

Abstract
Respiratory syncytial virus (RSV) belongs to the family Paramyxoviridae and is the single most important cause of serious lower respiratory tract infections in young children, yet no highly effective treatment or vaccine is available. Increased airway resistance and increased airway mucin production are two manifestations of RSV infection in children. RSV rA2-line19F infection induces pulmonary mucous production and increased breathing effort in BALB/c mice and provides a way to assess these manifestations of RSV disease in an animal model. In the present study, we investigated the effect of prophylactic treatment with the F(ab′)2 form of the anti-G protein monoclonal antibody (MAb) 131-2G on disease in RSV rA2-line19F-challenged mice. F(ab′)2 131-2G does not affect virus replication. It and the intact form that does decrease virus replication prevented increased breathing effort and airway mucin production, as well as weight loss, pulmonary inflammatory-cell infiltration, and the pulmonary substance P and pulmonary Th2 cytokine levels that occur in mice challenged with this virus. These data suggest that the RSV G protein contributes to prominent manifestations of RSV disease and that MAb 131-2G can prevent these manifestations of RSV disease without inhibiting virus infection.
INTRODUCTION
Respiratory syncytial virus (RSV), a pneumovirus of the family Paramyxoviridae, is a leading cause of lower respiratory tract disease in infants and young children worldwide (1). Despite ≥50 years of research, there is still no highly effective treatment for or licensed vaccine to prevent RSV disease. Immune prophylaxis, initially with immune globulin with a high titer of antibody against RSV (2) and later with an RSV anti-F protein-neutralizing monoclonal antibody (MAb) (3), was shown to be effective and is used to prevent complications of infection in high-risk infants and young children (4). Good infection control practices, the other available preventive measures, are effective in preventing nosocomial transmission in health care settings (5).
RSV encodes 11 proteins, and among them the F and G surface glycoproteins are important for inducing protective immunity (6, 7). The F protein induces higher titers of neutralizing antibodies and better cross-protection against different RSV strains in animal models (8–10). The G protein plays a substantial role in inducing and modulating the host immune response to infection (11), and some of the induced responses appear to contribute to disease. For example, during RSV infection, the presence of the RSV G protein is associated with a lower frequency of gamma interferon (IFN-γ)- and a higher frequency of interleukin-5 (IL-5)- and IL-6-expressing T cells (12) and alterations in the production of cytokines in mice (13). The G protein has also been associated with increased pulmonary eosinophilia in formalin-inactivated RSV vaccine-enhanced disease (12, 14), increased pulmonary levels of substance P (15), and decreased respiratory rates when given intravenously to mice (16). The CX3C chemokine motif in the central conserved region of the G protein has been shown to contribute to some of these activities (14, 16, 17). The G protein CX3C motif binds to the CX3C chemokine receptor CX3CR1 and mimics some activities of the only known CX3C chemokine, fractalkine (18). This motif, located between amino acids (aa) 182 and 186 of the G protein, includes two of the four evolutionarily conserved cysteines at aa 173, 176, 182, and 186, which form a cysteine noose structure.
Monoclonal antibody 131-2G (19, 20) binds to the central conserved region of the G protein, blocks binding to CX3CR1, and decreases several manifestations of disease in RSV-challenged mice, including increased pulmonary inflammation, G protein-induced depression of respiratory rates, and enhanced inflammation in RSV-challenged formalin-inactivated RSV (FI-RSV)-vaccinated mice (16, 21–23). Treatment with this MAb also neutralizes virus in vivo in an Fc-dependent fashion, but not in vitro (20). Importantly, 131-2G F(ab′)2 decreases pulmonary inflammation after both primary RSV challenge and challenge in FI-RSV-vaccinated mice without decreasing viral load (22, 23).
A recently described strain of RSV, line 19, induces airway reactivity and increased mucous production in mice. This antigenic subgroup A strain of RSV was isolated from a sick infant at the University of Michigan (24). The airway reactivity and increased mucous production induced by line 19 infection are associated with the F gene, and a virus derived from the RSV A2 strain that has the line 19 F gene, rA2-line19F, induces both disease manifestations in mice (25). rA2-line19F also induces the Th2 cytokine IL-13, a mediator of pulmonary mucous secretion (26–28) which has been reported to be present in RSV-specific T cells from infants with RSV bronchiolitis (29).
Since increased airway resistance and mucous production are associated with RSV infection in humans (30–32), their induction by RSV rA2-line19F provides additional clinically relevant outcomes to study in mice (25, 33). In this study, we chose to determine if the F(ab′)2 form of MAb 131-2G can block these features of disease with rA2-line19F infection and, consequently, determine if the G protein likely contributes to these manifestations of RSV disease. We used the F(ab′)2 form of 131-2G to eliminate decreased virus replication as a reason for the observed effects. Our results show that prophylaxis with the F(ab′)2 form of MAb 131-2G, as well as the intact form, decreased airway mucin production, pulmonary cellular infiltration, pulmonary substance P levels, and breathing effort. These data suggest that prevention of increased mucous production and airway dysfunction can be added to the potential benefits of binding G protein with antibody against the central conserved region of G protein.
MATERIALS AND METHODS
Animals.
Animal studies were performed according to a protocol approved by the Emory University (Atlanta, GA) Institutional Animal Care and Use Committee. Four- to 6-week-old, specific-pathogen-free female BALB/c mice (Charles River Laboratory, Wilmington, MA) were used in all experiments. Mice were housed in microisolator cages and fed sterilized water and food ad libitum. In study 1, mice were anesthetized by intraperitoneal administration of tribromoethanol (Avertin; 2% 2,2,2-tribromoethanol, 180 to 250 mg/kg in tertiary amyl alcohol) and challenged intranasally with 1 × 106 PFU of RSV A2 or rA2-line19F in serum-free minimal essential medium (MEM) (volume, 50 μl) with no treatment. In study 2, mice were intraperitoneally treated with 300 μg each of the intact and F(ab′)2 forms of anti-RSV G MAb 131-2G and the intact and F(ab′)2 forms of normal mouse IgG (nIgG; Pierce Protein Research Products, Rockford, IL). Two days later, they were challenged intranasally with 1 × 106 50% tissue culture infective dose (TCID50) of RSV rA2-line19F in serum-free MEM (volume, 50 μl). At each time point, 3 to 5 mice per group were examined (Fig. 1).
Fig 1.

Experimental schedule for animal studies. Days are numbered relative to the day of RSV challenge.
Virus preparation.
RSV A2 or rA2-line19F was propagated in HEp-2 cells. RSV A2 was included in some experiments to enhance comparisons of the results of this study to those of earlier studies with rA2-line19F. Briefly, 5 days after inoculation of HEp-2 cells with RSV A2 or rA2-line19F at a multiplicity of infection (MOI) of 0.01, loosely capped flasks containing the cells were transferred to a −80°C freezer overnight. After thawing at 4°C, cells were scraped down and the cell lysates were transferred to 50-ml conical tubes. The cell debris was removed by centrifugation at 3,000 rpm for 7 min at 4°C. The supernatants were pooled and purified by centrifugation on a sucrose cushion (20% sucrose) at 16,000 × g for 4 h. After centrifugation, the supernatant was removed and the pellet was dissolved in serum-free MEM. The viral pools were divided into aliquots, quick-frozen in liquid nitrogen, and stored at −80°C until they were used.
Virus infectivity titers were determined by microinfectivity assays as previously described (34). Briefly, serial dilutions of the virus preparation in minimal essential medium supplemented with fetal bovine serum (Fisher, Pittsburgh, PA), 2 mM l-glutamine (Gibco, Grand Island, NY), and 5,000 units/ml penicillin-streptomycin (Gibco, Grand Island, NY) were inoculated onto subconfluent HEp-2 cells in 96-well microtiter plates. After 2 h of adsorption at 37°C, 180 μl of tissue culture media was added, and 5 days later, the cells were fixed with 80% acetone in 1× phosphate-buffered saline (PBS). Replication of virus was determined by RSV enzyme-linked immunosorbent assay (ELISA) using plates with the acetone-fixed RSV-infected HEp-2 cells. The plates were blocked with 5% bovine serum albumin (BSA) in 1× PBS for 1 h at 37°C, and then goat anti-RSV antibody (Millipore, Billerica, MA) was added and the plates were incubated for 1 h at 37°C. The plates were washed with 1× PBS containing 0.05% Tween and then incubated for 1 h at 37°C with peroxidase-conjugated donkey anti-goat antibody (Jackson ImmunoResearch, West Grove, PA). After a second washing, color was developed with o-phenylenediamine (OPD) substrate (Sigma-Aldrich, St. Louis, MO) as indicated by the manufacturer. The enzyme-substrate reaction was terminated by the addition of a sulfuric acid solution, and absorbance was measured at a wavelength of 450 nm. The infectivity titer was determined using the Reed and Muench method (35).
F(ab′)2 fragment preparation and treatment.
Monoclonal antibody 131-2G F(ab′)2 fragments were generated by pepsin digestion (Sigma-Aldrich). Briefly, purified MAb 131-2G was digested with porcine pepsin overnight at 37°C, and digested MAb was passed through a protein G Sepharose column (GE Healthcare, Alpharetta, GA) to eliminate Fc fragments and undigested antibodies. Purified F(ab′)2 fragments were dialyzed and concentrated using a Centricon spin column (Millipore, Temecula, CA) with a 30-kDa cutoff. The purity of the F(ab′)2 fragments was determined by SDS-PAGE (Bio-Rad, Hercules, CA) under nonreducing conditions. The protein concentration was determined by bicinchoninic acid (BCA) protein assay (Pierce Protein Research Products, Rockford, IL). Endotoxin concentrations were determined using a Limulus amebocyte lysate chromogenic endpoint assay in accordance with the manufacturer's instructions (Lonza, Atlanta, GA). Intact 131-2G was included in some experiments to determine if it had effects on rA2-line19F disease similar to those of F(ab′)2 131-2G and if it would also decrease replication of rA2-line19F in mice.
Quantification of lung viral load.
