

Abstract
Previous studies have shown that the abundant herpes simplex virus 1 (HSV-1) tegument protein VP11/12, encoded by gene UL46, stimulates phosphatidylinositol 3-kinase (PI3-kinase)/Akt signaling: it binds the Src family kinase (SFK) Lck, is tyrosine phosphorylated, recruits the p85 subunit of PI3-kinase, and is essential for the activation of Akt during HSV-1 infection. The C-terminal region of VP11/12 contains tyrosine-based motifs predicted to bind the SH2 domains of SFKs (YETV and YEEI), p85 (YTHM), and Grb2 (YENV) and the phosphotyrosine-binding (PTB) domain of Shc (NPLY). We inactivated each of these motifs in the context of the intact viral genome and examined effects on binding and activation of Lck and recruitment of p85, Grb2, and Shc. Inactivating the p85, Grb2, or Shc motif reduced (p85) or eliminated (Grb2 and Shc) the interaction with the cognate signaling molecule without greatly affecting the other interactions or activation of Lck. Inactivating either SFK motif had only a minor effect on Lck binding and little or no effect on recruitment of p85, Grb2, or Shc. In contrast, inactivation of both SFK motifs severely reduced Lck binding and activation and tyrosine phosphorylation of VP11/12 and reduced (p85) or eliminated (Grb2 and Shc) binding of other signaling proteins. Overall, these data demonstrate the key redundant roles of the VP11/12 SFK-binding motifs in the recruitment and activation of SFKs and indicate that activated SFKs then lead (directly or indirectly) to phosphorylation of the additional motifs involved in recruiting p85, Grb2, and Shc. Thus, VP11/12 appears to mimic an activated growth factor receptor.
INTRODUCTION
Herpes simplex viruses 1 and 2 (HSV-1 and HSV-2) are members of the subfamily Alphaherpesvirinae and are common human pathogens with a wide host range (reviewed in reference 1). Infection with HSV causes substantial morbidity (2) and increases the risk for HIV transmission (3). Primary infection and viral replication occur in epithelial cells. The establishment of life-long latency occurs in sensory neurons following translocation of progeny viral particles (reviewed in reference 1). Periodic reactivation of latent virus leads to recurrent lesions at the initial site of infection, which allows transmission of the virus through intimate contact (reviewed in reference 1).
Most viruses manipulate a variety of host signaling pathways in order to promote viral growth and avoid the innate and adaptive immune responses. One of the many pathways that are often targeted is the class 1A phosphatidylinositol 3-kinase (PI3K)/Akt pathway, which promotes cell survival and cap-dependent translation (reviewed in reference 4). PI3K consists of a regulatory subunit, p85, and a catalytic subunit, p110. The p85 subunit harbors two Src homology 2 (SH2) domains and one SH3 domain, all of which are important protein-protein interaction interfaces. In general, this pathway is activated following stimulation of cell surface receptors, including the T-cell receptor (TCR), the insulin receptor, and the platelet-derived growth factor receptor (PDGFR). For example, following stimulation of the PDGFR, receptor dimerization leads to autophosphorylation of its cytoplasmic tail (5), including the tyrosine residues within two YXXM motifs, creating docking sites for the SH2 domains of p85 (6). Translocation to the plasma membrane brings PI3K in close proximity to its substrate, phosphatidylinositol(4,5)-bisphosphate (PIP2). Phosphorylation of PIP2 leads to generation of the second messenger phosphatidylinositol(3,4,5)-trisphosphate (PIP3) (reviewed in reference 7). PIP3 in turn associates with the pleckstrin homology (PH) domains of Akt and phosphoinositide-dependent protein kinase-1 (PDK1), recruiting them to the inner leaflet of the plasma membrane. PDK1 leads to partial activation of Akt by phosphorylating the critical T308 residue in the activation loop (8). In order to attain full activity, Akt must also be phosphorylated at S473, located in the hydrophobic domain, by the mammalian target of rapamycin (mTOR) complex 2 (mTORC2) (9) or other potential PDK2 members. Upon activation, Akt translocates from the cell membrane to other compartments throughout the cell in order to phosphorylate downstream substrates that regulate many cellular pathways, including those that regulate growth and proliferation (10). One major regulator of the PI3K/Akt-pathway is the phosphatase and tensin homolog (PTEN). PTEN converts PIP3 into PIP2 by dephosphorylation (11, 12) and therefore terminates the PI3K/Akt-signaling axis. Expression of PTEN, on the other hand, is regulated on a transcriptional level by PI3K (reviewed in reference 13).
Given that activation of the PI3K/Akt axis stimulates the infectivity of many viruses, it is not surprising that viruses like influenza A virus (14), human cytomegalovirus (15), hepatitis C virus (16, 17), Epstein-Barr virus (18), Kaposi's sarcoma-associated herpesvirus (19, 20), and varicella zoster virus (21) encode specific proteins that activate this pathway in order to create a beneficial growth environment. For example, the influenza A virus NS1 protein directly interacts with p85, leading to stimulation of PI3K (14), and a recombinant virus that is no longer able to interact with p85 fails to activate the Akt pathway and shows reduced infectivity (14). In the case of HSV-1, Benetti and Roizman (22) showed that S473 of Akt is phosphorylated postinfection and that phosphorylation occurs in a PI3K-dependent manner, indicating that HSV activates the PI3K/Akt pathway. Interestingly, the HSV US3 serine/threonine kinase phosphorylates certain Akt substrates on the same residues as Akt, thus serving as an Akt mimic (23). These findings suggest that HSV may exert redundant control over the Akt pathway.
Our laboratory previously showed that the HSV-1 tegument protein VP11/12 plays a key role in the activation of Akt during HSV infection (24–26). VP11/12 is encoded by the UL46 gene and is one of the most abundant tegument proteins (27). Despite its abundance, VP11/12 is not essential for viral replication in cell culture (27). VP11/12 orthologs are found in other alphaherpesviruses but not in beta- or gammaherpesviruses (28). During viral infection, VP11/12 is delivered into the cytoplasm of the infected cell along with other tegument proteins. Intracellular VP11/12 associates with both cellular membranes and viral capsids, although the precise mode of membrane association remains unclear (29). This protein enhances the activity of the viral transcription factor VP16 in transient-transfection assays (30, 31) and forms a complex with VP16 (30, 32); however, the relevance of these activities during productive infection remains unclear (26, 28).
VP11/12 is tyrosine phosphorylated by cellular Src family kinases (SFKs), strongly in lymphocytes and less so in fibroblasts (25, 26) and Vero cells (H. Eaton, H. Saffran, and J. R. Smiley, unpublished data). In Jurkat T cells, tyrosine phosphorylation is accomplished predominantly through the SFK Lck (26). VP11/12 interacts with Lck and is required for the activation of Lck that occurs during infection (24). VP11/12 also interacts with p85 in an SFK-dependent fashion and is required for activation of Akt during infection of Jurkat cells and fibroblasts (25). Together, the data indicate that VP11/12 modulates the Akt/PI3K pathway in a fashion analogous to that of activated cell surface receptors. Wagner and Smiley (24) proposed that VP11/12 recruits and activates Lck and other SFKs through the consensus SFK SH2 binding motif YEEI (24, 33) at Y624 and/or the YETV potential SFK SH2 motif at Y614, leading to phosphorylation of the predicted p85 SH2 domain-binding motif YTHM at Y519 (consensus YXXM). Here we describe experiments that tested this model, by examining the effects of mutations that inactivate the proposed Lck- and p85-binding motifs on the ability of VP11/12 to interact with and activate Lck and recruit p85 in Jurkat T cells.
MATERIALS AND METHODS
Cells.
Jurkat E6-1 cells and Vero cells were obtained from the ATCC. Cre-Vero cells (Vero cells constitutively expressing Cre recombinase) were a gift from David Leib (Dartmouth College). Jurkat E6-1 cells were grown in RPMI 1640 medium (Sigma) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Sigma). Vero and Cre-Vero cells were grown in Dulbecco's modified Eagle's medium (DMEM) (Gibco) supplemented with 5% heat-inactivated FBS. Cre-Vero cells were additionally maintained in 400 μg/ml hygromycin B to select for Cre recombinase expression.
Viruses.
The HSV-1 KOS recombinant KOS-G virus was described previously (34). HSV-1 strain KOS37 was derived from a bacterial artificial chromosome (BAC) bearing the entire HSV-1 KOS37 genome (35), and the KOS37-derived mutant ΔUL46 was described previously (26). All virus stocks were prepared from infected Vero cells at a multiplicity of infection (MOI) of 0.025.
In order to facilitate studies of interactions between VP11/12 and cellular proteins, we generated KOS37 derivatives encoding N- and C-terminally tagged VP11/12-EGFP fusion proteins (KOS37-GFP UL46 and KOS37-UL46 GFP, respectively), using en passant mutagenesis (36). First, we engineered a silent unique AclI restriction site into enhanced green fluorescent protein (EGFP) coding sequences in pEGFP-C1 via Quikchange II-mediated site-directed mutagenesis (Stratagene) using oligonucleotides JRS 684 and 685 (Table 1) according to the manufacturer's instructions, yielding pEGFP-C1-AclI. We then inserted the en passant mutagenesis selection marker I-SceI-aphAI into this AclI site. Briefly, the I-SceI-aphAI cassette was PCR amplified from pEPKanS using primers that included an AclI restriction site (JRS 704 and JRS 705) (Table 1). Following digestion with AclI, the amplimer was ligated into the AclI site of pEGFP-C1-AclI to generate pEGFP-C1-AclI-sm. In order to generate KOS37-GFP UL46, the EGFP cassette was PCR amplified from pEGFP-C1-AclI-sm using primers JRS 708 and JRS 709 (Table 1) and incorporated into the KOS37 BAC via en passant mutagenesis exactly as described previously (36). The resulting BAC contains EGFP codons 1 to 232 fused to VP11/12 codon 2. KOS36-UL46 GFP was similarly generated by incorporating an amplimer generated from pEGFP-C1-AclI-sm using primers JRS 787 and JRS 788 (Table 1). The resulting construct inserts EGFP codons 1 to 239 between UL46 codons 710 and 711, with an additional lysine codon (AAG) placed upstream of the insert, reconstructing the VP11/12-GFP fusion construct present in the HSV-1 KOS mutant GHSV-UL46 (37).
