

Abstract
Herpes simplex virus (HSV) entry and cell-cell fusion require glycoproteins gD, gH/gL, and gB. We propose that receptor-activated changes to gD cause it to activate gH/gL, which then triggers gB into an active form. We employed a dual split-protein (DSP) assay to monitor the kinetics of HSV glycoprotein-induced cell-cell fusion. This assay measures content mixing between two cells, i.e., fusion, within the same cell population in real time (minutes to hours). Titration experiments suggest that both gD and gH/gL act in a catalytic fashion to trigger gB. In fact, fusion rates are governed by the amount of gB on the cell surface. We then used the DSP assay to focus on mutants in two functional regions (FRs) of gB, FR1 and FR3. FR1 contains the fusion loops (FL1 and FL2), and FR3 encompasses the crown at the trimer top. All FL mutants initiated fusion very slowly, if at all. However, the fusion rates caused by some FL2 mutants increased over time, so that total fusion by 8 h looked much like that of the WT. Two distinct kinetic patterns, “slow and fast,” emerged for mutants in the crown of gB (FR3), again showing differences in initiation and ongoing fusion. Of note are the fusion kinetics of the gB syn mutant (LL871/872AA). Although this mutant was originally included as an ongoing high-rate-of-fusion control, its initiation of fusion is so rapid that it appears to be on a “hair trigger.” Thus, the DSP assay affords a unique way to examine the dynamics of HSV glycoprotein-induced cell fusion.
INTRODUCTION
Our goal is to understand the earliest events in herpes simplex virus (HSV) entry into susceptible host cells and the related process of cell-cell fusion. To gain entry, HSV must fuse its lipid envelope with a host membrane, either at the cell surface or in an endosome (reviewed in reference 1), thereby delivering the capsid and tegument to the target cell. HSV uses four glycoproteins for this process, gD, gB, and gH/gL, and a cell receptor, either herpesvirus entry mediator (HVEM) or nectin-1 (2). These complex events, also common to HSV glycoprotein-induced cell-cell fusion, occur in a stepwise temporal process that requires receptor activation of gD to a form that can activate the gH/gL heterodimer. Once gH/gL is activated, it upregulates the fusogenic activity of gB, a class III fusion protein. Solution of the 3-dimensional (3-D) structures of the entry glycoproteins and receptors for HSV has provided significant insights into the entry-fusion process, but major issues remain unresolved.
First, although HSV gB and Epstein-Barr virus (EBV) gB look remarkably alike and have the structural features of a fusion protein (3, 4), neither works in the absence of gH/gL. This additional requirement is ubiquitous among herpesviruses but distinct from the mechanisms of other class III fusion proteins, including baculovirus gp64 and vesicular stomatitis virus glycoprotein G (VSV G) (5, 6). Second, the solved structures of HSV gB (4) and EBV gB (3) resemble the postfusion form of VSV G (6). Although a prefusion form of VSV G is known (7), there are no ultrastructural data for any prefusion forms of gB. Thus, our knowledge of gB structure fails to provide an explanation for how it interacts with gH/gL to attain this unknown prefusion form. Nevertheless, a variety of observations support the idea that fusion by gB is regulated by both gD and gH/gL (8). The challenge is to uncover the mechanisms involved in each step.
Previously, we and others (1, 9) analyzed HSV glycoprotein-induced cell-cell fusion by one of two methods: (i) immunofluorescence or Giemsa staining assays of receptor-bearing cells that fuse to form giant cells (syncytia) after being transfected with plasmids for the glycoproteins and (ii) a firefly luciferase reporter endpoint fusion assay whereby one set of cells is transfected with HSV glycoproteins and a luciferase reporter gene under the control of the phage T7 promoter while the other set is transfected with a plasmid bearing T7 RNA polymerase. Coculture and subsequent fusion allow the synthesis of luciferase, and its activity is measured in cell lysates after a fixed time, usually hours.
Here, to measure the dynamics of cell-cell fusion, we used a dual split-Renilla luciferase (RL)-green fluorescent protein (GFP) kinetic assay (dual split-protein [DSP] assay) developed by Kondo et al. (10) to directly measure the fusion (content mixing) of intact cells over time. In this assay, the two DSP reporter plasmids are transfected into separate sets of cells along with the plasmids necessary for fusion. The activities of the two reporter proteins, RL and GFP, are reconstituted only when cell content mixing (also referred to as pore formation) occurs following cell-cell fusion (10, 11). The reconstituted RL catalyzes the conversion of the membrane-permeant substrate coelenterazine (EnduRen) to coelenteramide by creating a photon of light, but only in live cells. Thus, fusion, i.e., content mixing, can be monitored in intact cells as it occurs in real time, yielding kinetic data. This assay was successfully used to follow HIV Env-induced fusion for minutes to hours, thereby establishing a fusion rate induced by wild-type (WT) and mutant forms of Env (10). Concurrently, as a result of cell fusion, GFP is reconstituted, and fusion can be monitored by fluorescence microscopy to detect green syncytia.
Here we adapted the DSP assay to examine the role of gB in HSV glycoprotein-induced fusion. We found that the rate of fusion is constant when the WT proteins are utilized and is governed principally by the amount of gB on the cell surface. We then applied the DSP assay to investigate the fusion rates of a number of gB mutants.
We showed previously that trimeric gB (4) has four functional regions (FRs) in its ectodomain, based on the locations of epitopes for gB-specific virus-neutralizing monoclonal antibodies (MAbs) (12). While the rate of fusion for WT gB at optimal amounts was constant over minutes or hours following transfection, this was not the case for several mutants with mutations in the two regions of gB studied here: FR1 (structural domains I and V) and FR3 (structural domain IV) (4). Each protomer of FR1 contains two fusion loops: FL1 and FL2 (4, 13). Although previous studies showed that certain FL2 mutants were nearly WT in total (endpoint) fusion levels (13, 14), these mutants exhibited two different sets of kinetics in the DSP assay. One set exhibited a low initial rate of fusion which then accelerated over time, reaching levels close to those for the WT by 6 to 8 h after fusion was initiated (low-rate mutants). Thus, these residues are critical for fusion initiation. In contrast, certain mutants with mutations in FR3, within the crown, showed enhanced early rates (high-rate mutants), with fusion decelerating over time but still occurring faster than WT fusion. Of great interest was the remarkably fast initiation phenotype of a well-characterized syn mutant with mutations located in the cytoplasmic tail (15), the LL871/872AA mutant. Although it was thought that the Syn phenotype was due mostly to the amount of gB on the virion envelope (15), our results suggest that the phenotype is inherent in the mutation. The different rate phenotypes uncovered by the DSP assay highlight changes to gB that likely reflect the conformational changes in structure it undergoes as it carries out its fusion function.
MATERIALS AND METHODS
Cells and plasmids.
Mouse melanoma cells (B78H1) were grown in Dulbecco's modified Eagle medium with 5% fetal bovine serum (5% FBS–DMEM). For mouse melanoma cells expressing nectin-1 (designated C10), the medium was supplemented with 500 μg/ml G418 (16). Plasmids pPEP98 (gB), pEP99 (gD), pEP100 (gH), and pEP101 (gL) were gifts from P. G. Spear (17, 18). The glycoprotein B fusion loop mutants (with the W174K, F175K, G176K, H177A, Y179K, E260A, A261D, F262L, H263A, R264A, and Y265R mutations) have been published previously (13, 14). The construction of plasmids DSP1-7, DSP8-11, RLuc81-7, and RLuc88-11 has been described in detail previously (10, 11). The major differences between the two versions of each plasmid are the amino acid sequence and the split point in Renilla luciferase (RL). The RL with eight amino acid mutations, called Rluc8, was split between residues 155 and 156 (the original DSP plasmid was split between amino acids 229 and 230). The terms “1-7” and “8-11” refer to the names of the beta-strands of GFP included in each DSP plasmid.
Construction of gB mutants.
The QuikChange site-directed mutagenesis kit (Stratagene Cloning Systems, La Jolla, CA) was used to generate full-length mutant gB constructs (19). Primers designed to mutate individual gB residues were used to amplify the gB gene of plasmid pPEP98 (18) by PCR. The mutations were confirmed by sequencing of the entire gB gene. The 13 plasmids encoding the gB substitutions that were expressed on the cell surface were named as follows (with mutations in parentheses): pBH741 (W539F), pBH692 (V553A), pBH785 (V553L), pBH831 (Q584A), pBH690 (G594R), pBH875 (E607A), pBH836 (D608A), pBH837 (Q609A), pBH691 (F641Y), pBH731 (Y649A), pBH737 (H657R), pBH751 (H657A), and pJG974 (LL871/872AA). The mutants that were not expressed were named as follows (with mutations in parentheses): pBH740 (A561V), pBH832 (R588A), pBH833 (R592A), pBH834 (R605A), pBH793 (R638A), pBH838 (R639A), pBH731 (Y649A), and pBH791 (Y647A).
CELISA.
