

Abstract
Rift Valley fever virus (RVFV), an ambisense member of the family Bunyaviridae, genus Phlebovirus, is the causative agent of Rift Valley fever, an important zoonotic infection in Africa and the Middle East. Phlebovirus proteins are translated from virally transcribed mRNAs that, like host mRNA, are capped but, unlike host mRNAs, are not polyadenylated. Here, we investigated the role of PABP1 during RVFV infection of HeLa cells. Immunofluorescence studies of infected cells demonstrated a gross relocalization of PABP1 to the nucleus late in infection. Immunofluorescence microscopy studies of nuclear proteins revealed costaining between PABP1 and markers of nuclear speckles. PABP1 relocalization was sharply decreased in cells infected with a strain of RVFV lacking the gene encoding the RVFV nonstructural protein S (NSs). To determine whether PABP1 was required for RVFV infection, we measured the production of nucleocapsid protein (N) in cells transfected with small interfering RNAs (siRNAs) targeting PABP1. We found that the overall percentage of RVFV N-positive cells was not changed by siRNA treatment, indicating that PABP1 was not required for RVFV infection. However, when we analyzed populations of cells producing high versus low levels of PABP1, we found that the percentage of RVFV N-positive cells was decreased in cell populations producing physiologic levels of PABP1 and increased in cells with reduced levels of PABP1. Together, these results suggest that production of the NSs protein during RVFV infection leads to sequestration of PABP1 in the nuclear speckles, creating a state within the cell that favors viral protein production.
INTRODUCTION
First identified in Kenya in the 1930s, Rift Valley fever virus (RVFV) causes a zoonotic disease of medical and financial significance across Africa and the Middle East (1). RVFV primarily affects livestock, causing acute fever and spontaneous abortion in 80 to 100% of pregnant animals (2, 3). High rates of mortality are observed in animal infections, with the highest mortality in young animals (4, 5). Humans may become infected with RVFV from bites of infected mosquitoes or contact with infected animal tissues. RVF in humans typically presents as a mild febrile illness; however, serious complications occur in 1 to 3% of patients, including retinitis leading to blindness, encephalitis, acute hepatitis, hemorrhagic fever, and death (6, 7). Because of the potential threat that RVFV poses to both human and animal health, it is listed as an overlap select agent on the National Select Agent Registry. No effective interventional therapy for Rift Valley fever is currently available, and thus, a better understanding of the basic molecular biology of the infection might help establish targetable areas of the viral replication cycle.
RVFV, a member of the Phlebovirus genus in the family Bunyaviridae, is an enveloped virus with three RNA genome segments, termed L, M, and S. Within virions, each segment is complexed with the nucleocapsid protein (N) (8). The RVFV genome segments encode seven proteins, L and M in negative sense and S in ambisense. The L segment encodes the RNA-dependent RNA polymerase (9–11). The M segment encodes the two glycoproteins Gn and Gc, as well as a nonstructural protein, NSm, and a protein termed the 78-kDa protein (12, 13). The glycoproteins are essential for attachment and entry, as well as assembly (14). While NSm is dispensable for replication in cell culture and animal models, it has been shown to inhibit apoptosis in infected cells (15–18). Additionally, NSm has been shown to be important for RVFV infection and transmission in mosquitoes (19). The S segment encodes the nucleocapsid protein (N) in negative sense and a nonstructural protein, NSs, in positive sense (20). N has several functions that are important for viral replication. N binds along the length of the viral RNA as a ribonucleoprotein (RNP) complex. Binding of N to the viral RNA is required for viral transcription and replication (21). Additionally, during assembly, N is thought to interact with Gn to bring the viral RNPs and glycoprotein-containing membranes together prior to release from the cell (14). NSs is not absolutely required for replication in culture, but viruses lacking NSs are greatly attenuated in animals (22–24). NSs has several described functions. NSs directly targets host RNA synthesis by sequestering two components of the TFIIH transcription factor and degrading a third (25, 26). Additionally, NSs has been shown to have immunomodulatory functions. The expression of NSs leads to a block in the production of antiviral interferons and the degradation of protein kinase R (27–30).
Poly(A) binding protein 1 (PABP1) plays an important role in host protein translation. PABP1 binds the 3′ poly(A) tail found on most host mRNAs and interacts with the 5′ cap-binding eIF4F complex (31–33). Interactions between PABP1 and the eIF4F complex member eIF4G have a positive effect on protein translation by increasing eIF4F-cap binding affinity, enhancing the efficiency of ribosome initiation complex formation, and increasing the rate of protein translation (34–37).
Many viruses have developed protein translation strategies that allow them to translate viral proteins independently of PABP1. Many of these viruses have been shown to perturb PABP1's location and/or function, creating a cellular environment in which viral protein translation is favored. For example, the polio virus 3C protease cleaves PABP1, inhibiting translation of host mRNA (38, 39). Herpes simplex virus (HSV) proteins ICP27 and UL47 both bind to PABP1, leading to a decrease in PABP1's interaction with binding partners eIF4G and PABP-interacting protein 2 (40). Additionally, PABP1 partially relocalizes to the nucleus during HSV-1 infection (40, 41). Rotavirus protein NSP3A functionally replaces and evicts PABP1 from the eIF4F complex and leads to nuclear relocalization of PABP1 (42, 43). Kaposi's sarcoma-associated herpesvirus SOX protein expression leads to PABP1 relocalization to the nucleus (44). Each of the described perturbations of PABP1 has been hypothesized to be related to the shutoff of host protein synthesis.
Unlike most host mRNAs, phlebovirus mRNAs do not contain poly(A) tails (45, 46). Thus, the production of RVFV protein is likely independent of PABP1. To date, only one study has appeared in which PABP1 has been studied with a bunyavirus. In that study, infection with Bunyamwera virus (BUNV), a member of the Orthobunyavirus genus, was shown to result in relocalization of PABP1 to the nucleus (47). The investigators also showed that there was an association of BUNV N and PABP1 in the cytoplasm but did not determine whether the binding was specific or led to nuclear targeting of PABP1. Although BUNV and RVFV share some replication strategies, they differ both in the coding and the function of their NSs proteins. For BUNV, a small (11-kDa) NSs is encoded in an overlapping reading frame with N. For RVFV, ambisense coding produces a 31-kDa NSs. In addition, differing functions of the NSs proteins of orthobunyaviruses and phleboviruses have been identified in several studies (28, 48, 49). Consequently, it cannot be assumed that findings with BUNV also apply to bunyaviruses in other genera of the family, such as phleboviruses. Here, we have investigated the role of PABP1 in the replication of RVFV. Our findings are consistent with those reported earlier for BUNV but extend those results by more clearly defining a role for NSs in PABP1 perturbation during infection with RVFV, determining that the transcriptional inhibition activity of NSs mediates this phenomenon, and showing a negative correlation between RVFV replication and high levels of PABP1. Additionally, our observation that PABP1 accumulates in nuclear speckles suggests a role for mRNA in this phenomenon.
MATERIALS AND METHODS
Cells and viruses.
Unless otherwise noted, all experiments were performed with HeLa cells and the MP12 strain of RVFV (50). Hantaan virus (HTNV) (strain 76-118) and Andes virus (ANDV) (strain 808034) infections were performed in A549 cells. HeLa and A549 cells were maintained in modified essential medium (MEM) supplemented with 10% (vol/vol) fetal calf serum (FCS), 75 U/ml penicillin-streptomycin, and 2 mM l-glutamine. Infections were performed by adding virus to cell cultures. Following 1 h of incubation, the inoculum was removed and replaced with fresh MEM. Cells were infected at various multiplicities of infection (MOIs) ranging from 2 to 10.