Table 1.
Primers used for generating KOS37-UL46 GFP and KOS37-GFP UL46
| Primer | Sequence (5′–3′)a |
|---|---|
| JRS 684 | ctacaacagccaccaacgttatatcatggccgacaag |
| JRS 685 | cttgtcggccatgatataaacgttgtggctgttgtag |
| JRS 704 | ggcggcaacgtttatatcatggccgacaagcagaagaacggcatcaaggtgaacaacaagattagggataacagggtaatcgattt |
| JRS 705 | ggcggcaacgttgccagtgttacaaccaattaacc |
| KOS37-UL46 GFP JRS 787/JRS 788 | AACGACGGCCCGACCAACGTCGCCGCCCTGAGCGCCCTCCTGACCAAGCTTAAgatggtgagcaagggcgaggag |
| CGACGGCAGCACGGGCGGAGGCGTTCACCGGCTCCGGCGTCCTTCGCGTTTcttgtacagctcgtccatgcc | |
| KOS37-GFP UL46 JRS 708/JRS 709 | gacgcggcataactccgaCCGGCGGGTCCCGACCGAACGGGCGTCACCATGgtgagcaagggcgag |
| CGTCAGGCACCGCGCCAGCCGCAGGGAGCTCGCGCCGCGCGTCCGGCGCTGCttgtacagctcgtccatgc |
HSV-1 sequences are capitalized, and AcII sequences are in bold.
Mutations that inactivate the VP11/12 tyrosine-based motifs at Y509, Y613, Y624, Y633, and Y657 were generated by en passant mutagenesis in the context of the KOS37 and KOS37-UL46 GFP BACs. In all cases, the motifs were inactivated by converting the relevant tyrosine (Y) codon (TTA) to a phenylalanine (F) codon (TTC). The mutagenic primers used for amplification of the I-SceI-aphAI cassette from pEPkan-S (36) are listed in Table 2.
Table 2.
Primers used for generating point mutated KOS37-UL46 viruses
| Primer | Sequence (5′–3′)a |
|---|---|
| Y519F-JRS 812/JRS 813 | CCGAGCCCCCGCTGCGGCCACACAGCCGGCCACGTATTTCACGCACATGGGGGAGGTGCtagggataacagggtaatcgattt |
| ACGGGCCGGGAGGCGCGGGGGCACCTCCCCCATGTGCGTGAAATACGTGGCCGGCTGTGTGgccagtgttacaaccaattaacc | |
| Y613F-JRS 846/JRS 847 | CGCGAACGGCACGCCCCCTACGAGGACGACGAGTCAATATTCGAGACGGTGAGCGAGGACGtagggataacagggtaatcgattt |
| TTCCTCGTAGACACGCCCCCCGTCCTCGCTCACCGTCTCGAATATTGACTCGTCGTCCTCGgccagtgttacaaccaattaacc | |
| Y624F-JRS 687/RS 688 | GTCAATATACGAGACGGTGAGCGAGGACGGGGGGCGTGTCTTCGAGGAAATACCATGGATGCtagggataacagggtaatcgattt |
| GCAGACGTTTTCGTAGACCCGCATCCATGGTATTTCCTCGAAGACACGCCCCCCGTCCTCGgccagtgttacaaccaattaacc | |
| Y613F/Y624F-JRS 846/JRS 848 | CGCGAACGGCACGCCCCCTACGAGGACGACGAGTCAATATTCGAGACGGTGAGCGAGGACGtagggataacagggtaatcgattt |
| TTCCTCGAAGACACGCCCCCCGTCCTCGCTCACCGTCTCGAATATTGACTCGTCGTCCTCGgccagtgttacaaccaattaacc | |
| Y633F-JRS 760/JRS 761 | GGGGGGCGTGTCTACGAGGAAATACCATGGATGCGGGTCTTCGAAAACGTCTGCGTGAACAtagggataacagggtaatcgattt |
| GGCCGGCGCTGCATTCGCCGTGTTCACGCAGACGTTTTCGAAGTCCCGCATCCATGGTATTgccagtgttacaaccaattaacc | |
| Y657F-JRS 810/JRS 811 | GCGCCGGCCTCCCCGTACATTGAGGCGGAAAATCCCCTGTTCGACTGGGGGGGATCCGCCCtagggataacagggtaatcgattt |
| GCGGCCCGGGGGGGAAAATAGGGCGGATCCCCCCCAGTCGAACAGGGGATTTTCCGCCTCAgccagtgttacaaccaattaacc |
HSV-1 sequences are capitalized, point mutations are in bold, and I-SceI-aphAI sequences are in lowercase.
Infection.
Jurkat E6-1 cells were infected for 11 h or 13 h at an MOI of 10 in serum-free RPMI 1640 medium. Cells were harvested 11 h or 13 h postinfection (hpi) (see the figure legends).
Chemical inhibition of SFK activation.
Jurkat E6-1 cells were infected in serum-free RPMI 1640 supplemented with the SFK inhibitor PP2 or the inactive analogue PP3 (10 μM; both from Calbiochem) for 1 h. Cells were then maintained in RPMI 1640 medium supplemented with 10% heat-inactivated FBS and either PP2 or PP3 (10 μM).
Immunoprecipitation.
Cell extracts were harvested at 13 hpi as described previously (24) with the exception that cells were lysed at 4°C for 15 min. Immunoprecipitation was carried out using 90% of the lysate as described previously (24) with the following changes. Cell debris was spun down at 3,000 × g for 15 min, antibodies were incubated overnight with protein G-agarose (Roche) in phosphate-buffered saline (PBS) at 4°C, lysates and protein G-agarose-conjugated antibodies were immunoprecipitated at 4°C for 3.5 h, and precipitates were washed four times with lysis buffer. Ten percent of the lysate was run separately to provide a whole-cell lysate loading control.
Purification of GST fusion proteins.
pGEX-4T-1 was from GE Health Care. The following pGEX-4T-1-derived glutathione S-transferase (GST) fusion protein expression plasmids were generous gifts from R. Ingham: GST-Grb2 SH2 domain (human, originally from Tony Pawson), GST-Shc phosphotyrosine-binding (PTB) domain (human, originally from Tony Pawson) and GST-Lck SH2 domain (Mus musculus, originally from Andre Veillette). Plasmids were maintained in E. coli BL21 cells (Stratagene). Following overnight incubation of the culture at 37°C, the cultures were diluted in lysogeny broth (LB) and grown at 37°C to an optical density at 600 nm (OD600) of 0.6. Expression of GST fusion proteins was induced by adding 1.0 mM isopropylthio-β-galactoside (IPTG) (Sigma) prior to further incubation at 37°C for 3 h. Next, the cultures were centrifuged at 5,000 × g for 20 min at 4°C, and the pellets were lysed by incubation in lysis buffer (1% Triton X-100, 20 mM Tris [pH 8], 2 mM EDTA, 137 mM NaCl, 10% glycerol) supplemented with protease inhibitors (Roche) for 10 min on ice. Lysates were sonicated and clarified by centrifugation at 13,000 × g for 30 min at 4°C. Clarified lysates were incubated with glutathione-agarose (Sigma) at 4°C overnight. The beads were washed three times in lysis buffer, and the yield of purified GST fusion protein immobilized to glutathione-agarose was determined by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and subsequent staining with Ponceau S (Sigma).
GST pulldown assays.
Jurkat E6-1 cells were infected as indicated in the figure legends. At 13 hpi cells were lysed in 200 μl lysis buffer (1% Triton X-100, 20 mM Tris [pH 8], 2 mM EDTA, 137 mM NaCl) supplemented with protease inhibitors for 15 min at 4°C. The lysates (2 × 106 cells) were clarified by centrifugation at 3,000 × g for 15 min at 4°C and precleared by incubation with 75 μl glutathione-agarose for 2 h at 4°C. Precleared lysates were mixed with GST fusion proteins immobilized to glutathione-agarose (75 μl) at 4°C overnight. The beads were then washed three times with 200 μl lysis buffer at 4°C, resuspended in 100 μl lysis buffer with 20 μl 3× SDS loading dye supplemented with 2-mercaptoethanol (2-ME), and analyzed by SDS-PAGE followed by Western blotting.
Western blotting.
Western blotting was carried out as described previously (26) with the following change. For detection of tyrosine phosphorylation, samples were separated by electrophoresis through a 10% SDS-polyacrylamide gel. All other samples were separated by electrophoresis through an 8% SDS-polyacrylamide gel. Following Western blotting, the signals were visualized via either enhanced chemiluminescence (ECL) (Thermo Scientific Pierce) or infrared imaging on an Odyssey infrared imager (Licor), as indicated in the figure legends.