(i) To detect gB, gD, or gH/gL expression on the cell surface on the basis of the amount of DNA transfected, we used a modified cell-based enzyme-linked immunosorbent assay (CELISA) (20, 21). B78 cells growing in 96-well plates were transfected with all four glycoproteins as described above using Lipofectamine2000 (Invitrogen). Various amounts of the plasmid of interest—carrying gB, gD, or gH/gL—were used, while the other plasmids were kept at 125 ng. Cells were grown overnight at 37°C, fixed with 3% paraformaldehyde, and rinsed with phosphate-buffered saline (PBS) containing Ca2+ and Mg2+. The cells were incubated for 1 h with polyclonal antibody (Pab) IgGs for gD (R7) (19) or gH/gL (R137) (8). For gB, we used a cocktail of monoclonal antibodies (A22, SS10, and SS67) (12). In all cases, these antibodies were first diluted in 3% bovine serum albumin (BSA)-PBS and then incubated for 30 min with goat anti-rabbit antibodies coupled to horseradish peroxidase for gD and gH/gL, or with goat anti-mouse IgG coupled to horseradish peroxidase for gB, at room temperature. Cells were rinsed with 20 mM citrate buffer (pH 4.5); 2,2′-azino-di(3-ethylbenzthiazoline) sulfonic acid peroxidase substrate (Moss, Inc.) was added; and the absorbance at 405 nm was recorded using a BioTek plate reader. The absorbance values of cells that had not been transfected with the plasmids of interest were subtracted, and data were normalized by setting the absorbance value obtained with 125 ng of each plasmid at 100%.
(ii) To detect gB cell surface expression, shown in Fig. 5C, 6B, and 7A, B78 cells were transfected with 125 ng of gD, gH, and gL and with 375 ng gB by following the transfection and CELISA protocols described above. However, instead of using a cocktail of gB antibodies, cells were first incubated with Pab IgG for gB (R68) diluted in 3% BSA-PBS and were then incubated for 30 min with a goat anti-rabbit antibody coupled to horseradish peroxidase. The absorbance value of cells transfected with an empty vector instead of gB was subtracted, and data were normalized by setting the absorbance value obtained with WT gB at 100%.
Fig 5.

Functional region 3 (FR3) and low-rate-of-fusion mutations. (A) (Left) Ribbon structure of gB with hypothetical membrane. The site of the LL871/872AA mutation in the cytoplasmic tail is shown in a cartoon representation. (Right) Enlarged gB crown with color-coded residues. (B) Ribbon representation of a gB crown protomer showing residues altered in low-rate-of-fusion mutants and a low-rate-of-virus-entry mutant. (C) Expression of low-rate-of-fusion mutants on the cell surface. Data are normalized to WT levels. (D and E) All mutants show slower kinetics than the WT in either the long (D) or the short (E) time course.
Fig 6.

FR3 and high-rate-of-entry mutants. (A) Ribbon structure of the gB crown. Residues altered in fast-entry mutants are depicted as spheres. (B) All mutants were expressed at or near WT levels. (C and D) The kinetics of fusion were either WT (C) or enhanced over WT kinetics (D) and were maintained over a long period. (E) Both hyperfusogenic mutants exhibited a fast phenotype right from the initial stages of fusion.
Fig 7.
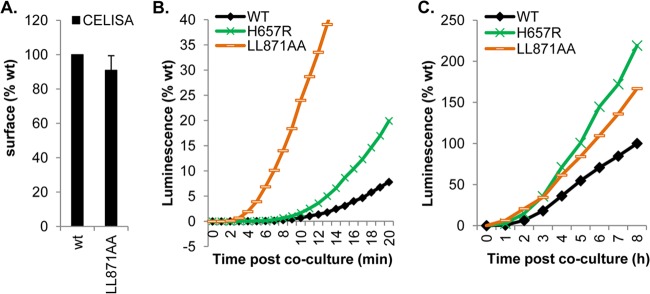
A mutant with alterations in the cytoplasmic tail of gB exhibits a superhigh initial fusion rate. (A) Surface expression of the LL871/872AA cytoplasmic tail mutant. (B and C) Kinetics of fusion of the LL871/872AA mutant over the first 20 min (B) or over 8 h (C) in comparison to those of WT gB and gB H657R.
Split-luciferase assay.
A total of 5 × 104 B78 cells (effector cells) were seeded on a 96-well luciferase plate treated for cell culture (Corning). A total of 2 × 105 C10 cells (target cells) were seeded on 24-well plates. Unless otherwise stated, on the following day, effector cells were transfected with a mixture containing 4 μl of Lipofectamine 2000 (Invitrogen) and 125 ng each of the gD, gH, gL, and DSP1–7 plasmids in a 250-μl final volume. gB was transfected at 375 ng. This master mix was divided over 3 wells. Target cells were transfected with 250 ng of the DSP8-11 plasmid per well. Twenty-four hours posttransfection, the target cells were detached with EDTA. Cells were washed with a fusion medium (DMEM without phenol red supplemented with 50 mM HEPES and 5% FBS) and were transferred to an Eppendorf tube. After spinning for 5 min at 4°C, cells were resuspended in the fusion medium. EnduRen substrate (Promega) was added at a final concentration of 1:1,000. Target cells (in a final volume of 60 μl) were transferred over the effector cells. Luciferase production was monitored with a BioTek plate reader at the time intervals indicated in the figures.
GFP fluorescence and counting of syncytia.
To determine the fusion rates, B78 cells cultured on coverslips were transfected with gB, gD, gH, gH, and DSP1–7 as described above. Fusion was triggered by overlaying the transfected B78 cells with C10 cells transfected with DSP8–11. At the indicated times, the coverslips were lifted. Cells were fixed with 3% paraformaldehyde for 30 min at room temperature (RT) and were then quenched with 50 mM NH4Cl for 10 min at RT. Cells were washed with PBS, incubated with 10% normal goat serum for 30 min at RT, and then labeled with anti-gH Pab R137 as described previously (8, 22, 23). After being washed with PBS, coverslips were incubated for 30 min with Alexa Fluor 594-conjugated goat anti-rabbit IgG (Invitrogen) diluted in 10% goat serum-PBS as a secondary antibody. The coverslips were rinsed three times with PBS and once with H2O and were mounted in ProLong Gold Antifade reagent (Invitrogen). Samples were examined by confocal microscopy with a Nikon TE-300 inverted microscope coupled to a PerkinElmer imaging system as described previously (8). A two-line argon krypton laser emitting at 488 and 568 nm was used to excite the fluorescence of Alexa Fluor 594. The levels of fusion were determined by multiplying the total number of green syncytia by the average number of nuclei per syncytium (8, 22–24).
Construction of a prefusion model of gB.
Starting from a recent crystal structure of HSV gB (3NWA), we aligned the known structural domains and functional regions to their structural homologues in the prefusion form of VSV G, using the modeling program UCSF Chimera and its associated alignment tool, MatchMaker (25). HSV gB domains I and II (4, 12) mapped onto the VSV G structure (7) with little ambiguity. Domain IV could be coarsely mapped onto the homologous domain of VSV G, but an improved alignment of segments of secondary structure resulted from breaking this domain into two pieces and mapping each independently. When this was done, the orientation of domain IV aligned well with that of VSV G. VSV G has no domain V, so this had to be omitted from our model. Residues from the central stalk of the postfusion form (domain III) were tentatively placed based on connectivity to the crown and domain II. It should be emphasized that the model was constructed independently of the phenotypes of the mutants (especially those in FR3).
RESULTS
Our first objective was to apply the DSP assay to WT forms of the four HSV glycoproteins and the gD receptor nectin-1 (Fig. 1A) in order to determine if this assay would work in this complicated system. To determine the rate of substrate conversion and cytoplasmic mixing, we cocultured the two cell populations for 12 h at 37°C, taking luminescence readings at 1-h intervals after substrate addition (Fig. 1B). The increase in product accumulation was linear between 2 h and 8 h after coculture and then plateaued from 8 to 12 h. It should be noted that this luciferase assay measures the rate of fusion of a cell population, but not fusion by single cells. As another measure of fusion rates, we took advantage of the presence of the two GFP halves on DSP1–7 and DSP8–11 and the re-formation of GFP as a fluorescent protein to monitor fusion by microscopy.
Fig 1.

Method and validation. (A) Schematic representation of the dual split-luciferase assay (DSP assay). B78 cells were transfected with the four essential glycoproteins and a plasmid that encodes the inactive N-terminal half of luciferase coupled to the N-terminal half of GFP. Nectin-1-expressing cells were transfected with the other half of the luciferase-GFP fusion construct. The major differences between the two versions are the amino acid sequence and the split point in Renilla luciferase (RL). The terms “1–7” and “8–11” refer to the names of the beta-strands of GFP included in each DSP plasmid. Fusion (content mixing) occurs upon coculture of the two cell populations, leading to the restoration of functional luciferase and intact GFP. The enzyme catalyzes the conversion of the membrane-permeant substrate EnduRen to a luminescent product. (B) Typical fusion time course using DSP plasmids. (C) To determine rates, luminescence values were normalized by considering the luminescence units at 7 h to be 100% and calculating the slope of the line between 3 and 7 h. (D) For GFP fluorescence, coverslips of target and effector cells were fixed and examined by confocal microscopy. Fusion events were calculated by multiplying the number of green syncytia by the number of nuclei per syncytium. Values were normalized by considering the 7th-hour counts to be 100%.
Rates determined by re-formation in the split luciferase assay versus restoration of GFP fluorescence.