Immunofluorescence.
RVFV-infected samples and accompanying mock-infected samples were fixed by submersion in 4% (wt/vol) paraformaldehyde (PFA) for 10 min. Hantaan and Andes virus-infected samples and accompanying mock-infected samples were fixed by submersion in 10% (vol/vol) formaldehyde for 24 h. After fixation, cells were permeabilized by submersion in ice-cold methanol for 5 min. Nonspecific binding sites were blocked for 1 h at room temperature in 5% (vol/vol) goat serum. Primary antibody incubation proceeded for 1 h at room temperature at antibody-specific optimized dilutions in 5% (vol/vol) goat serum. Cells were washed three times with 1× phosphate-buffered saline (PBS). Secondary antibodies (Alexa Fluor-conjugated goat anti-mouse and/or goat anti-rabbit secondary antibodies [Life Technologies]) were added at a dilution of 1:2,000 for 1 h at room temperature. Cells were then washed three times in PBS and mounted on slides with mounting medium containing diamidino-2-phenylindole (DAPI) (Prolong Gold, Life Technologies). Slides were allowed to cure overnight at room temperature prior to imaging.
As the fixation/permeabilization method can sometimes influence protein localization, a second permeabilization method was tested. For this, cells were fixed by submersion in 4% (wt/vol) PFA for 10 min and permeabilized by submersion in 0.2% (vol/vol) Triton X-100 in water for 10 min. The staining patterns were identical to those observed with methanol permeabilization (data not shown).
Surface sensing of translation (SUnSET) assay.
RVFV-infected and accompanying mock-infected samples were treated with MEM containing 5 μM puromycin for 30 min prior to cell lysate harvest. Cells were washed two times with PBS and harvested by the addition of lysis buffer (0.5% [wt/vol] sodium deoxycholate, 50 mM Tris, pH 7.5, 150 mM NaCl, 1% [vol/vol] Igepal) with protease inhibitor (Roche Complete Ultra tabs). The protein concentrations were normalized by using the bicinchoninic acid (BCA) assay. Samples were boiled in NuPAGE LDS (lithium dodecyl sulfate) sample buffer with reducing agent (Life Technologies), and proteins were separated by gel electrophoresis and electrotransferred to polyvinylidene fluoride (PVDF) membranes. The blots were probed with anti-puromycin antibody to detect newly translated protein.
Microscopy and image processing.
All fluorescence microscopy was performed with a Zeiss Axio Observer D1 microscope, with the exception of the images in Fig. 1A, which were obtained with a Nikon Eclipse E600. Contrast enhancement was performed equally on all areas and panels of Fig. 1B and C, 4D, and 5A and C and the puromycin blot shown in Fig. 3.
Fig 1.
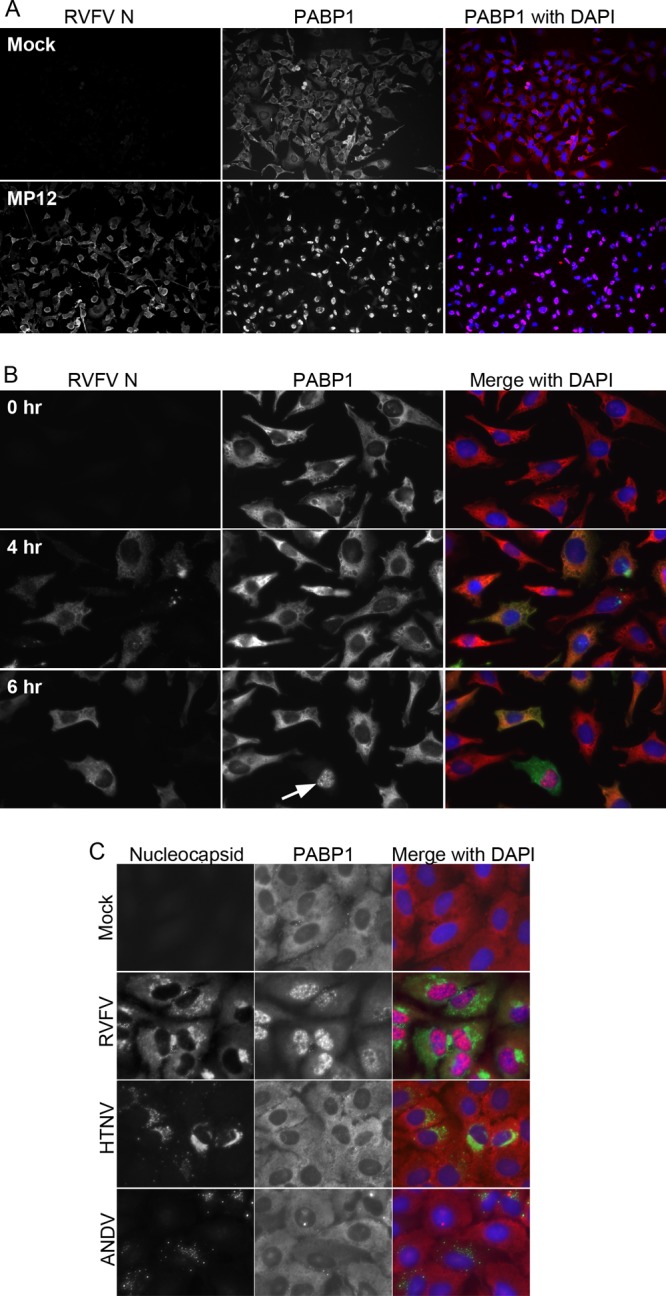
PABP1 localization during RVFV infection. (A) Immunofluorescence assay of cells infected with RVFV MP12 or mock infected for 24 h and then fixed and stained for PABP1 and RVFV N. (B) Immunofluorescence assay of RVFV MP12-infected HeLa cells. Cells were infected for 0, 4, or 6 h and then fixed and stained for PABP1 (red in overlay) and RVFV N (green in overlay). (C) Immunofluorescence assay of mock-infected, RVFV-infected, Hantaan virus-infected, or Andes virus-infected A549 cells fixed and stained for nucleocapsid and PABP1. RVFV-infected samples were fixed at 24 h postinfection. Mock-infected, Hantaan-infected, and Andes-infected samples were fixed at 72 h postinfection.
Fig 4.

PABP1 localization during overexpression of RVFV proteins. (A) Western blot analysis of HeLa cell lysates that were either untransfected, transfected with a plasmid encoding N, or transfected with a plasmid encoding the M segment. Untransfected cells gave no signal when probed with antibodies specific for Gn or N. Lysates of M-transfected but not N-transfected cells gave a positive signal when probed with antibody specific for Gn. The corresponding protein was between 51 and 64 kDa. Lysates of N-transfected but not M-transfected cells gave a positive signal when probed with antibody specific for N. The corresponding protein was just below 28 kDa. (B) Western blot analysis of lysates of HeLa cells that were untransfected or transfected with a plasmid encoding a FLAG-tagged partial NSm. Lysates of transfected but not untransfected cells gave a positive signal when probed with antibody specific for the FLAG epitope tag. The corresponding protein was well below the 28-kDa marker. (C) Western blot analysis of lysates of HeLa cells that were untransfected or transfected with a plasmid encoding a V5-tagged NSs. Lysates of transfected but not untransfected cells gave a positive signal when probed with antibody specific for the V5 epitope tag. The corresponding protein was between 28 and 39 kDa. (D) Immunofluorescence micrographs of HeLa cells individually expressing Gn, N, NSm, and NSs. Cells were transfected and stained for PABP1 and the specific viral protein being expressed. Arrow indicates nuclear accumulation of PABP1 in one NSs-expressing cell.