Antibodies.
Primary antibodies used for detection by Western blotting included goat anti-GFP (provided by L. Berthiaume), mouse anti-ICP27 (1:5,000; Virus Corporation), mouse anti-Lck (1:1,000; Santa Cruz), mouse anti-Shc (1:2,000; BD Transduction Laboratories), mouse anti-phosphotyrosine 4G10 (1:10,000; Millipore), mouse anti-VP16 (1:50; provided by Tony Minson), rabbit anti-actin (1:5,000; Sigma), rabbit anti-active SFK Y614 (1:1,000; Cell Signaling), rabbit anti-Grb2 (1:2,000; Cell Signaling), rabbit anti-p85 (1:10,000; Upstate), and rabbit HSV-2 anti-UL46 (1:10,000; provided by Yukihiro Nishiyama). Secondary antibodies included anti-mouse horseradish peroxidase (HRP) true blot (1:1,000 to 1:5,000; eBioscience), anti-rabbit HRP true blot (1:2,000 to 1:5,000; eBioscience), donkey anti-goat HRP (1:10,000; Jackson ImmunoResearch), donkey anti-mouse IR800 (1:10,000; Rockland, Inc.), goat anti-mouse HRP (1:1,000 to 1:5,000; Promega), goat anti-mouse Alexa Fluor 680 (1:10,000; Invitrogen), goat anti-rabbit Alexa Fluor 680 (1:10,000; Invitrogen), goat anti-rabbit HRP (1:2,000 to 1:5,000; Promega), and goat anti-rabbit IR800 (1:10,000; Rockland, Inc.). Immunoprecipitation was carried out using anti-rabbit IgG (4 μg; Sigma), goat anti-GFP (1 μg; provided by L. Berthiaume), mouse anti-Grb2 (2 μg; Santa Cruz), mouse anti-Lck (1 μg; Santa Cruz), rabbit anti-p85 (4 μl, serum; Millipore) and rabbit anti-Shc (2.5 μg; BD Transduction Laboratories).
RESULTS
VP11/12 interacts with Grb2 and Shc in addition to p85 and Lck.
VP11/12 contains an amino-terminal UL46 domain that is conserved in the UL46 orthologues of all alphaherpesviruses and a C-terminal region that is conserved only in HSV-1, HSV-2, and closely related members of the genus Simplexvirus (Fig. 1). Interestingly, the previously proposed putative Lck and p85 SH2 binding motifs are located in the C-terminal region (Fig. 1A). The p85 motif at Y519 (YTHM) and the Lck motif at Y624 (YEEI) are highly conserved in related members of the genus Simplexvirus, consistent with their being functionally significant, while the Y613 Lck motif (YETV) is less well conserved (Fig. 1B). As also shown in Fig. 1B, the C-terminal region of VP11/12 contains well-conserved tyrosine-based motifs predicted to bind the SH2 domain of Grb2 (YENV at Y633) and the phosphotyrosine-binding (PTB) domain of Shc (NPLY at Y657), raising the possibility that VP11/12 may interact with these additional host signaling proteins. We therefore examined the roles of the five tyrosine-based motifs described above in mediating interactions with Lck, p85, Grb2, and Shc.
Fig 1.
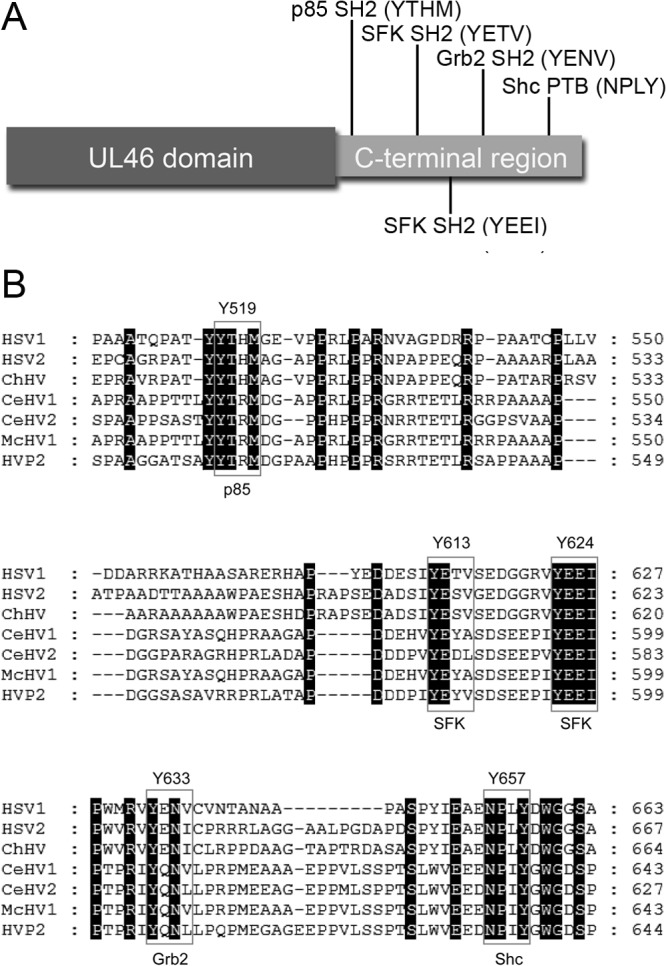
Schematic diagram of VP11/12. (A) The VP11/12 protein consists of a highly conserved N-terminal UL46 domain of unknown function found in all alphaherpesviruses and a less-conserved C-terminal region that harbors putative tyrosine-based signaling motifs predicted by the Scansite 2.0 algorithm to bind p85, SFKs, Grb2, and Shc. (B) The C-terminal region is conserved in HSV1/2 and closely related members of the genus Simplexvirus. Sequences were aligned using MUSCLE (http://www.ebi.ac.uk/Tools/msa/muscle/) and visualized using GeneDoc (http://www.nrbsc.org/gfx/genedoc/). HSV1/2, herpes simplex virus 1 and 2 (ACM62269.1; AEV91385.1); ChHV, chimpanzee alpha-1 herpesvirus (AFV26935.1); CeHV1/2, cercopithecine herpesvirus 1 and 2 (BAC58086.1; YP_164489.1); McHV1, macacine herpesvirus 1 (NP_851906.1); HVP2, papiine herpesvirus 2 (YP_443893.1).
In order to facilitate these studies, we tagged VP11/12 with EGFP at its N or C terminus in the KOS37 BAC (35) using en passant mutagenesis (36). A coimmunoprecipitation assay was used to confirm that these newly generated VP11/12 fusion proteins interact with Lck and p85 and also determine if they interact with Grb2 and Shc as predicted (Fig. 2). Jurkat E6-1 cells were either mock infected or infected with KOS37-UL46 GFP, KOS37-GFP UL46, or a KOS-derived virus expressing GFP (KOS-G) for 13 h. Cell extracts were then immunoprecipitated using an anti-GFP antibody, and the precipitates were analyzed by Western blotting using antibodies against p85, Lck, Shc, and Grb2 (Fig. 2). Analysis of the whole-cell lysates demonstrated that KOS37-GFP UL46 and KOS37-UL46 GFP gave rise to equivalent levels of VP11/12 fusion protein. Both VP11/12 fusion proteins coimmunoprecipitated all four cellular proteins, although the position of the GFP tag appeared to influence the coimmunoprecipitation efficiency in that the N-terminally tagged VP11/12 coprecipitated somewhat smaller amounts of p85 and Shc than the C-terminally tagged version. Importantly, the free GFP encoded by KOS-G did not coprecipitate any of the cellular proteins, indicating that the protein-protein interactions are mediated by the VP11/12 portion of the fusion proteins.
Fig 2.
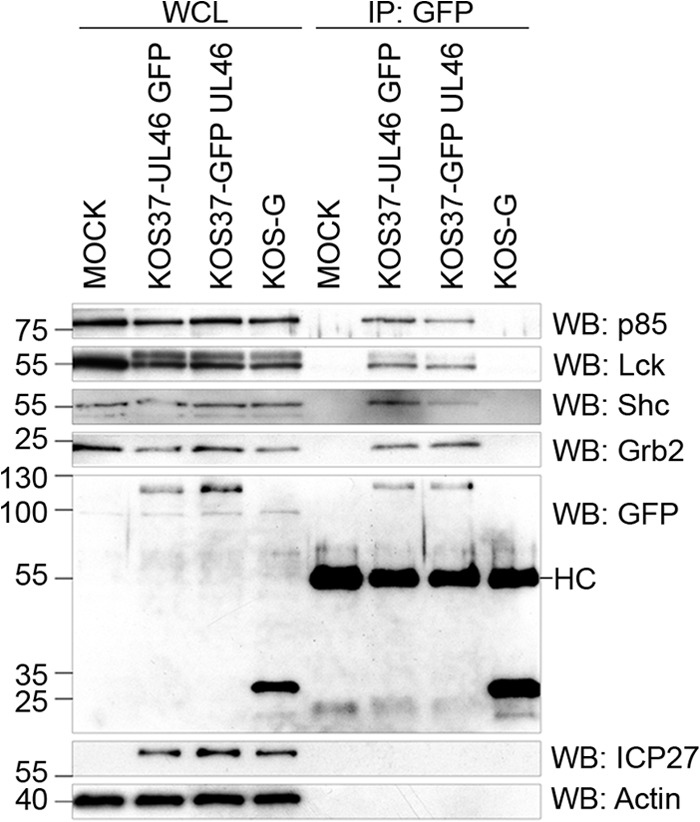
VP11/12 interacts with p85, Lck, Shc, and Grb2. Jurkat E6-1 cells were mock infected or infected with a KOS37-derived virus expressing C-terminally GFP-tagged VP11/12 (KOS37-UL46 GFP), a KOS37-derived virus expressing N-terminal tagged VP11/12 (KOS37-GFP UL46), or a KOS-derived virus expressing GFP (KOS-G) for 13 h. Whole-cell lysates (WCL) were immunoprecipitated using an antibody against GFP (IP: GFP) and analyzed via Western blotting (WB) for p85, Lck, Shc, Grb2, GFP, and ICP27 (HC, heavy chain).
