

Abstract
Preferential RNA packaging in coronaviruses involves the recognition of viral genomic RNA, a crucial process for viral particle morphogenesis mediated by RNA-specific sequences, known as packaging signals. An essential packaging signal component of transmissible gastroenteritis coronavirus (TGEV) has been further delimited to the first 598 nucleotides (nt) from the 5′ end of its RNA genome, by using recombinant viruses transcribing subgenomic mRNA that included potential packaging signals. The integrity of the entire sequence domain was necessary because deletion of any of the five structural motifs defined within this region abrogated specific packaging of this viral RNA. One of these RNA motifs was the stem-loop SL5, a highly conserved motif in coronaviruses located at nucleotide positions 106 to 136. Partial deletion or point mutations within this motif also abrogated packaging. Using TGEV-derived defective minigenomes replicated in trans by a helper virus, we have shown that TGEV RNA packaging is a replication-independent process. Furthermore, the last 494 nt of the genomic 3′ end were not essential for packaging, although this region increased packaging efficiency. TGEV RNA sequences identified as necessary for viral genome packaging were not sufficient to direct packaging of a heterologous sequence derived from the green fluorescent protein gene. These results indicated that TGEV genome packaging is a complex process involving many factors in addition to the identified RNA packaging signal. The identification of well-defined RNA motifs within the TGEV RNA genome that are essential for packaging will be useful for designing packaging-deficient biosafe coronavirus-derived vectors and providing new targets for antiviral therapies.
INTRODUCTION
Transmissible gastroenteritis coronavirus (TGEV) is a member of the Coronaviridae family of viruses with positive-sense RNA genomes of around 30 kb and a common genome organization (1, 2). TGEV is an enveloped virus whose envelope membrane includes the spike (S), the envelope (E), and the membrane (M) proteins. Inside the envelope, the internal core, composed of the nucleoprotein (N) and the 28.5-kb RNA genome forming the nucleocapsid, interacts with the carboxy terminus of the M proteins (3). During infection, the viral genome is replicated by continuous RNA synthesis. Genes located at the 3′ end of the genome are transcribed by discontinuous RNA synthesis, which leads to a collection of 3′-coterminal subgenomic mRNAs (sgmRNAs), each containing the leader sequence (L), which is located only once at the 5′ end of the genome. Therefore, the leader sequence must be added by a discontinuous transcription process that requires a recombination between the nascent negative RNA and a copy of the leader sequence. This high-frequency recombination step is assisted by the homology between the transcription-regulating sequences (TRS) located at the 3′ end of the leader and sequences preceding each gene, both including a conserved core sequence (CS) and variable flanking sequences (1, 4, 5). Additionally, transcription of viral genes is promoted by long-distance RNA-RNA interactions forming high-order structures that bring into physical proximity distant genome sequences involved in the recombination process (6, 7).
Genome packaging in RNA viruses is a specific process, since the genomic RNA (gRNA) is preferentially incorporated into the viral particle, in contrast to viral sgmRNAs or cellular RNAs, which are packaged with limited efficiency. Packaging specificity of gRNA may depend on different components. RNA packaging involves the recognition of cis-acting factors, mainly specific sequences exclusively present on gRNA, referred to as the packaging signal (PS). Frequently, the cis-acting PS forms stable RNA secondary structures required for optimal packaging. Virus genome PSs are highly variable in the extent of the sequence and localization. PSs may be located at a unique site in the viral genome or distributed at different positions along the gRNA. In some cases, PSs consist of a unique RNA motif with a short defined sequence, while in others, PSs include a large region of the viral genome. In the mouse hepatitis coronavirus (MHV), a PS consisting of a unique sequence of 69 nucleotides (nt) located within ORF1b, 21 kb from the 5′ end of the genome, has been described. This sequence is present in the gRNA, but not in sgmRNAs, and adopts a stem-loop (SL) secondary structure, which serves as the primary cis signal for MHV packaging (8). In other related positive-strand RNA viruses, such as the equine arterivirus (EAV), the PS consists of three genomic sequences located at the 5′ end of the genome (nt 1 to 589), the 3′ end (the last 1,068 nt), and internally in ORF1b (nt 8566 to 9149) (9). For TGEV, our previous studies with defective minigenomes have localized the packaging signals to the first 649 nt at the 5′ end and the last 494 nt at the 3′ end of the genome (10). These studies were performed with defective genomes rescued by a helper virus along several passages in cell culture. Since RNA rescue implies the amplification and packaging of the defective minigenomes, these studies could not determine the relevance of the sequences at the 3′ end of the genome or dissociate sequences necessary for packaging from those necessary for replication (11). In addition to the PS, trans-acting factors could also contribute to the specificity of the gRNA packaging. Viral factors as well as cellular proteins can bind to the PS and promote viral gRNA packaging (12). In TGEV, the N protein binds to the gRNA to form a ribonucleoprotein complex that gets included in virions. It has been proposed that the M protein specifically interacts with the PS, mediating the selective packaging of RNAs containing the PS even in the absence of the N protein (13). It has been shown that the extent and specificity of the RNA packaging in Brome mosaic virus (BMV) is in part regulated by the electrostatic interaction between the RNA and the coat protein (14).
In general, viral replication and packaging processes are coupled to favor the packaging of replication-competent RNA molecules in order to select for functional viral genomes. Coupling between replication and packaging processes has been demonstrated for polioviruses (15), flaviviruses (16), and alphaviruses (17). However, there are viruses that do not require the replication of their RNA genomes for packaging. In BMV, packaging in independent virions of the three RNAs comprising the tripartite genome is not replication dependent (18). Analysis of the requirement of replication for packaging was performed for the first time with poliovirus using replication inhibitors (19). An alternative approach to dissect replication and packaging focuses on the packaging of replicative and nonreplicative engineered viral RNAs by a helper virus.
In this paper, a dominant component of TGEV PS has been delimited to the first 598 nt from the 5′ end of its RNA genome, which includes the leader sequence (nt 1 to 99) and the downstream 499 nt extending over the 5′ untranslated region (UTR) and open reading frame (ORF) 1a sequences. Partial deletions of these viral sequences significantly decreased packaging efficiency, indicating that cis-acting packaging signals in TGEV extended across a relatively large genomic region. The relevance of the 3′-end genomic sequences in packaging and replication has been analyzed by using an experimental approach which consists of transfection of engineered viral RNAs in conjunction with infection with a helper virus. Comparative analysis of RNA synthesis in noninfected and helper virus-infected cells provided information on replication competence of RNAs, while analysis of RNA inside the virions allowed quantification of packaging efficiency. It has been shown that viral 3′-end genomic sequences were not strictly essential for packaging, although they contributed by improving the efficiency of the process. Interestingly, packaging of TGEV RNAs was independent of their replication by the helper virus.
MATERIALS AND METHODS
Cells and viruses.
Baby hamster kidney (BHK) cells stably transformed with the porcine amino peptidase N (pAPN) gene (20) were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 5% fetal calf serum (FCS). G418 (1.5 mg/ml) served as the selection agent for the pAPN gene. The TGEV PUR46-MAD strain (21) and recombinant TGEV viruses were grown in swine testis (ST) cells (22) with DMEM supplemented with 10% FCS. TGEV recombinant viruses were obtained using a reverse genetics system from an infective clone with the sequence of the TGEV PUR46-MAD isolate (23) and the spike gene from the Purdue type strain (PTV) (24). Virus titration was performed on ST cell monolayers as previously described (25).
Cell transfection.
ST or BHK cells were grown to 95% confluence on 35-mm-diameter plates in the absence of antibiotics. Cells were transfected with 4 μg of each cDNA encoding TGEV-defective minigenomes or recombinant viruses by using 12 μl of Lipofectamine 2000 (Invitrogen), according to the manufacturer's specifications.
For the reverse transfection, ST cells were grown to 80% confluence on 100-mm-diameter plates in the absence of antibiotics, trypsinized, and resuspended in DMEM-10% FCS. The suspension of cells was placed on 12-well or 60-mm-diameter plates and incubated with transfection complexes formed by mixing 1.6 μg of DNA and 4 μl of Lipofectamine 2000 (Invitrogen) or 8 μg of DNA and 20 μl of Lipofectamine, respectively.
The conditions in transfection experiments were strictly controlled. (i) The same number of cells per 35-mm-diameter, 12-well, or 60-mm-diameter plates was seeded (1.5 × 106 cells/plate, 8 × 105 cells/well, or 4 × 106 cells/plate, respectively). (ii) The same amount of cDNA was always transfected. (iii) cDNA was purified using a large-construct kit (Qiagen), including an exonuclease treatment to remove bacterial DNA contamination and damaged plasmids, thus providing ultrapure DNA plasmid for transfection or a plasmid Midi Kit (Qiagen) for the reverse transfection.
Plasmid constructs containing TGEV-derived defective minigenome sequences.
To obtain the cDNA of defective minigenome M26 (the number after the M indicates the size of the defective minigenome in hundreds of nucleotides), two PCR fragments containing 5′- and 3′-end TGEV genomic sequences were generated using the primer pair 5′-AAGCTTTATGATATCTTCGG-3′ and 5′-TTCAAATGATGAACC-3′, the primer pair 5′-ATGCTGTATTTATTACAG-3′ and 5′-CACGTGCTTACCATTC-3′ (restriction sites are underlined; i.e., AAGCTT for HindIII and CACGTG for Eco72I), and the plasmid pSL M33L (11) as the template. The first PCR product was digested with HindIII and EcoRI restriction enzymes, and the second product was EcoRI and Eco72I digested. Both fragments were ligated into a pcDNA vector, leading to pcDNA M26, containing the first 2,144 nt from the 5′ end and the last 494 nt from the 3′ of the TGEV genome separated by a 60-nt pGEM-T-derived linker.