Fig 5.
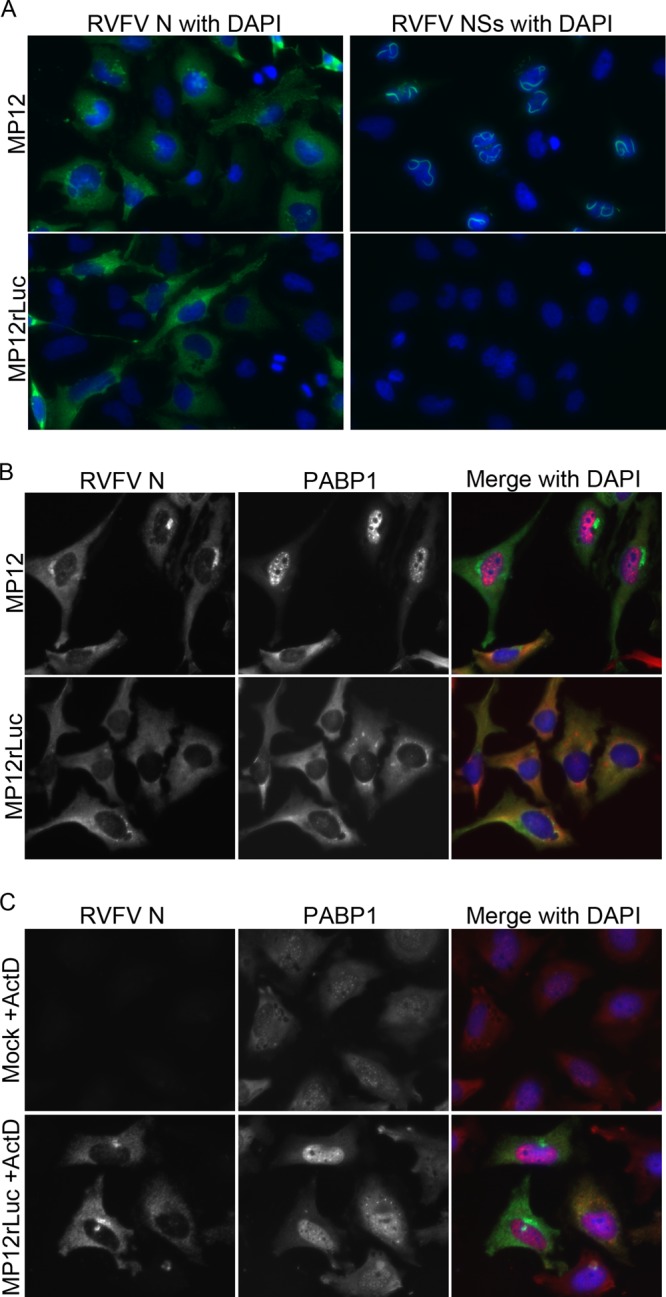
PABP1 localization in cells infected with NSs-deleted virus. (A, B) HeLa cells were infected with MP12 or MP12rLuc for 24 h and then fixed and stained. (A) Cells were stained for RVFV N or RVFV NSs. (B) Cells were stained for PABP1 and RVFV N. (C) HeLa cells were mock infected or infected with MP12rLuc and treated with actinomycin D (+ActD) for 18 h. Cells were then fixed and stained for PABP1 and RVFV N.
Fig 3.
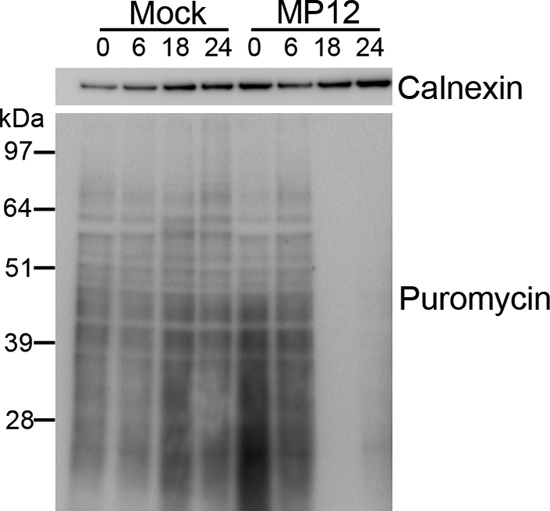
Protein translation shutdown during RVFV infection. SUnSET assay with Western blot analysis of puromycin incorporation as a marker of protein translation from mock- and MP12-infected HeLa cells harvested 0, 6, 18, and 24 h postinfection. Calnexin Western blot analysis is shown as a loading control for protein concentration.
Immunoprecipitations.
HeLa cells were grown to 80% confluence in 100-mm2 tissue culture dishes. The plates were either mock infected by the addition of noninfectious medium or infected with MP12 at an MOI of 2. Infection was allowed to proceed for 18 h at 37°C with 5% CO2. Cells were washed twice with 10 ml sterile PBS. Lysates were harvested by adding 1 ml of lysis buffer with protease inhibitor per plate, incubating the plates for 5 min on ice, and clarifying the lysates by centrifugation at 15,682 × g for 16 min at 4°C. For RNase treatment, RNase A was added to a final concentration of 0.75 μg/μl, and samples were incubated at room temperature for 1 h. An amount of 10 μg/ml RVFV N antibody was added, and samples were incubated with rotation overnight at 4°C. One-tenth volume of protein G-conjugated magnetic beads (Dynabeads-Life Technologies) were added and incubated with rotation for 30 min at 4°C. Beads were washed twice with 2 to 3 volumes lysis buffer and three times with PBS. Beads were boiled in NuPAGE LDS sample buffer with reducing agent (Life Technologies), and proteins were separated by gel electrophoresis, electrotransferred to PVDF membranes, and detected by Western blot assay.
Transfection.
HeLa cells were grown to 75% confluence in MEM supplemented with 10% (vol/vol) FCS. Plasmid DNA was diluted to 1 μg/50 μl in Opti-MEM. FuGene HD transfection reagent (Promega) was added at a FuGene-to-plasmid ratio of 3 μl to 2 μg. The FuGene-DNA solution was incubated at room temperature for 15 min and then added dropwise to cells. At 24 h posttransfection, sets of transfected cells were fixed for immunofluorescence assay (IFA) as described above or lysates were harvested for Western blot assay by the addition of lysis buffer.
Actinomycin D treatments.
HeLa cells were plated to allow for 75% confluence on the day of infection. Virus was added to cells diluted in growth medium. After 1 h of incubation, the inoculum was removed and replaced with growth medium containing 0.083 μg/ml actinomycin D (Sigma).
Flow cytometry.
Cells were washed, trypsinized, and suspended in fluorescence-activated cell sorting (FACS) buffer (1× PBS, 2.5% [vol/vol] FCS, 0.12% [wt/vol] sodium azide) at approximately 2 × 105 cells per sample. The cells were fixed in BD Cytofix buffer for 30 min at 4°C, washed in FACS buffer, and permeabilized in BD Perm/Wash buffer for 15 min at room temperature. The cells were stained in primary antibody diluted in BD Perm/Wash buffer for 30 min at 4°C, washed four times in BD Perm/Wash buffer, and then incubated with secondary antibody diluted in BD Perm/Wash buffer for 20 min at 4°C in the dark. The cells were again washed four times with BD Perm/Wash buffer and then were resuspended in FACS buffer and counted on a BD FACSCalibur instrument using Cellquest software (BD Biosciences). The data were formatted using FlowJo, version 8.8.2 (Tree Star).