These findings suggest that VP11/12 interacts either directly or indirectly with Shc and Grb2 in addition to the previously described interactions with Lck (24) and p85 (25). Based on the apparently more efficient coimmunoprecipitation of p85 and Shc by the C-terminally tagged VP11/12 derivative, KOS-UL46 GFP was used in the additional experiments described below.
VP11/12 interacts with the SH2 domain of Grb2 through the YENV motif at position 633.
To test if phosphorylation of the tyrosine of the putative Grb2 SH2 binding motif (YENV) at position 633 is essential for the VP11/12-Grb2 association, we converted the tyrosine codon (TTA) to a phenylalanine codon (TTC) in the context of the C-terminally tagged VP11/12 encoded by KOS37-UL46 GFP, generating KOS37-UL46 GFP Y633F. The effect of this mutation on the VP11/12-GRB2 interaction was then assessed using coimmunoprecipitation and GST pulldown assays (Fig. 3).
Fig 3.

Interaction between VP11/12 and Grb2 requires the YENV motif at position 633. (A) Jurkat E6-1 cells were mock infected or infected with the indicated viruses for 13 h. Whole-cell lysates (WCL) were immunoprecipitated using an antibody against GFP (IP: GFP) and analyzed by Western blotting (WB) for p85, Lck, Shc, Grb2, GFP, ICP27, and actin. (B) Jurkat E6-1 cells were mock infected or infected with KOS37-UL46 GFP or KOS37-UL46 GFP Y633F, and cell lysates were subjected to a GST pulldown assay with the indicated GST fusion proteins. Protein bound to the beads was then analyzed for GFP, ICP27, and actin by Western blotting.
Jurkat E6-1 cells were mock infected or infected with KOS37-UL46 GFP, KOS-G, or KOS37-UL46 GFP Y633F for 13 h. Lysates were immunoprecipitated with an anti-GFP antibody as for Fig. 2 and then analyzed by Western blotting for p85, Lck, Grb2, and Shc (Fig. 3A). We found that the Y633F mutation abolished the interaction between VP11/12 and Grb2 without affecting the interactions with p85, Lck, and Shc. The interaction between VP11/12 and Grb2 was also abolished in a reciprocal coimmunoprecipitation assay using an antibody against Grb2 for immunoprecipitation and Western blotting for GFP to visualize the interaction (data not shown).
To determine if the interaction between Grb2 and VP11/12 is mediated by the SH2 domain of Grb2, Jurkat E6-1 cells were infected as described above, and cell extracts were incubated with glutathione-agarose beads bearing immobilized GST-Grb2 SH2 fusion protein (Fig. 3B). Bound VP11/12 was then detected by Western blotting using an antibody to GFP (Fig. 3B). VP11/12 efficiently bound to beads bearing the GST-Grb2 SH2 domain fusion protein, while no interaction was observed with GST. In contrast, the free GFP encoded by KOS-G did not bind the SH2 domain of Grb2 (data not shown). These data indicate that VP11/12 binds the SH2 domain of Grb2. As expected, the Y633F mutation abolished the interaction. Taken together, these data suggest that VP11/12 interacts directly with the Grb2 SH2 domain through the YENV motif at Y633. Inasmuch as SH2 domains bind phosphorylated tyrosine residues, the interaction almost certainly requires phosphorylation of Y633.
The interaction between VP11/12 and Shc requires phosphorylation of the NPLY motif at position 657 and is PTB dependent.
We used the same approach to examine the relevance of the putative Shc PTB domain-binding motif at Y657 (NPLY) in the interaction between VP11/12 and Shc, by generating and analyzing KOS37-UL46 GFP Y657F. The Y657F mutation abolished the interaction between VP11/12 and Shc in the coimmunoprecipitation assay without affecting interactions with p85, Lck, and Grb2 (Fig. 4A). Furthermore, an interaction between VP11/12 and Shc was no longer detectable in a reverse immunoprecipitation assay using an anti-Shc antibody (data not shown).
Fig 4.
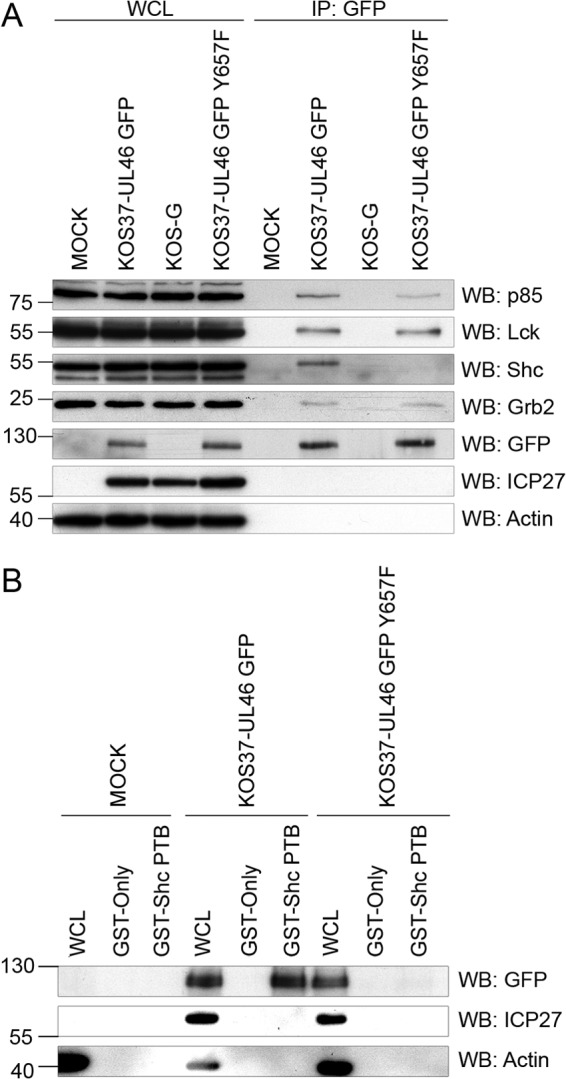
Association between VP11/12 and Shc requires the NPLY motif at position 657. (A) Extracts of Jurkat E6-1 cells infected with the indicated viruses (WCL) were precipitated with an anti-GFP antibody (IP: GFP) and analyzed by Western blotting (WB) as described for Fig. 3A. (B) Extracts of Jurkat E6-1 cells infected with KOS37-UL46 GFP or KOS37-UL46 GFP Y657F were analyzed in a GST pulldown assay using GST or GST-Shc PTB as for Fig. 3B.
To assess whether the interaction is dependent on the PTB domain of Shc, a GST pulldown assay was performed (Fig. 4B). VP11/12 bound to beads bearing GST fused to the PTB domain of Shc but not to beads bearing GST, and the interaction was abolished by the Y657F mutation. Moreover, the free GFP encoded by KOS-G did not bind the Shc PTB domain (data not shown). In summary, these data support a model of a direct PTB-mediated interaction between VP11/12 and Shc dependent on the NPLY motif at Y657.
Inactivation of the YTHM motif reduces but does not eliminate the VP11/12-p85 interaction.
A previous report from this laboratory showed that VP11/12 interacts with p85 in Jurkat cells and HEL fibroblasts and that in HEL cells this interaction is SKF dependent (25). Wagner and Smiley hypothesized that the YTHM motif, located at position 519, might be used by VP11/12 to interact with the SH2 domain of p85 to activate the Akt/PI3K-pathway (25). To determine the role of the YTHM motif we generated KOS37-UL46 GFP Y519F.
Inactivation of the YTHM motif greatly reduced but did not eliminate the interaction between p85 and VP11/12 without obviously affecting the interactions with Lck and Shc, as measured in the coimmunoprecipitation assay (Fig. 5). The interaction with Grb2 also was somewhat reduced, and further studies are required to test the significance of this observation. These data confirm that the YTHM motif contributes to the interaction with p85 and suggest that one or more additional interaction interfaces may also play a role.
Fig 5.
