stanat SC, Hemphill ML, Miller T, Drach JC, Townsend LB, Biron KK. 1998. Inhibition of human cytomegalovirus DNA maturation by a benzimidazole ribonucleoside is mediated through the UL89 gene product. J. Virol. 72:717–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Krosky PM, Underwood MR, Turk SR, Feng KW, Jain RK, Ptak RG, Westerman AC, Biron KK, Townsend LB, Drach JC. 1998. Resistance of human cytomegalovirus to benzimidazole ribonucleosides maps to two open reading frames: UL89 and UL56. J. Virol. 72:4721–4728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hwang JS, Kregler O, Schilf R, Bannert N, Drach JC, Townsend LB, Bogner E. 2007. Identification of acetylated, tetrahalogenated benzimidazole d-ribonucleosides with enhanced activity against human cytomegalovirus. J. Virol. 81:11604–11611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lozza L, Lilleri D, Percivalle E, Fornara C, Comolli G, Revello MG, Gerna G. 2005. Simultaneous quantification of human cytomegalovirus (HCMV)-specific CD4+ and CD8+ T cells by a novel method using monocyte-derived HCMV-infected immature dendritic cells. Eur. J. Immunol. 35:1795–1804. [DOI] [PubMed] [Google Scholar]
- 64.Reap EA, Morris J, Dryga SA, Maughan M, Talarico T, Esch RE, Negri S, Burnett B, Graham A, Olmsted RA, Chulay JD. 2007. Development and preclinical evaluation of an alphavirus replicon particle vaccine for cytomegalovirus. Vaccine 25:7441–7449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cosme RC, Martinez FP, Tang Q. 2011. Functional interaction of nuclear domain 10 and its components with cytomegalovirus after infections: cross-species host cells versus native cells. PLoS One 6:e19187. 10.1371/journal.pone.0019187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dolan A, Cunningham C, Hector RD, Hassan-Walker AF, Lee L, Addison C, Dargan DJ, McGeoch DJ, Gatherer D, Emery VC, Griffiths PD, Sinzger C, McSharry BP, Wilkinson GW, Davison AJ. 2004. Genetic content of wild-type human cytomegalovirus. J. Gen. Virol. 85:1301–1312. [DOI] [PubMed] [Google Scholar]
- 67.Reap EA, Dryga SA, Morris J, Rivers B, Norberg PK, Olmsted RA, Chulay JD. 2007. Cellular and humoral immune responses to alphavirus replicon vaccines expressing cytomegalovirus pp65, IE1, and gB proteins. Clin. Vaccine Immunol. 14:748–755. [DOI] [PMC free article] [PubMed] [Google Scholar]