Pulmonary viral load replication was assessed by measuring infectious virus in homogenized lung tissue. To homogenize the lungs, we used a BeadBeater (Biospec Products, Bartlesville, OK). Two-milliliter-deep 96-well plates (Axygen Scientific, Union City, CA) were loaded with 1 ml of zirconium beads (Biospec Products, Bartlesville, OK), sealed with a rubber lid (Axiomat; Axygen Scientific, Union City, CA), and autoclaved. Sterile basal MEM (500 μl) was added to each well, and the plates were briefly centrifuged and then kept on ice. For each mouse, the left lung lobe was harvested, weighed, cut into four to five pieces, and placed into a well of a 2-ml-deep 96-well plate on ice. An additional 400 μl of basal MEM was added to each well, and the plates were sealed with the Axiomat lids. The plates were shaken in the BeadBeater for 1 min, followed by a 1-min plunge in an ice and water slurry. This was repeated 12 times for a total of 12 1-min homogenizations. The plates were centrifuged at 2,000 rpm for 5 min at 4°C. Lung supernatants were divided into aliquots; one aliquot was used for virus infectivity titration and the remaining aliquots were stored at −80°C for future studies. Virus infectivity titers were determined as described above in the section “Virus Preparation.”
Viral RNA levels were determined by RSV real-time PCR.
Total RNA was extracted from bronchoalveolar lavage (BAL) fluid using a Qiagen total-RNA extraction kit (Qiagen, Valencia, CA) according to the manufacturer's instructions and stored at −80°C. Quantitative real-time PCR was performed by using an AgPath-ID one-step reverse transcription (RT)-PCR kit (Applied Biosystems, Foster City, CA) and an ABI 7700 sequence detection system (PE Applied Biosystems, Foster City, CA). Thermal cycling conditions included 45°C for 2 min and 95°C for 10 min, followed by 40 cycles of amplification at 95°C for 15 s and 55°C for 2 min for denaturing and annealing, respectively. The primers and probes for the RSV matrix (M) gene (forward primer, 5′-GGC AAA TAT GGA AAC ATA GCT GAA-3′; reverse primer, 5′-TCT TTT TCT AGG ACA TTG TAY TGA ACA G-3′; probe, 5′-6-carboxyfluorescein (FAM)-TGT CCG TCT TCT ACG CCC TCG TC- black hole quencher 1 (BHQ-1)-3′) (36) were obtained from Integrated DNA Technologies (IDT) (Coralville, IA). Threshold cycles (CT) for each sample were calculated. Assays were performed in duplicate for each experiment, and they were repeated for three different sets of mice.
BAL leukocyte specimens.
Mice were anesthetized and euthanized by exsanguination after severing of the left axillary artery. Leuckocytes were harvested by lavaging the lungs 3 times using 1 ml sterile 1× phosphate-buffered saline (PBS) for each wash. The BAL cells were stained for extracellular markers using microculture staining according to the protocol described by Tripp et al. (12). Briefly, BAL cells were blocked in Fc blocker (anti-CD16/32) in 1× PBS with 1% bovine serum albumin for 15 min at 4°C and then stained for 30 min at 4°C in the dark with the appropriate combinations of anti-CD3 (17A2) (T cells), anti-CD4 (GK1.5) (T cells), anti-CD8 (53-6.7) (T cells), anti-CD45R/B220 (RA3-6B2) (B cells), anti-CD11b (M1/70) (macrophages, dendritic cells, and monocytes), anti-mouse Ly-6G/Gr-1 (RB6-8C5) (polymorphonuclear cells [PMNs]), anti-mouse CD49b/integrin alpha 2 (DX5) (NK and NK T cells), and mouse isotype antibody controls (all from eBiosciences, San Diego, CA) diluted in staining buffer. The distributions and patterns of cell surface markers were determined for 10,000 lymphocyte-gated events analyzed on a BD LSRII flow cytometer (BD Biosciences, Mountain View, CA), and data were analyzed using FlowJo software (TreeStar, Ashland, OR).
Quantitation of substance P.
Substance P (SP) levels in cell-free BAL supernatant were analyzed by using a competitive ELISA kit (Cayman Chemical, Ann Arbor, MI) in accordance with the manufacturer's instructions and as previously described (37). The assay is based on the competition between free SP and an SP tracer for a limited number of SP-specific binding sites. The percent sample bound/maximum bound was calculated, and the SP concentration in each sample was determined based on the percent standard bound/maximum bound versus the standard SP concentration. The intra- and interassay coefficients of variation are 10%.
Pulse oximetry.
Pulsus paradoxus is an exaggeration of normal variation in the pulse volume with respiration that can be caused by labored breathing and the associated increased negative and positive intrathoracic pressures with inspiration and expiration, respectively. The breath-associated difference in pulse volume (difference between systole and diastole) was assessed by measuring the absorbance of light passing through tissue with a rodent pulse oximeter (MouseOx; Starr Life Sciences Corp., Oakmont, PA). The difference in light absorbance between systole and diastole was used to calculate changes in the effective path length of light passing through pulsating vessels or arterioles as an indicator of pulse volume, and differences in pulse volume between inspiration and expiration were estimated and reported as breath distention in microns (38). For these measurements, mice were anesthetized using an isoflurane (2.5%)-oxygen (2 liters/min) mixture provided by an anesthesia machine (VetEquip, Pleasanton, CA). Mice were anesthetized five at a time in a Plexiglas box connected to the anesthesia machine. One mouse at a time was removed from the Plexiglas box and placed on its back on a pad with its nose in an isoflurane-oxygen nosecone connected to the anesthesia machine. The anesthetized mouse's feet were restrained to a pad with tape, and the MouseOx sensor was applied to the mouse's thigh. The mouse was covered with a cloth to reduce ambient light, and its arterial O2 saturation, heart rate, pulse rate, pulse distension, and breath distention were recorded approximately every 0.1 s (MouseOx software, version 4.0) for 1 to 5 min. Only time points in which all of these physiological parameters were successfully measured were included in analyses, giving 30 s to 2 min of measurements per mouse. The average recorded breath-associated change in vessel wall distension (in microns) was used as the value for each mouse. Data were analyzed using Microsoft Excel and presented in microns.
Determination of Muc5AC protein expression.
Mucin-5AC protein (Muc5AC) concentrations in lung tissues were assayed with a mouse mucin-5 subtype AC ELISA kit (USCN Life Science, Inc., Wuhan, China) used according to the manufacturer's protocol. Briefly, an aliquot of homogenized lung tissue was thawed, and the total protein concentration was determined by a BCA protein assay kit (Thermo Scientific, Rockford, IL). Muc5AC standards and lung homogenates were added to designated microtiter plate wells precoated with anti-Muc5AC antibody. After 2 h of incubation at 37°C, biotin-conjugated polyclonal antibody was added and the wells were incubated for 1 h at 37°C. Next, avidin conjugated to horseradish peroxidase was added and the wells were incubated for 1 h at 37°C. After the wells were washed, color was developed with TMB substrate solution (3,3′,5,5′-tetramethylbenzidine). The enzyme-substrate reaction was terminated by the addition of a sulfuric acid solution, and absorbance was measured at a wavelength of 450 nm. The concentration of Muc5AC in the samples was determined by comparing the optical density (OD) of the samples to the standard curve generated and expressed as a fold value.
Histopathology.
Lungs were fixed in 10% formalin for 1 h, followed by washing with 70% ethanol and embedding in paraffin blocks. Five-micrometer sections of lung tissue were deparaffinized in xylene, rehydrated through a graded series of ethanol washes, and stained with a periodic acid-schiff (PAS) kit (Sigma-Aldrich, St. Louis, MO) to assess intracellular pulmonary mucin levels. PAS-stained slides were digitally scanned using a Hamamatsu NanoZoomer 2.0-HT slide scanner (Meyer Instruments, Houston, TX) with a 20× objective and analyzed using ImageJ software. Fifteen to 20 fields (20× magnification) per tissue section were examined.
For immunohistochemistry staining, deparaffinized sections were sequentially incubated at room temperature with blocking serum for 30 min; primary antibody, i.e., anti-Muc5AC (clone 45MI) (Santa Cruz, Dallas, TX), at 4°C overnight; biotinylated secondary antibody (Santa Cruz, South Carolina) for 30 min; 3% H2O2 for 5 min to eliminate endogenous hydrogen peroxidase activity; and horseradish peroxidase-streptavidin for 30 min. Final visualization of antigen was achieved by incubating sections with a diaminobenzidine-H2O2 solution for 5 min until the desired stain intensity developed. The sections were digitally scanned using a Hamamatsu NanoZoomer 2.0-HT slide scanner with a 20× objective and analyzed using ImageJ software. Fifteen to 25 fields in the tissue sections were analyzed.
For immunofluorescence staining, deparaffinized sections were sequentially incubated at room temperature with avidin-biotin blocking (Vector Laboratories, Burlingame, CA) for 30 min; primary antibody, i.e., goat polyclonal anti-RSV (Millipore, Burlingame, CA), for 1 h at room temperature; biotinylated rabbit anti-goat secondary antibody (Vector Laboratories) for 30 min; and fluorescein avidin DCS (Vector Laboratories) for 10 min in the dark. The slides were mounted with ProLong Gold antifade reagent with DAPI (4′,6-diamidino-2-phenylindole; Invitrogen, Grand Island, NY) at room temperature. The sections were visualized with an Amg Evos* fl digital inverted fluorescence microscope (Fisher Scientific, Pittsburgh, PA) with a 20× objective and analyzed using ImageJ software. Fifteen to 20 fields (20× magnification) per tissue section were examined.
Multiplex cytokine analysis.
Cytokine levels in lung homogenates were normalized to the protein present (in milligrams) in cell-free preparations of lung supernatants as measured by BCA assay used according to the manufacturer's protocol (Thermo Scientific, Rockford, IL). The lung homogenates were centrifuged, and the cell-free supernatants were tested by multiplex Luminex assays for IL-13, IL-4, IL-5, monocyte chemotactic protein 1 (MCP-1), interferon-inducible protein 10 (IP-10), and IL-12 (Invitrogen, Valencia, CA) according to the manufacturer's instructions (mouse cytokine 20-plex panel; Invitrogen, Valencia, CA) using an xMap 200 system (Invitrogen). The concentration of each cytokine was determined by comparison to the standard curve according to the manufacturer's instructions. The threshold of detection was 5 pg/ml.
Statistical analyses.
Unless otherwise indicated, groups were compared by one-way analysis of variance (ANOVA) and post hoc Tukey's honestly significant difference (HSD) test (P ≤ 0.05). A P value of ≤0.05 was considered statistically significant. All statistical analyses were performed using the statistical package R (R Developmental Core Team 2012). Data are shown as means ± standard errors of the means (SEMs).