As a target for quantification by quantitative PCR (qPCR), an 87-nt synthetic linker (GeneArt; Life Technologies) derived from the Musa species ribulose-1,5-bisphosphate carboxylase oxygenase (Rubisco) small subunit was inserted into cDNAs encoding TGEV-derived sequences. The synthetic Rubisco sequence (Ru) flanked by SbfI and KpnI restriction sites was inserted between 5′- and 3′-end viral sequences of plasmids pcDNA M26 and pcDNA L-R1, leading to pcDNA M26-Ru-3′wt and pcDNA L-R1-Ru-3′wt, respectively. pcDNA M26-Ru-3′wt contained the first 2,144 nt from the 5′ end and the last 494 nt from the 3′ end of the TGEV genome. pcDNA L-R1-Ru-3′wt included the first 598 nt from the 5′ end and the last 494 nt from the 3′ end of the TGEV genome. The Ru sequence flanked by SbfI and KpnI was ligated into vectors pcDNA M26-Δ171 and pcDNA L-R1-Δ171, leading to pcDNA M26-Ru-Δ171 and pcDNA L-R1-Ru-Δ171, respectively. In both plasmids, the Ru sequence was inserted between the viral 5′-end sequences described above and the last 494 nt from the 3′ end of the TGEV genome, with a 171-nt deletion (nt 28388 to 28559) containing cis-acting signals required for replication (11). The Ru sequence flanked by SbfI and KpnI sites was ligated into vector pcDNA L-R1-Δ3′, leading to pcDNA L-R1-Ru-Δ3′, containing the first 598 nt from the 5′ end and the Ru sequence and lacking the last 494 nt from the 3′ end of the TGEV genome.
To obtain pcDNA L-R1-Ru-GFP, a 494-nt sequence from the green fluorescence protein (GFP) gene was amplified with the primer pair 5′-TTTGGTACCATGGTGAGCAAGGGCGAGGAGC-3′ and 5′-TTGGTACCTTACTTGTACAGCTCGTCCATGCCG-3′ (the KpnI restriction site is underlined) using pIRES2 GFP (catalog no. 6029-1; Clontech) as the template. The PCR fragment digested with KpnI was ligated into the same site of the pcDNA L-R1-Ru-Δ3′ plasmid, leading to pcDNA L-R1-Ru-GFP, containing the first 598 nt from the 5′ end of the TGEV genome and a 494-nt sequence from the GFP gene, separated by the Ru sequence. The plasmid pcDNA L-R1ΔSL5-Ru-GFP was generated by inserting the 494-nt sequence from the GFP gene into the SbfI and BamHI sites of pcDNA L-R1ΔSL5 (results not shown), leading to a plasmid containing the first 598 nt from the 5′ end of the TGEV genome, including the deletion in the stem-loop SL5 (nt 106 to 136) and 494 nt from the GFP gene, separated by the Ru sequence.
The negative control for replication and packaging assays, pcDNA Ru-GFP, was obtained by blunt-end ligation of the Ru sequence and 494 nt from the GFP gene into the pcDNA plasmid. All plasmid sequences were confirmed by sequencing.
Plasmid constructs containing recombinant TGEV genomes.
Recombinant TGEV viruses were generated by inserting defective minigenome M26-derived fragments or the GFP gene replacing the nonessential 3a and 3b (3a/b) genes in the plasmid pBAC-TGEV (23), containing the TGEV genome (GenBank accession no. AJ271965). Recombinant cDNAs, including M26-derived sequences, were obtained by overlapping PCRs. In the first PCR, a fragment containing the 3′ end of the S gene and the TRS from the 3a gene (nt 24153 to 24883 of the TGEV genome) was amplified using the primers 5′-TTTGACCCCCAGAACTA-3′ and 5′-ATCAAGTTCGTCAAGTACAGCATCTAC-3′ and the plasmid pBAC-TGEV as the template. The first 80 nt from the 3a gene downstream of the CS were included, preceding defective minigenome M26-derived sequences as a common target for TaqMan probe detection. The second PCR product included the specific sequences of overlapping fragments R1 to R8 encompassing the M26 defective minigenome and was generated using specific primers (Table 1) and plasmid pcDNA M26 as the template. The overlapping PCR products were digested with the restriction enzymes PpuMI and BlpI and cloned into the same sites of an intermediate plasmid, pSL-TGEV-S-3ab-M-N-7, including TGEV 3′-end genomic sequences (nt 22073 to 28700). AvrII digestion products from the resulting plasmids, containing M26-derived sequences replacing the 3a/b genes, were cloned into the same sites of the pBAC-TGEV plasmid, resulting in a collection of plasmids, pBAC-TGEV-3a-R1 to pBAC-TGEV-3a-R8. Plasmid pBAC-TGEV-3a-GFP was generated as a negative control by introducing 400 nt from the GFP gene at the site of the 3a/b genes. Plasmid pBAC-TGEV-R1, lacking the 80-nt sequence from the 3a gene, was also engineered by overlapping PCR. The first PCR product included nt 24153 to 24803 from the TGEV genome and was obtained by using as the template the plasmid pBAC-TGEV and the primers 5′-TTTGACCCCCAGAACTA-3′ and 5′-GTTTAGTTCTTAAAGACTTTATTCATCATCC-3′. The second PCR product included the sequence of fragment R1 (nt 100 to 499 of the TGEV genome) and was obtained using the plasmid pcDNA M26 as the template and the primers 5′-GTCTTTAAGAACTAAACGAAATATTTGTCTTTTATGAAATC-3′ and 5′-CGGGCCGCTAAGCCCATTGCAGTTAGCTCTAAC-3′.
Table 1.
Oligonucleotides used for the construction of recombinant cDNAs of M26-derived sequences
| PCR fragment | Forward primer (5′→3′) | Reverse primer (5′→3′) | Genome position (nt) |
|---|---|---|---|
| R1 | GACGAACTTGATGAAATATTTGTCTTTCTATG | CGGGCCGCTAAGCCCATTGCAGTTAGCTCTAAC | 100–598 |
| R2 | GACGAACTTGATTTGGTGTTCTTGGTAACGGAG | CGGGCCGCTAAGCTTGACTGGTGGTGCTACATG | 499–898 |
| R3 | GACGAACTTGATTATATGTTGATCAATAATG | GGGCCCGCTTAGCTCAGAACCACACGGGCATC | 649–1048 |
| R4 | GACGAACTTGATATCAGCAAACTCTCTTTACCATTC | CGGGCCGCTAAGCTTGGCAAAGAACTTAACTCC | 799–1198 |
| R5 | GACGAACTTGATTCTTCGGATCACCCTTTATG | GGGCCCGCTTAGCGCATGCAACTAGTGATTTGC | 949–1348 |
| R6 | GACGAACTTGATTTCTGGCAAAGTTAAGGGTG | CGGGCCGCTAAGCTGCACCACACTTTTGTACAAAC | 1099–1498 |
| R7 | GACGAACTTGATATTGTATCATTACAGGAAAATTG | CGGGCCGCTAAGCACAAGTTTGACAAGTGCATTATT | 1399–1798 |
| R8 | GACGAACTTGATATGCCCCCACGCAGATTATC | CGGGCCGCTAAGCTTCAAATGATGAACCAAGTTTTG | 1699–2144 |
| GFP | GACGAACTTGATACAAGACCCGCGCCGAGGTG | CGGGCCGCTAAGCTTACTTGTACAGCTCGTCCATG |
R1 deletion mutants R1ΔD1, R1ΔD2, R1ΔD3, and R1ΔD4 were obtained by PCR-directed mutagenesis with specific primers (Table 2) and plasmid pBAC-TGEV as the template. The first PCR products for R1ΔD1, R1ΔD2, and R1ΔD3 were obtained with the forward primer S-4310 vs and a specific reverse primer (Table 2). The second PCR products for R1ΔD1, R1ΔD2, and R1ΔD3 were obtained with the specific forward primer (Table 2) and the reverse primer BlpI-R1 rs. To generate the R1ΔD4 fragment, a unique PCR product was generated using primers S-4310 vs and ΔD4-BlpI rs (Table 2). The final PCR products were digested with the restriction enzymes PpuMI and BlpI and cloned into the same sites of the intermediate plasmid pSL-TGEV-S-3ab-M-N-7, as described above. Finally, the AvrII digestion products were introduced into the same site of plasmid pBAC-TGEV, resulting in plasmids pBAC-TGEV-R1ΔD1 to pBAC-TGEV-R1ΔD4. R1-derived sequences including six point mutations inside the stem-loop SL5 (R1mutSL5) or a partial deletion in SL5 (R1ΔSL5) were obtained by overlapping PCR using specific primers (Table 2) and pBAC-TGEV-R1 as the template. In the first PCR, R1mutSL5-1 and R1ΔSL5-1 fragments were obtained with the forward primer S-4310 vs and the specific reverse primer (Table 1). In the second PCR, R1mutSL5-2 and R1ΔSL5-2 fragments were obtained with the specific forward primer and the reverse primer BlpI-R1 rs. The final PCR products were digested with the restriction enzymes PpuMI and BlpI and subcloned into an intermediate plasmid as described above. Viral sequences delimited by AvrII sites were introduced into the plasmid pBAC-TGEV, resulting in plasmids pBAC-TGEV-R1mutSL5 and pBAC-TGEV-R1ΔSL5. All plasmid sequences were confirmed by sequencing.
