

Abstract
Cytotoxic T cells (CTL) play a critical role in the clearance of respiratory viral infections, but they also contribute to disease manifestations. In this study, we infected mice with a genetically modified pneumonia virus of mice (PVM) that allowed visualization of virus-specific CTL and infected cells in situ. The first virus-specific T cells entered the lung via blood vessels in the scattered foci of PVM-infected cells, which densely clustered around the bronchi at day 7 after infection. At this time, overall pulmonary virus load was maximal, but the mice showed no overt signs of disease. On days 8 to 9, T cells gained access to the infected bronchial epithelium and to the lung interstitium, which was associated with a reduction in the number of virus-infected cells within the initial clusters but could not prevent further virus spread throughout the lung tissue. Interestingly, recruitment of virus-specific CTL throughout the parenchyma was still ongoing on day 10, when the virus infection was already largely controlled. This also represented the peak of clinical disease. Thus, disease was associated with an exuberant T cell infiltration late in the course of the infection, which may be required to completely eliminate virus at residual foci of infection. PVM-induced immunopathology may thus result from the need to generate widespread T cell infiltrates to complete the elimination of virus-infected cells in a large organ like the lung. This experimental model provides the first insights into the spatiotemporal evolution of pulmonary antiviral T cell immunity in vivo.
INTRODUCTION
Respiratory viral infections are among the most frequent infections in humans. As illustrated by patients with T cell deficiencies (1–3) and confirmed in a variety of animal models (4–7), the elimination of many pulmonary viral infections is dependent on the generation of an effective antiviral T cell response. However, antiviral T cell responses are not only beneficial for the infected host; T cell-mediated immunopathology contributes to disease manifestations in a variety of pulmonary viral infections (5, 6, 8, 9). A precise understanding of this dual role of T cells in virus control and immunopathology is crucial for targeted therapeutic interventions.
Infection of mice with pneumonia virus of mice (PVM) is a useful model for the experimental study of a respiratory viral infection, because PVM is a natural mouse pathogen that shows significant viral replication after intranasal infection with low virus doses (5, 10–12). PVM shares many molecular and biological features with respiratory syncytial virus (RSV) (13–15), the most important respiratory pathogen in human infants and children, and PVM-infected mice develop many clinical features of RSV-infected infants, such as bronchiolitis and pneumonia (13, 14, 16–18). Control of experimental murine PVM infection is mainly mediated by T cells which also critically contribute to virus-induced immunopathology (5). Thus, T cell-deficient mice fail to eliminate PVM and become virus carriers in the absence of the clinical or histopathological signs of pneumonia that occur after infection of control mice.
Because viral infections represent highly dynamic situations, the relative kinetics of virus replication versus the generation and recruitment of virus-specific T cells is decisive for the outcome of infection (19, 20). The analysis of the evolution and dynamics of pulmonary antiviral T cell responses in mouse models of respiratory virus infection has so far largely been restricted to the phenotypic and functional characterization of T cells extracted by bronchoalveolar lavage (BAL) or isolated from lung tissue (21–24). In parallel, viral titers have been quantified by plaque assays from lung homogenates. While these methods allow monitoring of the dose and time relationship between viral replication and the T cell response, they do not provide a picture of the spatiotemporal evolution of the antiviral effector T cell response in the infected tissue. In a large organ such as the lung, it is likely that there are regional differences with respect to the spread of virus and infection control. Visualization of the spatial development of the virus-specific T cell effector response in the infected tissue could add a “third dimension” of analysis allowing understanding the pulmonary antiviral T cell response in more detail.
In this study, we developed an experimental model allowing the visualization of virus-infected cells and virus-specific effector T cells on large sections of lung tissue in a mouse model of PVM infection. This approach provides the first analysis of the spatiotemporal evolution of a pulmonary antiviral effector T cell response in situ.
MATERIALS AND METHODS
Mice, infections, and adoptive cell transfer.
C57BL/6 mice were obtained from Charles River (Sulzfeld, Germany). P14.Thy1.1 mice were provided by H. Pircher (Institute for Medical Microbiology and Hygiene, Freiburg, Germany). Mice were kept in microisolator cages and infected intranasally (i.n.) at 6 to 8 weeks of age under ketamine and xylazine anesthesia with PVM in 80 μl of serum-free Eagle's minimal essential medium (EMEM). For adoptive-transfer experiments, total spleen cells containing Vα2+ Thy-1.1+ CD8+ T cells obtained from naive or memory P14.Thy1.1 mice were transfused intravenously (i.v.) into sex-matched C57BL/6 recipients. Animal experiments were approved by the local animal care commission (accreditation no. 35/9185.81/G-08/47).
Viruses and cell lines.
PVM strain 15 was originally derived from the American Type Culture Collection (ATCC) (12). rPVM-1 and rPVM.GFP were generated from this wild-type virus as described previously (11). rPVM-GFPgp33 was generated as described below from the parental virus rPVM-GFP with the GFPgp33 gene cassette at the same position as the green fluorescent protein (GFP) gene cassette, i.e., between the small hydrophobic (SH) and G gene on position 7. PVM viruses were propagated in BHK cells (ATCC; CCL-10) (25) and purified via a discontinuous sucrose gradient (PVM 15 and rPVM-1) or pelleted through a 30% sucrose layer (rPVM-GFPgp33 and rPVM-GFP). Virus titers were determined by immunostaining (plaque assay) on Vero cells (CCL-81) under 0.8% methylcellulose as described previously (11). RAW309Cr1 (ATCC; TIB-69) is a murine macrophage cell line expressing both H-2d and H-2b major histocompatibility complex class I (MHC-I) molecules that is permissive for PVM (5). Lymphocytic choriomeningitis virus (LCMV-WE) was provided by H. Pircher.
Construction of pPVM.GFPgp33 and recovery of recombinant virus.