RESULTS
Kinetics of rA2-line19F infection.
The effects of rA2-line19F and A2 challenges (1 × 106 TCID50) on weight loss, viral load, pulmonary inflammation, and pulsus paradoxus were determined on days 1, 3, 5, and 8. Both A2 and rA2-line19F infections caused noticeable weight loss compared to mock-challenged mice, with the peak difference on day 5 postinfection (p.i.) (P ≤ 0.05) (Fig. 2A). rA2-line19F caused greater weight loss, but the differences were not significant. Both viruses showed the peak virus titer in the lungs at day 5 p.i., with rA2-line19F giving a 0.42 ± 0.12 log TCID50 higher titer than A2 (Fig. 2B) (P ≤ 0.001). The kinetics of pulmonary inflammation paralleled the titers of virus in the lungs, with peak values on day 5 p.i. rA2-line19F-infected mice showed a significantly greater number (P ≤ 0.001) of inflammatory cells in the lung than A2-infected mice (Fig. 2C). rA2-line19F-infected mice had a 37.37% ± 16.13% increase in cell numbers in BAL fluid at day 5 p.i. Note also the difference in the types of cells in the BAL fluid (Table 1). For example, on day 5 p.i., relative to A2-infected mice, rA2-line19F-infected mice had a 74.98% ± 7.18% increase in CD3− CD45R/B220 (RA3-6B2)+ cells (B cells) and a 66.99% ± 8.42% increase in CD3− CD11b (M1/70)+ cells (macrophages, dendritic cells, and monocytes) compared to a 23.34% ± 4.28% increase in CD3− Ly-6G/Gr-1 (RB6-8C5)+cells (polymorphonuclear cells), a 54.26% ± 11.37% increase in CD3− CD49b/integrin alpha 2 (DX5)+ cells (NK cells), a 47.45% ± 14.18% increase in CD8 (53-6.7)+ cells (T cells), and a 56.62% ± 11.20% increase in CD4 (GK1.5)+ cells (T cells). By contrast, on day 8 p.i. the most prominent increase in a BAL cell type for rA2-line19F-infected mice compared to that for A2-infected mice was the 78.30% ± 6.59% increase in CD8 (53.6.7)+ cells (T cells). Pulsus paradoxus, or breathing effort, was used as our indicator of lung dysfunction, and similar to those in a previous study (25), rA2-line19F-challenged mice had a significant increase in breathing effort values compared to those for A2- and mock-challenged mice, with the peak increase on day 8 p.i (Fig. 2D). Based on these data, we used day 5 p.i. to assess the effect of F(ab′)2 131-2G MAb prophylaxis on weight loss, pulmonary viral load, and inflammation and day 8 p.i. to assess the effect on mucous production and breathing effort.
Fig 2.

Comparisons of challenges with two RSV strains, rA2-line19F and A2, on weight loss (n = 10 mice/group) (A), viral load (n = 3 mice/group) (B), pulmonary inflammation (n = 5 mice/group) (C), and pulsus paradaxus (n = 5 mice/group) (D). BALB/c mice were challenged with 1 × 106 TCID50 of either rA2-line19F or A2. Data points represent means ± SEMs. +, values for rA2-line19F are significantly different (P ≤ 0.001; ANOVA) from those for A2; #, values for rA2-line19F are significantly different (P ≤ 0.001; ANOVA) from those for mock infections; *, values forA2 are significantly different (P ≤ 0.001; ANOVA) from those for mock infections.
Table 1.
Pulmonary leukocyte trafficking after rA2-line19F and A2 RSV infectionsa
| Day postinfection | Phenotype | Mean total no. of A2 cells ± SE (103) | Mean total no. of rA2-line19F cells ± SE (103) | % increase, A2 to rA2-line19F |
|---|---|---|---|---|
| 1 | CD3 | 3.96 ± 0.57 | 4.96 ± 0.39** | 18.90 ± 7.13 |
| CD4 | 0.22 ± 0.05 | 0.35 ± 0.09 | 33.77 ± 16.89 | |
| CD8 | 0.24 ± 0.02 | 0.49 ± 0.08*** | 49.53 ± 5.65 | |
| B cell | 2.42 ± 0.50 | 3.61 ± 1.31 | 27.67 ± 8.60 | |
| Macrophage | 4.95 ± 0.64 | 8.63 ± 0.94*** | 42.01 ± 9.54 | |
| PMN | 3.15 ± 0.72 | 4.76 ± 0.94** | 40.62 ± 9.64 | |
| NK | 1.86 ± 0.75 | 3.71 ± 1.14** | 49.86 ± 9.30 | |
| 3 | CD3 | 1.36 ± 0.61 | 5.33 ± 0.45*** | 73.55 ± 13.58 |
| CD4 | 0.20 ± 0.14 | 1.36 ± 0.35*** | 83.81 ± 13.45 | |
| CD8 | 0.16 ± 0.06 | 0.94 ± 0.09*** | 83.05 ± 6.07 | |
| B cell | 1.94 ± 0.76 | 5.50 ± 0.57*** | 63.11 ± 17.94 | |
| Macrophage | 6.57 ± 1.75 | 10.23 ± 1.31** | 36.09 ± 14.15 | |
| PMN | 0.35 ± 0.31 | 0.66 ± 0.12 | 41.07 ± 10.01 | |
| NK | 2.04 ± 0.70 | 4.71 ± 0.22** | 56.77 ± 19.98 | |
| 5 | CD3 | 4.69 ± 1.72 | 7.45 ± 2.56 | 37.05 ± 2.06 |
| CD4 | 0.96 ± 0.39 | 1.67 ± 0.50** | 56.62 ± 11.20 | |
| CD8 | 1.22 ± 0.46 | 1.66 ± 0.72 | 47.45 ± 14.18 | |
| B cell | 3.20 ± 0.94 | 13.64 ± 5.63** | 74.98 ± 7.18 | |
| Macrophage | 7.70 ± 0.71 | 25.13 ± 8.08** | 66.99 ± 8.42 | |
| PMN | 0.56 ± 0.21 | 0.72 ± 0.26 | 23.34 ± 4.28 | |
| NK | 3.94 ± 0.69 | 7.65 ± 1.70** | 54.26 ± 11.37 | |
| 8 | CD3 | 5.94 ± 1.10 | 18.20 ± 2.13*** | 67.27 ± 5.05 |
| CD4 | 1.29 ± 0.10 | 3.86 ± 0.45*** | 66.27 ± 4.56 | |
| CD8 | 2.20 ± 0.74 | 10.15 ± 1.25*** | 78.30 ± 6.59 | |
| B cell | 2.44 ± 0.55 | 3.11 ± 0.20 | 21.82 ± 5.06 | |
| Macrophage | 7.47 ± 0.49 | 10.59 ± 0.62*** | 29.14 ± 8.10 | |
| PMN | 0.90 ± 0.11 | 1.96 ± 0.22*** | 53.19 ± 10.21 | |
| NK | 4.40 ± 0.57 | 6.73 ± 0.43*** | 34.35 ± 10.22 |
BALB/c mice were infected with 1 × 106 TCID50 PFU of A2 or rA2-line19F RSV. Data are mean total numbers of BAL cells by subtype at day 5 p.i. per lung. Subtypes: lymphocyte gate, anti-CD3+ (17A2) and anti-CD4+ (GK1.5) CD4 T cells; lymphocyte gate, CD3+ and anti-CD8+ (53-6.7)− CD8 T cells; CD3− and anti-CD45R/B220+ (RA3-6B2)− B cells; CD3− and anti-CD11b+ (M1/70)− macrophages, dendritic cells, and monocytes; CD3− and anti-mouse Ly06G/Gr-1 (RB6-8C5)− polymorphonuclear cells (PMNs); CD3− and anti-mouse CD49b/integrin alpha 2 (DX5)− NK cells. **, P ≤ 0.05, as determined by ANOVA; ***, P ≤ 0.001, a significant increase in total BAL cell types in the rA2-line19F-infected group compared to the A2-infected group. Results are representative of two independent experiments for rA2-line19F challenge and mock challenge and one experiment for A2 challenge. The data are from no fewer than five mice per experiment.
F(ab′)2 131-2G MAb treatment protects mice from RSV-induced weight loss but not pulmonary virus replication.
Treatment with the F(ab′)2 and intact forms of 131-2G prevented much of the weight loss seen in mice challenged with rA2-line19F (P ≤ 0.001) (Fig. 3). As expected, the intact form of anti-RSV G MAb 131-2G decreased pulmonary virus load (on day 5 p.i.; P ≤ 0.001), while the F(ab′)2 form had no effect on virus load (Fig. 4A). The RSV real-time PCR results for BAL specimens (5 mice/group) on day 5 p.i. also showed that 131-2G F(ab′)2 prophylaxis did not affect virus replication; i.e., the CT values were 34 ± 2 for untreated, nIgG antibody-treated, or 131-2G F(ab′)2-treated mice challenged with RSV (data not shown). Thus, the effects on disease of 131-2G F(ab′)2 prophylaxis are not the result of a decrease in virus replication but are presumably from binding to G and blocking G-induced host responses to infection.
Fig 3.
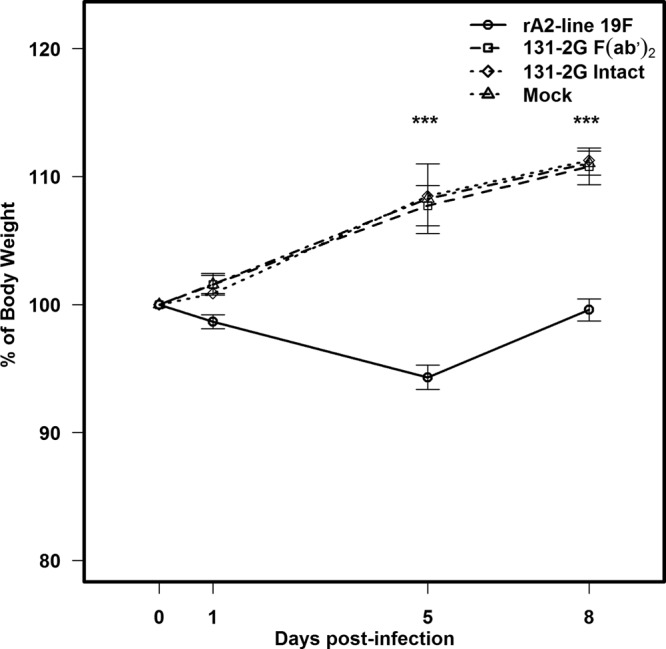
Effect of prophylactic administration of anti-RSV G protein MAb 131-2G F(ab′)2 on weight change in BALB/c mice. BALB/c mice were challenged with rA2-line19F (1 × 106 TCID50) with or without the administration of anti-RSV G protein MAb F(ab′)2 2 days earlier (n = 10 mice/group). Data are means ± SEMs. ***, values for r-A2-line19F were significantly lower (P ≤ 0.001; ANOVA) than those for the other groups. Results are representative of two independent experiments.