Table 2.
Oligonucleotides used for the construction of recombinant cDNAs of R1-derived sequencesa
| PCR fragment | Primer (5′→3′) |
|---|---|
| S-4310 vs | ATTACGAACCAATTGAAAAAGTGC |
| BlpI-R1 rs | CGGGCCGCTAAGCCCATTGCAGTTAGCTCTAAC |
| ΔD1 vs | CAAG(Δ)TGCCTAGTCTTCCTATTCG |
| ΔD1 rs | CTAGGCA(Δ)CTTGTCCTCTATGATTTC |
| ΔD2 vs | CGT(Δ)TTTCGTACCAGAATACTGTCG |
| ΔD2 rs | GTACGA(Δ)AAACGTTGACTTGATAGTCC |
| ΔD3 vs | CGT(Δ)AACGGAGTAAGTGATCTTAAACC |
| ΔD3 rs | CTCCGTT(Δ)ACGAAAACATAGCCCTC |
| ΔD4-BlpI rs | CGGGCCGCTAAGCTACCAAGAACACCAATG |
| mutSL5 vs | AGGACAAGCGTTGATTATTTC |
| mutSL5 rs | CGCTTGTCCT(Δ)AAAGACAAATATTTCGTT |
| ΔSL5 vs | CTCCTAGAGGACAAGCGTTG |
| ΔSL5 rs | CCTCTAGGAGTATGTATAAAGACAAATATTTCGTTTAG |
vs, forward oligonucleotides; rs, reverse oligonucleotides; (Δ), deleted region. The BlpI restriction site is underlined, and single-nucleotide mutations are indicated in bold.
Rescue of defective minigenomes in trans by a helper virus.
ST cells were transfected with pcDNA plasmids encoding TGEV-derived M33 (11)- and M26-defective minigenomes. Cellular RNA polymerase II transcribes defective minigenome RNA from cytomegalovirus (CMV) promoter. Six hours after transfection, cells were infected with the helper virus, TGEV PUR46-MAD, at a multiplicity of infection (MOI) of 5. At 18 h postinfection (hpi), one-third of passage zero supernatant was used to infect new confluent ST cells. After three consecutive passages in cell culture, total intracellular RNA was extracted at 16 to 18 hpi and the amount of defective minigenome RNAs was quantified by qPCR.
Reverse genetics system.
cDNAs encoding TGEV recombinant viruses were constructed using pBAC-TGEV (23) (GenBank accession number AJ271965), which includes the whole TGEV sequence, and the spike gene from strain PTV. BHK cells were transfected with the pBAC plasmids. The viral RNA is transcribed from the cDNA by cellular RNA polymerase II and translated to initiate the viral cycle. To rescue infectious viruses from cDNA clones, BHK cells were treated with trypsin at 6 h posttransfection (hpt) and plated over confluent ST cell monolayers. Viruses recovered from the supernatant were cloned three times by plaque purification (21).
Purification of TGEV virions.
TGEV virions were partially purified by concentration through a 15% (wt/vol) sucrose cushion. Initially, supernatants from TGEV-infected cells were clarified by centrifugation at 6,000 rpm for 20 min at 4°C. Virions were then sedimented by ultracentrifugation at 25,000 rpm for 90 min through a 15% (wt/vol) sucrose cushion in TEN buffer (10 mM Tris-HCl [pH 7.4], 1 mM EDTA, 1 M NaCl) with 0.2% Tween 20 (Sigma). This purification procedure was selected instead of the centrifugation through a continuous 15 to 42% sucrose gradient in order to concentrate in the same sample wild-type virions and defective minigenome-containing virions with lower density, as shown by previous results from our laboratory (26). Finally, pelleted virions were resuspended in TNE buffer (10 mM Tris-HCl [pH 7.4], 1 mM EDTA, 100 mM NaCl). Purified virions were disaggregated by sonication (six pulses at medium intensity in a Branson sonifier 450) and incubated with 0.5 μg of RNase A at 37°C for 30 min to degrade external contaminant RNA. RNA digestion was stopped with 100 U of RNase inhibitor RNasin (Promega). The viral RNA, protected inside the virions from RNase digestion, was extracted with an RNeasy Mini Kit (Qiagen). The concentration and purity of RNA were measured with an ND-1000 spectrophotometer (Nanodrop). Protein analysis of the purified TGEV virions by SDS-PAGE and silver staining showed the presence of the major viral proteins S, N, and M in addition to a discrete number of cell proteins (data not shown). This result confirmed the presence of TGEV virions in the samples used for quantification of RNA packaging efficiencies. This purification procedure was shown to provide results that were similar, in terms of viral RNA packaging, to those obtained with the procedure previously used in our laboratory (10), which included immunoprecipitation with anti-M antibodies. In addition, the new procedure led to higher RNA yields and more-reproducible quantitative results.
DNase treatment of RNAs from transfection experiments.
To remove transfected cDNA, encoding defective minigenome or recombinant TGEV viruses, from samples before qPCR analysis, 7 μg of each RNA in 100 μl was treated with 20 U of DNase I (Roche) for 30 min at 37°C. DNA-free RNAs were repurified using an RNeasy Mini Kit (Qiagen).
RNA analysis by RT-qPCR.
For packaging and replication assays, 60 ng of total cellular RNA and 20 ng of viral RNA from purified virions were used for cDNA synthesis at 37°C for 2 h with MultiScribe reverse transcriptase (high-capacity cDNA reverse transcription [RT] kit; Applied Biosystems) by following the manufacturer's specifications. One-sixteenth of the RT reaction product was used in the qPCR for quantitative analysis of genomic and subgenomic RNAs from infectious recombinant TGEVs and defective minigenomes. DNA products in qPCR were detected by TaqMan probes or SYBR green (Applied Biosystems). Oligonucleotides and probes used for qPCR were designed with Primer Express v2.0 software. Detection was performed with an ABI Prism 7500 using SDS version 1.2.3 software. The relative quantifications were performed using the 2−ΔΔCT method (27).
RNA analysis by semiquantitative RT-PCR.
Specific quantitative detection of sgmRNAs L-R1, L-R1ΔD2, L-R1ΔD3, and L-R1ΔD4 from TGEV recombinant viruses was not technically possible. Therefore, sgmRNAs were analyzed by semiquantitative RT-PCR using as a reference viral sgmRNAs previously quantified by qPCR. M26 and sgmRNA 3a RNAs were used as positive and negative controls for packaging, respectively. Sixty nanograms of total cellular RNA and 20 ng of virion-purified RNA were used for cDNA synthesis as described above. Four serial dilutions (1/4, 1/16, 1/64, and 1/256) of cDNAs from the RT reaction product were amplified by PCR with specific primers for genomic, defective minigenome M26 and subgenomic RNAs from recombinant TGEVs. PCR products were analyzed by electrophoresis in 1% agarose gels stained with Sybr-Safe DNA gel stain (Invitrogen). To visualize the PCR products, a ChemiDoc XRS+ with Image Lab software (Bio-Rad) was used. For semiquantitative RT-PCR analysis of sgmRNA L-R1 and L-R1ΔD2, L-R1ΔD3, and L-R1ΔD4 deletion mutants, several viral RNAs previously quantified by qPCR (M26, sgmRNAs 3a, and L-R1ΔD1) were used to make a standard curve relating the values for packaging efficiency determined by qPCR and semiquantitative RT-PCR (see Results). Packaging efficiency of sgmRNAs L-R1 and L-R1ΔD2, L-R1ΔD3, and L-R1ΔD4 deletion mutants was determined by interpolating their semiquantitative values on the standard curve.
RNA analysis by Northern blotting.
Total intracellular RNA and RNA from purified virions were analyzed by Northern blotting, as previously described (5). The 3′ UTR-specific single-stranded DNA probe used for detection was complementary to nt 28300 to 28544 of the TGEV strain PUR46-MAD genome (28).
Packaging assays.
Relative quantification of viral RNAs from cells and purified virions was performed by qPCR or semiquantitative RT-PCR. The packaging efficiency (Ψ) of a specific viral RNA was defined as a ratio of the amount of RNA inside the virions to the amount of this RNA in the cell: Ψ(sgmRNA) = [RNA sgmRNA (virion)]/[RNA sgmRNA (cell)].
This ratio was made relative to the packaging of viral gRNA, arbitrarily defined as 100%, as follows: Ψ(genome) = [RNA genome (virion)]/[RNA genome (cell)].
The packaging efficiency expressed as a percentage [Ψ(%)] was calculated as follows: ΨsgmRNA(%) = [Ψ(sgmRNA)]/[Ψ(genome)].
Replication assays.
Analysis of replication efficiency of TGEV-derived RNAs was performed by reverse transfecting ST cells as described above with plasmids encoding viral RNA sequences under the control of the CMV promoter. Transfected cells were divided and seeded in two separate wells. Three hours later, one well was infected with TGEV PUR46-MAD at an MOI of 5, and the other one remained uninfected. At 26 hpt, total intracellular RNA was extracted and virions from supernatants were purified for RNA extraction with an RNeasy Mini Kit (Qiagen). Replication of TGEV-derived RNAs was defined as the increase in the amount of RNA molecules in infected cells with respect to the amount of the same RNA in noninfected and transfected cells. The replication efficiency of TGEV-derived RNAs was expressed as a percentage relative to the replication of defective minigenome M26 RNA, which was considered to be 100%.