For construction of the plasmid encoding the LCMV-derived CTL epitope gp33-41, plasmid pPVM-GFP7 (11) was modified by addition of the LCMV sequence encoding amino acids (aa) 29 to 43 of the viral glycoprotein. For the introduction of the gp33 epitope into the enhanced GFP (eGFP) expression cassette, three consecutive mutagenesis PCRs were designed with reverse primers (available upon request) that led to the addition of 45 nucleotides (nt) corresponding to the sequence of aa 29 to 43 of the glycoprotein of LCMV. The final PCR fragment encoded the complete eGFP with gp33 directly fused to it, flanked by AgeI restriction sites. The AgeI restriction sites were used to insert the eGFPgp33 fragment at the site of the parental eGFP fragment into the full-length clone pPVM-GFP7. The integrity of the eGFPgp33 fragment in pPVM-GFPgp33 was checked by sequencing (GATC, Constance, Germany). rPVM-GFPgp33 was generated by transfection as previously described (11). To confirm the sequence of viral RNA, complete cellular RNA from infected BHK-21 cells was isolated with TRIzol and reverse transcribed with random hexamers using SuperScript II reverse transcriptase (Invitrogen, Karlsruhe, Germany). cDNA was amplified by PCR using PVM-specific primers spanning the inserted GFPgp33 fragment. The sequence of the obtained PCR fragment was confirmed by sequencing (GATC, Constance, Germany).
Flow cytometry.
BAL, isolation of pulmonary inflammatory cells, and flow cytometry were performed as described previously (26) using the following antibodies: CD3 (clone 145-2C11), CD8 (clone 53-6.7), Thy-1.1 (clone Ox-7), gamma interferon (IFN-γ) (clone XMG1.2), and an isotype control antibody (clone R3-43) from BD Pharmingen, San Diego, CA, and Vα2 T cell receptor (TCR) (clone B20.1) from eBioscience. For gp33 dimer staining, 1 μg of DimerX (mouse H-2Db:Ig; BD Pharmingen) was incubated with gp33 peptide (10−3 M; NeoMPS, Strasbourg, France) and 0.15 μg of β2 microglobulin (BD Pharmingen) overnight at 37°C. The gp33-loaded DimerX was incubated with anti-IgG1-phycoerythrin (PE) (BD Pharmingen; clone A85-1) for 1 h by room temperature and then used together with anti-CD3 (clone 145-2C11) and anti-CD8 (clone 53-6.7) in a total volume of 50 μl for 1.5 h to stain BAL fluid or lung lymphocytes, which had been blocked for 30 min in 50 μl of fluorescence-activated cell sorter (FACS) buffer containing naive mouse serum.
CFSE proliferation assay.
Spleen cells of a P14 mouse were isolated and incubated with 10 μl of carboxyfluorescein succinimidyl ester (CFSE) (0.5 mM; Sigma-Aldrich, St. Louis, MO) in 10 ml of phosphate-buffered saline (PBS; 37°C) for 15 min at 37°C. Thereafter, cells were washed with ice-cold PBS (containing 2% fetal calf serum [FCS]) and then with Iscove's modified Dulbecco's medium (IMDM), pressed through a cell strainer, and used as responder cells. RAW309Cr1 cells were used as stimulator cells, either loaded with gp33 peptide (positive control), infected with rPVM-GFPgp33, or infected with rPVM-1 (negative control) 40 h previously. Responder and stimulator cells were plated at a ratio of 20 to 1 and incubated at 37°C. At different time points, cells were harvested, blocked with naive mouse serum (1:100), and stained with antibodies against CD3 and Vα2 before analysis on a FACSort cytometer.
Tissue processing, cryosectioning, and immunostaining.
After preparation of the mouse, the lung was instilled with 0.8 ml of 4% paraformaldehyde (PFA) and the trachea was clamped off to retain the PFA in the lung. Thereafter, the lung was perfused via the right heart ventricle, removed, and stored for 2 h in 4% PFA at room temperature, followed by washing with PBS and incubation in 30% sucrose for 24 h at 4°C. The organ was embedded in OCT Tissue-Tek (Sakura Finetek, Zoeterwoude, Netherlands) and stored for an additional 24 h at 4°C, followed by slow-freezing in a polystyrene box covered with linen at −80°C. Sections 12 μm thick were cut with a cryostat and stored overnight at 4°C. Sections were fixed with 4% PFA for 20 min at room temperature, briefly incubated in PBS, and then blocked with Image-iT signal enhancer (Invitrogen, Molecular Probes, Eugene, OR) for 30 min. Thereafter, sections were stained with Thy-1.1 PE (1:100, clone Ox-7) and anti-PVM antibody (raised against the G protein of PVM in rabbits; 1:4,000) for 2 h and washed twice with PBS for 5 min, followed by incubation with a second-step antibody (polyclonal anti-rabbit IgG-Alexa 488, 1:200, A-11008; Invitrogen) for 1 h. After 5 washing steps in PBS, slides were dried, covered with Dako fluorescent mounting medium (Dako North America Inc., Carpinteria, CA), and analyzed.
ScanR analysis.
Lung tissue sections were analyzed using the Olympus ScanR high-content screening station, which is a combination of fluorescence microscopy with computer-based analysis. From stained individual lung sections (complete left lobe) an area of 12 mm2 (representing about 20% of the whole area of the left lung) was scanned at a 20-fold magnification using the Olympus ScanR acquisition software (version 2.1.0.16). This resulted in 84 images, some of which (at the border of the lung) did not contain tissue. The thresholds for green and red fluorescence signals, representing infected cells and virus-specific T cells, respectively, were defined subjectively by comparing specific signal to the background signal of noninfected tissue, after a general subtraction of the background. A minimum area and a maximum area of the fluorescence signal were additionally used to help identify individual positive cells. This definition was then applied uniformly to the whole scan.
Determination of absolute counts of virus-infected cells and virus-specific T cells per lung section.