Fig 4.
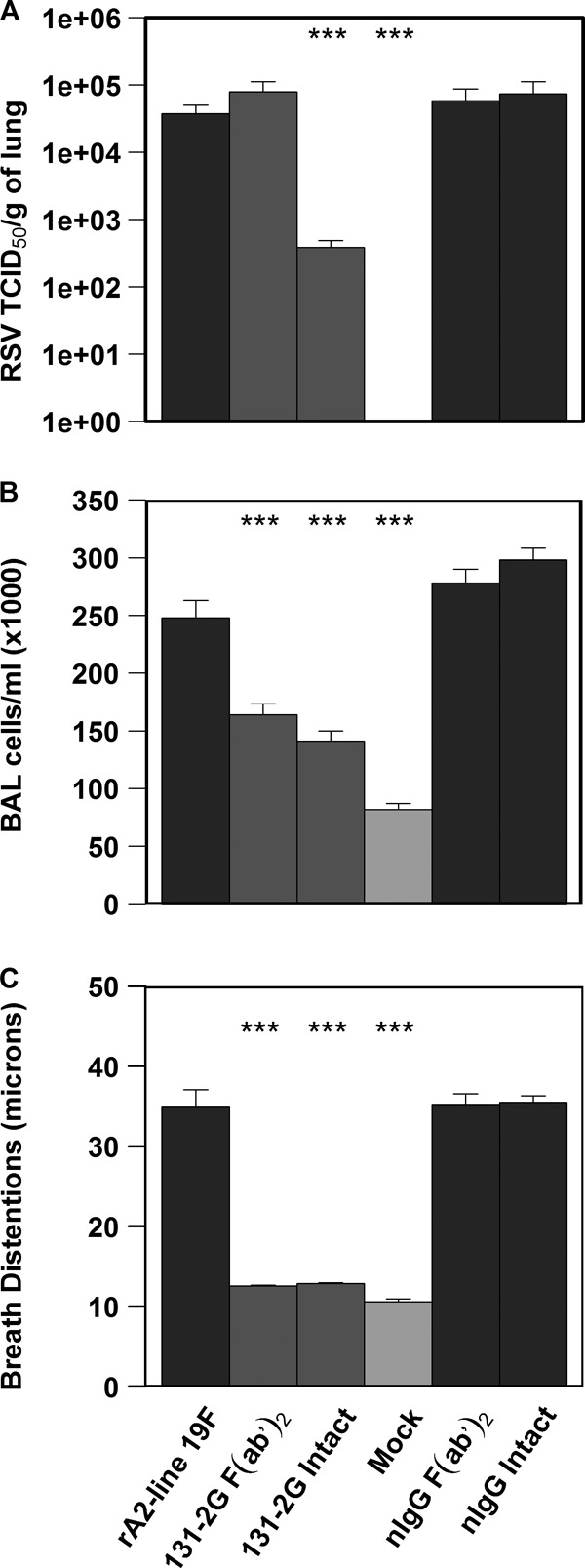
BALB/c mice were challenged with rA2-line19F (1 × 106 TCID50) with or without the administration of 131-2G F(ab′)2, intact 131-2G, or normal, or control, immune globulin (nIgG) 2 days earlier (n = 5 mice/group). (A) Effect of anti-RSV G protein MAb 131-2G treatment on changes in breathing effort in BALB/c mice. Breath-associated changes in the distension of peripheral arteries in microns (breathing effort) was measured at 8 days p.i. (B) Effect of prophylactic administration of anti-RSV G protein MAb 131-2G on viral load. (C) Effect of prophylactic administration of anti-RSV G protein MAb 131-2G on BAL cell numbers. Data are means ± SEMs. ***, significant decrease (P ≤ 0.001; ANOVA) in virus titer compared to that for challenged, untreated mice (rA2-line 19F). The results for 131-2G F(ab′)2 and rA2-line19F are representative of two independent experiments.
F(ab′)2 131-2G MAb treatment reduces pulmonary cell infiltrates and substance P levels.
Prophylactic treatment with both the F(ab′)2 and intact forms of 131-2G significantly decreased (P ≤ 0.001) pulmonary inflammation, as indicated by the total BAL cell numbers (33% and 43% reduction, respectively) and BAL cell types (Fig. 4B and Table 2). With the F(ab′)2 treatment, the most marked decrease in BAL cell types was for CD45R/B220 (RA3-6B2)+ cells (B cells; 81.63% ± 7.50%) and CD11b (M1/70)+ (macrophages, dendritic cells, and monocytes; 66.43% ± 10.30%) (Table 2).
Table 2.
Changes in the characteristics of the BAL cell infiltrate after prophylaxis with the intact anti-G protein MAb or F(ab′)2 form of 131-2Ga
| Phenotype | Untreated | F(ab′)2 131-2G-treated |
Intact 131-2G-treated |
||
|---|---|---|---|---|---|
| Mean no. of cells ± SE (103) | Mean no. of cells ± SE (103) | % reduction | Mean no. of cells ± SE (103) | % reduction | |
| CD3 | 6.46 ± 1.79 | 3.26 ± 0.51** | 43.47 ± 32.42 | 1.54 ± 0.51*** | 74.85 ± 8.60 |
| CD4 | 1.60 ± 0.46 | 0.65 ± 0.07** | 55.25 ± 8.23 | 0.29 ± 0.09*** | 78.79 ± 5.44 |
| CD8 | 1.66 ± 0.72 | 0.54 ± 0.04** | 59.95 ± 8.48 | 0.29 ± 0.12 | 77.70 ± 7.40 |
| B cell | 12.97 ± 5.16 | 2.18 ± 0.20*** | 81.63 ± 7.50 | 3.91 ± 0.77*** | 64.32 ± 19.01 |
| Macrophage | 23.36 ± 7.14 | 6.75 ± 3.90*** | 66.43 ± 10.30 | 10.96 ± 2.01*** | 53.71 ± 13.94 |
| PMN | 0.72 ± 0.23 | 0.44 ± 0.04** | 47.78 ± 14.02 | 0.30 ± 0.11*** | 62.58 ± 10.49 |
| NK | 7.33 ± 1.64 | 3.76 ± 2.22** | 52.54 ± 19.69 | 2.87 ± 0.69** | 56.38 ± 14.25 |
Mice were treated with 300 μg of the intact or F(ab′)2 form of 131-2G 2 days before RSV rA2-line19F (1 × 106 TCID50) challenge (n = 5 mice/group). Data are mean total numbers of BAL cells by subtype at day 5 p.i. per lung. Subtypes: lymphocyte gate, anti-CD3+ (17A2) and anti-CD4+ (GK1.5) CD4 T cells; lymphocyte gate, CD3+ and anti-CD8+ (53-6.7)− CD8 T cells; CD3− and anti-CD45R/B220+ (RA3-6B2)− B cells; CD3− and anti-CD11b+ (M1/70)− macrophages, dendritic cells, and monocytes; CD3− and anti-mouse Ly06G/Gr-1 (RB6-8C5)− polymorphonuclear cells (PMNs); CD3− and anti-mouse CD49b/integrin alpha 2 (DX5)− NK cells. The fold reduction is the decrease in the number of treated mice relative to the number of untreated mice for the cell type. **, P ≤ 0.05 as determined by ANOVA; ***, P ≤ 0.001, a significant decrease in the number of untreated mice compared to the number receiving the indicated treatment. Results are representative of two independent experiments for rA2-line19F challenge and F(a′)2 131-2G prophylaxis of rA2-line19F-challenged mice.
To determine a potential role for substance P in rA2-line19F-associated disease, we tested and compared SP levels in the BAL specimens of rA2-line19F-, A2-, and mock-challenged mice (Table 3). The levels of SP in BAL specimens after mock challenge ranged from 238 to 278 pg/ml. SP levels after A2 challenge, as previously noted (13), were significantly increased. rA2-line19F challenge gave the highest pulmonary SP levels, and these levels were significantly above those for A2 on day 5 p.i. (Table 3). Prophylaxis with F(ab′)2 131-2G, as well as with intact 131-2G, significantly decreased pulmonary SP (42.52% ± 4.09% and 43.79% ± 6.13%, respectively, compared to that for untreated rA2-line19F-challenged mice) at day 5 p.i., to levels below that for A2-challenged mice (Table 3).
Table 3.
Differences between SP levels in BAL cells of A2- and rA2-line19F-challenged mice and between untreated and MAb 131-2G-treated mice challenged with rA2-line19Fa
| Treatment | Substance P level in BAL cells (pg/ml ± SE) |
|---|---|
| A2 | 432.58 ± 54.34 |
| rA2-line19F** | 620.91 ± 30.30 |
| rA2-line19F+ F(ab′)2 131-2G*** | 355.85 ± 10.95 |
| rA2-line19F+ intact 131-2G*** | 349.92 ± 47.37 |
| Mock | 258.21 ± 20.86 |
| rA2-line19F+ F(ab′)2 nIgG | 628.36 ± 17.31 |
| rA2-line19F+ intact nIgG | 617.52 ± 8.97 |
BALB/c mice were infected with 1 × 106 TCID50 of rA2-line19F or A2 RSV. Mice were treated with 300 μg of the intact or F(ab′)2 form of 131-2G or control immunoglobulin (nIgG) 2 days before RSV rA2-line19F challenge (n = 5 mice/group). Data are means ± SEMs. BAL samples were collected on day 5 p.i. **, P ≤ 0.05 (determined by ANOVA), a significant difference between the levels of substance P in rA2-line19F-challenged and A2-challenged mice; ***, P ≤ 0.001 (determined by ANOVA), a significant difference between the levels of substance P in untreated mice and rA2-line19F-challenged mice receiving indicated treatment. Results are representative of two independent experiments for untreated and F(ab′)2 131-2G-treated rA2-line19F-challenged mice. The data for intact-131-2G-treated mice and A2-challenged mice are from one experiment.