To validate that luminescence measurements truly represent content mixing due to fusion, we performed the luminescence assay head to head with a fluorescence assay (syncytium counting) to look for the reconstitution of GFP from the two halves encoded by DSP plasmids within syncytia. A master transfection mixture containing HSV-1 glycoproteins gB, gD, gH, and gL and reporter plasmid DSP1-7 was divided in two. One half was used to transfect B78 cells in a 96-well plate for luciferase product measurements (Fig. 1C), and the other half was used to transfect B78 cells on coverslips in order to count nuclei in GFP fluorescence-containing syncytia (Fig. 1D). Both sets of cells were overlaid with nectin-1-bearing C10 cells to trigger fusion. For GFP analysis, coverslips were fixed at various times and were examined by confocal microscopy. The number of fusion events was normalized to total fusion events at 7 h (Fig. 1D). Similarly, the WT luminescence value observed at the 7th hour was used to normalize the other values. The rates of fusion measured in each assay were similar to each other as well as to those shown in Fig. 1B, revealing that green syncytia form at a rate that compares favorably with the rate determined by luminescence. Thus, for the complex HSV system, the values obtained by the DSP assay are, as found for HIV (10), a true representation of content mixing due to cell-cell fusion.
DNA titration: gB is the rate-limiting protein.
To optimize the DSP assay for fusion, we needed to establish the optimal amount of DNA for each wild-type form of the glycoprotein that would yield the highest fusion rate. To address this, B78 cells were transfected with varying DNA concentrations for one glycoprotein while the other three were kept constant. (In the case of gH/gL, the DNA concentrations for both glycoproteins were varied.) In this case, rates were normalized to the luminescence achieved by a control using 125 ng of each DNA (Fig. 2A, filled bars). We found for gD that substantial fusion occurred by use of an amount of DNA as small as 5 ng and that the level of fusion reached the 100% plateau between 30 to 60 ng (Fig. 2A). For gH/gL (Fig. 2A), the concentration profile was similar, i.e., substantial fusion occurred with 5 ng DNA, and the fusion level reached the 100% plateau by 30 ng. However, for gB, there was little fusion at 5 ng or even 30 ng of DNA (Fig. 2A), even though the plasmids for the other glycoproteins were clearly in excess (at 125 ng). In fact, gB fusion rates reached a maximum plateau only at 250 ng DNA. In a separate experiment, we determined the amount of each glycoprotein on the surface by CELISA (Fig. 2B).The amounts of gB, gD, and gH/gL increased steadily with increasing amounts of DNA transfected. Thus, for both gD and gH/gL, even small amounts of protein were sufficient to yield maximal fusion rates. We believe these data show that both gD and gH/gL act in a catalytic manner as opposed to a stoichiometric fashion to trigger fusion by gB. We conclude that the rate of fusion is determined and limited by the amount of gB on the cell surface. Based on the titration data, we performed all future experiments using 375 ng gB and 125 ng each of gD, gH, and gL, unless otherwise noted.
Fig 2.
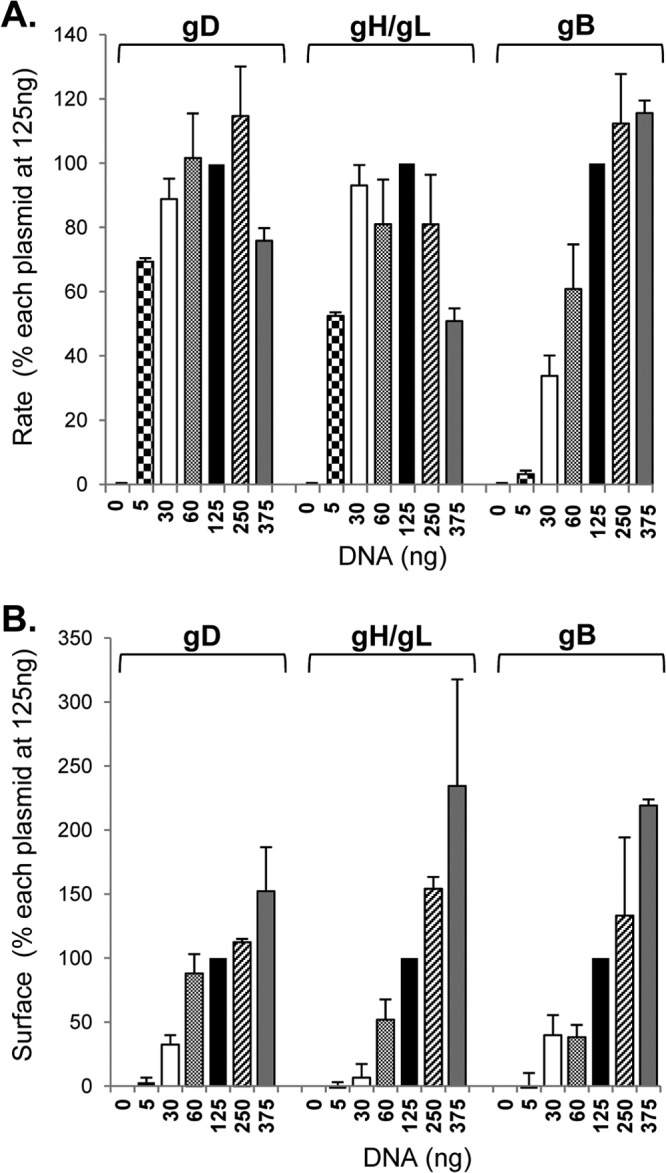
gB is the rate-limiting protein. (A) B78 cells were transfected with varying amounts of DNA corresponding to one viral glycoprotein, while the other three were maintained at 125 ng along with one of the DSP plasmids. Fusion was triggered with receptor-bearing target cells carrying the second DSP half as shown in Fig. 1A. Data were normalized to the rates observed for cells transfected with 125 ng of each glycoprotein (filled bars). To determine rates, luminescence values were normalized by considering the luminescence units at 7 h to be 100% and calculating the slope of the line between 3 and 7 h. (B) B78 cells were transfected the same way as for panel A, and surface expression of gB, gD, and gH/gL was measured using CELISA. Data were normalized by considering the absorbance value for cells transfected with 125 ng of each glycoprotein (filled bars) to be 100%. Data from 3 independent experiments were averaged, and the curve was considered linear when the R2 value was equal to or higher than 0.96.
Determining the fusion rates of wild-type HSV-1 glycoproteins.
Figure 1B and C show that there is a lag (2 to 3 h) after the mixing of the cells until the signal-to-background ratio becomes significant. To improve the quality of the signal, a second generation of the DSP plasmids (RLuc8) was employed that increases the sensitivity of the luciferase signal 100-fold (11) and can be used to define when fusion starts (termed “initiation”). Here we compared the two sets of DSP plasmids to establish the time frame during which each assay was linear for the wild-type versions of each HSV glycoprotein and to determine whether we could establish when and how fast initiation of fusion could be detected.
Over an 8-h time period, the kinetic profiles of fusion were similar regardless of which set of plasmids (DSP or RLuc8) was used (Fig. 3A and B). However, the new RLuc8 plasmids enabled us to detect significant luminescence within minutes after the mixing of the two cell populations (Fig. 3C and D). The results also show that for the WT proteins, the rates of fusion with both sets of plasmids are constant over a wide time range. Thus, from multiple experiments, we have defined the kinetics of fusion of the four WT glycoproteins under optimal conditions: initiation of fusion occurs at 7 min, and the rate is constant for 7 h. The value of examining the fusion rates over an extended period will become evident when mutant forms of gB are compared.
Fig 3.
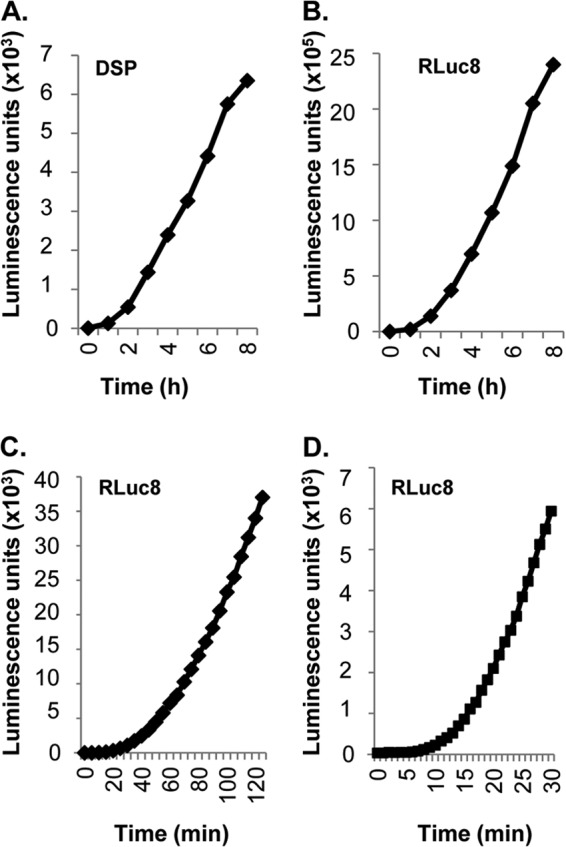
The rate of fusion with the WT HSV glycoproteins is constant over a wide time frame. Two sets of plasmids (DSP or RLuc8) were used to measure fusion rates over a short and a long time course using the WT versions of each HSV glycoprotein. (A and B) The rates measured over the same period for cells transfected with DSP plasmids (A) and cells transfected with RLuc8 plasmids (B) are similar. The sensitivity is markedly enhanced with RLuc8 plasmids. (B and C and D) Raw data are shown for comparison. RLuc8 plasmids yield robust luminescence readings taken every 5 min for 2 h (C) or every minute for 30 min (D).