In silico analysis.
RNA secondary structure predictions were performed using the Mfold web server for nucleic acid folding and hybridization prediction (http://mfold.rna.albany.edu/?q=mfold/RNA-Folding-Form) (29). The analysis of DNA sequences was performed using DNASTAR Lasergene software 7.0.
RESULTS
Delimitation of TGEV genome sequences required for defective minigenome rescue by a helper virus.
TGEV-derived defective minigenome M33 (3.3 kb) was the smallest analyzed sequence efficiently rescued (i.e., replicated and packaged) by a helper virus after eight consecutive cell passages (11). Other TGEV-derived defective minigenomes smaller than defective minigenome M33, such as M24, M22, M19, M17, and M15 (the number after the M indicates the size of the defective minigenome in hundreds of nucleotides), could not be significantly rescued by the helper virus after five consecutive cell passages (results not shown). Therefore, M33 was the smallest defective minigenome containing the sequences required for efficient replication and packaging. The M33 defective minigenome consisted of the first 2,144 nt of the TGEV genome, two discontinuous regions comprising nt 12195 to 12762 and nt 20357 to 20372, and the last 494 nt of the 3′ end of the genome (Fig. 1A). To delimit the minimal sequences necessary for defective minigenome rescue by a helper virus, the internal discontinuous regions of the M33 defective minigenome were deleted, resulting in defective minigenome M26 (2.6 kb), which includes 2,144 nt from the 5′ end and 494 nt from the 3′ end of the TGEV genome (Fig. 1A). The two defective minigenomes included a common 60-nt sequence between the two genomic 5′ and 3′ fragments of TGEV RNA. This sequence was inserted to introduce a common reporter sequence for detection by qPCR with SYBR green (Fig. 1A). To compare the rescue efficiencies of the M33 and M26 defective minigenomes, ST cells were transfected with cDNAs encoding M33 and M26 sequences and were infected at 6 hpt with the helper virus, TGEV PUR46-MAD, which provided in trans the proteins required for replication and packaging. After three consecutive passages in cell culture, total intracellular RNA was extracted and the amount of defective minigenome RNAs was quantified by qPCR. The relative abundance of defective minigenome M33 and M26 intracellular RNA with respect to viral genome RNA was around 80% (Fig. 1B), indicating that defective minigenome M26 was rescued by the helper virus as efficiently as was defective minigenome M33. Therefore, the sequences required for the replication and packaging of TGEV RNAs by a helper virus were tentatively located within 2,144 nt of the 5′ end and 494 nt of the 3′ end of the TGEV genome. These M26 5′-end sequences were the starting point to identify the TGEV packaging sequences.
Fig 1.

Rescue efficiency of defective minigenome M26. (A) Scheme of TGEV genomic RNA (gRNA) indicating the name of the genes. L, leader sequence; 1a and 1b, replicase genes; S, spike protein gene; 3ab, 3a and 3b accessory genes; E, envelope protein gene; M, membrane protein gene; N, nucleocapsid protein gene; 7, accessory protein gene; An, poly(A). Numbers below the boxes indicate the nucleotide position that delimit genomic sequences included in defective minigenomes. Below is represented the scheme of defective minigenomes M33 and M26, showing with shadows the genomic fragments from which they are derived. Linker, heterologous sequence used for defective minigenome quantification. M33 consisted of the first 2,144 nt of the TGEV genome, two discontinuous regions of 468 and 16 nt, comprising nt 12195 to 12762 and nt 20357 to 20372, respectively, and the last 494 nt of the 3′ end of the genome. M26 consisted of 2,144 nt from the 5′ end and 494 nt from the 3′ end of the TGEV genome. Genome size is shown to the right. Arrows below the boxes represent the oligonucleotides used for quantitative PCR analysis. Oligonucleotides for gRNA detection hybridize at positions 4829 to 4853 and 4884 to 4909 of the TGEV genome and defective minigenomes M33 and M26. Oligonucleotides for detection of minigenomes M33 and M26 hybridize at the linker sequence and nt 28200 to 28232 of the TGEV genome. (B) Rescue efficiency of defective minigenomes M33 and M26 by the helper virus after three passages in cell culture. qPCR analysis of the intracellular amount of RNA from defective minigenomes (mgRNA) M33 and M26 relative to that of genomic RNA (mgRNA/gRNA). The relative quantifications were performed using the 2−ΔΔCT method. Genome, rescue efficiency for gRNA, defined as 100%. Mock, value from cells infected with the helper virus without the defective minigenome. The data are the averages from three independent experiments, and error bars represent standard deviations.
Packaging efficiency of M26-derived sequences.
To delimit the minimal sequences in the M26 defective minigenome necessary for RNA packaging, we initially focused our attention on the 5′-end sequences from the TGEV genome (nt 1 to 2144). This RNA fragment was divided into eight overlapping fragments, named R1 to R8, that were 400 to 500 nt in size (Fig. 2A). To analyze the packaging of the fragment, a novel experimental approach was designed, consisting of TGEV recombinant viruses transcribing engineered sgmRNAs which included M26 sequences from the TRS of gene 3a. In this experimental approach, both the helper virus and the RNA sequences involved in our packaging efficiency studies are encoded by a single viral RNA, in contrast to the two independent RNA components used to perform previous RNA rescue experiments. The first fragment, R1, comprised nt 100 to 598 of the TGEV genome. The nucleotides flanking fragments R2 to R8, with a size similar to that of R1, are indicated in Fig. 2A and Table 1. Each fragment, R1 to R8, or the GFP gene as a negative control, was inserted into the TGEV genome in the location of the nonessential 3a/b genes. In addition, at the 5′ end, the first 80 nt downstream of the CS of gene 3a were inserted preceding each fragment, R1 to R8, as a common target sequence for RNA quantification using TaqMan probes in a qPCR assay (Fig. 2A). During infection, M26-derived sequences were transcribed by the virus as sgmRNAs. As in the standard synthesis of viral sgmRNAs, the leader sequence, present only once at the 5′ end of the genome (nt 1 to 99), was added to the 5′ end of each sgmRNA, and the sequences of downstream genes (3b, E, M, N, and 7 and the 3′ UTR) were included at the 3′ end of the sgmRNA. Total cellular RNA from ST cells infected with the recombinant viruses was extracted at 16 hpi and analyzed by RT-qPCR. All the recombinant viruses expressed the expected sgmRNAs containing M26-derived sequences. In the case of the rTGEV-3a-R1 virus, in addition to the expected sgmRNA L-3a-R1, a smaller sgmRNA lacking the 3a sequence was detected (data not shown). This sgmRNA contained the sequence of fragment R1 directly preceded by the leader sequence (sgmRNA L-R1). The loss of the 3a sequence preceding R1 fragment was probably caused by a recombination event between sgmRNA L-3a-R1 and the genome leader region. To obtain a unique sgmRNA including the continuity between the leader and the R1 sequence, present in the native virus, a new recombinant virus was engineered by deleting the first 80 nt of the 3a gene previously inserted between the leader and R1 (Fig. 2A). To analyze the packaging efficiency of sgmRNAs containing M26-derived sequences, ST cells were infected with recombinant viruses and intracellular as well as virion-packaged RNAs were analyzed by qPCR or semiquantitative RT-PCR at 16 hpi. The packaging efficiency of each sgmRNA was determined in relation to the total intracellular sgmRNA. In addition, to normalize the data, this packaging efficiency was made relative to the packaging of the viral genome, according to the formula described in Materials and Methods. The packaging efficiency of defective minigenome M26, used as the positive control, was approximately 40% with respect to that of the full-length RNA genome, which is assumed to be packaged at 100% efficiency. In these experiments, the packaging efficiency of the viral sgmRNA 3a and the sgmRNA L-3a-GFP was below 10%. The sgmRNAs including the R1 to R8 M26-derived sequences were packaged with efficiencies ranging from 10 to 20%, with the exception of the sgmRNA L-R1, which contains the fragment R1 directly linked to the leader sequence (L-R1) (Fig. 2B). This sgmRNA L-R1 showed a packaging efficiency similar to that of the defective minigenome M26 (Fig. 2B), indicating that viral sequences in fragment R1 (nt 100 to 598) were necessary for efficient packaging. In addition, the direct continuity between the leader and R1 was also required for packaging, since the packaging efficiency of sgmRNA L-R1 was significantly higher than that of sgmRNA L-3a-R1, including 80 nt from the 3a gene between the leader and R1 sequences. Analysis of the RNA from purified virions by Northern blotting confirmed the presence of genomic RNA inside viral particles. Interestingly, only sgmRNA L-R1 was efficiently packaged into virions (Fig. 2C). Other sgmRNAs were not detected in the virions with the exception of sgmRNA N. sgmRNA N is the most abundant viral mRNA in infected cells and is probably unspecifically packaged. The slower migration of the abundant sgmRNA N might be the origin of the smearing RNA signal observed above the sgmRNA N band in Northern blot experiments with intracellular and virion RNA samples (Fig. 2C). In fact, quantification by qPCR of viral sgmRNAs included in purified virions showed that sgmRNA N was packaged with an 18% efficiency compared to gRNA, while the packaging efficiencies of sgmRNAs S, M, E, and 7 were around 5% (data not shown). All these results suggested that the packaging of the TGEV genome required a continuous sequence from the genomic 5′ end (nt 1 to 598), including the leader (nt 1 to 99) and downstream sequences (nt 100 to 598) from the 5′ UTR and ORF1a. In these experiments, the relevance of packaging of the TGEV genome 3′ end sequences was not assessed, as these sequences are present in all analyzed sgmRNAs.