F(ab′)2 MAb 131-2G inhibits rA2-line19F-induced airway dysfunction in BALB/c mice.
Airway dysfunction, manifested by signs of increased airway resistance, is the hallmark feature of RSV bronchiolitis. The increase in breathing effort based on pulsus paradoxus is an indicator of airway dysfunction. As noted in Fig. 2, the increase in breathing effort with rA2-line19F-challenged mice peaked on day 8 p.i., and values for day 8 p.i. were used for this analysis. Prophylaxis with either form of MAb 131-2G markedly decreased (P ≤ 0.001) RSV rA2-line19F-induced breathing effort compared to that for untreated and nIgG antibody-treated mice challenged with rA2-line19F (Fig. 4C). The values for MAb 131-2G-treated mice were similar to those for mock-challenged mice. The decrease in breathing effort with F(ab′)2 131-2G prophylaxis shows that this decrease is independent of a decrease in virus replication.
F(ab′)2 MAb 131-2G reduces rA2-line19F-induced pulmonary mucin production in BALB/c mice.
An increase in airway mucous is one feature of RSV disease in children (30, 31). Similar to increased pulsus paradoxus, increased mucous production was documented 8 days after challenge, and values for day 8 were used for these analyses. There was an increase in Muc5AC protein expression in the lungs of mice challenged with rA2-line19F but not in the lungs of A2-challenged mice (Fig. 5A), which was similar to the results of other studies (25). In a fashion similar to that for increased pulsus paradoxus, prophylactic treatment with both the F(ab′)2 and intact forms of 131-2G significantly decreased (P ≤ 0.001) Muc5AC levels compared to those in untreated and nIgG antibody-treated mice challenged with rA2-line19F (Fig. 5B). The ability of F(ab′)2 131-2G to so effectively decrease mucin production shows that this effect is also independent of decreasing virus replication.
Fig 5.

Effect of different virus strains and anti-RSV G protein MAb 131-2G treatment on mucin production in BALB/c mice. (A) BALB/c mice were challenged with mock-infected tissue culture supernatant (mock) or 1 × 106 TCID50 of rA2-line19F or A2 RSV(n = 5mice/group). Lungs were harvested 8 days p.i., and lung homogenates were tested for Muc5AC levels by ELISA. Data are means ± SEMs. ***, significant difference (P ≤ 0.001; ANOVA) between values for rA2-line19F-challenged mice and other indicated groups. (B) BALB/c mice were treated 2 days before rA2-line19F challenge with the intact or F(ab′)2 form of either 131-2G or control antibody (nIgG) (n = 5mice/group). Lungs were harvested 8 days p.i., and lung homogenates were tested for Muc5AC levels. Data are means ± SEMs. ***, significant difference (P ≤ 0.001; ANOVA) between values for untreated rA2-line19F-challenged mice and other indicated groups. Results are representative of two independent experiments for untreated and F(ab′)2 131-2G-treated mice challenged with rA2-line19F. Data for challenge with A2 and for prophylaxis with intact 131-2G are from one experiment.
Differences in pulmonary mucous production between A2 and rA2-line19F infections and between treatment with either form of MAb 131-2G and no treatment before rA2-line19F infection were evident in lung histopathology studies (Fig. 6A). Lung sections harvested 8 days p.i. showed PAS (a staining method used to detect intracellular pulmonary mucin levels)-stained cells for untreated, or control MAb-treated, and rA2-line19F-challenged mice. A2-challenged mice and MAb 131-2G-treated, rA2-line19F-challenged mice did not show PAS-stained cells. As expected, A2-challenged mice and rA2-line19F-challenged mice treated with the F(ab′)2 form of 131-2G were positive for RSV antigens by immunofluorescent staining (Fig. 6B).
Fig 6.

Mucin production and RSV replication associated with RSV rA2-line19F challenge and anti-G protein MAb 131-2G prophylaxis. BALB/c mice were challenged with mock-infected tissue culture supernatant (mock) or 1 × 106 TCID50 of A2 or rA2-line19F and were untreated or treated 2 days before challenge with the intact or F(ab′)2 form of both intact 131-2G and control immune globulin (nIgG) (n = 5 mice/group). Lungs were harvested 5 days p.i. for RSV antigen immunofluorescence and 8 days p.i. for PAS staining. (A) Representative photomicrographs of PAS-stained lung tissue collected at 8 days p.i. Reddish-purple color (e.g., evident in rA2-line19F-labeled panel) indicates PAS-positive cells indicative of mucin (n = 3 mice/group). (B) Representative photomicrographs of immunofluorescence staining for RSV from the groups of mice noted above at day 5 p.i. Green indicates RSV proteins in the cells (e.g., evident in panels labeled A2 and rA2-line19F; n = 3 mice/group). Results are representative of two independent experiments for untreated or 131-2G F(ab′)2-treated mice challenged with rA2-line19F.
Immunohistochemical staining (IHC) for Muc5AC production analyzed using ImageJ software (see Materials and Methods) was used to quantitate differences between treatment groups. Consistent with PAS-staining results, the percentages of Muc5AC-positive stained areas in the total epithelial area were 32.8% after rA2-line19F challenge, 13.2% after A2 challenge, 9.6% after mock challenge, and 10.8% and 11.6% for rA2-line19F-challenged mice given prophylaxis with intact and F(ab′)2 forms of MAb 131-2G.
Cytokine responses.
To address a potential mechanism for the decrease in pulsus paradoxus and mucin production after prophylaxis with the anti-RSV G protein MAb 131-2G, we determined the levels of pulmonary cytokines and chemokines in lung homogenates. As noted with earlier studies, elevated levels of Th2 cytokines, such as IL-4, IL-13, IL-5, and MCP-1, have been associated with the increased airway resistance, mucous production, and bronchoconstriction seen in patients with asthma. At 8 days p.i., significant increases (P ≤ 0.001) in the levels of IL-4, IL-5, IL-13, and MCP-1 were detected in untreated, nIgG antibody-treated, and untreated rA2-line19F-challenged mice (Fig. 7). These increases were significantly lower in rA2-line19F-challenged mice treated with either form of MAb 131-2G and in A2-challenged mice. In addition, substantially higher levels of IP-10 and IL-12 were detected in lung homogenates after prophylaxis with either form of the 131-2G MAb than in untreated or nIgG antibody-treated, rA2-line19F-challenged mice. A2-challenged mice also had higher levels of IL-12 and IP-10 (Fig. 7).
Fig 7.

Effect of anti-G protein MAb 131-2G prophylaxis on pulmonary cytokine levels in rA2-line19F RSV-challenged mice. BALB/c mice were challenged with mock-infected tissue culture supernatant (mock) or 1 × 106 TCID50 of rA2-line19F RSV with or without treatment (2 days before challenge) with the intact or F(ab′)2 form of 131-2G and normal immune globulin (nIgG). Lungs were harvested 8 days p.i., and lung homogenates were tested by Luminex multiplex assay for IL-13 (A), IL-5 (B), IL-4 (C), MCP-1 (D), IP-10 (E), or IL-12 (F). All values are expressed as pg/mg of protein. Data are means ± SEMs. ***, significant difference (P ≤ 0.001; ANOVA) between values for untreated and treated (as indicated) challenged mice. Results are representative of two independent experiments for untreated and 131-2G F(ab′)2-treated mice. Data for challenge with A2 and for prophylaxis with intact 131-2G are from one experiment.
DISCUSSION
Development of an effective vaccine or treatment for RSV infection has not yet been achieved, though passive administration of an immune globulin with high titers of RSV antibodies or a neutralizing MAb is effective when given prophylactically. One possible explanation for success with antibody prophylaxis (4) and limited success with treatment of active infection could be that the virus-induced host inflammatory response is important to the disease process and is only partially responsive to stopping replication once infection is established (39, 40). Consequently, effective treatment of active infection may require both anti-inflammatory and antiviral components. RSV vaccines or prophylaxis may also be more effective if they have both anti-inflammatory and antiviral effects. There is increasing evidence for RSV G protein being an important contributor to RSV inflammation and disease pathogenesis, and thus it is a potential target for the anti-inflammatory component of vaccine and antiviral drug development strategies (22, 23, 41–44).
Since immune prophylaxis with palivizumab is about 50% effective in decreasing RSV hospitalizations (4), it is possible that binding G and blocking G-associated disease might improve the effectiveness of immune prophylaxis. The RSV G protein, G peptides, and secreted G have been associated with a number of immune and inflammatory responses. For example, vaccination with intact or secreted G and several peptides in or proximal to the central nonglycosylated region of G induce a Th2-biased memory response resulting in increased pulmonary inflammation and eosinophilia after RSV challenge (45–50). In other studies, the G protein has been associated with suppressing some immune responses, such as Toll-like receptor 3 (TLR3) or TLR4 induction of IFN-β (48), proinflammatory responses of lung epithelial cells (51), lymphoproliferation (52), and a number of other innate responses, including activation of dendritic cells (53, 54), enhanced cytotoxic T cell responses (55), and downregulation of type I IFN production by inducing suppression of cytokine signaling (SOCS) (56). The G protein has also been associated with depression of the respiratory rate (16), increased production of pulmonary substance P (15), and antibody decoy activity (57).
Earlier studies showed that binding the G protein with MAb 131-2G prevented pulmonary inflammation after primary infection, pulmonary inflammation, and eosinophilia in FI-RSV-vaccinated mice and weight loss after primary infection or infection in FI-RSV-vaccinated mice (21–23). The availability of the rA2-line19F virus allowed us to demonstrate that MAb 131-2G prophylaxis also decreases breathing effort and pulmonary mucous production. These findings suggest additional clinical benefits of treatment or prophylaxis with this or a comparable anti-G protein MAb. These findings also expand the possible contributions of the G protein to the pathogenesis of RSV disease.