Effects of mutations to the fusion loops of gB on fusion rates.
We previously carried out a mutational analysis of the fusion loops of gB (two per protomer and six per trimer) by making single and double amino acid changes (13, 14). We found that the fusion loops were critical for fusion and infection (13, 14, 26). Many of the mutations that converted an uncharged or hydrophobic amino acid to a charged one had a deleterious, sometimes fatal effect on gB function. These effects on fusion were documented by a standard (endpoint) fusion assay, as well as by null virus complementation (13, 14). None of these mutations had a significant effect on overall structure, as measured by cell surface expression and epitope analysis, although the crystal structures of the W174R and Y179S mutants (which were null mutants) exhibited local changes in the structure of the fusion loop (27). Other mutations, mostly those converting a charged residue to alanine, had an “in-between” or wild-type fusion phenotype when examined in these traditional assays.
With the wealth of data about the effect of mutations on fusion, we used the DSP assay to reexamine the capacities of these proteins to cause fusion (Fig. 4A) from the time of initiation of the event and throughout the fusion process. First, using CELISA, all of the mutants were detected at WT or nearly WT levels on the cell surface (data not shown), as reported previously (13, 14). Four FL1 mutants (the W174K, F175K, G176K, and Y179K mutants) that were previously reported to be severely impaired for fusion and virus complementation showed low rates of fusion at both early and late times of fusion (compare Fig. 4B and C and Table 1). The fifth FL1 mutant, with the H177A mutation, exhibited a reduced but constant rate of fusion over the entire time course. This reduction correlated with the reduced levels of H177A protein expressed on the cell surface (data not shown) (13), suggesting that this mutation primarily affects the structure and transport of gB. We conclude that for FL1, the fusion phenotype of each of the mutants previously studied is due to failure to initiate the fusion event.
Fig 4.

Functional region 1 (FR1) with fusion loops. (A) Ribbon representation of a gB protomer. The amino acid sequences of fusion loop 1 (FL1) (blue) and FL2 (cyan) are shown. The color scheme for the gB protomer is as published previously (4). The fusion loops are enlarged (box) to highlight individual amino acids. The colors of these residues correspond to the colors of the curves in panels B to E. (B) The DSP plasmids were used to measure the rates of fusion for FL1 mutants over 8 h. (C) The RLuc8 plasmids were used to measure the rates of fusion for FL1 mutants over 120 min. The same phenotypes as those for the 8-h time course are seen over 2 h. (D) The DSP plasmids were used to measure the rates of fusion for FL2 mutants over 8 h. (E) The RLuc8 plasmids were used to measure the rates of fusion for FL2 mutants over 120 min. For panels B and D, luminescence values we normalized to the 8th-hour reading for the WT. For panels C and E, luminescence values were normalized to the 2-h reading for the WT.
Table 1.
Rates of fusion for gB fusion loop mutantsa
| Mutant | Rate of fusion (% of WT) at: |
Total fusion at 18 h (% of WT)b | |
|---|---|---|---|
| 40–90 min | 3–8 h | ||
| WT | 100 | 100 | 100 |
| W174K | 0 | 0 | 0 |
| F175K | 16 ± 3 | 11 ± 2 | 26 |
| G176K | 0 | 0 | 0 |
| H177A | 54 ± 4 | 56 ± 19 | 63 |
| Y179S | 0 | 0 | 0 |
| E260A | 26 ± 4 | 66 ± 32 | 74 |
| A261D | 0 | 0 | 9 |
| F262L | 19 ± 1 | 43 ± 14 | 70 |
| H263A | 6 ± 1 | 18 ± 3 | 51 |
| R264A | 1.8 ± 1 | 2.1 ± 1 | 31 |
| Y265R | 24 ± 4 | 61 ± 6 | 93 |
Rates were calculated from the linear portions of the luminescent curves between 40 and 90 min (RLuc8) or between 3 and 8 h in a long time course (DSP) based on raw data. Values are expressed as percentages of WT rates and represent averages for at least three independent experiments, each carried out in duplicate, ± standard deviations.
For mutations in FL2, we detected a different pattern of fusion rates at early and late times after coculture. When the rates were calculated between 3 and 8 h after coculture, four mutants (the E260A, F262L, H263A, and Y265R mutants) had reduced rates or behaved like the WT (Fig. 4D; Table 1). Two others (the A261D and R264A mutants) were null. Because the DSP assay is a kinetic assay and the firefly luciferase assay is an endpoint assay, direct comparison of results obtained with the two assays is not possible. However, we could compare the fusion levels at the 2nd (Fig. 4E) or 8th (Fig. 4D) hour from the DSP assay with the total fusion from the luciferase assay (Table 1) (13, 14). We found that the total fusion levels measured at the 8th or 18th hour correlated very well. However, when we compared kinetic profiles from 0 to 2 h (Fig. 4E) to those measured over a longer period (Fig. 4D), all of the FL2 mutants showed severely impaired initial fusion rates (Table 1). We conclude that for the E260A, F262L, H263A, and Y265R mutants, the mutations impair gB function at an early stage of fusion, but at later times, these forms of gB function at higher rates, allowing them to “catch up” to WT levels by 8 h, as exemplified by the E260A mutant (Fig. 4D; Table 1). These unexpected data show that all of the mutations in FL2 have a deleterious effect on gB, many on the initiation of fusion—even those previously thought not to be impaired (13, 14). These results highlight the critical role of even charged amino acids, such as E260, in fusion loop function, since a mutation can impede the ability of gB to initiate fusion. The results also emphasize the advantage of using the DSP assay over the endpoint luciferase assay. While both assays allow one to determine the effect of a mutation on the function of the protein, only the DSP assay allows the dissection of the fusion process to determine where the inhibition takes place.
gB crown mutants (FR3).
Previous studies showed that a soluble form of gB, gB730t, could inhibit virus infection and interact with the surfaces of different cells and that the interaction could be blocked with certain neutralizing monoclonal antibodies (28). In particular, MAb SS55, which maps to FR1 (13), blocks the association of gB with lipid membranes (29) and the interaction between gB and gH/gL required for the initiation of fusion (8, 23). However, SS10 (12), which maps to residues 640 to 670 in the crown of gB (FR3), blocks the interaction of gB with the cell surface (28), suggesting that it blocks the interaction of gB with a cellular receptor (23). Furthermore, SS10 does not interfere with gB-lipid association (29) or with the gB–gH/gL interaction (23). Thus, FR3 plays an important role in the fusion process (28).
Several rate-of-virus-entry mutations (30–34), as well as linker insertion mutations (35), map to the crown of gB. We have created a series of point mutations (a total of 21) with the intention of identifying residues within the crown that are essential for gB function (Fig. 5A). From the structure of gB, we chose residues that are exposed at the crown tips, e.g., E607, D608, and Q609. Another group of mutants (the H657 mutant and others) line up near a crevice between protomers of the crown (Fig. 5A). Lastly, other residues correspond to rate-of-entry mutants, such as the G594R (identified by monoclonal antibody selection) and V553A (found in the syncytial HSV strain ANG) mutants (30, 31). We also created additional mutations at certain sites (V553L, H657R).
Initially, we prepared expression plasmids for the point mutants and assessed expression on the cell surface (Fig. 5C and 6B) and fusion using both the endpoint luciferase assay and the DSP assay (Table 2). Nine mutants (the A561V, R588A, R592A, Y604A, R605A, R638A, R639A, Y649A, and Y647A mutants) were not expressed on the cell surface and were not studied further. Of the 12 remaining mutants, all had WT or close-to-WT levels of fusion in the endpoint luciferase assay. The Q584A mutant was the exception (Table 2).
Table 2.