Gates distinguishing high and low PABP1 expressors were drawn based on nontargeting small interfering RNA (siRNA)-transfected controls and PABP1 siRNA-transfected controls. In multiple experiments, siRNA-treated samples did not give two clearly distinct populations. Therefore, gates were drawn that placed a majority of untransfected cells in the PABP1 high gate and a majority of siRNA-transfected cells in the PABP1 low gate. These gates were then applied to all samples.
To validate that the percentage of RVFV N-positive cells as determined by our flow cytometry assay correlated with the MOI, we performed infections at MOIs of 1 and 0.1. The percentages of RVFV-positive cells adjusted roughly with the MOI (data not shown).
Antibodies.
The following primary antibodies and antibody concentrations were used: RVFV N R3-1D8-1-1a (mouse monoclonal antibody) (Western blotting, 1:1,000; IFA, 1:1,000; flow cytometry, 1:500) (J. Smith, United States Army Medical Research Institute of Infectious Diseases [USAMRIID]) and antibodies to PABP1 (Western blotting, 1:250; IFA, 1:100) (sc32318; Santa Cruz), PABP1 (Western blotting, 1:1,000; IFA, 1:1,000; flow cytometry, 1:250) (ab21060; Abcam), PABP2 (IFA, 1:500) (ab75855; Abcam), glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Western blotting, 1.5:1,000; flow cytometry, 1:250) (14C10; Cell Signaling), SC35 (IFA, 1:1,000) (ab11826; Abcam), PML (promyelocytic leukemia protein) (IFA, 1:200) (ab53773; Abcam), histone H3 (IFA, 1:200) (D1H2; Cell Signaling), FLAG (Western blotting, 1:1,000; IFA, 1:1,000) (F1804; Sigma), V5 (Western blotting, 1:5,000; IFA, 1:200) (P/N 46-0705; Invitrogen), Gn 4D4 (mouse monoclonal antibody) (Western blotting, 1:1,000; IFA, 1:500) (51), NSs 3C3 (mouse monoclonal antibody) (IFA, 1:1,000) (52), actin (Western blotting, 1:1,000) (612656; BD), calnexin (Western blotting, 1:200) (ab22595; Abcam), and puromycin (Western blotting, 1:20,000) (MABE343 clone 12D10; Millipore). Alexa Fluor-conjugated secondary antibodies were all used at 1:2,000 for IFA and 1:500 for flow cytometry (Alexa Fluor 594 goat anti-mouse, Alexa Fluor 594 goat anti-rabbit, Alexa Fluor 488 goat anti-mouse, Alexa Fluor 488 goat anti-rabbit, and Alexa Fluor 647 goat anti-rabbit antibodies; Life Technologies), and horseradish peroxidase (HRP)-conjugated secondary antibodies were both used at 1:12,500 for Western blotting (donkey anti-rabbit and sheep anti-mouse antibodies; GE Healthcare).
Plasmids.
A plasmid encoding the open reading frame of the RVFV ZH-501 N protein (pCAGGS RVFV ZH-501 N) was provided by Stuart Nichol (Centers for Disease Control and Prevention, Atlanta, GA). A V5-tagged NSs plasmid was provided by Sheli Radoshitzky, Julie Costantino, and Sina Bavari (United States Army Medical Research Institute of Infectious Diseases). A plasmid encoding the RVFV M segment was provided by Robert Doms (University of Pennsylvania). This construct was codon-optimized for expression in human cells and only contained the coding region of the M segment starting at the fourth ATG start codon, which omits the NSm coding region. For production of the plasmid encoding FLAG-tagged NSm (ZH-501 codon-optimized M segment, amino acids 15 to 130), the NSm region was amplified by PCR and introduced into the pDEST737 destination vector (a gift of Dom Esposito, NCI-Frederick) by standard Gateway cloning approaches. The final construct has a 3× FLAG tag fused in-frame with the N terminus of the NSm domain.
siRNA.
HeLa cells were plated in 24-well plates in antibiotic-free MEM with 10% (vol/vol) FCS at 50% confluence 24 h prior to transfection. A lipid transfection reagent (DharmaFect 1) was used to deliver targeted siRNA (Ambion Silencer select) and nontargeting controls at a final concentration of 25 nM (sense sequences; the targeted control proteins were PABP1 [CCUAAAUGAUCGCAAAGUATT], nucleolin [GGAUAGUUACUGACCGGGATT], and 14-3-3 epsilon [GGCAAAUGGUUGAGACUGATT]). For transfections, DharmaFect was diluted 1:10 in Opti-MEM. siRNA stocks (5 μM) were diluted 1:20 in Opti-MEM. Following a 5-min incubation at room temperature, dilute DharmaFect and dilute siRNA were combined 1:1 and incubated at room temperature for 20 min. Four volumes of MEM supplemented with 10% (vol/vol) FCS were added to each DharmaFect-siRNA solution, and the solutions were added to cells. At 24 h posttransfection, the medium was replaced with fresh MEM. At 48 h after transfection, cells were infected with RVFV MP12 at an MOI of 2. At 24 h postinfection, the cells were harvested for flow cytometry analysis.
RESULTS
PABP1 relocalizes to nuclear speckles during RVFV infection.
We evaluated PABP1 location and protein associations to determine whether PABP1 was altered during infection with RVFV. We detected neither a degradation of PABP1 nor any change in PABP1's association with eIF4F complex members during infection of HeLa cells with RVFV (data not shown), both of which have been shown to occur during viral infections (38, 39, 43). To determine whether there was a change in the localization of PABP1 during RVFV infections, we examined infected cells for PABP1 and RVFV N at various times postinfection by immunofluorescence microscopy, with RVFV N staining serving as a marker of infected cells. We first looked at cells 18 to 24 h postinfection and found that PABP1 had largely relocalized to the nucleus, with 89% of cells counted exhibiting nuclear PABP1 staining (Fig. 1A). To determine when PABP1 relocalization began, we looked at early times postinfection. Newly synthesized RVFV N was first observed at 4 h postinfection (Fig. 1B). At 4 and 6 h postinfection, RVFV N and PABP1 colocalized in the cytoplasm of infected cells (Fig. 1B). By 6 h postinfection, we began to detect PABP1 in the nucleus of select cells (Fig. 1B, arrow). Nuclear PABP1 did not costain with RVFV N.
In light of these findings and those of a previous study showing PABP1 relocalization during infection with the orthobunyavirus Bunyamwera virus (BUNV), we sought to determine whether PABP1 relocalization also occurred in infections with two viruses in the hantavirus genus of the family Bunyaviridae, Hantaan virus (HTNV) and Andes virus (ANDV). We experimentally infected A549 cells with RVFV, HTNV, or ANDV. While PABP1 relocalized during RVFV infection of A549 cells, PABP1 did not relocalize during either of the hantavirus infections. These results indicate that PABP1 relocalization is not a bunyavirus-wide phenomenon (Fig. 1C).