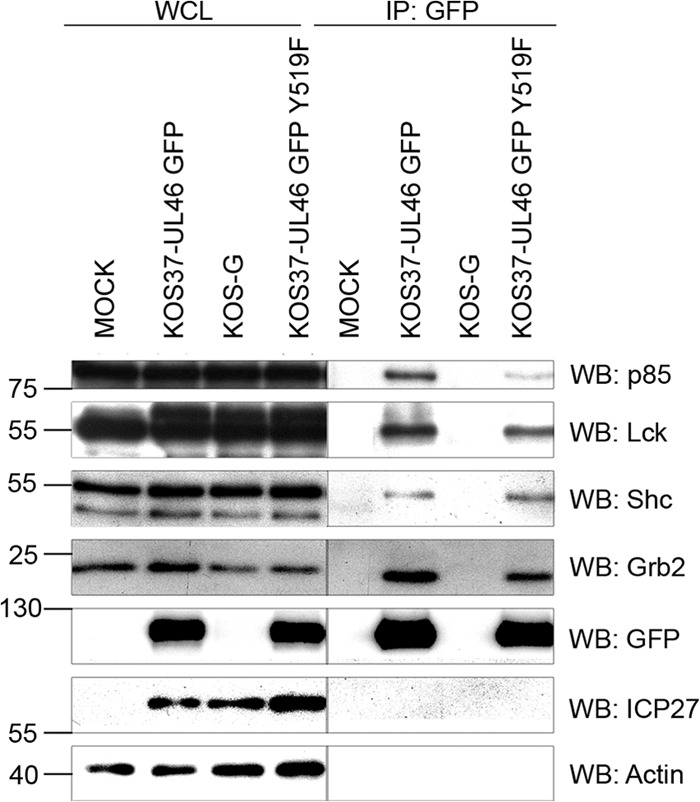
Inactivation of the YTHM motif at position 519 reduces but does not eliminate the VP11/12-p85 interaction. Extracts of Jurkat E6-1 cells infected with the indicated viruses (WCL) were precipitated with an anti-GFP antibody (IP: GFP) and analyzed by Western blotting (WB) as described for Fig. 3A.
The VP11/12 tyrosine-based motifs at Y613 and Y624 both contribute to interactions with Lck and Lck activation.
Wagner and Smiley (24, 25) hypothesized that VP11/12 interacts with Lck and other SFKs via the tyrosine-based motifs at Y613 (YETV) and/or Y624 (YEEI). We tested this hypothesis by generating viruses in which these motifs were inactivated either singly or in combination (KOS37-UL46 GFP Y613F, KOS37-UL46 GFP Y624F, and KOS37-UL46 GFP Y613F/Y624F). We found that inactivating either motif had relatively little effect on the ability of VP11/12 to coimmunoprecipitate Lck or active SFKs (Fig. 6A) and reduced but did not eliminate the interaction with the Lck SH2 domain in the GST pulldown assay (Fig. 6B). In contrast, inactivating both motifs virtually eliminated these interactions. These findings were confirmed in the reciprocal coimmunoprecipitation experiment, where Lck was immunoprecipitated and VP11/12 was detected in the immunoprecipitates (data not shown). These data suggest that the interaction between VP11/12 and Lck is mediated by the SH2 domain of Lck and requires phosphorylation of either the YEEI or the YETV motif.
Fig 6.
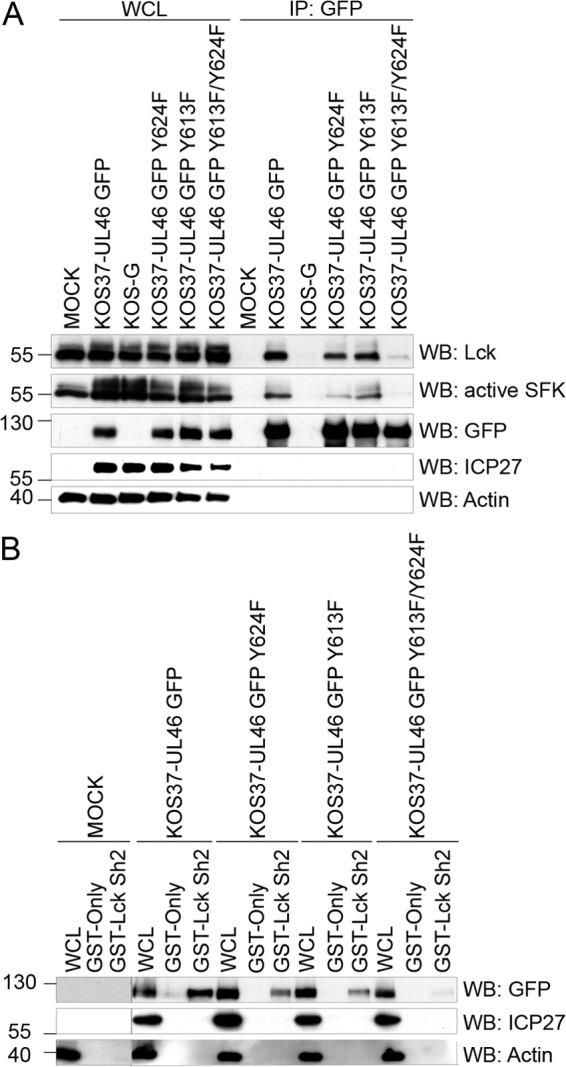
The interaction between Lck and VP11/12 requires both the YETV motif at position 613 and the YEEI motif at position 624. (A) Extracts of Jurkat E6-1 cells infected with the indicated viruses (WCL) were precipitated with an anti-GFP antibody (IP: GFP) and analyzed by Western blotting (WB) as described for Fig. 3A. (B) Extracts of Jurkat E6-1 cells infected with the indicated viruses were analyzed in a GST pulldown assay using GST or GST-Lck SH2 as for Fig. 3B.
We next examined the effects of inactivating the various tyrosine-based motifs on the ability of HSV to trigger activation of Lck during infection of Jurkat T cells. Cell lysates harvested at 11 hpi were analyzed by Western blotting for total Lck and for SFKs phosphorylated on the activation loop tyrosine (Lck residue Y394), and the results were quantified on an Odyssey infrared imager (Fig. 7A and B). The activation loops of SFKs are highly conserved, and hence the antibody used reacts with the active forms of all SFKs. However, our previous results have shown that the signal observed with the active SFK antibody in HSV-infected Jurkat cells arises predominantly if not entirely from Lck (24). As shown previously, Lck runs as a doublet with apparent molecular masses of ca. 56 and 60 kDa in both control and infected cells, and both species react with the active SFK antibody (i.e., the mobility shift is not due to phosphorylation of the activation loop). Although HSV infection does not alter the total Lck signal, it does increase the proportion running at ca. 60 kDa (24) (Fig. 7). Also as previously reported, HSV KOS37 induces a ca. 3 to 4 increase in the total active SFK signal (summed over the 56- and 60-kDa bands), an effect that is eliminated by the VP11/12-null mutation in ΔUL46. We found that inactivating the Grb2 (Y633) and Shc (Y657) binding motifs had little or no effect on Lck activation, while inactivating the p85 motif at Y519 induced a small but statistically significant increase. Inactivating the YEEI-Lck motif significantly reduced but did not eliminate Lck activation, while mutating the Y613 motif had no significant effect. Most importantly, inactivating both of the Lck SH2 binding motifs eliminated Lck activation, producing a phenotype similar to that of the VP11/12 null mutant ΔUL46. However, the Y613F/Y624F mutant consistently displayed lower levels of the ca. 60-kDa Lck species than ΔUL46, perhaps indicating that this form of VP11/12 actively interferes with the function of another viral or cellular protein(s) that induces the mobility shift. Overall, these results indicate that the predicted Lck SH2 domain-binding motifs at Y613 and Y624 both contribute to Lck activation, with the Y624 motif playing a more dominant role.
Fig 7.
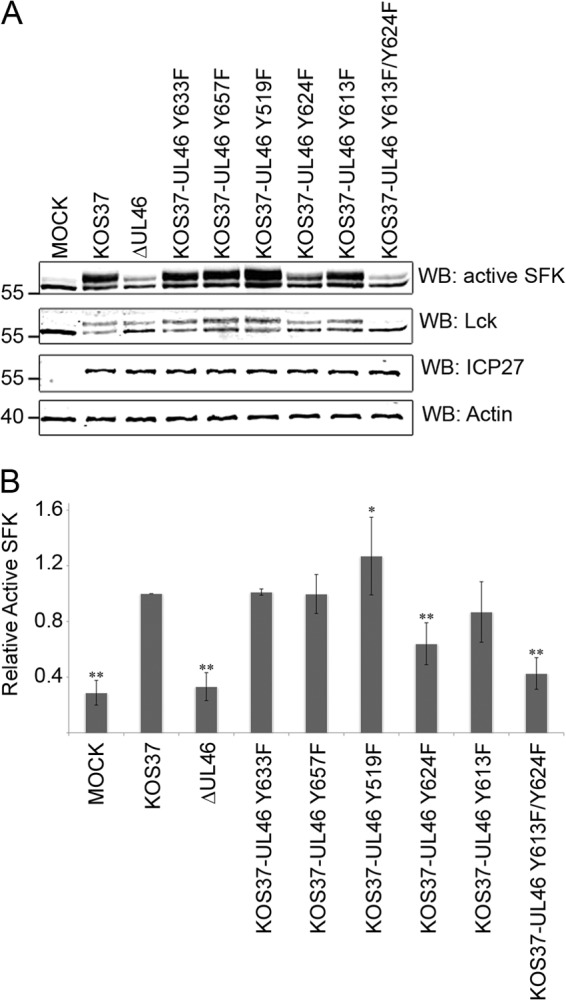
The YETV and YEEI motifs both contribute to Lck activation. Jurkat E6-1 cells were infected for 11 h with the indicated viruses. (A) Extracts were analyzed by Western blotting (WB) using antibodies directed against the indicated proteins and the results were collected using an Odyssey infrared imager. (B) Quantification of the results obtained in four independent experiments. The signal obtained with the active SFK antibody was summed over the ca. 56-kDa and ca. 60-kDa bands, divided by the total Lck signal, followed by normalization to the ratio obtained with KOS37, which was set to 1.0. The statistical significance of the differences between KOS37 and the other samples were evaluated using a two-tailed t test (*, P < 0.05; **, P < 0.01).