To determine the number of virus-infected cells and virus-specific T cells per scanned lung area, each of the 84 images was randomly subdivided into 30 fields (main objects), resulting in 2,520 fields per scan. The ScanR analysis software (version 1.1.0.9) then calculated the absolute number of green and red events within each field. Obviously, each of the scanned lung areas differed in the number of vessels and bronchi within the scanned area. Also, some areas included the border of the lung and thus contained fields that did not represent lung tissue. To account for these differences, each of the 84 images was manually analyzed for fields that did not contain lung parenchyma. These fields were subtracted from the total 2,520 fields, resulting in the number of fields covering lung parenchyma (Nlung). The number of red or green signals corrected for the area actually containing lung parenchyma was then calculated as follows: (total fields [n = 2,520]/Nlung) × number of red or green signals counted for the whole lung area. The mean of two sections for each mouse lung was used to determine the counts for a given mouse.
Analysis of the relative distribution of virus-infected cells and virus-specific CTL in lung tissue.
To analyze the relative distribution of red (Thy-1.1+ virus-specific T cells), and green (virus-infected) cells within the lung tissue, we used the “gallery view” tool, where all 84 images taken from one lung section were integrated into one large overview image. Since the red and green signals in these gallery views were quite small and of different intensities, we used the ScanR analysis software to extract the distribution of red and green signals within a given section. The data obtained for the red and green signals (original ScanR data) was overlaid by using Adobe Photoshop cs software (version 8.0.1), and each dot was enlarged to a standard size that simplified judging the distribution of green and red signals (virus-infected cells and virus-specific cytotoxic T lymphocytes [CTL]) within the analyzed section.
Statistical analysis.
Data were analyzed by using Student's t test in the case of a normal distribution of raw data and equality of standard deviations. In cases where standard deviations differed significantly, the Welch t test was used. The nonparametric Mann-Whitney test was performed if the data did not follow a Gaussian distribution. All data were analyzed with GraphPad InStat software program, version 3.06. Differences were considered significant at a P value of less than 0.05.
RESULTS
rPVM-GFPgp33 is attenuated but retains pathogenicity in vivo.
In order to create an experimental setup that allows visualizing virus-infected cells and virus-specific CTL in situ, we generated a recombinant PVM expressing the H-2Db-restricted CTL epitope gp33-41 (gp33) of the lymphocytic choriomeningitis virus (LCMV) fused to the C terminus of eGFP (Fig. 1A). This marker CTL epitope was introduced to allow tracing of virus-specific T cells after adoptive transfer of splenocytes from P14.Thy1.1 mice, expressing a transgenic TCR specific for gp33 (27), into Thy1.2 recipients. Furthermore, although there is a solid overall CTL response (5), the individual endogenous PVM-specific CTL epitopes identified so far do not induce prominent responses (28). Recombinant PVM expressing the gp33 epitope (rPVM-GFPgp33) was generated by introducing a PVM transcription cassette consisting of the coding sequence of LCMVgp29-43 fused to the C-terminal end of eGFP that was flanked by PVM-specific transcriptional start and stop sequences into the full-length clone pPVM between the SH and the G gene (Fig. 1A). A previously described recombinant PVM expressing GFP (rPVM-GFP) represented the parental wild-type virus (11). Infectious virus was obtained by transfection of BSR-T7/5 cells with the full-length plasmid containing the gene cassette (pPVM-GFPgp33) together with support plasmids expressing the viral proteins essential for genome replication and transcription as described previously (11).
Fig 1.

(A) Generation and characterization of recombinant PVM expressing the LCMV-derived CTL epitope gp33 fused to GFP. Shown are the GFPgp33 gene cassette and its placement into the PVM genome. The sequence encoding LCMV gp29-43 was fused to the C terminus of GFP. The cassette represents a PVM pseudogene encoding a single open reading frame for expression of a GFP-gp33 fusion protein flanked by PVM-specific transcriptional start (gene start) and stop (gene stop) sequences. (B) Multistep growth curve of rPVM-GFPgp33 in comparison to the recombinant parental virus rPVM-GFP and its parental wild-type PVM15. BHK-21 cells were infected with the respective viruses (multiplicity of infection of 0.01 PFU/cell) at day 0. Cells and supernatants were harvested in duplicates at the indicated time points, and virus titers were determined by plaque assay. The growth kinetics were determined twice, with similar results. (C) C57BL/6 mice were intranasally infected with 100 to 200 PFU of the indicated PVM isolate, and virus titers were determined in the lung at the indicated time points after infection. n.s., not significant; **, P < 0.05. (D) Mice were weighed prior to infection and daily thereafter. The percentage (mean ± SD) of the initial weight is indicated for 3 to 20 mice per time point. ***, significant differences between PVM15- and rPVM-GFPgp33-infected groups and between PVM15- and rPVM-GFP-infected groups.
rPVM-GFPgp33 replicated as efficiently as the parental virus rPVM-GFP and the biological wild-type virus PVM15 in BHK-21 cells in vitro (Fig. 1B). After intranasal infection of C57BL/6 mice, the replication of the gp33-expressing virus was similar to that of the parental rPVM-GFP virus, and 7 days after infection with 100 to 200 PFU, peak titers above 104 PFU per lung could be retrieved (Fig. 1C). However, both GFP-expressing viruses were attenuated compared to PVM15 and showed slower and delayed replication kinetics due to the additional gene as previously described (11). Consistent with delayed replication, weight loss as a rough measure of PVM-induced pathology started 1 to 2 days later but showed a similar pattern as in PVM15-infected mice (Fig. 1D). Moreover, rPVM-GFPgp33 induced significant levels of inflammatory cytokines in the lung on days 10, 12, and 15 after infection and retained its ability to induce lethal pneumonia after infection with a high dose, 5,000 PFU (data not shown). Thus, the modified virus was attenuated but retained most of its biological properties in vivo, including its ability to cause lethal immunopathology.
rPVM-GFPgp33 is immunogenic in vitro and in vivo.