To determine whether nuclear PABP1 was localized to a specific subnuclear compartment, we infected HeLa cells with RVFV and immunofluorescently colabeled the cells for PABP1 and various markers of nuclear subdomains. In parallel, we stained cells for RVFV N to confirm that 100% of cells in the monolayer were infected in this experiment (Fig. 2A). Two nuclear speckle components, SC35 and PABP2, colocalized with nuclear PABP1, while markers of paraspeckles (PSPC1), PML bodies (PML), and histones (histone h3) did not (Fig. 2B). These observations confirm that during infection with RVFV, PABP1 relocalized to nuclear speckles.
Fig 2.
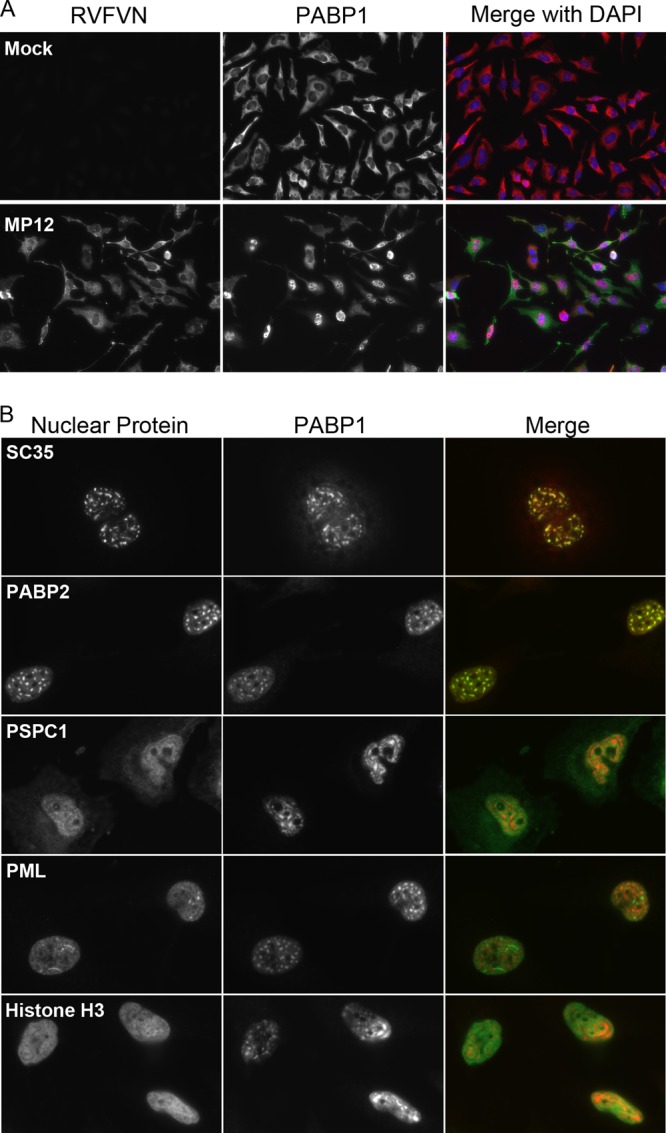
PABP1 relocalization to nuclear speckles during RVFV infection. (A) Immunofluorescence assay of RVFV MP12-infected or mock-infected HeLa cells stained for PABP1 (red in overlay) and RVFV N (green in overlay), demonstrating 100% infection at 24 h postinfection. (B) Immunofluorescence assay of RVFV MP12-infected HeLa cells stained for PABP1 and one of five markers of nuclear subdomains (SC35, PABP2, PSPC1, PML, and histone H3) at 24 h postinfection. Note the colocalization of PABP1 with SC35 and PABP2.
PABP1 relocalization correlates with decreased host protein translation.
It has previously been shown that relocalization of PABP1 to the cell nucleus correlates with a decrease in protein synthesis (53). This indicates a loss of PABP1 function upon relocalization to the nucleus. We sought to determine whether PABP1 relocalization by RVFV similarly correlated with decreased host protein translation. For this, a time course was performed with mock-infected or MP12-infected cells treated with the addition of puromycin for the detection of protein synthesis by SUnSET assay (54). Samples were harvested at 0, 6, 18, and 24 h postinfection and probed with anti-puromycin antibody. The 18-h and 24-h MP12-infected samples showed marked decreases in active protein translation compared to the levels of translation at both earlier time points and in mock-infected cells (Fig. 3). The blots were probed for calnexin as a loading control (Fig. 3). We noted above that, at 18 h to 24 h postinfection, PABP1 is relocalized to the nucleus in the majority of MP12-infected cells (Fig. 1A). These observations confirm that the RVFV-mediated nuclear sequestration of PABP1 corresponds to a decrease in host protein translation.
NSs is responsible for PABP1 relocalization.
To determine whether a specific RVFV protein was responsible for the PABP1 relocalization, we individually expressed sequences representing the RVFV M and S segments in HeLa cells and then assessed PABP1 localization by immunofluorescence. In parallel with the immunofluorescence studies, we harvested cell lysates to confirm the expression of each transfected protein by Western blot assay (Fig. 4A to C). A plasmid containing the codon-optimized M segment coding sequence from the fourth start codon was transfected, and expression was detected with an anti-Gn monoclonal antibody. As Gn and Gc are encoded in a continuous open reading frame within the M segment, we assumed that if Gn was detected, Gc was also being produced. We detected a perinuclear staining pattern, which is consistent with the expected Golgi complex localization of Gn and Gc (Fig. 4D). We did not detect colocalization of PABP1 and Gn or relocalization of PABP1. In contrast, RVFV N gene expression resulted in a cytoplasmic distribution that partially colocalized with PABP1 (Fig. 4D). No PABP1 relocalization was observed.
To specifically examine the potential roles of the nonstructural proteins, we made use of a partial NSm gene that produced a FLAG-tagged NSm protein. The NSs plasmid used produced a V5-tagged NSs. Immunostaining with anti-FLAG antibodies revealed a diffuse staining pattern for the NSm gene product and colocalization with PABP1 in the cytoplasm but did not result in PABP1 relocalization (Fig. 4D). Because NSm was so abundant throughout the micrograph, it is likely that the NSm-PABP1 colocalization was due to NSm ubiquity rather than to a specific interaction. In contrast, although only a few cells with NSs were detected by immunostaining with anti-V5 antibodies, those that were observed displayed faint cytoplasmic staining coupled with filamentous nuclear aggregates (Fig. 4D). Nuclear filaments are consistent with the expected localization of authentic NSs in RVFV-infected cells (55–57). PABP1 accumulated in the nuclei of several but not all cells expressing NSs. The relocalization observed upon NSs expression was similar to the PABP1 relocalization seen in RVFV-infected cells (Fig. 4D, arrow). These results indicated that NSs alone could affect PABP1's localization pattern.
To confirm the involvement of NSs in the observed nuclear PABP1 relocalization, we infected cells with either the MP12 vaccine strain of RVFV or with RVFV MP12rLuc, which was genetically engineered to replace the NSs gene with a luciferase gene (23). Cells were individually immunostained for N or NSs and assessed by fluorescence microscopy to confirm infection and the presence or absence of NSs protein. Both the RVFV MP12-infected and RVFV MP12rLuc-infected cells exhibited N staining in the cytoplasm (Fig. 5A). RVFV MP12-infected but not RVFV MP12rLuc-infected cells exhibited NSs staining, observed as nuclear filaments (Fig. 5A). The absence of NSs staining in RVFV MP12rLuc-infected cells is consistent with the lack of NSs gene in MP12rLuc. Next, we stained RVFV MP12-infected and RVFV MP12rLuc-infected cells with antibodies specific for RVFV N and PABP1 and assessed PABP1 location by immunofluorescence microscopy. Consistent with previous experiments, RVFV MP12-infected cells showed PABP1 relocalization to the nuclei (Fig. 5B, top). However, PABP1 remained predominantly cytoplasmic during infection with RVFV MP12rLuc (Fig. 5B, bottom). These results suggest that, in the absence of NSs expression, RVFV infection does not affect PABP1 localization. Taken together, our results indicate that NSs expression is responsible for the PABP1 relocalization during RVFV infection.