The potential importance of these findings to human RSV disease is highlighted by wheezing, often resulting in a diagnosis of bronchiolitis, being the hallmark clinical feature of RSV infection in children (32, 58). Wheezing is associated with increased airway resistance, which can result from bronchoconstriction or plugging of the airways with sloughed airway epithelial and inflammatory cells, other debris, and/or mucous. With rA2-line19F, we see increased pulsus paradoxus indicative of increased breathing effort, which indicates airway dysfunction. The data in this report and data from Moore et al. (25) are consistent with one or several mechanisms causing the increased breathing effort; i.e., infection with rA2-line19F is associated with the increased pulmonary inflammation, Th2 cytokine levels, and mucous production that parallel the increase in breathing effort. In addition, the data in this report show a decrease in breathing effort with F(ab′)2 MAb 131-2G treatment that correlates with decreases in BAL cell infiltrates (Fig. 5 and Table 2), pulmonary mucous production (Fig. 6), pulmonary Th2 cytokines (Fig. 7), and substance P levels (Table 3). The cellular infiltration and/or mucous production can plug airways, leading to increased breathing effort, and Th2 cytokines and substance P have been associated with bronchoconstriction, which would also lead to increased breathing effort.
Airway mucus hypersecretion is an important feature of chronic airway inflammatory diseases and has been detected in some autopsy specimens from children who died with severe RSV disease (59–61). Goblet cell hyperplasia plays a crucial role in mucus hypersecretion, and Muc5AC is a marker for increased goblet cell mucin production during airway inflammation (62). Hashimoto et al. demonstrated that RSV infection augmented Muc5AC and Gob-5 gene expression and staining in the ovalbumin allergy model of lung inflammation in mice (43). In the present study, rA2-line19F induced mucous production, the Th2-type cytokines IL-13, IL-4, and IL-5, and the chemokine MCP-1. Prophylaxis with MAb 131-2G decreased both mucous production and the expression of these cytokines and the chemokine. As noted with earlier studies, elevated levels of Th2 cytokines, such as IL-4 and IL-13, have been associated with the increased airway resistance, mucous production, and bronchoconstriction seen in patients with asthma (63–67). Blocking IL-13 and IL-4 has been shown to significantly inhibit mucus overproduction in a murine model, and local administration of recombinant IL-13 to nonimmunized mice induces goblet cell hyperplasia (43). The Th2 cytokine IL-5 is associated with allergic asthmatic responses in the lung (68). MCP-1 was also elevated and regulates migration and infiltration of monocytes/macrophages, which could indirectly contribute to mucous secretion (69) and airway dysfunction (70).
The increase in IL-12 and IP-10 with F(ab′)2 MAb 131-2G prophylaxis is consistent with a shift toward a Th1 response from a Th2 response with the decrease in lung disease and suggests that the virus is actively suppressing the production of these mediators. IL-12 has been associated with a pathophysiologically milder response to the infection (71). Production of IL-12 is thought to favor differentiation and function of (Th1) T cells while inhibiting the differentiation of Th2 cells. In many viral infections, IL-12 promotes viral clearance and host recovery from infection (71–74).
We chose to test for pulmonary substance P levels because the G protein has been associated with elevated levels in BAL fluid and substance P has been linked to increased inflammation and airway resistance (15). Our finding that F(ab′)2 MAb 131-2G prophylaxis decreased pulmonary substance P levels is consistent with previous work linking G to induction of substance P in the lung, as noted above, as well as a study suggesting that substance P participates in G downregulation of the respiratory rate in mice (16). Substance P has been suggested as an important contributor to bronchospasm, mucus secretion, and inflammation in asthma and various infections (75).
We hypothesize that some of the disease-modifying effects of MAb 131-2G noted in this study are linked to G binding to CX3CR1 through its CX3C chemokine motif (18). Several studies using a virus with a Cys-to-Arg mutation at amino acid position 186 in the CX3C motif (CX3R) and anti-CX3CR1 antibodies suggest a link between G binding to CX3CR1 and RSV disease. In one study, FI-RSV-vaccinated mice challenged with a virus that lacked G or the CX3R virus or were given prophylactic treatment with anti-CX3CR1 antibody before challenge had a marked decrease in the enhanced pulmonary inflammation otherwise seen in FI-RSV-vaccinated mice (14). In another study, mice challenged with the CX3R virus had significantly more effector T cells trafficking into the lungs and a higher percentage of T cells producing IFN-γ than mice challenged with the wild-type virus (17). In a third study, mice given the RSV G protein intravenously had a decrease in respiratory rate but mice given the CX3R G protein or mice treated with anti-CX3CR1 antibody before G was given did not show decreased respiratory rates (16). In this study, binding substance P with antibody also eliminated G's reduction in respiratory rates, raising the possibility that the effect of the G protein, CX3CR1, and substance P on RSV disease may be linked. Interestingly, a recent study noted that fractalkine and CX3CR1 contribute to inflammation and a Th2-biased response in a mouse model of allergic inflammation (76). We have initiated studies to determine if, in fact, the CX3C motif in G and G binding to CX3CR1 play a role in the manifestations of RSV disease noted in the present study.
Importantly, as noted in earlier studies, results with the F(ab′)2 form of the MAb show that the impact of G on disease is independent of virus replication. Binding G with the F(ab′)2 form of 131-2G does not decrease virus replication, which is consistent with the previous findings with this MAb (22) and another nonneutralizing anti-G protein MAb, IC2 (77). One chimera of Mab, IC2 with an intact γ chain, prevented virus replication, while another chimera with a glycosyl mutation that did not activate complement or binding to the Fc receptor was less effective in preventing virus replication. These findings are consistent with virus neutralization by antibody-dependent cell-mediated cytotoxicity (ADCC)-dependent or complement-mediated mechanisms. Intact 131-2G, as noted in the present study and previous studies (22, 23), effectively neutralizes RSV in vivo and thus in the mouse impacts RSV disease both by downregulating G-induced inflammation and immune modulation and by decreasing virus replication. If it impacts both in humans, it might prove more effective in decreasing disease than antiviral treatment that only decreases virus replication.
The ability of an anti-G protein MAb to so effectively decrease pulmonary mucous production and breathing effort is somewhat surprising, since the difference between RSV A2 and rA2-line19F is five amino acid changes in the F protein and no differences in the G protein. Consequently, both proteins must be involved in rA2-line19F infection-associated mucous production and airway reactivity. The G and F proteins do interact during infection, and this may explain a joint role in these manifestations of infection (78). Alternatively, the role of each may have independent effects that in concert result in mucous overproduction and increased breathing effort. We are beginning studies to better understand the role of F and G in these manifestations of RSV disease.
Two limitations need to be considered in interpreting the data presented in the report. First, pulsus paradoxus, or breathing effort, is an indication of airway dysfunction but does not indicate the location of this dysfunction. Previous work, however, has shown that increases in pulsus paradoxus, or breathing effort, in the mouse do correspond to increases in airway hyperresponsiveness (AHR) in mice challenged with RSV rA2-line19F. AHR was measured using a small animal ventilator and methacholine challenge (38), and the correspondence between this measurement and increased breathing effort supports that breathing effort in this model indicates dysfunction in the lung. We chose to use breathing effort because it is less invasive than measuring AHR and it allows serial measures in the same mouse. Second, it is important to recognize that findings in mice may, or may not, apply to humans. Thus, although these data suggest a substantial role for the G protein in the pathogenesis of human RSV disease, this suggestion needs to be confirmed by studies with humans.
Together, the data presented in this report identify additional roles for the RSV G protein in disease pathogenesis, i.e., mucous production and airway dysfunction. These results and the results of other studies of G suggest that understanding G's role in the pathogenesis of disease is likely essential to understanding RSV disease and likely important in the approach to its treatment and prevention. Finally, if G's impact on human RSV disease is similar to its impact on disease in mice, binding G with a MAb or antiviral drug with activities similar to those of 131-2G MAb could improve the effectiveness of RSV prophylaxis and/or treatment. The data also suggest that inducing antibodies with activity similar to that of MAb 131-2G might improve a vaccine's ability to prevent disease with later RSV infection.
ACKNOWLEDGMENTS
This work was supported by NIH grant 1U19AI095227 awarded to M.L.M. and L.J.A., funding from Children's Healthcare of Atlanta, support from the Immunology Core of Emory-Children's Pediatric Research Center, and Emory Vaccinology Training Grant (VTP) T32 5T32AI074492-03. Martin L. Moore was supported by NIH 1R01AI087798 and NIH 1U19AI095227.
We thank Carla J. Shoffeitt for histology slide preparations and Sica L. Gabriel for histology slide analysis.
The findings and conclusions in this report are those of the authors and do not necessarily represent the views of the Centers for Disease Control and Prevention.