To determine expression and presentation of gp33 in virus-infected cells, rPVM-GFPgp33-infected RAW309Cr1 macrophages were compared to gp33 peptide-loaded (positive control) or rPVM-infected (negative control) macrophages with respect to their ability to induce proliferation of CFSE-labeled T cells from P14 mice expressing a gp33-specific Vα2Vβ8 TCR. T cell proliferation was more prominent after stimulation by rPVM-GFPgp33-infected cells than by peptide-loaded cells, which may be explained by the limited half-life of the peptide. Cells infected with the control virus could not elicit proliferation (Fig. 2A). To analyze the ability of rPVM-GFPgp33 to induce gp33-specific CTL in vivo, pulmonary inflammatory cells were eluted by bronchoalveolar lavage (BAL) on days 10 and 12 after infection and CD8+ T cells were stained with MHC-I gp33 dimer or restimulated with gp33 peptide before intracellular staining for IFN-γ. On day 12 after infection, 2 to 6% of BAL fluid CTL was specific for gp33 (Fig. 2B and C). Moreover, LCMV-immune but not naive C57BL/6 mice eliminated an infection with 5,000 PFU of the gp33-expressing recombinant PVM within 7 days (Fig. 2D) without signs of disease, while the parental control virus replicated to similar titers (compare Fig. 2D and Fig. 1C) and caused severe disease in both groups of mice. Overall, these experiments provided clear evidence for functional expression and presentation of the gp33 peptide by cells infected with rPVM-GFPgp33 in vivo.
Fig 2.
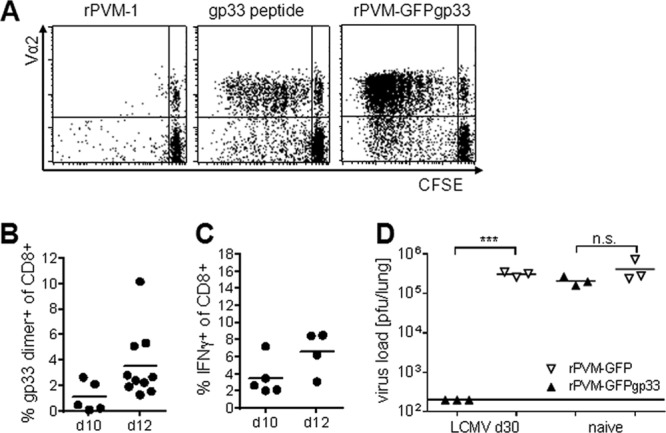
rPVM-GFPgp33-infected cells express gp33 in vitro and in vivo. (A) CFSE proliferation assay. RAW309Cr1 macrophages were infected with rPVM-1 (left), loaded with gp33 peptide (middle), or infected with rPVM-GFPgp33 (right) and incubated for 6 days with CFSE-labeled P14 spleen cells. CFSE dilution of Vα2-positive CD3+ CD8+ T cells is shown. Dot plots are representative of two independent experiments. (B and C) C57BL/6 mice were infected with 100 to 200 PFU of rPVM-GFPgp33 and at the indicated time points, BAL fluid cells were analyzed. (B) Percentage of gp33 dimer-positive cells among total CD3+ CD8+ T cells. Data were pooled from two independent experiments. (C) Percentage of cells producing IFN-γ after gp33 peptide stimulation among total CD3+ CD8+ lymphocytes. Data were pooled from two independent experiments. (D) LCMV-immune or naive C57BL/6 mice were intranasally infected with 5,000 PFU of rPVM-GFPgp33 or rPVM-GFP. Seven days after infection, lung viral titers were determined by plaque assay. n.s., not significant; ***, P < 0.001.
Adoptive-transfer system to colocalize virus-infected cells and virus-specific CTL in the lung.
In order to be able to detect virus-specific T cells in the lung, we adoptively transfused Thy1.1+ spleen cells from P14 donor mice into C57BL/6 (Thy1.2+) recipient mice, referred to as P14 chimeric mice in this paper (Fig. 3A). Following infection of these mice with rPVM-GFP33, gp33-specific P14 donor T cells expanded, were recruited to the lungs, and were identified in the lungs of recipient mice by their Thy1.1 expression (Fig. 3E). We first performed titration experiments and established that transfer of spleen cells containing 1 × 106 naive P14 T cells followed by infection with 200 PFU of rPVM-GFPgp33 3 days later represented the optimal conditions to allow significant expansion of gp33-specific Thy1.1+ donor CTL without changing the basic parameters of the viral infection. Under these conditions, the replication kinetics of rPVM-GFPgp33 in P14 chimeric and C57BL/6 mice were similar and resulted in comparable weight loss kinetics in the two groups (Fig. 3B and C). The percentage of gp33-specific CTL in the BAL fluid of infected P14 chimeric mice was 5-fold higher than in infected nontransfused mice (compare Fig. 3D with Fig. 2B). IFN-γ release assays revealed that about 50% of these virus-specific CD8+ T cells in the BAL fluid and 35% in the lung parenchyma were Thy1.1+ P14 donor CTL, while the residual T cells originated from the nontransgenic recipient mice (Fig. 3E). Thus, using the Thy1.1 marker to stain virus-specific CTL in P14 chimeric mice, we detected about 2-fold more cells in the lung parenchyma than in C57BL/6 mice, and these cells represented about 35% of the total gp33-specific response in these mice. Overall, about 85% of the pulmonary CTL were host derived, including T cells specific for gp33, endogenous PVM epitopes, and bystander cells recruited by the pulmonary inflammation. Importantly, the kinetics of virus replication, CTL recruitment, and disease were largely similar in P14 chimeric and C57BL/6 mice.
Fig 3.

An adoptive-transfer model to detect virus-specific T cells in the lung. (A) Illustration of the adoptive-transfer model. P14.Thy1.1 spleen cells containing 1 × 106 naive Vα2+ CD8+ CD3+ T cells were adoptively transfused into Thy1.2+ C57BL/6 recipients (P14 chimeric mice) which were infected 3 days later (d0) with 100 to 200 PFU of rPVM-GFPgp33. (B) Weight curves after infection of C57BL/6 and P14 chimeric mice with rPVM-GFPgp33. Data are means and standard deviations of 4 to 14 mice per time point pooled from 3 independent experiments. (C) Pulmonary virus titers at the indicated time points after infection. Data are from C57BL/6 mice and P14 chimeric mice gained in two different experiments. (D) Percentage of gp33 dimer-positive cells among total CD3+ CD8+ BAL fluid cells of P14 chimeric mice that had been infected with 100 to 200 PFU of rPVM-GFPgp33 8 to 12 days previously. (E) Percentage of Thy 1.1-positive cells among total BAL fluid or lung parenchyma CD3+ CD8+ T cells responding with IFN-γ production to stimulation with gp33 peptide. The means and SEMs from pooled data of three independent experiments are shown. n.s., not significant; ***, P < 0.0001.