Inhibition of transcription leads to PABP1 relocalization.
We next sought to determine whether the known transcriptional inhibition activity of NSs was responsible for PABP1 relocalization. We used actinomycin D, a transcription inhibitor, as a proxy for the transcriptional inhibition activity of NSs. HeLa cells were treated with actinomycin D concurrent with infection by RVFV MP12rLuc virus or mock infection. Following infection and actinomycin D treatment, cells were fixed and immunostained with antibodies specific for RVFV N and PABP1. RVFV MP12rLuc-infected actinomycin D-treated cells exhibited nuclear PABP1 localization that was similar to the relocalization previously seen in cells infected with RVFV MP12 without actinomycin D (Fig. 5C, bottom). Additionally, low-level PABP1 relocalization was observed in mock-infected cells that were treated with actinomycin D alone (Fig. 5C, top). Mock-infected actinomycin D-treated cells exhibited punctate nuclear and diffuse cytoplasmic PABP1 staining. The nuclear accumulation of PABP1 in actinomycin D-treated mock-infected cells was less robust than that seen in either MP12 infections or MP12rLuc actinomycin D-treated cells. These results indicate that transcriptional inhibition in the context of infection is sufficient to cause PABP1 relocalization and that transcriptional inhibition alone can lead to redistribution of PABP1 to the nucleus.
PABP1 precipitates with N but not NSs or NSm.
PABP1's relocalization following actinomycin D treatment was suggestive of an indirect effect of NSs on PABP1. However, we wanted to determine if we could detect an interaction between NSs and PABP1. For this, we performed high-resolution immunofluorescence microscopy of MP12-infected cells to determine whether any PABP1-NSs colocalization could be detected. MP12-infected and mock-infected cells were stained for PABP1 and NSs. In MP12-infected cells, both proteins were found in the nucleus; however, no colocalization was detected and PABP1 appeared to be displaced in some areas where NSs filaments were present (Fig. 6A). We next wanted to determine whether an interaction could be detected by immunoprecipitation. We used the plasmids described above to overexpress RVFV NSs, partial NSm, and N in 293T cells. The proteins were immunoprecipitated with antibodies specific for V5, FLAG, or N. The resultant immune complexes were immunoblotted with PABP1-specific antibodies to determine if PABP1 was present in the precipitates. The blots were also probed with GAPDH-specific antibodies as a loading control. NSs, NSm, and N were all detected by Western blotting in transfected lysates and transfected precipitated lysates but not in untransfected lysates (Fig. 6B to D). No PABP1 was detected in NSs or NSm precipitates (Fig. 6B and C). This indicates that PABP1 does not interact with NSs or NSm and provides further evidence that the influence of NSs on PABP1 is indirect. We did find PABP1 present in samples precipitated with antibodies specific for N (Fig. 6D).
Fig 6.
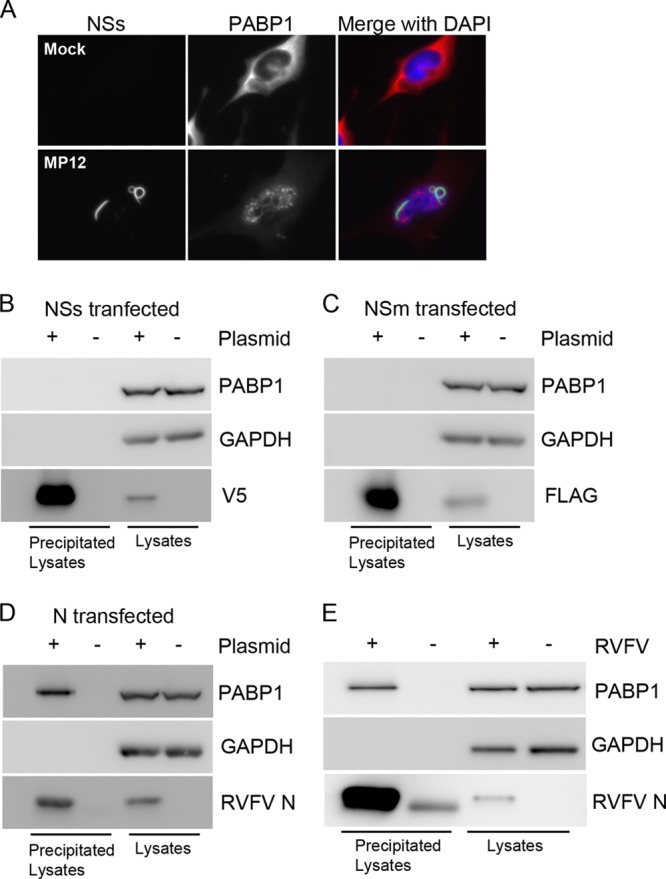
PABP1 precipitates with N but not NSs or NSm. (A) Immunofluorescence assay of nuclear NSs and PABP1. (B) Western blot analysis of lysates of NSs-transfected 293T cells. Lysates were precipitated with antibodies specific for V5. Blots were probed for PABP1, V5, and GAPDH. (C) Western blot analysis of lysates of NSm-transfected 293T cells. Lysates were precipitated with antibodies specific for FLAG. Blots were probed for PABP1, FLAG, and GAPDH. (D) Western blot analysis of lysates of N-transfected 293T cells. Lysates were precipitated with antibodies specific for N. Blots were probed for PABP1, N, and GAPDH. (E) Western blot analysis of lysates of RVFV MP12-infected and mock-infected HeLa cells harvested 18 h postinfection. Lysates were precipitated with antibodies specific for RVFV N. Blots were probed for PABP1, RVFV N, and GAPDH as a loading control.
To confirm that the observed PABP1-N interaction was not merely an artifact of overexpression, we immunoprecipitated proteins from MP12-infected and mock-infected cell lysates with antibodies specific for RVFV N. Lysates were harvested 18 h postinfection. The resultant immune complexes were immunoblotted with PABP1-specific antibodies to determine if PABP1 was present in precipitates. The blots were also probed with GAPDH-specific antibodies as a loading control. Western blots of lysates that had not been immunoprecipitated appeared similar with respect to the presence of PABP1 and GAPDH (Fig. 6E). RVFV N was present in precipitated and unprecipitated infected lysates but not in uninfected lysates (Fig. 6E). PABP1 coprecipitated with RVFV N from RVFV-infected lysates but not mock-infected lysates (Fig. 6E). A band of slightly lower molecular weight than N was observed in precipitated mock-infected lysates and is presumed to be IgG light chain, which would be expected to be present because of the immune precipitation step performed prior to Western blotting. Additionally, similar results were obtained with lysates harvested 6 h postinfection (data not shown). Coprecipitation of RVFV N and PABP1 is consistent with our immunofluorescence staining results showing colocalization of PABP1 and RVFV N. Both results suggest these two proteins interact during infection. The PABP1-RVFV N interaction is likely not related to the observed nuclear relocalization of PABP1, as RVFV N overexpression had no effect on PABP1 localization (Fig. 4D). The significance of the RVFV N-PABP1 interaction is yet to be determined.