Footnotes
Published ahead of print 24 July 2013
REFERENCES
- 1.Nair H, Nokes DJ, Gessner BD, Dherani M, Madhi SA, Singleton RJ, O'Brien KL, Roca A, Wright PF, Bruce N, Chandran A, Theodoratou E, Sutanto A, Sedyaningsih ER, Ngama M, Munywoki PK, Kartasasmita C, Simoes EA, Rudan I, Weber MW, Campbell H. 2010. Global burden of acute lower respiratory infections due to respiratory syncytial virus in young children: a systematic review and meta-analysis. Lancet 375:1545–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Groothuis JR, Simoes E, Levin MJ, Hall CB, Long CE, Rodriguez WJ, Arrobio J, Meissner HC, Fulton DR, Welliver RC, Tristram DA, Siber GR, Prince GA, Van Raden M, Hemming VG. 1993. Prophylactic administration of respiratory syncytial virus immune globulin to high-risk infants and young children. Respiratory Syncytial Virus Immune Globulin Study Group. N. Engl. J. Med. 329:1524–1530. [DOI] [PubMed] [Google Scholar]
- 3.IMpact-RSV Study Group. 1998. Palivizumab, a humanized respiratory syncytial virus monoclonal antibody, reduces hospitalization from respiratory syncytial virus infection in high-risk infants. Pediatrics 102:531–537. [PubMed] [Google Scholar]
- 4.Geevarghese B, Simões EA. 2012. Antibodies for prevention and treatment of respiratory syncytial virus infections in children. Antivir. Ther. 17:201–211. [DOI] [PubMed] [Google Scholar]
- 5.Siegel JD, Rhinehart E, Jackson M, Chiarello L. 2007. 2007 Guideline for isolation precautions: preventing transmission of infectious agents in health care settings. Am. J. Infect. Control 35(Suppl 2):S65–S164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Collins PL, Melero JA. 2011. Progress in understanding and controlling respiratory syncytial virus: still crazy after all these years. Virus Res. 162:80–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Graham BS. 2011. Biological challenges and technological opportunities for respiratory syncytial virus vaccine development. Immunol. Rev. 239:149–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Connors M, Collins PL, Firestone CY, Murphy BR. 1991. Respiratory syncytial virus (RSV) F, G, M2 (22K), and N proteins each induce resistance to RSV challenge, but resistance induced by M2 and N proteins is relatively short-lived. J. Virol. 65:1634–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Olmsted RA, Elango N, Prince GA, Murphy BR, Johnson PR, Moss B, Chanock RM, Collins PL. 1986. Expression of the F glycoprotein of respiratory syncytial virus by a recombinant vaccinia virus: comparison of the individual contributions of the F and G glycoproteins to host immunity. Proc. Natl. Acad. Sci. U. S. A. 83:7462–7466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stott EJ, Taylor G, Ball LA, Anderson K, Young KK, King AM, Wertz GW. 1987. Immune and histopathological responses in animals vaccinated with recombinant vaccinia viruses that express individual genes of human respiratory syncytial virus. J. Virol. 61:3855–3861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tripp RA. 2004. Pathogenesis of respiratory syncytial virus infection. Viral Immunol. 17:165–181. [DOI] [PubMed] [Google Scholar]
- 12.Tripp RA, Moore D, Jones L, Sullender W, Winter J, Anderson LJ. 1999. Respiratory syncytial virus G and/or SH protein alters Th1 cytokines, natural killer cells, and neutrophils responding to pulmonary infection in BALB/c mice. J. Virol. 73:7099–7107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tripp RA, Jones L, Anderson LJ. 2000. Respiratory syncytial virus G and/or SH glycoproteins modify CC and CXC chemokine mRNA expression in the BALB/c mouse. J. Virol. 74:6227–6229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haynes LM, Jones LP, Barskey A, Anderson LJ, Tripp RA. 2003. Enhanced disease and pulmonary eosinophilia associated with formalin-inactivated respiratory syncytial virus vaccination are linked to G glycoprotein CX3C-CX3CR1 interaction and expression of substance. P. J. Virol. 77:9831–9844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tripp RA, Moore D, Winter J, Anderson LJ. 2000. Respiratory syncytial virus infection and G and/or SH protein expression contribute to substance P, which mediates inflammation and enhanced pulmonary disease in BALB/c mice. J. Virol. 74:1614–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tripp RA, Dakhama A, Jones LP, Barskey A, Gelfand EW, Anderson LJ. 2003. The G glycoprotein of respiratory syncytial virus depresses respiratory rates through the CX3C motif and substance. P. J. Virol. 77:6580–6584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harcourt J, Alvarez R, Jones LP, Henderson C, Anderson LJ, Tripp RA. 2006. Respiratory syncytial virus G protein and G protein CX3C motif adversely affect CX3CR1+ T cell responses. J. Immunol. 176:1600–1608. [DOI] [PubMed] [Google Scholar]
- 18.Tripp RA, Jones LP, Haynes LM, Zheng H, Murphy PM, Anderson LJ. 2001. CX3C chemokine mimicry by respiratory syncytial virus G glycoprotein. Nat. Immunol. 2:732–738. [DOI] [PubMed] [Google Scholar]
- 19.Anderson LJ, Hierholzer JC, Stone YO, Tsou C, Fernie BF. 1986. Identification of epitopes on respiratory syncytial virus proteins by competitive binding immunoassay. J. Clin. Microbiol. 23:475–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anderson LJ, Bingham P, Hierholzer JC. 1988. Neutralization of respiratory syncytial virus by individual and mixtures of F and G protein monoclonal antibodies. J. Virol. 62:4232–4238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haynes LM, Caidi H, Radu GU, Miao C, Harcourt JL, Tripp RA, Anderson LJ. 2009. Therapeutic monoclonal antibody treatment targeting respiratory syncytial virus (RSV) G protein mediates viral clearance and reduces the pathogenesis of RSV infection in BALB/c mice. J. Infect. Dis. 200:439–447. [DOI] [PubMed] [Google Scholar]
- 22.Miao C, Radu GU, Caidi H, Tripp RA, Anderson LJ, Haynes LM. 2009. Treatment with respiratory syncytial virus G glycoprotein monoclonal antibody or F(ab′)2 components mediates reduced pulmonary inflammation in mice. J. Gen. Virol. 90:1119–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Radu GU, Caidi H, Miao C, Tripp RA, Anderson LJ, Haynes LM. 2010. Prophylactic treatment with a G glycoprotein monoclonal antibody reduces pulmonary inflammation in respiratory syncytial virus (RSV)-challenged naive and formalin-inactivated RSV-immunized BALB/c mice. J. Virol. 84:9632–9636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lukacs NW, Moore ML, Rudd BD, Berlin AA, Collins RD, Olson SJ, Ho SB, Peebles RS., Jr 2006. Differential immune responses and pulmonary pathophysiology are induced by two different strains of respiratory syncytial virus. Am. J. Pathol. 169:977–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moore ML, Chi MH, Luongo C, Lukacs NW, Polosukhin VV, Huckabee MM, Newcomb DC, Buchholz UJ, Crowe JE, Jr, Goleniewska K, Williams JV, Collins PL, Peebles RS., Jr 2009. A chimeric A2 strain of respiratory syncytial virus (RSV) with the fusion protein of RSV strain line 19 exhibits enhanced viral load, mucus, and airway dysfunction. J. Virol. 83:4185–4194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hershey GK. 2003. IL-13 receptors and signaling pathways: an evolving web. J. Allergy Clin. Immunol. 111:677–690. [DOI] [PubMed] [Google Scholar]
- 27.Walter DM, McIntire JJ, Berry G, McKenzie AN, Donaldson DD, DeKruyff RH, Umetsu DT. 2001. Critical role for IL-13 in the development of allergen-induced airway hyperreactivity. J. Immunol. 167:4668–4675. [DOI] [PubMed] [Google Scholar]
- 28.Zhu Z, Homer RJ, Wang Z, Chen Q, Geba GP, Wang J, Zhang Y, Elias JA. 1999. Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities, and eotaxin production. J. Clin. Invest. 103:779–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Waal L, Koopman LP, van Benten IJ, Brandenburg AH, Mulder PG, de Swart RL, Fokkens WJ, Neijens HJ, Osterhaus AD. 2003. Moderate local and systemic respiratory syncytial virus-specific T-cell responses upon mild or subclinical RSV infection. J. Med. Virol. 70:309–318. [DOI] [PubMed] [Google Scholar]
- 30.Lugo RA, Nahata MC. 1993. Pathogenesis and treatment of bronchiolitis. Clin. Pharm. 12:95–116. [PubMed] [Google Scholar]
- 31.Quinn SF, Erickson S, Oshman D, Hayden F. 1985. Lobar collapse with respiratory syncytial virus pneumonitis. Pediatr. Radiol. 15:229–230. [DOI] [PubMed] [Google Scholar]
- 32.Hall CB, Weinberg GA, Iwane MK, Blumkin AK, Edwards KM, Staat MA, Auinger P, Griffin MR, Poehling KA, Erdman D, Grijalva CG, Zhu Y, Szilagyi P. 2009. The burden of respiratory syncytial virus infection in young children. N. Engl. J. Med. 360:588–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moore ML, Peebles RS., Jr 2006. Respiratory syncytial virus disease mechanisms implicated by human, animal model, and in vitro data facilitate vaccine strategies and new therapeutics. Pharmacol. Ther. 112:405–424. [DOI] [PubMed] [Google Scholar]
- 34.Anderson LJ, Hierholzer JC, Bingham PG, Stone YO. 1985. Microneutralization test for respiratory syncytial virus based on an enzyme immunoassay. J. Clin. Microbiol. 22:1050–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reed LJ, Muench H. 1938. A simple method of estimating fifty percent endpoints. Amer. J. Hyg. 27:493–497. [Google Scholar]
- 36.Kodani M, Yang G, Conklin LM, Travis TC, Whitney CG, Anderson LJ, Schrag SJ, Taylor TH, Jr, Beall BW, Breiman RF, Feikin DR, Njenga MK, Mayer LW, Oberste MS, Tondella ML, Winchell JM, Lindstrom SL, Erdman DD, Fields BS. 2011. Application of TaqMan low-density arrays for simultaneous detection of multiple respiratory pathogens. J. Clin. Microbiol. 49:2175–2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tripp RA, Barskey A, Goss L, Anderson LJ. 2002. Substance P receptor expression on lymphocytes is associated with the immune response to respiratory syncytial virus infection. J. Neuroimmunol. 129:141–153. [DOI] [PubMed] [Google Scholar]