VP11/12-induced SFK activation is required for binding of Grb2, Shc, and p85.
Wagner and Smiley previously showed that chemical inhibition of SFK activity strongly inhibited the interaction between VP11/12 and p85 in HEL fibroblasts (25), leading to the hypothesis VP11/12-induced SFK activation leads to phosphorylation of the p85-binding YTHM motif (25). In order to determine if SFK activity is similarly required for the interactions between Grb2 and Shc, we analyzed the effects of the SFK inhibitor PP2 in a coimmunoprecipitation assay (Fig. 8A). As expected, PP2 had no effect on the interaction between VP11/12 and Lck but strongly inhibited tyrosine phosphorylation of VP11/12 and the association with p85; in contrast the inactive analogue PP3 had no effect. PP2 also essentially eliminated the interactions between VP11/12 and Grb2 and Shc, consistent with the hypothesis that SFK activity is required for the phosphorylation of the Grb2 and Shc binding tyrosine-based motifs. If this is indeed the case, then one predicts that mutations that prevent SFK binding and activation should reduce the interaction with p85 and eliminate those with Grb2 and Shc. In accord with this hypothesis, the Y613F/Y624F double substitution had the predicted effects on these protein-protein interactions, while each of the single substitutions had only a relatively minor effect (Fig. 8B). Taken in combination, these data indicate that binding of p85, Grb2, and Shc requires VP11/12-induced activation of SFKs. These findings suggest that the activated SFK then leads (either directly or indirectly) to phosphorylation of the tyrosine-based motifs at Y519, Y633, and Y657.
Fig 8.
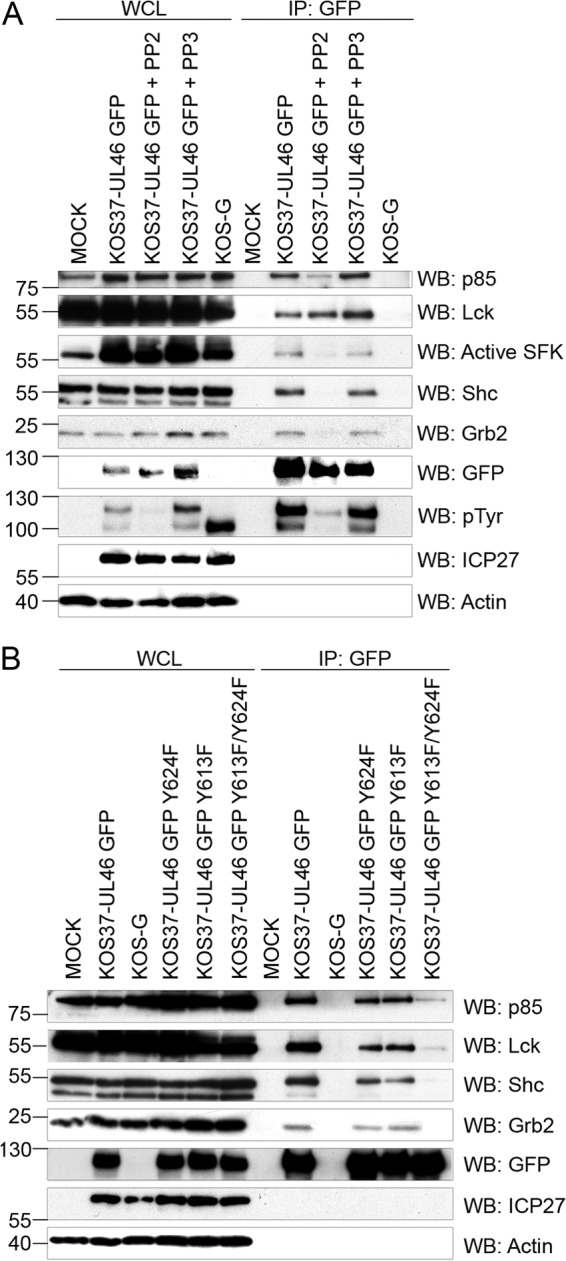
Role of Lck in the interactions between VP11/12 and p85, Grb2, and Shc. (A) Extracts (WCL) of Jurkat E6-1 cells infected with the indicated viruses in the presence and absence of PP2 or PP3 were precipitated with an anti-GFP antibody (IP: GFP) and analyzed by Western blotting (WB) for the indicated proteins as described for Fig. 3A (pTyr, tyrosine phosphorylation). (B) Extracts of Jurkat E6-1 cells infected with the indicated viruses (WCL) were precipitated with an anti-GFP antibody (IP: GFP) and analyzed by Western blot (WB) for the indicated proteins as for Fig. 3A.
VP11/12-induced SFK activation is necessary for the global induction of tyrosine phosphorylation during infection of Jurkat cells.
Zahariadis et al. (26) demonstrated that HSV infection of Jurkat cells triggers a dramatic increase in total phosphotyrosine levels in a VP11/12-dependent fashion, with VP11/12 being one of the major phosphotyrosine-reactive species. To evaluate the hypothesis that this striking increase in phosphotyrosine stems from VP11/12-induced activation of SFKs, including Lck, we infected Jurkat E6-1 cells with viruses bearing mutations that inactivate each of the VP11/12 tyrosine-based motifs in the context of untagged VP11/12. Cells were harvested at 11 hpi, and lysates were analyzed for total tyrosine phosphorylation through Western blotting (Fig. 9). Consisted with previous data (26), cells infected with KOS37 displayed strikingly enhanced tyrosine phosphorylation of three prominent species relative to uninfected cells: a ca. 110-kDa band that corresponds to tyrosine phosphorylated VP11/12, a doublet migrating at ca. 56 and 60 kDa that likely corresponds to Lck, and a ca. 40-kDa species that has yet to be identified. In contrast, the VP11/12-null mutant ΔUL46 displayed a pattern similar to that obtained with uninfected cells, with the exception that the ca. 60-kDa putative Lck species increased in abundance while the ca. 56-kDa form correspondingly declined. Inactivating the Y633 (Grb2) or the Y657 (Shc) motif had little effect on the overall pattern of tyrosine phosphorylation, while inactivating the Y519 (p85) motif consistently led to enhanced tyrosine phosphorylation. Most strikingly, simultaneous inactivation of the Y613 and Y624 motifs virtually eliminated enhanced tyrosine phosphorylation, giving rise to a pattern similar to uninfected cells, while each of the single mutants displayed an intermediate phenotype, with the Y624F mutant showing a greater decline than the Y613 mutant. Overall, these data support the hypothesis that VP11/12-induced activation of SFKs, including Lck, is required for the enhanced tyrosine phosphorylation observed in HSV-1-infected Jurkat T cells.
Fig 9.
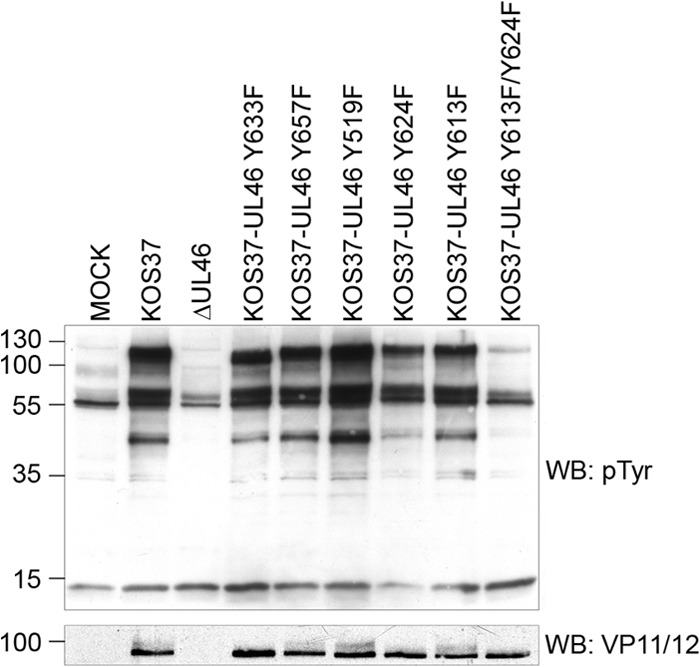
Role of VP16-induced activation of Lck in the global stimulation of tyrosine phosphorylation following HSV infection of Jurkat T cells. Extracts of Jurkat E6-1 infected for 11 h with the indicated viruses were analyzed via Western blotting for tyrosine phosphorylation (WB: pTyr).
DISCUSSION
As reviewed in the introduction, this laboratory has previously proposed that VP11/12 utilizes specific tyrosine-based motifs in its C-terminal region (Y624 and/or Y613) to recruit and activate SFKs, including Lck (24), leading (directly or indirectly) to phosphorylation of the YTHM motif at Y519, recruitment of PI3K, and activation of Akt (25). More recently, we noted that the C-terminal region of VP11/12 also contains tyrosine-based motifs predicted to bind the SH2 domain of Grb2 and the PTB domain of Shc (Fig. 1), and we confirmed in this study that VP11/12 indeed interacts with these additional signaling proteins in coimmunoprecipitation assays (Fig. 2). The major goal of the current study was to test the predicted roles of the five tyrosine-based motifs depicted in Fig. 1. in the interactions between VP11/12 and SFKs, p85, Grb2, and Shc. We elected to conduct these studies in Jurkat E6-1 cells, which support substantially higher levels of VP11/12 tyrosine phosphorylation than fibroblasts (24, 26) or Vero cells (data not shown).