RVFV nucleocapsid production is independent of PABP1.
To address the consequences of PABP1 nuclear relocalization for RVFV infection, we used siRNA to selectively remove PABP1 from HeLa cells. After siRNA treatment and RVFV infection, we costained cells for PABP1 and RVFV N and then assessed PABP1 knockdown by Western blot assay and measured RVFV N production using flow cytometry. Blots prepared from untreated cells or cells treated with nontargeting control siRNA, transfection reagent alone, or siRNA specific for control proteins (nucleolin, 14-3-3 epsilon, and GAPDH) all appeared to have similar levels of PABP1, whereas the PABP1-specific-siRNA-treated cells showed reduced PABP1 (Fig. 7A).
Fig 7.
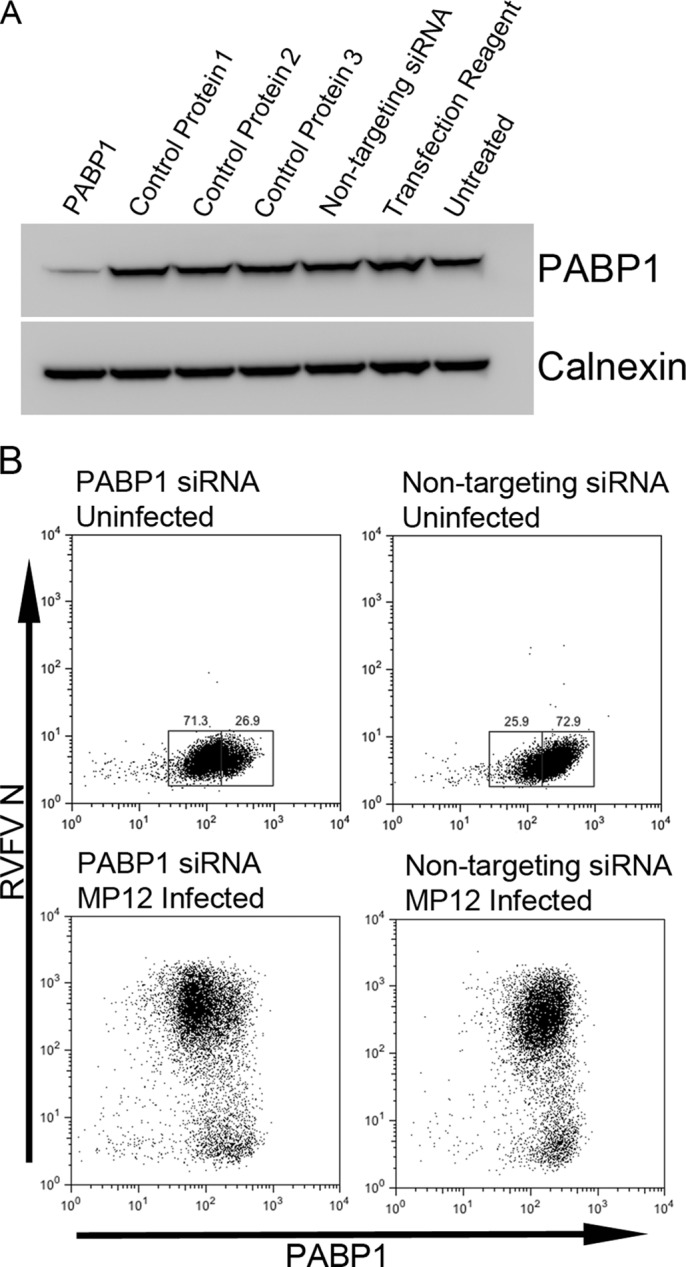
SiRNA knockdown of PABP1. (A) Western blot analysis of lysates of cells transfected with siRNA specific for PABP1 or a control protein (1, nucleolin; 2, 14-3-3 epsilon; 3, GAPDH), nontargeting siRNA, or transfection reagent alone or of untreated cells. Blots were probed for PABP1 or for calnexin as a loading control. (B) Flow cytometry plots of cells stained for PABP1 and RVFV N. Top panels, cells were uninfected; bottom panels, cells were MP12 infected (24 h infection); left panels, cells were treated with PABP1 siRNA; right panels, cells were treated with nontargeting siRNA.
Flow cytometry revealed a downward shift in PABP1 fluorescence intensity upon PABP1 siRNA treatment (Fig. 7B, top left versus top right). Gates were drawn that placed a majority of the PABP1 siRNA-treated cells in the box at the lower fluorescence intensity and the majority of nontargeting-siRNA-treated cells in the box at the higher fluorescence intensity. Unfortunately, some overlap of the two populations was unavoidable. We have termed the lower-intensity population PABP1low and the higher-intensity population PABP1high (Fig. 7B, top). PABP1low cells are presumed to be the cells in which PABP1 production was knocked down, while PABP1high cells are assumed to be those expressing physiologic levels of PABP1. Both nontargeting-siRNA-treated RVFV MP12-infected cells and PABP1 siRNA-treated infected cells displayed large populations of cells that were predominantly RVFV N positive and small populations of cells that were RVFV N negative (Fig. 7B, bottom).
To investigate the role of PABP1 during RVFV infection, we measured the percentages of RVFV N-positive cells present in populations treated with PABP1-specific siRNA or nontargeting siRNA. The histograms showed two peaks for RVFV N. The lower-fluorescence-intensity peak present to the left in Fig. 8A represents cells that are not expressing N, and the higher-fluorescence-intensity peak to the right represents cells expressing N. We found that PABP1 knockdown did not alter the overall percentage of RVFV N-positive cells as measured by flow cytometry (Fig. 8A and B). These results indicated that PABP1 was expendable for RVFV replication, as reflected by the production of N. To directly compare RVFV replication in cells expressing reduced levels of PABP1, we gated on the previously designated PABP1low or PABP1high cell populations. The PABP1high population consistently had a lower percentage of RVFV N-positive cells than the PABP1low population (Fig. 8C, left). Combined experiments showed a statistically significant difference between the percentages of RVFV N-positive cells in PABP1high and PABP1low cells following PABP1 siRNA treatment but no significant difference following nontargeting-siRNA treatment (Fig. 8C, right, and Fig. 8D). These results indicate that there is a negative correlation between RVFV replication and high levels of PABP1.
Fig 8.