Fig 2.
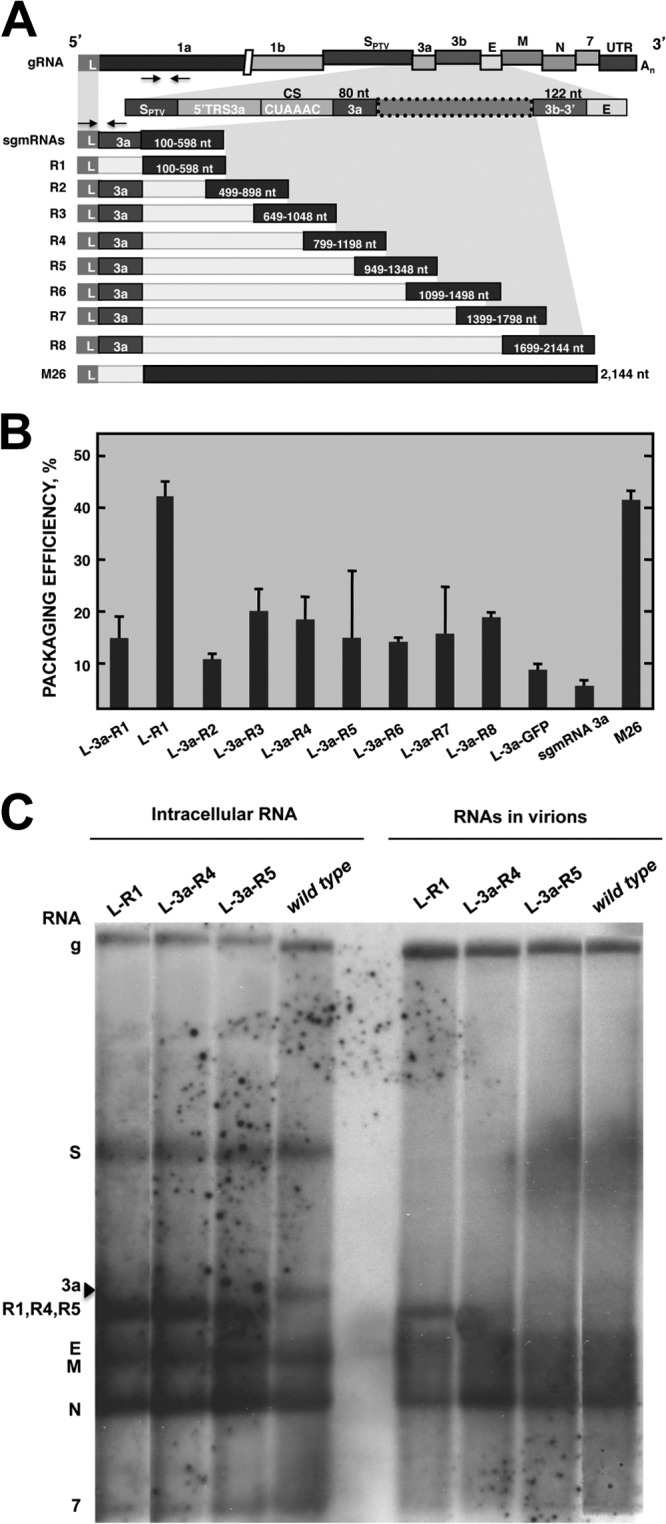
Packaging efficiency of M26-derived sequences expressed by recombinant TGEV viruses as sgmRNAs. (A) Scheme of TGEV gRNA indicating viral genes. Arrows below gene 1a represent the oligonucleotides used for gRNA quantification by semiquantitative RT-PCR (hybridizing at nt 1745 to 1772 and nt 3054 to 3072) or qPCR (hybridizing at nt 4829 to 4853 and nt 4884 to 4909). The genomic region where the M26-derived sequences were inserted (box with dotted line) is shown under the shadowed area. This region includes the TRS of the 3a gene, comprising the 5′ TRS of the 3a gene (5′TRS 3a), the conserved core sequence (CS), and the first 80 nt of the 3a gene used for sgmRNA quantification (3a). Arrows above sgmRNA represent the oligonucleotides used for quantitative PCR analysis of sgmRNAs that include the 3a sequence (hybridizing on the leader sequence, at nt 25 to 56, and the 3a gene sequence, at nt 24863 to 24889). The scheme of sgmRNAs including R1 to R8 overlapping fragments encompassing the M26 defective minigenome is shown. The sequence extent of fragments R1 to R8 is indicated inside the boxes. All sgmRNAs include the leader (L) sequence and the 3a sequence, with the exception of sgmRNA L-R1 (second line) that directly links the R1 fragment to the leader. sgmRNA L-R1 was quantified by semiquantitative RT-PCR, as described in Materials and Methods. sgmRNAs including R1 to R8 sequences contain TGEV 3′-end genomic sequences consisting of the last 122 nt of the 3b gene, the E, M, N, and 7 downstream genes, and the 3′UTR. SPTV, spike gene of the Purdue type strain. (B) Packaging efficiency of sgmRNAs represented in panel A, including M26-derived fragments R1 to R8. The M26 defective minigenome was used as a positive control. sgmRNAs L-3a-GFP and sgmRNA 3a, transcribed from the wild-type virus, were used as negative controls for packaging. The data are the averages from three independent experiments, and error bars represent standard deviations. (C) Northern blot analysis of viral intracellular RNA and RNAs inside the virions purified from supernatants of cells infected with recombinant viruses (rTGEV) expressing M26-derived sequences. The presence of genomic RNA (gRNA) and sgmRNAs with M26-derived sequences (sgmRNAs) inside the virions was detected with a probe complementary to the 3′ end of the genome. The names of rTGEV viruses expressing specific sgmRNAs are indicated above each lane. gRNA (g) or sgmRNAs (S, 3a, R1, R4, R5, E, M, N, and 7) expressed by rTGEV viruses are indicated to the left. The arrowhead indicates the sgmRNA 3a from wild-type virus.
Packaging efficiency of L-R1-derived sequences.
There is experimental evidence supporting the idea that genome packaging in some RNA viruses depends on the secondary structure of the PS (8, 30, 31). It has been shown that the L-R1 sequence was responsible for packaging in TGEV. To identify specific RNA structures involved in TGEV packaging, the secondary structure of fragment L-R1 was predicted with Mfold software (http://mfold.rna.albany.edu/?q=mfold/RNA-Folding-Form) (29). Five stem-loops (SL1 to -5) in the first 135 nt of the TGEV genome and four domains (D1 to -4), comprising nt 137 to 598, were identified (Fig. 3A). Stem-loops SL1 (nt 5 to 52), SL2 (nt 53 to 67), and SL3 (nt 72 to 83) were not considered RNA signals responsible for packaging because they are part of the leader sequence and, therefore, are present in all the sgmRNAs, which are not packaged (10). Stem-loop SL4 (nt 89 to 101) was also excluded because it was formed by the 3′-most leader sequence, comprising the CS and 3′-flanking sequences identical to those present in sgmRNA 7 (Fig. 3), which was not efficiently packaged (10). Stem-loop SL5 (nt 106 to 136) is highly stable and is present only in the R1 fragment. Furthermore, the stem-loop SL5 is conserved among different genera of coronavirus (CoV), such as human CoV 229E (HCoV-229E) and HCoV-NL63 from the Alphacoronavirus genus, bovine CoV (BCoV), MHV, and severe acute respiratory syndrome CoV (SARS-CoV) from the Betacoronavirus genus, and infectious bronchitis CoV (IBV) from the Gammacoronavirus genus, according to Mfold secondary structure predictions of 5′-end genomic regions. Therefore, SL5 was a candidate for leading L-R1 packaging. Likewise, the other structural domains in R1, D1 (nt 137 to 366), and D4 (nt 514 to 598), containing more than one stem-loop, and domains D2 (nt 367 to 439) and D3 (nt 446 to 512), containing only one long stem-loop (Fig. 3A), were also candidates for directing L-R1 packaging.
Fig 3.