Rates of fusion for gB crown mutantsa
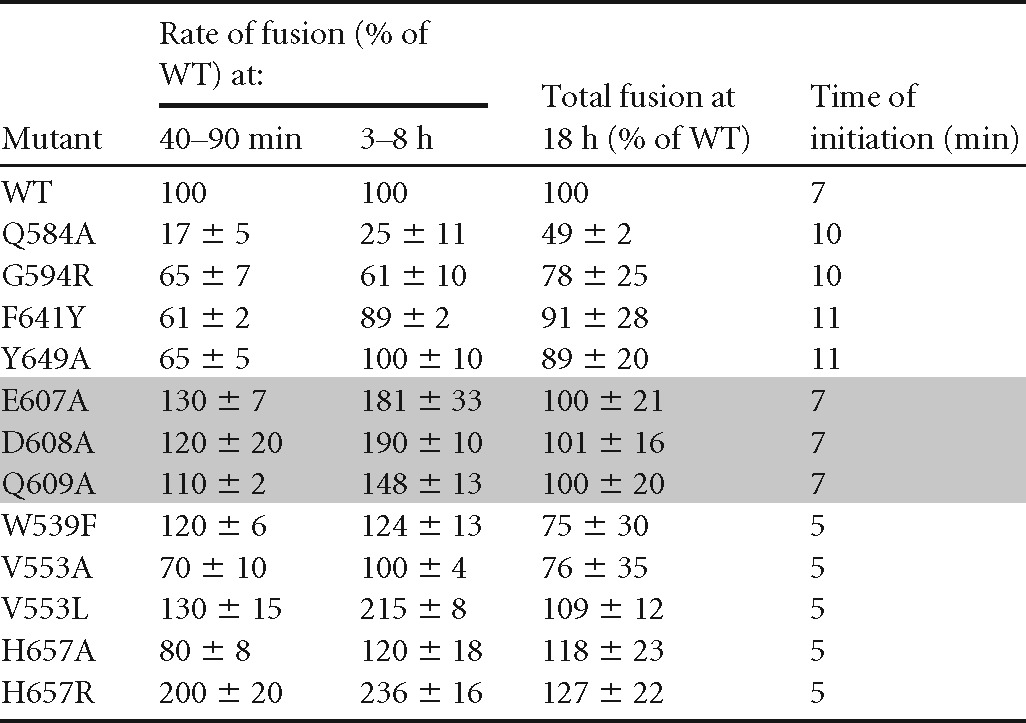
Rates were calculated from the linear portions of the luminescent curves between 40 and 90 min (RLuc8) or between 3 and 8 h in a long time course (DSP). Total fusion was determined by the standard endpoint luciferase assay. The data represent averages for at least three independent experiments, each carried out in duplicate, ± standard deviations. The time of initiation of fusion was defined as the time needed to detect a luminescent signal at least twice the background level.
Next, we examined these mutations using both versions of the DSP plasmids. Based on early times (0 to 120 min) (Fig. 5E and 6E; Table 2), these mutants were grouped into three categories: low-rate mutants (the Q584A, G594R, F641Y, and Y649A mutants), WT-like mutants (V553A, H657A mutants), and high-rate mutants (W539F, V553L, E607A, D608A, Q609A, H657R mutants). We have also recreated the LL871/872AA double mutant, with mutations in the tail of gB (15), as a control for the group of mutants with higher fusion rates. This mutant is examined in more detail below.
(i) Slow FR3 mutants.
The low-rate FR3 mutants (the Q584A, G594R, F641Y, and Y649A mutants) localize to the same region of the crown (Fig. 5A and B; Table 2). All were expressed at 50 to 80% of the WT level (Fig. 5C), and all showed lower rates of fusion than WT gB (Fig. 5D and E; Table 2), particularly when fusion was measured during the first 2 h (Fig. 5E). For two mutants, the G594R and Y649A mutants, the slower kinetics may be attributed to the presence of somewhat less gB on the cell surface (Fig. 5C). The G594R mutant was originally identified as a virus mutant that exhibits a low rate of entry (31), which may be due to the presence of less gB in the virion envelope. For the F641Y mutant, the level of expression on the cell surface was essentially the same as that of the WT, but the initial rate was only 60% of the WT rate, indicating an impairment in gB function. The fourth, the Q584A mutant, appears to be a “true” low-rate mutant, since it was expressed at 60% of the WT level and its kinetics were markedly slow (25% of the WT rate), regardless of when it was measured (Fig. 5D and E; Table 2). Thus, the low rate of fusion for gB carrying Q584A at both the initiation of fusion and continuing fusion steps is likely due to a functional alteration of the structure of gB.
(ii) Wild-type-like and fast FR3 mutants.
We studied three sets of mutations: the “tip” mutants (the E607A, D608A, and Q609A mutants), the junction mutants (the H657A and H657R mutants), and the rate-of-virus-entry mutations (the V553 and V553L mutants). Figure 6B shows that each of these mutants was expressed on the cell surface at wild-type levels. All three “tip” mutants initiated fusion at 7 min, like the WT (Table 2), but had an enhanced rate of fusion within the first 120 min, which continued over the 8-h period. Figure 6D shows the overall rate for the Q609A mutant, representative of the three tip mutants.
A junction mutation (H657A) and a rate-of-entry mutation (V553A) (30) had no effect on the ongoing rate of fusion (Fig. 6C; Table 2). However, fusion was detectable at 5 min, as opposed to 7 min for the WT (Table 2), indicating a difference in fusion initiation. To investigate these mutations further, we altered these residues again. The alanine mutation V553A did not add steric bulk to the residue but simply reduced the size to a single methyl group, so we considered an alternative mutation (leucine) that would increase the steric bulk while still maintaining the neutral change. For H657A, we asked whether mutating this residue to a positively charged arginine instead of the neutral alanine would alter the dynamics of fusion. When V553 was mutated to leucine (V553L), the mutant protein exhibited a hyperfusogenic phenotype (Fig. 6D and E). Similarly, the H657R mutant, with its additional methyl group, showed a marked enhancement of the fusion rate at both early and late times after coculture (Fig. 6D and E). Thus, we were able to further disrupt the interaction of H657 within the crown of gB, indicating that residues in FR3, the crown of gB, play an important role in the initiation and regulation of fusion.
(iii) The syn mutant with alterations in the cytoplasmic tail.
The LL871/872AA mutations (Fig. 5A), like others generated in the cytoplasmic tail of gB, were first identified in HSV strains with syncytial phenotypes (15). Initially, it was created as a control for fast-fusion mutants (36). However, the phenotype associated with this mutant warranted its placement in a category by itself (Fig. 7). Although this mutant has a hyperfusogenic phenotype (Table 2; Fig. 6), the mutations are located in the cytoplasmic tail, distant from the crown in the postfusion structure (Fig. 5A). The higher fusion activity of the LL871/872AA mutant was initially considered to be due to an increase in surface expression (15). However, Fig. 7A shows that the LL871/872AA protein is expressed on the cell surface at the same level as the WT protein.
To study the LL871/872AA mutant, we collected luminescence readings every minute for 20 min, using the H657R mutant and the WT as controls (Fig. 7B). As before, we found that for WT gB and the H657R mutant, initiation of fusion was detected at 7 min and 5 min after coculture, respectively (Table 2). In multiple experiments, the initiation of fusion by the LL871/872AA mutant was detected at 3 min. During the first 20 min, the fusion rate of the LL871/872AA mutant was significantly higher than that of the H657R mutant and 300 to 400% higher than that measured for WT gB (Fig. 7B). Figure 7C shows that by 3 to 4 h post-fusion triggering, the fusion kinetics of the LL871/872AA mutant slow down but are still faster than those for WT gB (Fig. 7C). We suggest that gB LL871/872AA is on a “hair trigger,” initiating fusion almost immediately after coculture.
In summary, the data confirm that the crown of gB plays a critical role in gB function and confirm the validity of our original identification of the crown as a functional region, which was based on mapping of virus-neutralizing antibodies (12). We also conclude that W539F, V553L, E607A, D608A, Q609A, H657R, and LL871/872AA, which are in different regions of the crown are all hyperfusogenic mutations but that the specific mechanisms by which they induce fusion are unknown but are likely to differ. While the change in the cytoplasmic tail alters its ability to modulate the activity of the ectodomain (37), the crown mutations might create local rearrangements of the ectodomain itself that also favor faster conversion of gB from a prefusion to a postfusion state.
DISCUSSION
In this report, we used a split-luciferase reporter assay (DSP assay) (10, 11, 38) to study the effects of mutations in HSV gB on the time of initiation and the overall rate of fusion. The assay allowed us to examine fusion within minutes of initiation and to continue to follow these events over hours. Because of the construction of the reporter, the half-luciferase genes are joined to half-GFP genes, which are re-formed during cytoplasmic mixing. We concomitantly followed syncytium formation by luciferase and GFP fluorescence, thereby confirming the validity of the DSP assay for studying cell-cell fusion caused by the four HSV glycoproteins, gD, gH/gL, and gB.
We showed previously that gB has four functional regions (FRs), based on the locations of epitopes for gB-specific virus-neutralizing monoclonal antibodies (12). Each of these regions was postulated to be involved in a different phase of function, such as interaction with gH/gL (23), binding to the cell surface (28), and membrane association (13, 14, 29, 39). Three of these regions are within the solved structure, while the fourth (FR4), which encompasses the first 100 N-terminal residues, is not present in the structure. Here, we focused on the effects of mutations on the rates of fusion for two regions: (i) FR1, which includes the two internal fusion loops (FL1 and FL2) within each protomer, and (ii) FR3, which forms the crown at the top of the gB trimer in the postfusion form (13, 14). For WT gB, initiation of cell fusion could be detected by 7 min, and the rate was constant for 8 h. For FL1 mutants, the original phenotypes reported using the standard endpoint fusion assay are now readily explained by the examination of the rate of fusion by each mutant. We found that, except for H177A, all these mutations were associated with severely impaired rates of fusion at both early and late times, which might explain the null complementation and reduced total fusion, as measured by the endpoint luciferase assay, found previously (13, 14). This suggests that these mutants are damaged at one of the earliest stages of fusion loop insertion (association), which is essential for gB function (26). Unexpectedly, however, several FL2 mutants that we had made, whose total-fusion levels were minimally affected (13, 14), were now found to be impaired in their ability to initiate fusion. For several of these mutants, we propose that fusion loop association with the lipid membrane is inefficient at the start of fusion but the damage seems to be less important as fusion progresses. The data highlight the importance of these residues in both FLs in FR1 for an early step of gB function, i.e., the proposed insertion of the fusion loops into the target membrane. Although we refer to the event as “insertion,” the mechanism of lipid association is still unknown. For FR3 (crown), we created a large panel of mutants with single amino acid changes, beginning with mutations that cause a documented rate-of-entry or fusion phenotype (30, 31). Other FR3 mutants were chosen by using the solved structure of gB as a guide. The DSP assay identified FR3 mutants with an enhanced rate of fusion, including mutants previously shown to have a high rate of virus entry (31, 33) and others that resembled the WT in endpoint fusion assays. Thus, a number of residues in different areas of gB FR3 contribute significantly to the optimal rate of fusion. Finally, we examined the rate of fusion induced by a double mutant with alterations in the cytoplasmic tail of gB that corresponds to one of the syn mutants, which form large syncytia (15). The size of syncytia and the endpoint luciferase assay have been used in the past to study herpesvirus cytoplasmic tail mutants (32, 37, 40). We found that these mutations markedly speed up the time of initiation (3 min for the syn mutant) and the initial fusion kinetics, suggesting that the mutations have deregulated gB so that it now functions on a “hair trigger.” This rate phenotype helps explain the syncytial phenotype of the virus and predicts that other gB syn mutants within the cytoplasmic tail will also exhibit similar fast fusion kinetics. Thus, by using the DSP assay, we have obtained significant insight into the role played by residues in two FRs of gB that help to drive HSV glycoprotein-induced cell-cell fusion.