In situ evolution of an antiviral CD8+ T cell response in the lung.
We then proceeded to immunohistological analysis. Since the GFP autofluorescence was too low for reliable identification after fixation of the tissue, the fluorescence of virus-infected cells was enhanced by staining with a PVM G-specific antiserum and an Alexa 488-conjugated secondary antibody. Lung sections from P14 chimeric mice 10 days after P14 spleen cell transfer in the absence of virus infection completely lacked Thy1.1-positive cells, demonstrating that any cells observed in the following experiments were recruited to the lung in the context of the viral infection. To analyze the spatiotemporal pattern of virus distribution and the virus-specific T cell response, we used the Olympus ScanR high-content screening station, which allowed visualization and automated quantification of infected cells (green fluorescence) and virus-specific T cells (red fluorescence) in sections of 12 mm2, covering about 20% of the area of a lung lobe of an infected mouse. Figure 4A shows the mean absolute counts of virus-infected cells per section, with a peak at day 7 and virus elimination by day 11, and the counts of PVM-specific T cells, which are low at day 7, followed by an increase up to day 10 and a decrease after day 12. These counts mirrored the results obtained by plaque assay and flow cytometry (compare Fig. 3C and D), validating this methodology.
Fig 4.

ScanR microscopy used for a detailed analysis of virus infection and the resulting T cell response. P14 chimeric mice were infected with 100 to 200 PFU of rPVM-GFPgp33 (d0). At the indicated time points, lung sections were stained for PVM-infected cells (green signal) and Thy1.1-positive CTL (red signal) and images were taken at a 20-fold magnification. Gallery views were generated from 84 individual images covering a lung area of 12 mm2 as described in Materials and Methods. Images from 2 to 5 mice per time point obtained in 2 independent experiments produced similar results. (A) Total number of virus-infected cells (green bars) and virus-specific CTL (red bars) as counted by the ScanR analysis software corrected for the number of fields actually containing lung tissue. Bars represent the mean value of 3 to 5 mice per time point, and whiskers represent the standard deviations. (B) Representative example of a gallery view taken at day 7 after infection as an original picture. (C) Gallery view from panel B transformed to a schematic view, where red and green dots were enlarged to a similar size. (D to I) The images show an overlay of the of red and green fluorescence signals obtained by the ScanR analysis after transformation to a schematic view. Large bronchi were stained manually in pale gray. The overlay pictures shown are representative of similar pictures obtained from 2 to 5 mice per time point.
An example of the view of a lung section obtained 7 days after rPVM-GFPgp33 infection of P14 chimeric mice is shown in Fig. 4B. Since red and green signals showed different sizes and intensities, we extracted this information from the histological slide by using the ScanR analysis software, adapted the signals to equal size and intensity, and thus obtained a schematic view of the virus distribution and the virus-specific T cell response (Fig. 4D to I). Seven days after infection, PVM had spread around several bronchi, forming dense clusters of infected cells (Fig. 4D). Closer magnification revealed the most intensive infection of bronchial epithelial cells in areas where the infection also extended to lung parenchymal cells. At this time point, initial infiltration of virus-specific CTL was also observed, with a preferential accumulation around vessels in peribronchial areas within clusters of virus-infected cells. However, in contrast to the viral infection, the virus-specific CTL did not yet spread into the lung parenchyma (Fig. 5A, d7, and Fig. 4D). Notably, mice did not show any signs of disease at this time point of maximal overall pulmonary virus load. By day 8, the clusters of virus-infected cells appeared less dense, potentially due to the antiviral activity of the recruited CTL (Fig. 4E and 5A, d8). Particularly, some specific T cells showed direct contact with infected cells (Fig. 5B) within infected areas. Nevertheless, the infection had spread further throughout the lung parenchyma and appeared to be surrounded by virus-specific CTL that had now significantly increased in number. This pattern became more pronounced by day 9 (Fig. 4F and 5A, d9), when the number of virus-infected cells was significantly reduced and clusters of parenchymal CTL were mainly found in areas where no virus-infected cells were detectable. Between day 7 and day 9, the mice developed significant weight loss (Fig. 3B). On day 10, PVM-specific CTL reached peak levels and were spread throughout the parenchyma (Fig. 4G and Fig. 5A, d10), while the overall virus infection was already largely controlled. However, there were still some areas with infected cells that showed no infiltrating antiviral CTL. By day 11, the CTL had evenly spread to all areas of the lung and only single PVM-infected cells remained (Fig. 4H). This pattern could be seen until complete resolution of the infection by day 12 (Fig. 5A, d12). Residual T cells remained detectable up to day 15, the latest time point analyzed (Fig. 4I and 5A, d15). Interestingly, weight loss was maximal on day 10 (Fig. 3B), when virus titers had already significantly declined, but CTL infiltrates had reached their maximum. The mice then gained weight again between days 10 and 12, a time period in which the overall CTL infiltrate remained constant. These observations, summarized in Table 1, suggest that disease was associated with neither virus load nor the presence of a large number of virus-specific CTL per se but that the continuous stimulation of infiltrating CTL by virus-infected cells was key for the disease manifestations.
Fig 5.

Coevolution of virus distribution and the antiviral T cell response in the PVM-infected mouse lung. P14 chimeric mice were infected with 100 to 200 PFU of rPVM-GFPgp33, and at the indicated time points, lungs were prepared for histological analysis. Lung sections (12 μm) were stained for PVM (Alexa 488 signal) and Thy-1.1 (PE signal) and images were taken with an Apo Tome fluorescence microscope (10-fold magnification) (A) or ScanR microscope (20-fold magnification) (B). Representative images for different time points after infection are shown. Images 1 and 2 are enlargements of sections illustrating colocalization of intraalveolar virus-specific CTL and virus-infected pulmonary epithelial cells.