Packaging efficiency of sgmRNA L-R1 with deletions of structural domains. (A) Mfold-predicted secondary structure of the 5′-most 598 nt of the TGEV genome included in the fragment L-R1. Secondary structure of L-R1 includes five stem-loops at the 5′ end named SL1 to SL5, and four domains, D1 to D4. (B) Mfold-predicted secondary structure of the first 140 nt of the TGEV genome, consisting of stem-loops SL1 to SL5 included in R1. The mutated (R1mutSL5) and deleted (R1ΔSL5) sequences of SL5 are also shown. Nucleotides corresponding to the leader sequence are shown in light gray. (C) Quantification of sgmRNAs with deletions in R1. The schemes of sgmRNAs L-R1 and ΔD1 are shown at the top. Arrows indicate the oligonucleotides used for PCR. sgmRNA L-R1 and agmRNAs ΔD2-ΔD4 were quantified by semiquantitative RT-PCR with oligonucleotides hybridizing on the leader sequence (nt 25 to 56) and the 3b gene (nt 25803 to 25824). sgmRNA-ΔD1 was quantified by qPCR with oligonucleotides hybridizing on the leader sequence (nt 82 to 108) and the R1 sequence (nt 376 to 399). Below is represented the correlation curve between the packaging efficiency of sgmRNAs determined by qPCR and semiquantitative RT-PCR (sqPCR). The diamond symbols represent sgmRNAs (M26, sgmRNA 3a, and sgmRNA L-R1ΔD1) quantified by qPCR and sqPCR. These values were used to generate the standard curve. The square symbols represent sgmRNAs L-R1, L-R1ΔD2, L-R1ΔD3, and L-R1ΔD4 as quantified by sqRT-PCR. These values were interpolated in the standard curve to estimate their packaging efficiency. (D) Analysis by qPCR of the packaging efficiency of sgmRNAs transcribed from rTGEVs with mutations (L-R1mutSL5) or a deletion (L-R1ΔSL5) in the SL5 sequence. (E) Analysis by qPCR of the packaging efficiency of sgmRNAs transcribed from rTGEVs including the R1 sequence with deletions of domains D1 to D4 (R1ΔD1 to R1ΔD4). L-R1, sgmRNA with wild-type sequence used as a positive control. sgmRNA L-3a-GFP and sgmRNA 3a, including GFP and the 3a gene transcribed from the wild-type virus, were used as negative controls. The data are the averages from three independent experiments, and error bars represent standard deviations.
To evaluate the relevance of SL5 in packaging, two different recombinant viruses were generated, both disrupting the secondary structure of SL5 contained in R1 by different strategies. In the R1mutSL5 recombinant virus, six point mutations were engineered to disrupt the upper part of the stem, thus opening a larger loop. In R1ΔSL5, the most apical 14 nt of SL5 were deleted, leading to a smaller stem-loop (Fig. 3B). The packaging efficiency of both sgmRNAs including the modified R1 sequence was similar to that of the negative controls, and it was significantly lower than that of sgmRNA L-R1 with the complete R1 sequence (Fig. 3D). Therefore, stem-loop SL5 was necessary for RNA packaging in TGEV.
To analyze the relevance of R1 structural domains D1 to D4 in packaging, regions D1 to D4 were deleted from R1. Each R1 deletion mutant was inserted in recombinant TGEV viruses, replacing genes 3a/b, immediately downstream of gene 3a CS. At 18 hpi, RNA from cells and virions was analyzed by qPCR (sgmRNAs L-R1ΔD1, 3a, and L-3a-GFP) or semiquantitative RT-PCR (sgmRNAs L-R1, L-R1ΔD2, L-R1ΔD3, and L-R1ΔD4) (Fig. 3C) when quantitative assays were not technically feasible because of too-large amplicon sizes. The packaging efficiency of all deletion mutants (R1ΔD1 to R1ΔD4) was similar to that of the negative controls, and only sgmRNA L-R1, containing the complete R1 sequence, was efficiently packaged, as expected (Fig. 3E). In conclusion, all the predicted D1 to D4 domains in R1, comprising nt 137 to 598, were required for efficient packaging of TGEV RNAs. All together, these results suggested that the complete R1 sequence comprising nt 100 to 598 of the 5′ end of the TGEV genome was necessary to direct RNA packaging, since any modifications of these sequences abolished the process.
Relevance of replication on packaging of M26- and R1-derived defective minigenomes.
Analysis of 5′ genome sequences required for packaging has been addressed by using TGEV recombinant viruses transcribing sgmRNAs that contained the potential packaging signal. However, this experimental system could not be used to address the question of whether the replication was required in the packaging process. For this reason, a new experimental approach was designed, consisting of the expression of virus-derived RNAs from transfected plasmids followed by infection with a helper virus. This approach allowed the introduction of modifications to the 3′-end genomic sequences to prevent replication. ST cells were transfected with plasmids containing TGEV-derived defective minigenome sequences. Transfected cells were divided and seeded in two separate plates. At 3 hpt, one of the plates was infected with the helper virus, TGEV PUR46-MAD. At 23 hpi, total intracellular RNA from infected and noninfected cells, as well as RNA from purified virions, was extracted and analyzed by qPCR. The replication efficiency of each defective minigenome was calculated as the ratio of the amount of defective minigenome RNA in transfected and infected cells to the amount of RNA in noninfected cells. In noninfected cells, the abundance of RNA was dependent only on the activity of the cellular polymerase II, whereas in infected cells, RNA was additionally amplified in trans by the helper virus polymerase, provided that the defective minigenome contained signals for replication. The packaging efficiency of TGEV-derived RNAs from transfected and infected cells was calculated as the relative amount of defective minigenome RNA inside the virions in relation to the intracellular amount of this RNA (see Materials and Methods). For detection of virus-derived RNAs by qPCR with the same TaqMan assay, a common sequence derived from the gene encoding the Rubisco enzyme was introduced in all the constructs (Fig. 4). As a negative control for replication and packaging, a defective minigenome construct carrying the Ru sequence linked to GFP sequences (Ru-GFP) was engineered. Defective minigenome M26-Ru-3′wt was efficiently replicated and packaged by the helper virus in rescue experiments performed as described above, and the values were used as the positive controls for both viral processes. To evaluate the requirement of RNA replication for its packaging, a nonreplicating version of defective minigenome M26, M26-Ru-Δ171, was constructed by deleting nt 28388 to 28559 of the TGEV genome; these nucleotides were included within the last 494 nt of the genomic 3′ end. This deletion prevents defective minigenome replication by the helper virus (11). The requirement of replication for packaging was also analyzed using defective minigenome L-R1-Ru-3′wt, containing the first 5′-end 598 nt and the last 3′-end 494 nt of the genome, and L-R1-Ru-Δ171, a deletion mutant lacking the 3′-end 171-nt region essential for replication (Fig. 4). The replication efficiency of defective minigenome M26-Ru-Δ171 was 18% relative to that of M26-Ru-3′wt (Fig. 4), confirming the expected reduction in replication competence mediated through deletion of the 3′ end of the defective minigenome. Defective minigenomes L-R1-Ru-3′wt and L-R1-Ru-Δ171, containing the first 598 nt of the 5′ end of the TGEV genome, had replication efficiencies below 5%, similar to that of the negative control that is not replicated by the helper virus and is transcribed only from the CMV promoter, since it lacks cis-acting replication signals. Nevertheless, the packaging efficiencies of defective minigenomes M26-Ru-Δ171, L-R1-Ru-3′wt, and L-R1-Ru-Δ171 were very similar, around 45% relative to the packaging of gRNA (Fig. 4), indicating that packaging was not affected by replication activity. These results indicated that replication of defective minigenomes by the helper virus was not necessary for packaging. Therefore, TGEV packaging seems to be a replication-independent process.
Fig 4.

Requirement of replication for packaging of M26- and R1-derived defective minigenomes. A schematic comparison of the structures of defective minigenomes analyzed in replication and packaging assays is represented on the left. Positions in the TGEV genome of the sequences included in defective minigenomes are indicated with numbers on the top. The size of each defective minigenome is indicated below its name. L, leader sequence; Ru, Rubisco sequence used for detection by qPCR. Size in nucleotides is indicated below the arrow. An, poly(A). The last 494 nt of the 3′ end of the TGEV genome are divided into two boxes, representing the 7 gene (light gray box) and the 3′ UTR (dark gray box). The white box represents the Δ171 deletion (nt 28388 to 28559) in the 3′ end of the TGEV genome, including sequences required for replication. To the right of the figure is represented the analysis by qPCR of the defective minigenome replication activity and packaging efficiencies, calculated as described in Materials and Methods. The data are the averages from five independent experiments, and error bars represent standard deviations.
Requirement of the TGEV genome 3′-end sequences for packaging.
To analyze the requirement of the TGEV genome 3′-end sequences for packaging, defective minigenome L-R1-Ru-Δ3′ was engineered by deleting the last 494 nt of the 3′ end of L-R1-Ru-3′wt (Fig. 5). The replication and packaging efficiencies of L-R1-Ru-3′wt and L-R1-Ru-Δ3′ defective minigenomes were determined using transfection and infection experiments, as described in Materials and Methods. Packaging efficiency of L-R1-Ru-3′wt was around 45%, while L-R1-Ru-Δ3′ packaged to a lower extent, around 30%. However, the packaging efficiency of both defective minigenomes was significantly higher than that of the negative control, which was around 5% (Fig. 5). The decrease in L-R1-Ru-Δ3′ packaging efficiency with respect to that of L-R1-Ru-3′wt suggested that the 3′-end sequences of the TGEV genome contributed to optimize packaging. Nevertheless, since the packaging efficiency of L-R1-Ru-Δ3′ did not decrease to the levels of the negative control, the 3′ end of the TGEV genome was not absolutely necessary for packaging, although it made a positive contribution to the process.
Fig 5.

Contribution of 3′-end sequences of the TGEV genome to defective minigenome packaging and packaging of the GFP heterologous sequence by TGEV L-R1 sequences. The schematic comparison of the structure of defective minigenomes analyzed in replication and packaging assays is represented on the left. The nucleotide positions in the TGEV genome of the sequences included in defective minigenomes are indicated with numbers on the top. The size of each defective minigenome is indicated below its name. L, leader sequence; Ru, Rubisco sequence used for detection in qPCR. Size in nucleotides is indicated below the arrow. GFP, green fluorescence protein; An, poly(A). The last 494 nt of the 3′ end of the TGEV genome are divided into two boxes, as indicated in Fig. 4. The white box (Δ3′) represents the deletion of the last 494 nt of the 3′ end of the TGEV genome. The vertical line in the L-R1ΔSL5-Ru-GFP defective minigenome indicates the partial deletion of the SL5. On the right is represented the analysis by qPCR of the replication activity and packaging efficiencies, calculated as described in Materials and Methods. The data are the averages from five independent experiments, and error bars represent standard deviations.