Overall, our results confirm that specific tyrosine-based motifs play key roles in the interaction of VP11/12 with Lck (Y613 and Y624) (Fig. 6), p85 (Y519) (Fig. 5), Grb2 (Y633) (Fig. 3), and Shc (Y657) (Fig. 4). The interactions with Grb2 and Shc appear to be entirely dependent on the cognate tyrosine-based motifs and are mediated by the SH2 and PTB domains, respectively, suggesting that the interactions are direct (Fig. 3 and 4). In contrast, the interaction with p85 appears to be more complex, as inactivating the YTHM motif at Y519 reduced but did not eliminate the interaction (Fig. 5). Consistent with this result, chemical inhibition of SFKs or disruption of the interaction between Lck and VP11/12 had effects similar to inactivating the YTHM motif (Fig. 8). Taken together, these data indicate that the interaction between VP11/12 and p85 does not solely depend on binding of the p85 SH2 domain to the YTHM motif, implying that one or more additional protein-protein interfaces contribute. The influenza A virus NS1 protein contains an SH2-binding motif and two SH3-binding motifs and all three motifs contribute to binding p85, although interactions via the SH3-binding motifs dominate (14). Inactivation of all three motifs produces a recombinant virus that is no longer able to interact with p85 or activate the Akt pathway (14). VP11/12 contains numerous proline-rich potential SH3 binding motifs, and we are currently testing if VP11/12 uses a similar mechanism to interact with p85.
Similarly, our data suggest that the interaction between VP11/12 and Lck is more complex than initially predicted. Based on the observations that phospho-YEEI is the optimal ligand for the SH2 domain of Lck (33) and that hamster polyomavirus middle T antigen uses a SH2-YEEI interaction mechanism to activate the SFK FYN (38), we previously hypothesized that the interaction likely depends on the YEEI motif at Y624 (24–26). However, our current data indicate that the adjacent YETV motif at Y613 also contributes and that both must be inactivated to severely impair the interaction with Lck (Fig. 6), Lck activation (Fig. 7), and recruitment of p85, Grb2, and Shc (Fig. 8). Thus, the YETV and YEEI motifs appear to be largely redundant.
Our results strongly support the hypothesis that VP11/12-dependent recruitment and activation of SFKs occur upstream of, and is required for, recruitment of p85, Grb2, and Shc, as chemical inhibition of SFKs and the Y613F/Y624F double substitution mutation has comparable inhibitory effects (Fig. 7). However, these results do not necessarily imply that the activated SFKs themselves directly phosphorylate the additional tyrosine-based motifs required for the interactions with p85, Grb2, and Shc. In this context, it is interesting that the seven-amino-acid-residue separation between the critical YETV and YEEI sequences required for all VP11/12 signaling activity recapitulates that found in immunoreceptor tyrosine-based activation motifs (ITAMs). ITAMs are located in the cytoplasmic tails of a variety of immune receptors and link them to downstream signaling cascades (reviewed in reference 39). Briefly, they consist of two YXXL/I sequences separated by six to eight residues. Following phosphorylation of the tyrosine residues by SFKs, the motif recruits members of the tandem SH2 domain containing Syk family of tyrosine kinases (Zap-70 in T cells), engaging additional signaling events (reviewed in reference 39). Although the YETV sequence does not match the YXXL/I consensus, it is conceivable that valine can functionally substitute for leucine or isoleucine, as all three have similar hydrophobic side chains. Although we have not yet been able to detect an interaction between VP11/12 and Zap-70 or Syk in coimmunoprecipitation assays (data not shown), it is nevertheless possible that one or both of these kinases contribute to phosphorylating the p85, Grb2, and Shc motifs. Further studies are warranted to test this possibility. Several viruses express proteins that contain functional ITAM motifs, for example, Epstein-Barr virus (EBV) LMP2a (40), Kaposi's sarcoma-associated herpesvirus K1 (41), and hepatitis C virus NS5A (42). Interestingly, the EBV LMP2a ITAM sequesters Syk, thereby attenuating signaling through the BCR (40). It will be interesting to learn whether the VP11/12 ITAM-like sequence plays an immunomodulatory role in T cells or other immune effector cells.
Previous work linked the ability of VP11/12 to recruit p85 to activation of Akt (14), and the varicella-zoster virus (VZV) orthologue ORF12 has recently been shown to display similar activity (21). In contrast, the biological consequences of the interactions between VP11/12 and Grb2 and Shc remain to be determined. Grb2 and Shc are multifunctional signaling adaptors that play key roles in a variety of signaling pathways (43–45). They are perhaps best characterized as positive effectors of the Ras/mitogen-activated protein kinase (MAPK)/ERK pathway downstream of receptor tyrosine kinases. The VZV ORF12 orthologue stimulates phosphorylation of ERK1/2 in transient-transfection assays, but HSV-1 VP11/12 does not (46). Moreover, HSV-1 infection suppresses rather than activates ERK1/2 phosphorylation in a range of cell types (47–49). We find that VP11/12 is not required for this effect in fibroblasts (data not shown), and Chuluunbaatar and colleagues have instead implicated the viral serine/threonine kinase US3 (49). Perhaps VP11/12 serves as a functionally redundant or cell-type-specific inhibitor of ERK activation; alternatively, the Grb2 and Shc interactions described here may modulate other pathways, including PI3K/Akt signaling. Further studies are required to examine these possibilities.
Viruses have adopted diverse strategies to stimulate cellular signaling pathways, in some cases acting at the apex of the pathway by stimulating the activity of cellular growth factor receptors or serving as activated receptor mimics. For example, the bovine papillomavirus E5 protein binds the transmembrane domain of PDGFR, forcing receptor dimerization and ligand-independent activation of signaling (reviewed in reference 50). In contrast, polyomavirus middle T antigen activates SFKs, leading to phosphorylation of key tyrosine motifs in middle T antigen that serve as docking sites for cellular signaling molecules, thus serving as an activated growth factor receptor mimic (reviewed in reference 51). Our data strongly argue that VP11/12 acts in a fashion analogous to that of polyomavirus middle T antigen to recruit cellular signaling proteins. Further work is required to clarify the role of VP11/12-induced signaling in virus replication, pathogenesis, and latency.
ACKNOWLEDGMENTS
We thank Rob Ingham for gifts of plasmids and helpful advice and Deborah Burshtyn for pointing out the similarity between the spacing of the Y613 and Y624 motifs and an ITAM.
This work was supported by an operating grant from the Canadian Institute for Health Research (FRN 12172). J.R.S. holds a Tier I Canada Research Chair in Molecular Virology.