Table 1.
Correlation of pulmonary virus and antiviral T cell localization with weight loss over timea
| Day | Overall viral load | Virus localization | Overall PVM-specific CTL | Localization of specific CTL | Weight loss |
|---|---|---|---|---|---|
| 7 | ↑↑↑ | Infectious foci around bronchi | (↑) | Around blood vessels in areas of infected bronchi | — |
| 8 | ↑↑ | Scattered throughout the lung parenchyma | ↑ | Interstitium, alveoli in infected areas | ↑ |
| 9 | ↑ | Some areas free of virus | ↑↑ | Interstitium, alveoli in infected and noninfected areas | ↑↑ |
| 10 | (↑) | Increasing areas with T cell clusters free of virus | ↑↑↑ | Interstitium, alveoli in infected and noninfected areas | ↑↑↑ |
| 11 | (↑) | Scattered single infected cells remaining throughout the lung parenchyma | ↑↑ | Scattered throughout the lung parenchyma | ↑↑ |
Day, day after virus infection; —, no difference compared to uninfected mice; (↑), minimal increase; ↑, mild increase; ↑↑, moderate increase; ↑↑↑, strong increase.
DISCUSSION
In this report, we provide analysis of the spatiotemporal evolution of a pulmonary antiviral CTL effector response. Viral pneumonia is obviously not a homogenous process but a highly dynamic disease with a heterogenous evolution in different sections of the lung. Therefore, determination of the overall virus load in the infected lung or the extraction of all T cells from the inflamed tissue can only sum up the net effect of infection and immunity at a given time point. A look at the spatial distribution of infection and T cell response in parallel adds a third dimension to the analysis, allowing the study of different processes occurring at the same time point. We therefore set up an experimental system where both virus-specific CTL and virus-infected cells could be visualized in the same section of the infected lung.
We chose the PVM model because it would allow us to study the course of productive virus replication in its natural host and the evolution of an antiviral T cell response. To enable identification of virus-specific CTL, the well-characterized H-2Db-restricted gp33 epitope of LCMV was introduced into the viral genome as an additional gene cassette expressing an eGFP-gp33 fusion protein. This approach was chosen since the pathogenesis of the parental rPVM-GFP is well characterized and comparable to that of the biological wild-type virus despite minor attenuation of replication due to the additional gene (11). In addition, fusion of gp33 to GFP rather than to a viral protein was chosen to avoid the possible interference with viral proteins necessary for the PVM replication cycle. The use of P14 Thy1.1 transgenic mice enabled the specific detection of gp33-specific CTL and, at the same time, enhanced the gp33-specific pulmonary T cell response about 5-fold. On the other hand, CTL specific for autologous PVM-derived CTL epitopes could not be visualized in that system. Overall, the total number of virus-specific CTL identified in the analyzed sections is probably higher than in a normal C57BL/6 mouse. Moreover, the overall pattern of the CD8 T cell response to the heterologous LCMV peptide may not fully reflect the response to natural PVM epitopes. Nevertheless, the main parameters of disease, including weight loss and pulmonary inflammatory cytokines, were largely maintained in P14 chimeric mice. Therefore, we believe that the observations on the distribution of virus-infected cells and virus-specific CTL made after rPVM-GFPgp33 infection of P14 chimeric mice to a large extent reflected the situation in PVM-infected wild-type mice.
After rPVM-GFPgp33 infection, weight loss reflecting clinical disease started at day 8 after infection, reached a maximum at day 10, and diminished thereafter (Table 1). Pulmonary cytokine homeostasis was not restored before day 15 after infection. It is interesting to note that the extensive viral infection of pulmonary epithelial cells reaching maximal virus titers at day 7 was not yet associated with measurable disease manifestations. At that time point, there were dense focal clusters of infected cells around the bronchi and some initial CTL infiltrates in these areas. However, many areas of the lung were still free of virus. By day 8 after infection, the dense peribronchial clusters of virus-infected cells had largely thinned out, and this was associated with more CTL infiltrates reaching further into the parenchyma. However, this did not prevent virus spread throughout the lung tissue—at day 8, the beginning of disease manifestations, virus-infected cells still clearly outnumbered the virus-specific CTL in the parenchyma. At day 9, antiviral T cells had spread to most infected areas in the lung, leading to the accumulation of T cell clusters in some areas which were mostly free of virus. Day 10 represented the maximum of the clinical disease, and this coincided with the maximum of the antiviral T cell response. At this time point, virus-infected cells were significantly reduced and large numbers of CTL accumulated in areas with thickened alveolar walls, indicating histological pathology. Clinical recovery occurred more rapidly than the eventual resolution of this antiviral T cell infiltrate, which persisted until day 12 (Table 1). Some T cells remained scattered throughout the lung parenchyma even at day 15, when cytokines had returned to normal, but they were found outside the immediate peribronchial areas, where they had entered on day 7.