The titration studies suggest that gB is rate-limiting for fusion, and there is no evidence for a stoichiometric relationship between gB and gH/gL.
Several reports have suggested that gB and gH/gL form a complex that carries out the fusion process (41, 42). As such, the gB trimer and the gH/gL heterodimer would be predicted to bind to each other in a stoichiometric fashion, e.g., one gH/gL heterodimer per gB protomer or one gH/gL heterodimer per gB trimer. Indeed, Vanarsdall et al. (43) have shown that cytomegalovirus (CMV) gB can be coimmunoprecipitated with gH/gL from transfected cell extracts. A similar claim has been made for the HSV homologues (41). In bimolecular complementation (BiMC) assays, we showed that gH/gL must interact with gB in order for fusion to occur but that they do not interact before fusion is triggered by the addition of soluble gD (22, 23). Our current results using the DSP assay indicate that the rate of fusion is governed by the amount of gB on the cell surface, i.e., very small amounts of gD and gH/gL are sufficient to trigger a substantial rate of fusion provided that the amount of gB on the surface is optimal. We suggest that there is no need for the formation of a tight complex between gH/gL and gB of a certain stoichiometry. However, that does not exclude the possibility that in some cases, as with CMV, a more stable complex can and does form. It is noteworthy that published studies by our lab and others indicate that fusion occurs when gB and gH/gL are in separate cells (in trans), as shown for both CMV and HSV (8, 43). Moreover, fusion can also be triggered by a combination of soluble gD and soluble gH/gL (gH with no transmembrane anchor) (8). Together, the data indicate that gH/gL need not be stably bound to gB in order to be activated and may in fact act in a catalytic manner (8), i.e., one molecule of gH/gL can activate more than one trimer of gB. The data also show that the major rate-limiting factor in cell-cell fusion is the amount of functional gB on the cell surface and potentially in the virion envelope. Furthermore, the optimal WT rate can be enhanced or inhibited by specific changes to gB. This would not exclude the possibility that the rate would also be affected, positively or negatively, by mutations in gH, gL, or even gD. Investigations of how mutations in these proteins alter fusion rates is under study.
How do syn mutants alter fusion rates?
syn mutants are fascinating in their effects on the fusion of HSV-infected cells. As is well known, HSV does not normally cause syncytia during human infections. Many of the syn mutants with large-plaque phenotypes have mutations in the cytoplasmic domain of gB (15, 30, 31, 44, 45). Many other mutants with deletions of residues in the cytoplasmic tail or other single or double amino acid changes have a similar phenotype (35, 37, 40, 57). The LL871/872AA syn mutant that we studied in this report, though in a cell-cell fusion assay, showed remarkably enhanced fusion over that of WT gB by an early time of initiation (3 min). Indeed, as mentioned above, this mutant appears to be poised to trigger fusion as soon as receptor-expressing cells are added, and although the kinetics slowed by 2 h after initiation, the final rate of fusion was still higher than that for WT gB. Thus, it appears that this mutant and perhaps other hyperfusogenic mutants may be at the same energy level, poised to initiate fusion. We suspect that these mutants may be able to carry out fusion in the presence of gH/gL and gD in the absence of a gD receptor. A precedent for this scenario has been described for mutant HSVs that can infect cells in the absence of a gD receptor (34, 46, 47). These viruses have compensatory mutations in gB, gD, and gH/gL. The question of whether hyperfusogenic mutants can operate in the absence of a gD receptor warrants further investigation.
The question of how a mutation in the cytoplasmic domain of gB alters the rate of fusion, which occurs on the cell surface, has been raised numerous times. Silverman et al. (37) showed that the cytoplasmic tail of gB is trimeric and that the trimeric structure is maintained through the transmembrane region and cytoplasmic parts of gB. Muggeridge et al. (32) also suggested that changes to the cytoplasmic domain may alter the structure of the ectodomain. However, both labs, using conformation-dependent MAbs, failed to detect structural differences between WT and mutant gB. This does not exclude the possibility that the conformational changes in gB during fusion would involve the movement of entire domains that preserve the epitopes for conformational MAbs, rather than extended local rearrangements.
Rates of fusion may help explain rate-of-entry mutants.
A number of rate-of-entry mutations that map to the ectodomain of gB have been identified. It is striking that many map specifically to FR3 (domain IV of Heldwein et al. [4]). These include the G594R mutation, which slows entry (31), and V553A, which speeds up entry (30). We found a similar defect in the rate of fusion for G594R and detected significantly faster cell fusion when V553 was changed to leucine. Recently, Uchida et al. reported that a double mutation in gB, D285N A549T, markedly enhanced the rate of virus entry (34). Residue D285 is in FR1, and A549 is in FR3. Of the two mutations, the phenotype was ascribed mostly to A549T, since this mutation alone was sufficient to enhance infectivity regardless of the cell type or route of entry. We would predict that A549T, located near W539 and V553 (both with hyperfusogenic mutants), will lead to fast fusion kinetics and that the D285N A549T double mutation may further enhance the rate of fusion. Conversely, it would be interesting to examine the effects of these high-rate-of-fusion crown mutations on the rate of virus entry and plaque formation.
A model for prefusion gB provides a basis for understanding the complex kinetics of gB FR3 mutants.
Uchida et al. (34) point out that structural domain III of gB is predicted by models of prefusion EBV gB to undergo critical changes in conformation as the protein transitions from a prefusion to a postfusion conformation (3). The rate-of-virus-entry mutations that map to FR3 (V553A) and other mutations in FR3 that positively affect fusion rates also highlight the potential importance of this region in determining the rate of change from a pre- to a postfusion conformation. In the absence of a structure for prefusion gB, we constructed a “prefusion model” of this protein based on the arrangement of the homologous domains of VSV G in its known prefusion form (7) and the hypothetical model constructed for EBV gB (3). Because both forms of VSV G are known, we speculate that as in VSV G, fusion in HSV is driven primarily by changes in the positions of the five structural domains of gB relative to each other, rather than by changes within individual domains. However, the relative position of the same amino acid in separate protomers may also change. If our in silico model approximates reality, then FR3 (and domain III) residues located on the outside of the postfusion structure likely start out positioned in the center of prefusion gB. In this model, three copies of H657 sit at the interface of three protomers pointing toward the central stalk of another protomer, but each is still solvent exposed (Fig. 8). If this residue were mutated to arginine, which is essentially always positively charged, one can imagine that it would cause repulsion between protomers. This would provide enough force to destabilize this portion of FR3, open it, and drive it more rapidly into intermediate forms in which this altered residue is more readily accommodated. Similarly, mutation of residues E607, D608, and Q609 might destabilize a FR2–FR3 interface, accounting for the faster kinetics of fusion. However, the initiation of fusion by these mutants would be very similar to that for WT gB (Table 2), because these residues should be more accessible than H657. We acknowledge that our VSV G-based model of prefusion gB is far from perfect, due to differential requirements for accessory proteins (e.g., gH/gL and gD for HSV gB) and pH dependence (absolute for VSV G but cell type dependent for HSV gB). Moreover, we cannot model gB domain V, because it has no counterpart in VSV G. However, we believe that this model provides a framework for understanding the phenotypes of some of our mutants with altered fusion rates.
Fig 8.