Packaging of a nonviral sequence by the R1 genomic region.
It has been shown that R1, comprising nt 1 to 598 from the TGEV genome, was an essential component required for packaging. To determine whether R1 sequences were sufficient to direct the packaging of a heterologous sequence, such as that of GFP gene, two defective minigenomes were constructed, including GFP sequences preceded either by R1 (L-R1-Ru-GFP) or by R1 with a deletion in stem-loop SL5 to abolish packaging (L-R1ΔSL5-Ru-GFP) (Fig. 5). Both defective minigenomes were replication deficient, as were the negative control and defective minigenomes L-R1-Ru-3′wt and L-R1-Ru-Δ3′. Analysis of packaging in transfection-infection assays showed that packaging efficiency of L-R1-Ru-GFP was 13%, an intermediate value between that of L-R1-Ru-Δ3′ and those of the negative controls (L-R1ΔSL5-GFP and Ru-GFP) (Fig. 5). These results suggested that viral R1 region, including RNA motifs necessary for packaging, could not efficiently direct the packaging of the heterologous GFP sequence. Therefore, R1 TGEV region is necessary but not sufficient for packaging of heterologous sequences.
DISCUSSION
Previous work from our laboratory (10) had shown, by using rescue experiments consisting of the replication and packaging of recombinant defective minigenomes by a helper virus, that essential TGEV packaging motifs are located within the first 649 nt of the genomic 5′ end. Rescue experiments, involving two components, the defective minigenome and the helper virus, facilitate the recovery of virus-derived RNAs after serial passages in cell culture. Therefore, rescued RNAs must contain cis-acting signals for replication and packaging by the helper virus. In the present study, a major motif of TGEV PS has been delimited to the first 598 nt of the genomic 5′ end using a single-component system consisting of recombinant TGEV viruses that transcribed sgmRNAs, including potential packaging sequences. The continuity between the leader sequence (nt 1 to 99) and downstream sequences (nt 100 to 598) in the PS was required for packaging, since the insertion of ectopic viral sequences between the leader and R1 in sgmRNA L-3a-R1 led to a significant reduction in the packaging efficiency compared to that of sgmRNA L-R1. This continuity might be required to maintain the correct secondary structure of the PS, since structural requirements frequently determine packaging in RNA viruses (8, 30, 31). Further deletions along the TGEV PS prevented packaging of viral RNAs, indicating that the whole genomic region was necessary for its function. The SL5, a stable stem-loop located immediately downstream of the leader sequence and conserved among CoVs of different genera, was essential for packaging, since minor deletions and mutations within the SL5 sequence prevented RNA packaging. Therefore, the TGEV major PS motif extends over the first 598 nt of the viral genome. This packaging sequence, L-R1, was present in gRNA and not in sgmRNAs, therefore providing a packaging selectivity advantage to gRNA and not to sgmRNAs (10). Although the location of PSs in RNA viruses is variable, they are most frequently located at the 5′ end of the genome (32, 33), similarly to what we have observed for TGEV. Alternatively, in other RNA viruses, such as the MHV coronavirus (8), the PS has been delimited to a 69-nt stem-loop located internally in the genome within ORF1b, around 20 kb from the genomic 5′ end. In addition, a 291-nt region in ORF1b, showing homology with the MHV sequence, was also described as the PS of BCoVs, belonging to the same genus, Betacoronavirus, as MHV. However, recent work on MHV PS has shown that this RNA motif is not required per se for RNA packaging and virus viability but is essential in the selection of gRNA for packaging into virions (34).
To delimit the minimal PS, recombinant viruses were constructed by transcribing sgmRNAs that contained potential packaging signals. However, all sgmRNAs also included the genomic 3′ end. Therefore, to analyze the function of the 3′ end in packaging and to dissect replication and packaging functions, a new experimental approach was designed, consisting of a comparative analysis of the intracellular and virion RNAs levels of the engineered RNAs in noninfected and helper virus-infected cells. Since RNAs were first expressed by cellular polymerase II from transfected plasmids, nonreplicating viral RNAs could be packaged by the helper virus. This approach allowed the uniform quantification of all the defective minigenomes using a common heterologous sequence, derived from the plant Rubisco gene, for quantification using qPCR with TaqMan probes. Analysis of replication and packaging of mutant defective minigenomes showed for the first time in CoVs that efficient packaging was a replication-independent process. Previously, the packaging of nonreplicating RNA was described for MHV CoV, although the efficiency of this process was not quantified in comparison to that of genome packaging (35). In fact, although the L-R1-Ru-3′wt defective minigenome, containing the PS at the first 598 nt at the genomic 5′ end and the 3′-end genome sequences, showed undetectable replication levels, it was efficiently packaged to a level similar to that of replicating defective minigenome M26. Defective minigenomes L-R1-Ru-3′wt and L-R1-Ru-Δ171, containing the 598 nt from the 5′ end and different 3′-end sequences, were not competent for replication but were proficient in packaging, confirming previous work in our laboratory that defined the minimal sequences required for rescue as the first 5′-end 1,348 nt and the last 3′-end 492 nt (11). Interestingly, we have shown that the last 494 nt of the TGEV genome 3′ end were not necessary for packaging, although these sequences positively contributed to the packaging process. At this point, we cannot rule out the possibility that 5′-3′-end interaction promotes the packaging of complete gRNA molecules. Defective minigenome L-R1-Ru-Δ3′, lacking the genomic 3′ end, was packaged, although less efficiently than was L-R1-Ru-Δ171. This difference in packaging efficiency indicated that genomic 3′-end sequences other than the deleted 171 nt (nt 28388 to 28559) had a positive contribution to packaging.
In all our experiments, with different experimental approaches, packaging efficiency of virus-derived RNAs did not reach the levels seen for gRNA. The maximum packaging efficiency of defective minigenome M26- and R1-derived sequences was around 45% with respect to gRNA. These results suggested that in addition to the main PS described, other factors contribute to packaging. It is possible that additional viral sequences distributed along the genome might also be involved in TGEV packaging, reinforcing the idea that the TGEV PS is complex. In fact, although the cis-acting PS is required for packaging, other RNA factors have been involved in genome packaging in other viral systems. It has been noted that for turnip crinkle virus, from the Tombusviridae family, the size of packaged RNA is a critical factor (36), and perhaps small RNAs with a subgenomic size like those of defective minigenomes or sgmRNAs might not be favored for packaging. Therefore, the packaging of TGEV gRNA is a complex process and might require additional unknown cis and trans factors.
We have shown that the identified major PS motif of TGEV was necessary for packaging. However, it was not sufficient to efficiently package a heterologous sequence like that of the GFP gene. We expected the packaging efficiency of L-R1-Ru-GFP to be similar to that of L-R1-Ru-Δ3′, since both defective minigenomes contained similar viral sequences (nt 1 to 598). However, a (2.5-fold) lower packaging efficiency was observed for L-R1-Ru-GFP. It is possible that the GFP RNA sequence could interfere with the formation of a correct PS secondary structure, thereby affecting its optimal function. In addition, the heterologous GFP sequence might interfere with different processes required for packaging. In the related MHV CoV, the PS was described as necessary and sufficient to efficiently direct the packaging of the chloramphenicol acetyltransferase (CAT) reporter gene (35). Nevertheless, those results were obtained by Northern blotting, a less sensitive and accurate technique than the qPCR used in this paper. Furthermore, the packaging of CAT directed by the MHV PS was analyzed qualitatively, without comparing its efficiency with that of genome packaging. However, recent work on MHV PS has shown that this RNA motif confers selectivity to gRNA packaging, although it is not essential either for RNA packaging or virus viability (34).
The identification of the TGEV PS could be of interest for the development of biosafe packaging-deficient viral vectors used in vaccines and gene therapy. Biosafety of viral vectors, such as those derived from lentiviruses, has been improved by separating the packaging signal into different cassettes (37). Additionally, the identified PS might represent a new target for antiviral therapies, as described for HIV (38–40) and hepatitis B virus (HBV) (41).
In this paper, we have provided evidence supporting the concept of the high complexity of TGEV packaging. An essential component of the TGEV PS has been defined in the first 598 nt of the 5′ end of the genome. In addition, it has been shown that the 3′ end of the genome is not necessary for packaging but makes a positive contribution to packaging efficiency. Furthermore, TGEV packaging is a replication-independent process. However, further studies are needed to obtain insight into the viral and cellular factors contributing to the specificity and efficiency of viral gRNA packaging.
ACKNOWLEDGMENTS
This work was supported by grants from the Ministry of Science and Innovation of Spain (BIO2010-167075) and the European Community's Seventh Framework Programme (FP7/2007-2013), under the project PoRRSCon (EC grant agreement 245141). L.M. and P.A.M.-G. received predoctoral fellowships from the Ministry of Science and Innovation of Spain (BES-2011-043489 and BES-2008-001932, respectively). L.D.P. received a predoctoral fellowship from the National Institute of Health (ISCIII) of Spain (FI08/00109).
We gratefully acknowledge C. M. Sanchez, M. Gonzalez, and S. Ros for technical assistance.