While many studies on T cell responses to pulmonary virus infections focus on cells obtained by bronchoalveolar lavage, our analysis allowed us to capture tissue resident cells and their distribution in the infected tissue, adding an additional dimension to the view of a viral infection. The spatiotemporal evolution of the specific CTL response relative to the viral infection and the disease manifestations illustrates several interesting points. First, disease manifestations correlated with the widespread distribution and accumulation of antiviral CTL in the lung parenchyma and not with the extent of the virus infection (5). Disease was maximal when only a few virus-infected cells remained, but the virus-specific T cell infiltrate was substantial. On the other hand, clinical recovery started when virus was fully eliminated and T cell infiltrates were still high. This may suggest that the CTL-mediated immunopathology is dependent on contact with virus-infected cells, while in the absence of antigenic stimulation, the pulmonary CTL are rapidly silenced (29). Second, spread of infection on the one hand and T cell infiltration into infected areas associated with reduction of infection on the other hand occurred at the same time in different areas of the lung. Between days 7 and 8, the newly recruited antiviral CTL were highly active in reducing the peribronchial clusters of virus-infected cells but could not prevent further spread of the infection to other areas of the lung parenchyma. In addition to tissue resident CTL, individual alveolar CTL could be detected in close contact with the luminal side of infected alveolar cells. However, the single plane of analysis and the probable loss of CTL during lung preparation for histology make a quantitative assessment of the contribution of alveolar CTL to virus clearance unreliable. Overall, the acute viral pneumonia was characterized by the parallel occurrence of virus control in some areas and additional viral spread in other areas. Third, the number of pulmonary virus-specific CTL (although rather overestimated in this model) never reached the number of virus-infected cells in the mouse lung and the bulk of reduction in the number of virus-infected cells occurred at a time point when T cells made up no more than 5% of the virus-infected cells. Although recirculation of T cells has to be considered, this makes it likely that one virus-specific effector CTL acts on several virus-infected cells in the lung. On the other hand, reduction of virus-infected cells mainly occurred in areas infiltrated by virus-specific CTL, while de novo infection of noninfiltrated areas occurred in parallel, suggesting that CTL can act only in a limited area. Unfortunately, the effector mechanisms used by CTL to control PVM infection still remain elusive. Neither perforin, IFN-γ, TNF (5), nor granzymes (30), individually, are required for virus elimination. This does not exclude the possibility that a combination of these effector mechanisms is active. For example, target cell lysis by a combination of Fas- and perforin-mediated mechanisms may represent the main CTL effector mechanism in murine influenza virus infection (31). In the PVM model, however, it is difficult to envisage how soluble, widespread, cytolytic effector mechanisms could be compatible with sufficient lung function. Contact-dependent, short-range cytokine-mediated virus clearance appears to be most compatible with our morphological observations. Fourth, recruitment and spread of virus-specific CTL throughout the parenchyma were still ongoing when the virus infection was already largely controlled. This eventually resulted in an excessive response, i.e., 5- to 10-fold more virus-specific T cells than virus-infected cells at day 10 after infection. It can be speculated that this excess of T cells is required to get completely rid of widely scattered infected cells as fast as possible, since each can serve as a “factory” for virus production. It is apparently not possible to direct and limit the antiviral T cells only to those areas of the lung where productive infection is still ongoing, compatible with a random search mechanism with local recognition.
In summary, this study provides a new perspective on the effector phase of an antiviral CTL response in the mouse lung. While the priming phase of antiviral T cells by dendritic cells in lymph nodes has been analyzed in some detail by two-photon microscopy (32, 33), to our knowledge this is the first attempt to characterize the “geography” of the effector phase of an antiviral CTL response in the infected lung. Elaboration of this experimental model will help to further elucidate the protective and pathogenic roles of antiviral T cells in pulmonary virus infections—a key prerequisite for developing a targeted therapeutic approach to viral pneumonia.
ACKNOWLEDGMENTS
This work was supported by the Bundesministerium für Bildung und Forschung (BMBF 01 EO 0803) and the Deutsche Forschungsgemeinschaft (DFG EH145/4-2 and KR2964/1-2). P.C. was supported by the Intramural Research Program of NIAID, NIH.
We acknowledge the excellent technical assistance of Nadja Goos.
S.F., M.F., and C.K. performed research and analyzed data, H.P. and P.C. contributed reagents and analytical tools, and S.E. designed research, analyzed data, and drafted the manuscript.
The authors declare no conflict of interest.
Footnotes
Published ahead of print 14 August 2013
REFERENCES
- 1.El Saleeby CM, Suzich J, Conley ME, DeVincenzo JP. 2004. Quantitative effects of palivizumab and donor-derived T cells on chronic respiratory syncytial virus infection, lung disease, and fusion glycoprotein amino acid sequences in a patient before and after bone marrow transplantation. Clin. Infect. Dis. 39:e17–e20. 10.1086/421779. [DOI] [PubMed] [Google Scholar]
- 2.Fishaut M, Tubergen D, McIntosh K. 1980. Cellular response to respiratory viruses with particular reference to children with disorders of cell-mediated immunity. J. Pediatr. 96:179–186. [DOI] [PubMed] [Google Scholar]
- 3.Hall CB, Powell KR, MacDonald NE, Gala CL, Menegus ME, Suffin SC, Cohen HJ. 1986. Respiratory syncytial viral infection in children with compromised immune function. N. Engl. J. Med. 315:77–81. [DOI] [PubMed] [Google Scholar]
- 4.Doherty PC, Topham DJ, Tripp RA, Cardin RD, Brooks JW, Stevenson PG. 1997. Effector CD4+ and CD8+ T-cell mechanisms in the control of respiratory virus infections. Immunol. Rev. 159:105–117. [DOI] [PubMed] [Google Scholar]