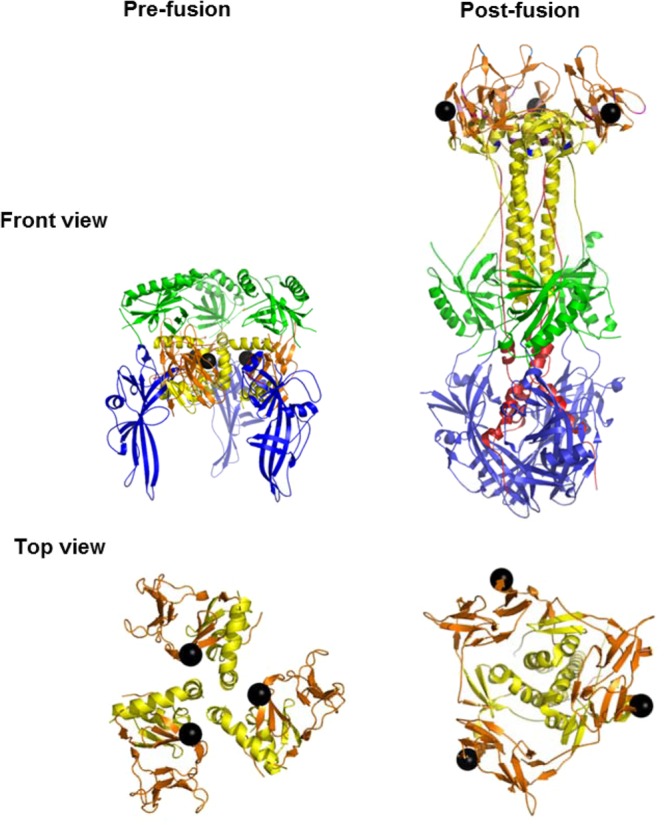
Proposed model of prefusion gB. The prefusion gB structure was generated using the MatchMaker program (UCSF Chimera) by aligning structural domains of postfusion HSV gB to the known structural domains of the prefusion form of VSV G. H657 (black spheres) is shown. This residue is located in the center of the prefusion model (front view), covered by FR2 (green), and on the outside of each protomer in the postfusion form. The relationships of H657 with other residues in the crown (brown) and stalk (yellow) change as gB transitions from a pre- to a postfusion structure; this is more evident in the top views, where only domains III (yellow) and IV (brown) are shown.
What are the intermediate steps in fusion going from a trimeric prefusion form to the postfusion structure we know?
Recently, Albertini et al. (48) presented data suggesting that in contrast to its crystal structure, VSV G assumes a monomeric structure in solution. Indeed, as they point out, it is well established that class II fusion proteins “transit from a (homo- or hetero-) pre-fusion dimer to a post-fusion trimer through an intermediate monomer” (reference 48 and references therein; see also references 49–52, and 53). Based on their own data and on earlier reports about VSV G, they proposed that VSV G on the virion surface is present in a structure similar to that of the crystallized prefusion trimer (48). Once VSV virions contact a cell and are internalized into an acidic endosome, the trimers dissociate into monomers and then, at an even lower pH (pH 6 or below), transition into the postfusion form as fusion is triggered between an endosomal membrane and the virion envelope containing VSV G. As we know, the gB trimer can dissociate into monomers at low pH values (27, 29, 54), and a mechanism similar to that for VSV G might be at work for gB when HSV is taken up by endocytosis. However, in some cells, e.g., Vero cells, HSV fusion occurs at the plasma membrane in a pH-independent manner (55), suggesting that the fusion pathway is different (i.e., there is no monomeric intermediate). Alternatively, there might be local pH changes or compensatory environmental cues on these cells that in some way make up for the low pH of the endosome.
Clearly, many of the tantalizing questions raised by the studies reported here can be answered only when we know more about the prefusion structure of gB. Although recent attempts to stabilize such a form have not succeeded (56), our hypothesis remains that prefusion gB will bear some resemblance to prefusion VSV G. In any event, however, the dual split-reporter assay has been very useful in identifying new phenotypes. Although this assay does not offer details about hemifusion, pore formation, or pore enlargement (steps preceding content mixing), its superior ability to measure fusion (as defined by content mixing) of live cells in real time should make it the standard for using luciferase-based assays to study HSV-induced cell fusion.
ACKNOWLEDGMENTS
The research reported in this publication was supported by NIH grants R01-AI-076231 and R01-AI-056045 to R.J.E. and R37-AI-18289 to G.H.C. J.R.G. was supported by NIH training grant T32-AI-07234. Z.M. was supported by a contract research fund from the Ministry of Education, Culture, Sports, Science and Technology for Program of Japan Initiative for Global Research Network on Infectious Diseases.
We thank all the members of our laboratory for helpful advice. We particularly thank Leslie King, School of Veterinary Medicine, for wonderful editorial input.
The content of this paper is solely the responsibility of the authors and does not represent the official views of the National Institutes of Health.
Footnotes
Published ahead of print 14 August 2013
REFERENCES
- 1.Eisenberg RJ, Atanasiu D, Cairns TM, Gallagher JR, Krummenacher C, Cohen GH. 2012. Herpes virus fusion and entry: a story with many characters. Viruses 4:800–832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Spear PG, Eisenberg RJ, Cohen GH. 2000. Three classes of cell surface receptors for alphaherpesvirus entry. Virology 275:1–8 [DOI] [PubMed] [Google Scholar]
- 3.Backovic M, Longnecker R, Jardetzky TS. 2009. Structure of a trimeric variant of the Epstein-Barr virus glycoprotein B. Proc. Natl. Acad. Sci. U. S. A. 106:2880–2885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heldwein EE, Lou H, Bender FC, Cohen GH, Eisenberg RJ, Harrison SC. 2006. Crystal structure of glycoprotein B from herpes simplex virus 1. Science 313:217–220 [DOI] [PubMed] [Google Scholar]
- 5.Kadlec J, Loureiro S, Abrescia NG, Stuart DI, Jones IM. 2008. The postfusion structure of baculovirus gp64 supports a unified view of viral fusion machines. Nat. Struct. Mol. Biol. 15:1024–1030 [DOI] [PubMed] [Google Scholar]
- 6.Roche S, Bressanelli S, Rey FA, Gaudin Y. 2006. Crystal structure of the low-pH form of the vesicular stomatitis virus glycoprotein G. Science 313:187–191 [DOI] [PubMed] [Google Scholar]
- 7.Roche S, Rey FA, Gaudin Y, Bressanelli S. 2007. Structure of the prefusion form of the vesicular stomatitis virus glycoprotein G. Science 315:843–848 [DOI] [PubMed] [Google Scholar]
- 8.Atanasiu D, Saw WT, Cohen GH, Eisenberg RJ. 2010. Cascade of events governing cell-cell fusion induced by herpes simplex virus glycoproteins gD, gH/gL, and gB. J. Virol. 84:12292–12299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Connolly SA, Jackson JO, Jardetzky TS, Longnecker R. 2011. Fusing structure and function: a structural view of the herpesvirus entry machinery. Nat. Rev. Microbiol. 9:369–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kondo N, Miyauchi K, Meng F, Iwamoto A, Matsuda Z. 2010. Conformational changes of the HIV-1 envelope protein during membrane fusion are inhibited by the replacement of its membrane-spanning domain. J. Biol. Chem. 285:14681–14688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ishikawa H, Meng F, Kondo N, Iwamoto A, Matsuda Z. 2012. Generation of a dual-functional split-reporter protein for monitoring membrane fusion using self-associating split GFP. Protein Eng. Des Sel. 25:813–820 [DOI] [PubMed] [Google Scholar]
- 12.Bender FC, Samanta M, Heldwein EE, de Leon MP, Bilman E, Lou H, Whitbeck JC, Eisenberg RJ, Cohen GH. 2007. Antigenic and mutational analyses of herpes simplex virus glycoprotein B reveal four functional regions. J. Virol. 81:3827–3841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hannah BP, Heldwein EE, Bender FC, Cohen GH, Eisenberg RJ. 2007. Mutational evidence of internal fusion loops in herpes simplex virus glycoprotein B. J. Virol. 81:4858–4865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hannah BP, Cairns TM, Bender FC, Whitbeck JC, Lou H, Eisenberg RJ, Cohen GH. 2009. Herpes simplex virus glycoprotein B associates with target membranes via its fusion loops. J. Virol. 83:6825–6836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beitia Ortiz de Zarate I, Cantero-Aguilar L, Longo M, Berlioz-Torrent C, Rozenberg F. 2007. Contribution of endocytic motifs in the cytoplasmic tail of herpes simplex virus type 1 glycoprotein B to virus replication and cell-cell fusion. J. Virol. 81:13889–13903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miller CG, Krummenacher C, Eisenberg RJ, Cohen GH, Fraser NW. 2001. Development of a syngenic murine B16 cell line-derived melanoma susceptible to destruction by neuroattenuated HSV-1. Mol. Ther. 3:160–168 [DOI] [PubMed] [Google Scholar]
- 17.Okuma K, Nakamura M, Nakano S, Niho Y, Matsuura Y. 1999. Host range of human T-cell leukemia virus type I analyzed by a cell fusion-dependent reporter gene activation assay. Virology 254:235–244 [DOI] [PubMed] [Google Scholar]
- 18.Pertel PE, Fridberg A, Parish ML, Spear PG. 2001. Cell fusion induced by herpes simplex virus glycoproteins gB, gD, and gH-gL requires a gD receptor but not necessarily heparan sulfate. Virology 279:313–324 [DOI] [PubMed] [Google Scholar]
- 19.Connolly SA, Landsburg DJ, Carfi A, Wiley DC, Cohen GH, Eisenberg RJ. 2003. Structure-based mutagenesis of herpes simplex virus glycoprotein D defines three critical regions at the gD-HveA/HVEM binding interface. J. Virol. 77:8127–8140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Geraghty RJ, Jogger CR, Spear PG. 2000. Cellular expression of alphaherpesvirus gD interferes with entry of homologous and heterologous alphaherpesviruses by blocking access to a shared gD receptor. Virology 268:147–158 [DOI] [PubMed] [Google Scholar]
- 21.Milne RS, Connolly SA, Krummenacher C, Eisenberg RJ, Cohen GH. 2001. Porcine HveC, a member of the highly conserved HveC/nectin 1 family, is a functional alphaherpesvirus receptor. Virology 281:315–328 [DOI] [PubMed] [Google Scholar]