- 38.Stokes KL, Chi MH, Sakamoto K, Newcomb DC, Currier MG, Huckabee MM, Lee S, Goleniewska K, Pretto C, Williams JV, Hotard A, Sherrill TP, Peebles RS, Jr, Moore ML. 2011. Differential pathogenesis of respiratory syncytial virus clinical isolates in BALB/c mice. J. Virol. 85:5782–5793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Empey KM, Peebles RS, Kolls JK. 2010. Pharmacologic advances in the treatment and prevention of respiratory syncytial virus. Clin. Infect. Dis. 50:1258–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ottolini MG, Curtis SJ, Porter DD, Mathews A, Richardson JY, Hemming VG, Prince GA. 2002. Comparison of corticosteroids for treatment of respiratory syncytial virus bronchiolitis and pneumonia in cotton rats. Antimicrob. Agents Chemother. 46:2299–2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Andersson C, Liljestrom P, Stahl S, Power UF. 2000. Protection against respiratory syncytial virus (RSV) elicited in mice by plasmid DNA immunisation encoding a secreted RSV G protein-derived antigen. FEMS Immunol. Med. Microbiol. 29:247–253. [DOI] [PubMed] [Google Scholar]
- 42.Collarini EJ, Lee FE, Foord O, Park M, Sperinde G, Wu H, Harriman WD, Carroll SF, Ellsworth SL, Anderson LJ, Tripp RA, Walsh EE, Keyt BA, Kauvar LM. 2009. Potent high-affinity antibodies for treatment and prophylaxis of respiratory syncytial virus derived from B cells of infected patients. J. Immunol. 183:6338–6345. [DOI] [PubMed] [Google Scholar]
- 43.Hashimoto K, Graham BS, Ho SB, Adler KB, Collins RD, Olson SJ, Zhou W, Suzutani T, Jones PW, Goleniewska K, O'Neal JF, Peebles RS., Jr 2004. Respiratory syncytial virus in allergic lung inflammation increases Muc5ac and gob-5. Am. J. Respir. Crit. Care Med. 170:306–312. [DOI] [PubMed] [Google Scholar]
- 44.Kauvar LM, Harcourt JL, Haynes LM, Tripp RA. 2010. Therapeutic targeting of respiratory syncytial virus G-protein. Immunotherapy 2:655–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bembridge GP, Garcia-Beato R, Lopez JA, Melero JA, Taylor G. 1998. Subcellular site of expression and route of vaccination influence pulmonary eosinophilia following respiratory syncytial virus challenge in BALB/c mice sensitized to the attachment G protein. J. Immunol. 161:2473–2480. [PubMed] [Google Scholar]
- 46.Hancock GE, Speelman DJ, Heers K, Bortell E, Smith J, Cosco C. 1996. Generation of atypical pulmonary inflammatory responses in BALB/c mice after immunization with the native attachment (G) glycoprotein of respiratory syncytial virus. J. Virol. 70:7783–7791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Johnson TR, Graham BS. 1999. Secreted respiratory syncytial virus G glycoprotein induces interleukin-5 (IL-5), IL-13, and eosinophilia by an IL-4-independent mechanism. J. Virol. 73:8485–8495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shingai M, Azuma M, Ebihara T, Sasai M, Funami K, Ayata M, Ogura H, Tsutsumi H, Matsumoto M, Seya T. 2008. Soluble G protein of respiratory syncytial virus inhibits Toll-like receptor 3/4-mediated IFN-beta induction. Int. Immunol. 20:1169–1180. [DOI] [PubMed] [Google Scholar]
- 49.Sparer TE, Matthews S, Hussell T, Rae AJ, Garcia-Barreno B, Melero JA, Openshaw PJ. 1998. Eliminating a region of respiratory syncytial virus attachment protein allows induction of protective immunity without vaccine-enhanced lung eosinophilia. J. Exp. Med. 187:1921–1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tebbey PW, Hagen M, Hancock GE. 1998. Atypical pulmonary eosinophilia is mediated by a specific amino acid sequence of the attachment (G) protein of respiratory syncytial virus. J. Exp. Med. 188:1967–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Arnold R, Konig B, Werchau H, Konig W. 2004. Respiratory syncytial virus deficient in soluble G protein induced an increased proinflammatory response in human lung epithelial cells. Virology 330:384–397. [DOI] [PubMed] [Google Scholar]
- 52.Ray R, Hoft DF, Meyer K, Brown R, Lagging LM, Belshe RB. 2001. Immunoregulatory role of secreted glycoprotein G from respiratory syncytial virus. Virus Res. 75:147–154. [DOI] [PubMed] [Google Scholar]
- 53.Johnson TR, McLellan JS, Graham BS. 2012. Respiratory syncytial virus glycoprotein G interacts with DC-SIGN and L-SIGN to activate ERK1 and ERK2. J. Virol. 86:1339–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Polack FP, Irusta PM, Hoffman SJ, Schiatti MP, Melendi GA, Delgado MF, Laham FR, Thumar B, Hendry RM, Melero JA, Karron RA, Collins PL, Kleeberger SR. 2005. The cysteine-rich region of respiratory syncytial virus attachment protein inhibits innate immunity elicited by the virus and endotoxin. Proc. Natl. Acad. Sci. U. S. A. 102:8996–9001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bukreyev A, Serra ME, Laham FR, Melendi GA, Kleeberger SR, Collins PL, Polack FP. 2006. The cysteine-rich region and secreted form of the attachment G glycoprotein of respiratory syncytial virus enhance the cytotoxic T-lymphocyte response despite lacking major histocompatibility complex class I-restricted epitopes. J. Virol. 80:5854–5861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Oshansky CM, Krunkosky TM, Barber J, Jones LP, Tripp RA. 2009. Respiratory syncytial virus proteins modulate suppressors of cytokine signaling 1 and 3 and the type I interferon response to infection by a toll-like receptor pathway. Viral Immunol. 22:147–161. [DOI] [PubMed] [Google Scholar]
- 57.Bukreyev A, Yang L, Fricke J, Cheng L, Ward JM, Murphy BR, Collins PL. 2008. The secreted form of respiratory syncytial virus G glycoprotein helps the virus evade antibody-mediated restriction of replication by acting as an antigen decoy and through effects on Fc receptor-bearing leukocytes. J. Virol. 82:12191–12204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hall CB. 2001. Respiratory syncytial virus and parainfluenza virus. N. Engl. J. Med. 344:1917–1928. [DOI] [PubMed] [Google Scholar]
- 59.Aherne W, Bird T, Court SD, Gardner PS, McQuillin J. 1970. Pathological changes in virus infections of the lower respiratory tract in children. J. Clin. Pathol. 23:7–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Johnson JE, Gonzales RA, Olson SJ, Wright PF, Graham BS. 2007. The histopathology of fatal untreated human respiratory syncytial virus infection. Mod. Pathol. 20:108–119. [DOI] [PubMed] [Google Scholar]
- 61.Welliver TP, Garofalo RP, Hosakote Y, Hintz KH, Avendano L, Sanchez K, Velozo L, Jafri H, Chavez-Bueno S, Ogra PL, McKinney L, Reed JL, Welliver RC., Sr 2007. Severe human lower respiratory tract illness caused by respiratory syncytial virus and influenza virus is characterized by the absence of pulmonary cytotoxic lymphocyte responses. J. Infect. Dis. 195:1126–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rose MC, Voynow JA. 2006. Respiratory tract mucin genes and mucin glycoproteins in health and disease. Physiol. Rev. 86:245–278. [DOI] [PubMed] [Google Scholar]
- 63.Cockcroft DW, Davis BE. 2006. Mechanisms of airway hyperresponsiveness. J. Allergy Clin. Immunol. 118:551–559. [DOI] [PubMed] [Google Scholar]
- 64.Holgate ST, Polosa R. 2008. Treatment strategies for allergy and asthma. Nat. Rev. Immunol. 8:218–230. [DOI] [PubMed] [Google Scholar]
- 65.Perkins C, Yanase N, Smulian G, Gildea L, Orekov T, Potter C, Brombacher F, Aronow B, Wills-Karp M, Finkelman FD. 2011. Selective stimulation of IL-4 receptor on smooth muscle induces airway hyperresponsiveness in mice. J. Exp. Med. 208:853–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rosenberg HF, Phipps S, Foster PS. 2007. Eosinophil trafficking in allergy and asthma. J. Allergy Clin. Immunol. 119:1303–1310. [DOI] [PubMed] [Google Scholar]
- 67.Vock C, Hauber HP, Wegmann M. 2010. The other T helper cells in asthma pathogenesis. J. Allergy 2010:519298. 10.1155/2010/519298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Franova S, Joskova M, Sadlonova V, Pavelcikova D, Mesarosova L, Novakova E, Sutovska M. 2013. Experimental model of allergic asthma. Adv. Exp. Med. Biol. 756:49–55. [DOI] [PubMed] [Google Scholar]
- 69.Deshmane SL, Kremlev S, Amini S, Sawaya BE. 2009. Monocyte chemoattractant protein-1 (MCP-1): an overview. J. Interferon Cytokine Res. 29:313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Foster WM, Adler KB, Crews AL, Potts EN, Fischer BM, Voynow JA. 2010. MARCKS-related peptide modulates in vivo the secretion of airway Muc5ac. Am. J. Physiol. Lung Cell. Mol. Physiol. 299:L345–L352. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 71.Tekkanat KK, Maassab H, Berlin AA, Lincoln PM, Evanoff HL, Kaplan MH, Lukacs NW. 2001. Role of interleukin-12 and stat-4 in the regulation of airway inflammation and hyperreactivity in respiratory syncytial virus infection. Am. J. Pathol. 159:631–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bortolatto J, Borducchi E, Rodriguez D, Keller AC, Faquim-Mauro E, Bortoluci KR, Mucida D, Gomes E, Christ A, Schnyder-Candrian S, Schnyder B, Ryffel B, Russo M. 2008. Toll-like receptor 4 agonists adsorbed to aluminium hydroxide adjuvant attenuate ovalbumin-specific allergic airway disease: role of MyD88 adaptor molecule and interleukin-12/gamma interferon axis. Clin. Exp. Allergy 38:1668–1679. [DOI] [PubMed] [Google Scholar]
- 73.Romani L, Puccetti P, Bistoni F. 1997. Interleukin-12 in infectious diseases. Clin. Microbiol. Rev. 10:611–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rudd BD, Schaller MA, Smit JJ, Kunkel SL, Neupane R, Kelley L, Berlin AA, Lukacs NW. 2007. MyD88-mediated instructive signals in dendritic cells regulate pulmonary immune responses during respiratory virus infection. J. Immunol. 178:5820–5827. [DOI] [PubMed] [Google Scholar]
- 75.Douglas SD, Leeman SE. 2011. Neurokinin-1 receptor: functional significance in the immune system in reference to selected infections and inflammation. Ann. N. Y. Acad. Sci. 1217:83–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mionnet C, Buatois V, Kanda A, Milcent V, Fleury S, Lair D, Langelot M, Lacoeuille Y, Hessel E, Coffman R, Magnan A, Dombrowicz D, Glaichenhaus N, Julia V. 2010. CX3CR1 is required for airway inflammation by promoting T helper cell survival and maintenance in inflamed lung. Nat. Med. 16:1305–1312. [DOI] [PubMed] [Google Scholar]
- 77.Mekseepralard C, Toms GL, Routledge EG. 2006. Protection of mice against human respiratory syncytial virus by wild-type and aglycosyl mouse-human chimaeric IgG antibodies to subgroup-conserved epitopes on the G glycoprotein. J. Gen. Virol. 87:1267–1273. [DOI] [PubMed] [Google Scholar]
- 78.Low KW, Tan T, Ng K, Tan BH, Sugrue RJ. 2008. The RSV F and G glycoproteins interact to form a complex on the surface of infected cells. Biochem. Biophys. Res. Commun. 366:308–313. [DOI] [PubMed] [Google Scholar]