- 5.Frey S, Krempl CD, Schmitt-Graff A, Ehl S. 2008. Role of T cells in virus control and disease after infection with pneumonia virus of mice. J. Virol. 82:11619–11627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Graham BS, Bunton LA, Wright PF, Karzon DT. 1991. Role of T lymphocyte subsets in the pathogenesis of primary infection and rechallenge with respiratory syncytial virus in mice. J. Clin. Invest. 88:1026–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kolli D, Bataki EL, Spetch L, Guerrero-Plata A, Jewell AM, Piedra PA, Milligan GN, Garofalo RP, Casola A. 2008. T lymphocytes contribute to antiviral immunity and pathogenesis in experimental human metapneumovirus infection. J. Virol. 82:8560–8569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Openshaw PJ. 1995. Immunity and immunopathology to respiratory syncytial virus. The mouse model. Am. J. Respir. Crit. Care Med. 152:S59–S62. [DOI] [PubMed] [Google Scholar]
- 9.Xu L, Yoon H, Zhao MQ, Liu J, Ramana CV, Enelow RI. 2004. Cutting edge: pulmonary immunopathology mediated by antigen-specific expression of TNF-alpha by antiviral CD8+ T cells. J. Immunol. 173:721–725. [DOI] [PubMed] [Google Scholar]
- 10.Buchholz UJ, Ward JM, Lamirande EW, Heinze B, Krempl CD, Collins PL. 2009. Deletion of nonstructural proteins NS1 and NS2 from pneumonia virus of mice attenuates viral replication and reduces pulmonary cytokine expression and disease. J. Virol. 83:1969–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krempl CD, Wnekowicz A, Lamirande EW, Nayebagha G, Collins PL, Buchholz UJ. 2007. Identification of a novel virulence factor in recombinant pneumonia virus of mice. J. Virol. 81:9490–9501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krempl CD, Collins PL. 2004. Reevaluation of the virulence of prototypic strain 15 of pneumonia virus of mice. J. Virol. 78:13362–13365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Easton AJ, Domachowske JB, Rosenberg HF. 2004. Animal pneumoviruses: molecular genetics and pathogenesis. Clin. Microbiol. Rev. 17:390–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rosenberg HF, Domachowske JB. 2008. Pneumonia virus of mice: severe respiratory infection in a natural host. Immunol. Lett. 118:6–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Collins PL, Crowe JE. 2007. Respiratory syncytial virus and metapneumovirus, p 1601–1646. In Knipe PM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 16.Domachowske JB, Bonville CA, Easton AJ, Rosenberg HF. 2002. Differential expression of proinflammatory cytokine genes in vivo in response to pathogenic and nonpathogenic pneumovirus infections. J. Infect. Dis. 186:8–14. [DOI] [PubMed] [Google Scholar]
- 17.Domachowske JB, Bonville CA, Easton AJ, Rosenberg HF. 2002. Pulmonary eosinophilia in mice devoid of interleukin-5. J. Leukoc. Biol. 71:966–972. [PubMed] [Google Scholar]
- 18.Graham BS, Perkins MD, Wright PF, Karzon DT. 1988. Primary respiratory syncytial virus infection in mice. J. Med. Virol. 26:153–162. [DOI] [PubMed] [Google Scholar]
- 19.Barchet W, Oehen S, Klenerman P, Wodarz D, Bocharov G, Lloyd AL, Nowak MA, Hengartner H, Zinkernagel RM, Ehl S. 2000. Direct quantitation of rapid elimination of viral antigen-positive lymphocytes by antiviral CD8(+) T cells in vivo. Eur. J. Immunol. 30:1356–1363. [DOI] [PubMed] [Google Scholar]
- 20.Ehl S, Klenerman P, Zinkernagel RM, Bocharov G. 1998. The impact of variation in the number of CD8(+) T-cell precursors on the outcome of virus infection. Cell. Immunol. 189:67–73. [DOI] [PubMed] [Google Scholar]
- 21.Belz GT, Wodarz D, Diaz G, Nowak MA, Doherty PC. 2002. Compromised influenza virus-specific CD8(+)-T-cell memory in CD4(+)-T-cell-deficient mice. J. Virol. 76:12388–12393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huck B, Neumann-Haefelin D, Schmitt-Graeff A, Weckmann M, Mattes J, Ehl S, Falcone V. 2007. Human metapneumovirus induces more severe disease and stronger innate immune response in BALB/c mice as compared with respiratory syncytial virus. Respir. Res. 8:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mo XY, Sangster M, Sarawar S, Coleclough C, Doherty PC. 1995. Differential antigen burden modulates the gamma interferon but not the immunoglobulin response in mice that vary in susceptibility to Sendai virus pneumonia. J. Virol. 69:5592–5598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vallbracht S, Jessen B, Mrusek S, Enders A, Collins PL, Ehl S, Krempl CD. 2007. Influence of a single viral epitope on T cell response and disease after infection of mice with respiratory syncytial virus. J. Immunol. 179:8264–8273. [DOI] [PubMed] [Google Scholar]
- 25.Krempl CD, Lamirande EW, Collins PL. 2005. Complete sequence of the RNA genome of pneumonia virus of mice (PVM). Virus Genes 30:237–249. [DOI] [PubMed] [Google Scholar]
- 26.Ostler T, Hussell T, Surh CD, Openshaw P, Ehl S. 2001. Long-term persistence and reactivation of T cell memory in the lung of mice infected with respiratory syncytial virus. Eur. J. Immunol. 31:2574–2582. [DOI] [PubMed] [Google Scholar]
- 27.Brändle D, Brduscha-Riem K, Hayday AC, Owen MJ, Hengartner H, Pircher H. 1995. T cell development and repertoire of mice expressing a single T cell receptor alpha chain. Eur. J. Immunol. 25:2650–2655. [DOI] [PubMed] [Google Scholar]
- 28.Claassen EA, van der Kant PA, Rychnavska ZS, van Bleek GM, Easton AJ, van der Most RG. 2005. Activation and inactivation of antiviral CD8 T cell responses during murine pneumovirus infection. J. Immunol. 175:6597–6604. [DOI] [PubMed] [Google Scholar]
- 29.Vallbracht S, Unsold H, Ehl S. 2006. Functional impairment of cytotoxic T cells in the lung airways following respiratory virus infections. Eur. J. Immunol. 36:1434–1442. [DOI] [PubMed] [Google Scholar]
- 30.Bem RA, van Woensel JB, Lutter R, Domachowske JB, Medema JP, Rosenberg HF, Bos AP. 2010. Granzyme A- and B-cluster deficiency delays acute lung injury in pneumovirus-infected mice. J. Immunol. 184:931–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Topham DJ, Tripp RA, Doherty PC. 1997. CD8+ T cells clear influenza virus by perforin or Fas-dependent processes. J. Immunol. 159:5197–5200. [PubMed] [Google Scholar]
- 32.Khanna KM, Aguila CC, Redman JM, Suarez-Ramirez JE, Lefrancois L, Cauley LS. 2008. In situ imaging reveals different responses by naive and memory CD8 T cells to late antigen presentation by lymph node DC after influenza virus infection. Eur. J. Immunol. 38:3304–3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Norbury CC, Malide D, Gibbs JS, Bennink JR, Yewdell JW. 2002. Visualizing priming of virus-specific CD8+ T cells by infected dendritic cells in vivo. Nat. Immunol. 3:265–271. [DOI] [PubMed] [Google Scholar]