- 22.Atanasiu D, Whitbeck JC, Cairns TM, Reilly B, Cohen GH, Eisenberg RJ. 2007. Bimolecular complementation reveals that glycoproteins gB and gH/gL of herpes simplex virus interact with each other during cell fusion. Proc. Natl. Acad. Sci. U. S. A. 104:18718–18723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Atanasiu D, Whitbeck JC, de Leon MP, Lou H, Hannah BP, Cohen GH, Eisenberg RJ. 2010. Bimolecular complementation defines functional regions of herpes simplex virus gB that are involved with gH/gL as a necessary step leading to cell fusion. J. Virol. 84:3825–3834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Atanasiu D, Cairns TM, Whitbeck JC, Saw WT, Rao S, Eisenberg RJ, Cohen GH. 2013. Regulation of herpes simplex virus gB-induced cell-cell fusion by mutant forms of gH/gL in the absence of gD and cellular receptors. mBio 4(2):e00046–13. 10.1128/mBio.00046-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. 2004. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25:1605–1612 [DOI] [PubMed] [Google Scholar]
- 26.Wright CC, Wisner TW, Hannah BP, Eisenberg RJ, Cohen GH, Johnson DC. 2009. Fusion between perinuclear virions and the outer nuclear membrane requires the fusogenic activity of herpes simplex virus gB. J. Virol. 83:11847–11856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stampfer SD, Lou H, Cohen GH, Eisenberg RJ, Heldwein EE. 2010. Structural basis of local, pH-dependent conformational changes in glycoprotein B from herpes simplex virus type 1. J. Virol. 84:12924–12933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bender FC, Whitbeck JC, Lou H, Cohen GH, Eisenberg RJ. 2005. Herpes simplex virus glycoprotein B binds to cell surfaces independently of heparan sulfate and blocks virus entry. J. Virol. 79:11588–11597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cairns TM, Whitbeck JC, Lou H, Heldwein EE, Chowdary TK, Eisenberg RJ, Cohen GH. 2011. Capturing the herpes simplex virus core fusion complex (gB-gH/gL) in an acidic environment. J. Virol. 85:6175–6184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bzik DJ, Fox BA, DeLuca NA, Person S. 1984. Nucleotide sequence of a region of the herpes simplex virus type 1 gB glycoprotein gene: mutations affecting rate of virus entry and cell fusion. Virology 137:185–190 [DOI] [PubMed] [Google Scholar]
- 31.Highlander SL, Dorney DJ, Gage PJ, Holland TC, Cai W, Person S, Levine M, Glorioso JC. 1989. Identification of mar mutations in herpes simplex virus type 1 glycoprotein B which alter antigenic structure and function in virus penetration. J. Virol. 63:730–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Muggeridge MI, Grantham ML, Johnson FB. 2004. Identification of syncytial mutations in a clinical isolate of herpes simplex virus 2. Virology 328:244–253 [DOI] [PubMed] [Google Scholar]
- 33.Saharkhiz-Langroodi A, Holland TC. 1997. Identification of the fusion-from-without determinants of herpes simplex virus type 1 glycoprotein B. Virology 227:153–159 [DOI] [PubMed] [Google Scholar]
- 34.Uchida H, Chan J, Goins WF, Grandi P, Kumagai I, Cohen JB, Glorioso JC. 2010. A double mutation in glycoprotein gB compensates for ineffective gD-dependent initiation of herpes simplex virus type 1 infection. J. Virol. 84:12200–12209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lin E, Spear PG. 2007. Random linker-insertion mutagenesis to identify functional domains of herpes simplex virus type 1 glycoprotein B. Proc. Natl. Acad. Sci. U. S. A. 104:13140–13145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Connolly SA, Longnecker R. 2012. Residues within the C-terminal arm of the herpes simplex virus 1 glycoprotein B ectodomain contribute to its refolding during the fusion step of virus entry. J. Virol. 86:6386–6393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Silverman JL, Greene NG, King DS, Heldwein EE. 2012. Membrane requirement for folding of the herpes simplex virus 1 gB cytodomain suggests a unique mechanism of fusion regulation. J. Virol. 86:8171–8184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kondo N, Miyauchi K, Matsuda Z. 2011. Monitoring viral-mediated membrane fusion using fluorescent reporter methods. Curr. Protoc. Cell Biol. Chapter 26:Unit 26.9. 10.1002/0471143030.cb2609s50 [DOI] [PubMed] [Google Scholar]
- 39.Bender FC, Whitbeck JC, Ponce de Leon M, Lou H, Eisenberg RJ, Cohen GH. 2003. Specific association of glycoprotein B with lipid rafts during herpes simplex virus entry. J. Virol. 77:9542–9552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Garcia NJ, Chen J, Longnecker R. 2013. Modulation of Epstein-Barr virus glycoprotein B (gB) fusion activity by the gB cytoplasmic tail domain. mBio 4(1):e00571–12. 10.1128/mBio.00571-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Avitabile E, Forghieri C, Campadelli-Fiume G. 2009. Cross talk among the glycoproteins involved in herpes simplex virus entry and fusion: the interaction between gB and gH/gL does not necessarily require gD. J. Virol. 83:10752–10760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fuller AO, Santos RE, Spear PG. 1989. Neutralizing antibodies specific for glycoprotein H of herpes simplex virus permit viral attachment to cells but prevent penetration. J. Virol. 63:3435–3443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vanarsdall AL, Ryckman BJ, Chase MC, Johnson DC. 2008. Human cytomegalovirus glycoproteins gB and gH/gL mediate epithelial cell-cell fusion when expressed either in cis or in trans. J. Virol. 82:11837–11850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baghian A, Huang L, Newman S, Jayachandra S, Kousoulas KG. 1993. Truncation of the carboxy-terminal 28 amino acids of glycoprotein B specified by herpes simplex virus type 1 mutant amb1511-7 causes extensive cell fusion. J. Virol. 67:2396–2401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gage PJ, Levine M, Glorioso JC. 1993. Syncytium-inducing mutations localize to two discrete regions within the cytoplasmic domain of herpes simplex virus type 1 glycoprotein B. J. Virol. 67:2191–2201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Uchida H, Chan J, Shrivastava I, Reinhart B, Grandi P, Glorioso JC, Cohen JB. 2013. Novel mutations in gB and gH circumvent the requirement for known gD receptors in HSV-1 entry and cell-to-cell spread. J. Virol. 87:1430–1442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Uchida H, Shah WA, Ozuer A, Frampton AR, Jr, Goins WF, Grandi P, Cohen JB, Glorioso JC. 2009. Generation of herpesvirus entry mediator (HVEM)-restricted herpes simplex virus type 1 mutant viruses: resistance of HVEM-expressing cells and identification of mutations that rescue nectin-1 recognition. J. Virol. 83:2951–2961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Albertini AA, Merigoux C, Libersou S, Madiona K, Bressanelli S, Roche S, Lepault J, Melki R, Vachette P, Gaudin Y. 2012. Characterization of monomeric intermediates during VSV glycoprotein structural transition. PLoS Pathog. 8:e1002556. 10.1371/journal.ppat.1002556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barth BU, Wahlberg JM, Garoff H. 1995. The oligomerization reaction of the Semliki Forest virus membrane protein subunits. J. Cell Biol. 128:283–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Harrison SC. 2008. Viral membrane fusion. Nat. Struct. Mol. Biol. 15:690–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kielian M, Rey FA. 2006. Virus membrane-fusion proteins: more than one way to make a hairpin. Nat. Rev. Microbiol. 4:67–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wahlberg JM, Bron R, Wilschut J, Garoff H. 1992. Membrane fusion of Semliki Forest virus involves homotrimers of the fusion protein. J. Virol. 66:7309–7318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zagouras P, Rose JK. 1993. Dynamic equilibrium between vesicular stomatitis virus glycoprotein monomers and trimers in the Golgi and at the cell surface. J. Virol. 67:7533–7538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dollery SJ, Wright CC, Johnson DC, Nicola AV. 2011. Low-pH-dependent changes in the conformation and oligomeric state of the prefusion form of herpes simplex virus glycoprotein B are separable from fusion activity. J. Virol. 85:9964–9973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Milne RS, Nicola AV, Whitbeck JC, Eisenberg RJ, Cohen GH. 2005. Glycoprotein D receptor-dependent, low-pH-independent endocytic entry of herpes simplex virus type 1. J. Virol. 79:6655–6663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vitu E, Sharma S, Stampfer SD, Heldwein EE. 2013. Extensive mutagenesis of the HSV-1 gB ectodomain reveals remarkable stability of its postfusion form. J. Mol. Biol. 425:2056–2071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chowdary TK, Heldwein EE. 2010. Syncytial phenotype of C-terminally truncated herpes simplex virus type 1 gB is associated with diminished membrane interactions. J. Virol. 84:4923–4935 [DOI] [PMC free article] [PubMed] [Google Scholar]