Footnotes
Published ahead of print 14 August 2013
REFERENCES
- 1.Roizman B, Knipe DM, Whitley RJ. 2007. Herpes simplex viruses, p 2501–2601. In Knipe DM, Howley PM. (ed), Fields virology, 5th ed. Lippincott, Williams and Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Corey L, Wald A, Celum CL, Quinn TC. 2004. The effects of herpes simplex virus-2 on HIV-1 acquisition and transmission: a review of two overlapping epidemics. J. Acquir. Immune Defic. Syndr. 35:435–445. [DOI] [PubMed] [Google Scholar]
- 3.Wald A, Schacker T, Corey L. 1997. HSV-2 and HIV: consequences of an endemic opportunistic infection. STEP Perspect. 9:2–4. [PubMed] [Google Scholar]
- 4.Manning BD, Cantley LC. 2007. AKT/PKB signaling: navigating downstream. Cell 129:1261–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kelly JD, Haldeman BA, Grant FJ, Murray MJ, Seifert RA, Bowen-Pope DF, Cooper JA, Kazlauskas A. 1991. Platelet-derived growth factor (PDGF) stimulates PDGF receptor subunit dimerization and intersubunit trans-phosphorylation. J. Biol. Chem. 266:8987–8992. [PubMed] [Google Scholar]
- 6.Wu H, Windmiller DA, Wang L, Backer JM. 2003. YXXM motifs in the PDGF-beta receptor serve dual roles as phosphoinositide 3-kinase binding motifs and tyrosine-based endocytic sorting signals. J. Biol. Chem. 278:40425–40428. [DOI] [PubMed] [Google Scholar]
- 7.Liu P, Cheng H, Roberts TM, Zhao JJ. 2009. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat. Rev. Drug Discov. 8:627–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, Cohen P. 1997. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Bα. Curr. Biol. 7:261–269. [DOI] [PubMed] [Google Scholar]
- 9.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. 2005. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307:1098–1101. [DOI] [PubMed] [Google Scholar]
- 10.Andjelkovic M, Alessi DR, Meier R, Fernandez A, Lamb NJ, Frech M, Cron P, Cohen P, Lucocq JM, Hemmings BA. 1997. Role of translocation in the activation and function of protein kinase B. J. Biol. Chem. 272:31515–31524. [DOI] [PubMed] [Google Scholar]
- 11.Stambolic V, Suzuki A, de la Pompa JL, Brothers GM, Mirtsos C, Sasaki T, Ruland J, Penninger JM, Siderovski DP, Mak TW. 1998. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell 95:29–39. [DOI] [PubMed] [Google Scholar]
- 12.Maehama T, Dixon JE. 1998. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 273:13375–13378. [DOI] [PubMed] [Google Scholar]
- 13.Carracedo A, Pandolfi PP. 2008. The PTEN-PI3K pathway: of feedbacks and cross-talks. Oncogene 27:5527–5541. [DOI] [PubMed] [Google Scholar]
- 14.Shin YK, Liu Q, Tikoo SK, Babiuk LA, Zhou Y. 2007. Influenza A virus NS1 protein activates the phosphatidylinositol 3-kinase (PI3K)/Akt pathway by direct interaction with the p85 subunit of PI3K. J. Gen. Virol. 88:13–18. [DOI] [PubMed] [Google Scholar]
- 15.Johnson RA, Wang X, Ma XL, Huong SM, Huang ES. 2001. Human cytomegalovirus up-regulates the phosphatidylinositol 3-kinase (PI3-K) pathway: inhibition of PI3-K activity inhibits viral replication and virus-induced signaling. J. Virol. 75:6022–6032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mannova P, Beretta L. 2005. Activation of the N-Ras-PI3K-Akt-mTOR pathway by hepatitis C virus: control of cell survival and viral replication. J. Virol. 79:8742–8749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Street A, Macdonald A, McCormick C, Harris M. 2005. Hepatitis C virus NS5A-mediated activation of phosphoinositide 3-kinase results in stabilization of cellular beta-catenin and stimulation of beta-catenin-responsive transcription. J. Virol. 79:5006–5016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Portis T, Longnecker R. 2004. Epstein-Barr virus (EBV) LMP2A mediates B-lymphocyte survival through constitutive activation of the Ras/PI3K/Akt pathway. Oncogene 23:8619–8628. [DOI] [PubMed] [Google Scholar]
- 19.Montaner S, Sodhi A, Pece S, Mesri EA, Gutkind JS. 2001. The Kaposi's sarcoma-associated herpesvirus G protein-coupled receptor promotes endothelial cell survival through the activation of Akt/protein kinase B. Cancer Res. 61:2641–2648. [PubMed] [Google Scholar]
- 20.Tomlinson CC, Damania B. 2004. The K1 protein of Kaposi's sarcoma-associated herpesvirus activates the Akt signaling pathway. J. Virol. 78:1918–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu X, Cohen JI. 2013. Varicella-zoster virus ORF12 protein activates the phosphatidylinositol 3-kinase/Akt pathway to regulate cell cycle progression. J. Virol. 87:1842–1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Benetti L, Roizman B. 2006. Protein kinase B/Akt is present in activated form throughout the entire replicative cycle of ΔU(S)3 mutant virus but only at early times after infection with wild-type herpes simplex virus 1. J. Virol. 80:3341–3348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chuluunbaatar U, Roller R, Feldman ME, Brown S, Shokat KM, Mohr I. 2010. Constitutive mTORC1 activation by a herpesvirus Akt surrogate stimulates mRNA translation and viral replication. Genes Dev. 24:2627–2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wagner MJ, Smiley JR. 2009. Herpes simplex virus requires VP11/12 to induce phosphorylation of the activation loop tyrosine (Y394) of the Src family kinase Lck in T lymphocytes. J. Virol. 83:12452–12461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wagner MJ, Smiley JR. 2011. Herpes simplex virus requires VP11/12 to activate Src family kinase-phosphoinositide 3-kinase-Akt signaling. J. Virol. 85:2803–2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zahariadis G, Wagner MJ, Doepker RC, Maciejko JM, Crider CM, Jerome KR, Smiley JR. 2008. Cell-type-specific tyrosine phosphorylation of the herpes simplex virus tegument protein VP11/12 encoded by gene UL46. J. Virol. 82:6098–6108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang Y, McKnight JL. 1993. Herpes simplex virus type 1 UL46 and UL47 deletion mutants lack VP11 and VP12 or VP13 and VP14, respectively, and exhibit altered viral thymidine kinase expression. J. Virol. 67:1482–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Y, Sirko DA, McKnight JL. 1991. Role of herpes simplex virus type 1 UL46 and UL47 in alpha TIF-mediated transcriptional induction: characterization of three viral deletion mutants. J. Virol. 65:829–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Murphy MA, Bucks MA, O'Regan KJ, Courtney RJ. 2008. The HSV-1 tegument protein pUL46 associates with cellular membranes and viral capsids. Virology 376:279–289. [DOI] [PubMed] [Google Scholar]
- 30.Kato K, Daikoku T, Goshima F, Kume H, Yamaki K, Nishiyama Y. 2000. Synthesis, subcellular localization and VP16 interaction of the herpes simplex virus type 2 UL46 gene product. Arch. Virol. 145:2149–2162. [DOI] [PubMed] [Google Scholar]
- 31.McKnight JL, Pellett PE, Jenkins FJ, Roizman B. 1987. Characterization and nucleotide sequence of two herpes simplex virus 1 genes whose products modulate alpha-trans-inducing factor-dependent activation of alpha genes. J. Virol. 61:992–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vittone V, Diefenbach E, Triffett D, Douglas MW, Cunningham AL, Diefenbach RJ. 2005. Determination of interactions between tegument proteins of herpes simplex virus type 1. J. Virol. 79:9566–9571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Songyang Z, Shoelson SE, Chaudhuri M, Gish G, Pawson T, Haser WG, King F, Roberts T, Ratnofsky S, Lechleider RJ. 1993. SH2 domains recognize specific phosphopeptide sequences. Cell 72:767–778. [DOI] [PubMed] [Google Scholar]
- 34.Minaker RL, Mossman KL, Smiley JR. 2005. Functional inaccessibility of quiescent herpes simplex virus genomes. Virol. J. 2:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gierasch WW, Zimmerman DL, Ward SL, Vanheyningen TK, Romine JD, Leib DA. 2006. Construction and characterization of bacterial artificial chromosomes containing HSV-1 strains 17 and KOS. J. Virol. Methods 135:197–206. [DOI] [PubMed] [Google Scholar]
- 36.Tischer BK, Smith GA, Osterrieder N. 2010. En passant mutagenesis: a two step markerless red recombination system. Methods Mol. Biol. 634:421–430. [DOI] [PubMed] [Google Scholar]
- 37.Willard M. 2002. Rapid directional translocations in virus replication. J. Virol. 76:5220–5232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dunant NM, Messerschmitt AS, Ballmer-Hofer K. 1997. Functional interaction between the SH2 domain of Fyn and tyrosine 324 of hamster polyomavirus middle-T antigen. J. Virol. 71:199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Isakov N. 1997. Immunoreceptor tyrosine-based activation motif (ITAM), a unique module linking antigen and Fc receptors to their signaling cascades. J. Leukoc. Biol. 61:6–16. [DOI] [PubMed] [Google Scholar]
- 40.Fruehling S, Longnecker R. 1997. The immunoreceptor tyrosine-based activation motif of Epstein-Barr virus LMP2A is essential for blocking BCR-mediated signal transduction. Virology 235:241–251. [DOI] [PubMed] [Google Scholar]
- 41.Lee H, Guo J, Li M, Choi JK, DeMaria M, Rosenzweig M, Jung JU. 1998. Identification of an immunoreceptor tyrosine-based activation motif of K1 transforming protein of Kaposi's sarcoma-associated herpesvirus. Mol. Cell. Biol. 18:5219–5228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Inubushi S, Nagano-Fujii M, Kitayama K, Tanaka M, An C, Yokozaki H, Yamamura H, Nuriya H, Kohara M, Sada K, Hotta H. 2008. Hepatitis C virus NS5A protein interacts with and negatively regulates the non-receptor protein tyrosine kinase Syk. J. Gen. Virol. 89:1231–1242. [DOI] [PubMed] [Google Scholar]
- 43.Jang IK, Zhang J, Gu H. 2009. Grb2, a simple adapter with complex roles in lymphocyte development, function, and signaling. Immunol. Rev. 232:150–159. [DOI] [PubMed] [Google Scholar]
- 44.Tari AM, Lopez-Berestein G. 2001. GRB2: a pivotal protein in signal transduction. Semin. Oncol. 28:142–147. [DOI] [PubMed] [Google Scholar]
- 45.Wills MK, Jones N. 2012. Teaching an old dogma new tricks: twenty years of Shc adaptor signalling. Biochem. J. 447:1–16. [DOI] [PubMed] [Google Scholar]
- 46.Liu X, Li Q, Dowdell K, Fischer ER, Cohen JI. 2012. Varicella-zoster virus ORF12 protein triggers phosphorylation of ERK1/2 and inhibits apoptosis. J. Virol. 86:3143–3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McLean TI, Bachenheimer SL. 1999. Activation of cJUN N-terminal kinase by herpes simplex virus type 1 enhances viral replication. J. Virol. 73:8415–8426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sloan DD, Han JY, Sandifer TK, Stewart M, Hinz AJ, Yoon M, Johnson DC, Spear PG, Jerome KR. 2006. Inhibition of TCR signaling by herpes simplex virus. J. Immunol. 176:1825–1833. [DOI] [PubMed] [Google Scholar]
- 49.Chuluunbaatar U, Roller R, Mohr I. 2012. Suppression of extracellular signal-regulated kinase activity in herpes simplex virus 1-infected cells by the Us3 protein kinase. J. Virol. 86:7771–7776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Talbert-Slagle K, DiMaio D. 2009. The bovine papillomavirus E5 protein and the PDGF beta receptor: it takes two to tango. Virology 384:345–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schaffhausen BS, Roberts TM. 2009. Lessons from polyoma middle T antigen on signaling and transformation: A DNA tumor virus contribution to the war on cancer. Virology 384:304–316. [DOI] [PMC free article] [PubMed] [Google Scholar]