Footnotes
Published ahead of print 21 August 2013
REFERENCES
- 1.Enjuanes L, Almazan F, Sola I, Zuniga S. 2006. Biochemical aspects of coronavirus replication and virus-host interaction. Annu. Rev. Microbiol. 60:211–230 [DOI] [PubMed] [Google Scholar]
- 2.Enjuanes L, Gorbalenya AE, de Groot RJ, Cowley JA, Ziebuhr J, Snijder EJ. 2008. The Nidovirales, p 419–430 In Mahy BWJ, Van Regenmortel M, Walker P, Majumder-Russell D. (ed), Encyclopedia of virology, 3rd ed. Elsevier Ltd., Oxford, United Kingdom [Google Scholar]
- 3.Escors D, Ortego J, Laude H, Enjuanes L. 2001. The membrane M protein carboxy terminus binds to transmissible gastroenteritis coronavirus core and contributes to core stability. J. Virol. 75:1312–1324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zúñiga S, Sola I, Alonso S, Enjuanes L. 2004. Sequence motifs involved in the regulation of discontinuous coronavirus subgenomic RNA synthesis. J. Virol. 78:980–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sola I, Moreno JL, Zúñiga S, Alonso S, Enjuanes L. 2005. Role of nucleotides immediately flanking the transcription-regulating sequence core in coronavirus subgenomic mRNA synthesis. J. Virol. 79:2506–2516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mateos-Gómez PA, Zuñiga S, Palacio L, Enjuanes L, Sola I. 2011. Gene N proximal and distal RNA motifs regulate coronavirus nucleocapsid mRNA transcription. J. Virol. 85:8968–8980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mateos-Gomez PA, Morales L, Zuñiga S, Enjuanes L, Sola I. 2013. Long-distance RNA-RNA interactions in the coronavirus genome form high-order structures promoting discontinuous RNA synthesis during transcription. J. Virol. 87:177–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fosmire JA, Hwang K, Makino S. 1992. Identification and characterization of a coronavirus packaging signal. J. Virol. 66:3522–3530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Molenkamp R, Rozier BCD, Greve S, Spaan WJM, Snijder EJ. 2000. Isolation and characterization of an arterivirus defective interfering RNA genome. J. Virol. 74:3156–3165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Escors D, Izeta A, Capiscol C, Enjuanes L. 2003. Transmissible gastroenteritis coronavirus packaging signal is located at the 5′ end of the virus genome. J. Virol. 77:7890–7902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Izeta A, Smerdou C, Alonso S, Penzes Z, Mendez A, Plana-Duran J, Enjuanes L. 1999. Replication and packaging of transmissible gastroenteritis coronavirus-derived synthetic minigenomes. J. Virol. 73:1535–1545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mouland AJ, Mercier J, Luo M, Bernier L, DesGroseillers L, Cohen EA. 2000. The double-stranded RNA-binding protein Staufen is incorporated in human immunodeficiency virus type 1: evidence for a role in genomic RNA encapsidation. J. Virol. 74:5441–5451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Narayanan K, Makino S. 2001. Cooperation of an RNA packaging signal and a viral envelope protein in coronavirus RNA packaging. J. Virol. 75:9059–9067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ni P, Wang Z, Ma X, Das NC, Sokol P, Chiu W, Dragnea B, Hagan M, Kao CC. 2012. An examination of the electrostatic interactions between the N-terminal tail of the Brome mosaic virus coat protein and encapsidated RNAs. J. Mol. Biol. 419:284–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nugent CI, Johnson KL, Sarnow P, Kirkegaard K. 1999. Functional coupling between replication and packaging of poliovirus replicon RNA. J. Virol. 73:427–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Khromykh AA, Varnavski AN, Sedlak PL, Westaway EG. 2001. Coupling between replication and packaging of flavivirus RNA: evidence derived from the use of DNA-based full-length cDNA clones of Kunjin virus. J. Virol. 75:4633–4640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Volkova E, Gorchakov R, Frolov I. 2006. The efficient packaging of Venezuelan equine encephalitis virus-specific RNAs into viral particles is determined by nsP1-3 synthesis. Virology 344:315–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Annamalai P, Rao AL. 2005. Replication-independent expression of genome components and capsid protein of brome mosaic virus in planta: a functional role for viral replicase in RNA packaging. Virology 338:96–111 [DOI] [PubMed] [Google Scholar]
- 19.Jacobson MF, Baltimore D. 1968. Morphogenesis of poliovirus. I. Association of the viral RNA with coat protein. J. Mol. Biol. 33:369–378 [DOI] [PubMed] [Google Scholar]
- 20.Delmas B, Gelfi J, Sjöström H, Noren O, Laude H. 1993. Further characterization of aminopeptidase-N as a receptor for coronaviruses. Adv. Exp. Med. Biol. 342:293–298 [DOI] [PubMed] [Google Scholar]
- 21.Sánchez CM, Jiménez G, Laviada MD, Correa I, Suñé C, Bullido MJ, Gebauer F, Smerdou C, Callebaut P, Escribano JM, Enjuanes L. 1990. Antigenic homology among coronaviruses related to transmissible gastroenteritis virus. Virology 174:410–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McClurkin AW, Norman JO. 1966. Studies on transmissible gastroenteritis of swine. II. Selected characteristics of a cytopathogenic virus common to five isolates from transmissible gastroenteritis. Can. J. Comp. Med. Vet. Sci. 30:190–198 [PMC free article] [PubMed] [Google Scholar]
- 23.Almazán F, González JM, Pénzes Z, Izeta A, Calvo E, Plana-Durán J, Enjuanes L. 2000. Engineering the largest RNA virus genome as an infectious bacterial artificial chromosome. Proc. Natl. Acad. Sci. U. S. A. 97:5516–5521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sánchez CM, Izeta A, Sánchez-Morgado JM, Alonso S, Sola I, Balasch M, Plana-Durán J, Enjuanes L. 1999. Targeted recombination demonstrates that the spike gene of transmissible gastroenteritis coronavirus is a determinant of its enteric tropism and virulence. J. Virol. 73:7607–7618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiménez G, Correa I, Melgosa MP, Bullido MJ, Enjuanes L. 1986. Critical epitopes in transmissible gastroenteritis virus neutralization. J. Virol. 60:131–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Méndez A, Smerdou C, Izeta A, Gebauer F, Enjuanes L. 1996. Molecular characterization of transmissible gastroenteritis coronavirus defective interfering genomes: packaging and heterogeneity. Virology 217:495–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔC(T) method. Methods 25:402–408 [DOI] [PubMed] [Google Scholar]
- 28.Penzes Z, Gonzalez JM, Calvo E, Izeta A, Smerdou C, Méndez A, Sanchez CM, Sola I, Almazan F, Enjuanes L. 2001. Complete genome sequence of transmissible gastroenteritis coronavirus PUR46-MAD clone and evolution of the Purdue virus cluster. Virus Genes 23:105–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zuker M. 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 31:3406–3415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mansky LM, Wisniewski RM. 1998. The bovine leukemia virus encapsidation signal is composed of RNA secondary structures. J. Virol. 72:3196–3204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lu K, Heng X, Summers MF. 2011. Structural determinants and mechanism of HIV-1 genome packaging. J. Mol. Biol. 410:609–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tchatalbachev S, Flick R, Hobom G. 2001. The packaging signal of influenza viral RNA molecules. RNA 7:979–989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lever A, Gottlinger H, Haseltine W, Sodroski J. 1989. Identification of a sequence required for efficient packaging of human immunodeficiency virus type 1 RNA into virions. J. Virol. 63:4085–4087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kuo L, Masters PS. 2013. Functional analysis of the murine coronavirus genomic RNA packaging signal. J. Virol. 87:5182–5192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Woo K, Joo M, Narayanan K, Kim KH, Makino S. 1997. Murine coronavirus packaging signal confers packaging to nonviral RNA. J. Virol. 71:824–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Qu F, Morris TJ. 1997. Encapsidation of turnip crinkle virus is defined by a specific packaging signal and RNA size. J. Virol. 71:1428–1435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Escors D, Breckpot K. 2010. Lentiviral vectors in gene therapy: their current status and future potential. Arch. Immunol. Ther. Exp. (Warsz.) 58:107–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chadwick DR, Lever AM. 2000. Antisense RNA sequences targeting the 5′ leader packaging signal region of human immunodeficiency virus type-1 inhibits viral replication at post-transcriptional stages of the life cycle. Gene Ther. 7:1362–1368 [DOI] [PubMed] [Google Scholar]
- 39.Dorman NM, Lever AM. 2001. Investigation of RNA transcripts containing HIV-1 packaging signal sequences as HIV-1 antivirals: generation of cell lines resistant to HIV-1. Gene Ther. 8:157–165 [DOI] [PubMed] [Google Scholar]
- 40.Dietz J, Koch J, Kaur A, Raja C, Stein S, Grez M, Pustowka A, Mensch S, Ferner J, Moller L, Bannert N, Tampe R, Divita G, Mely Y, Schwalbe H, Dietrich U. 2008. Inhibition of HIV-1 by a peptide ligand of the genomic RNA packaging signal Psi. ChemMedChem 3:749–755 [DOI] [PubMed] [Google Scholar]
- 41.Feng H, Beck J, Nassal M, Hu KH. 2011. A SELEX-screened aptamer of human hepatitis B virus RNA encapsidation signal suppresses viral replication. PLoS One 6:e27862. 10.1371/journal.pone.0027862 [DOI] [PMC free article] [PubMed] [Google Scholar]