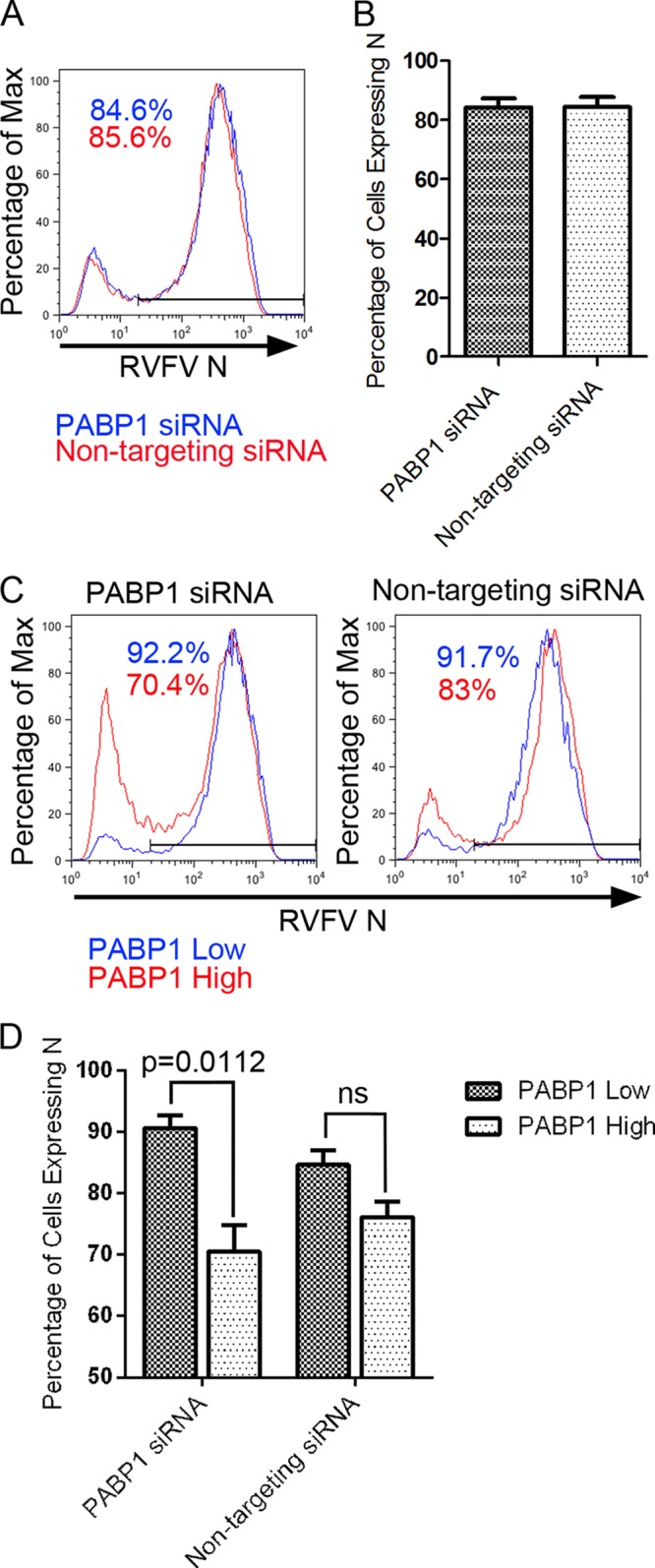
Effect of siRNA knockdown of PABP1 on RVFV N expression at 24 h postinfection with RVFV MP12. (A) Overlay of representative histograms showing percentages of RVFV N-positive cells for PABP1 siRNA- and nontargeting siRNA-treated populations. The data are normalized and presented as percentages of maximum. (B) Graph of percentages of cells expressing N. Bars represent means of triplicate experiments. (C) Overlay of representative histograms depicting percentages of RVFV N-positive cells. Results for PABP1 siRNA-treated cells and nontargeting-siRNA-treated cells are shown. The data are normalized and presented as percentages of maximum. (D) Graph of percentages of cells expressing N. Bars represent means of independent replicate samples from duplicate experiments. For PABP1 siRNA-treated samples, a statistically significant difference (P = 0.0112) was seen between PABP1low and PABPhigh cells as determined by Welch's t test.
DISCUSSION
Viruses replicate within living cells and depend upon host machinery for their own replication. At the same time, many viruses target host processes to increase the efficiency of viral replication and evade host immune responses. For example, inhibiting various facets of host protein synthesis can both help to subvert an immune response and also free up resources for the virus to use to produce its own proteins. RVFV has previously been shown to inhibit global protein transcription by targeting the TFIIH transcription factor in the nucleus (25, 26). Here, we have investigated the role of another key factor in host protein production, PAPB1, as it relates to RVFV infection.
PABP1 is a host protein that binds both the eIF4F complex and the 3′ poly(A) tail of mRNAs to increase the efficiency of protein translation (31–37). Several viruses have been shown to target PABP1 as a means to inhibit host translation and to facilitate viral mRNA translation. RVFV mRNAs are not polyadenylated, making PABP1 a logical target for RVFV. With this in mind, we sought to determine whether PABP1 was altered during infection with RVFV.
Within the family Bunyaviridae, one member of the Orthobunyavirus genus, BUNV, was previously found to perturb PABP1 by causing relocalization of PABP1 to the host cell nucleus (47). In this study, we showed that RVFV, a member of the Phlebovirus genus of the family Bunyaviridae, also causes relocalization of PABP1 to the host cell nucleus. In contrast, infection with viruses in the Hantavirus genus did not result in PABP1 relocalization. This finding is consistent with earlier observations that both RVFV and BUNV inhibit host protein synthesis, while hantaviruses do not (58–60). Relocalization of PABP1 to the host cell nucleus following stress has been shown to correlate with decreases in host protein synthesis (53), indicating a loss of PABP1 function upon relocalization. We showed here that RVFV's sequestration of PABP1 in the host cell nucleus corresponded to a sharp decrease in translation of host proteins.
Also similar to results from the earlier study with BUNV, we showed that RVFV does not require PABP1 for replication. The absence of a requirement for PABP1 was evidenced by continued RVFV protein production when PABP1 was selectively removed from cells by siRNA treatment. Furthermore, when we directly compared cells producing reduced amounts of PABP1 to those producing physiologic levels of PABP1, the cells producing physiologic levels of PABP1 exhibited a significantly lower percentage of RVFV N-positive cells than the PABP1-depleted cells. The observed change in infection suggests that lower cellular levels of PABP1 are beneficial to the virus. While RVFV infection itself does not lower cellular levels of PABP1, nuclear sequestration of PABP1 during infection functionally removes PABP1, presumably creating a similar benefit for the virus. We speculate that the beneficial effect of PABP1 removal is the result of reduced competition between the RVFV mRNAs and host mRNAs for translational machinery when PABP1 is scarce. Our studies do not rule out the possibility that the seemingly preferential infection of cells producing small amounts of PABP1 is due to a dampening of the antiviral response related to reduced host protein translation.
To attempt to uncover the underlying mechanism by which RVFV causes PABP1 to relocalize to the nucleus, we studied the effects of expressing genes encoding individual M and S segment structural and nonstructural proteins. We found that only the NSs gene product had a discernible effect on PABP1 subcellular localization. Likewise, a recombinant RVFV in which the NSs gene was replaced with a luciferase gene did not cause PABP1 relocalization. These results differ from those obtained with a genetically engineered BUNV without an NSs gene, which was still able to cause nuclear relocalization of PABP1. Such a difference is probably not surprising given the drastically different NSs proteins of orthobunyaviruses and phleboviruses, with the smaller BUNV NSs encoded in an overlapping reading frame with N and the RVFV NSs encoded in an ambisense subgenomic mRNA.
As previously noted, one of the most striking activities of RVFV's NSs is its ability to shut down general host transcription. We used actinomycin D to test whether exogenous inhibition of transcription could restore PABP1 relocalization during infection with RVFV lacking NSs. It did, and additionally, transcription inhibition in the absence of infection led to PABP1 relocalization. We noted a more robust relocalization in cells that were both infected with RVFV MP12rLuc and treated with actinomycin D. As various cell stressors have been shown to lead to PABP1 relocalization, it is possible that factors related to the stress of infection also contribute to relocalization of PABP1 (41, 61, 62). We concluded that PABP1 relocalization is likely secondary to the NSs host transcription block.
While predominantly cytoplasmic, PABP1 is a dynamic protein that shuttles between the cytoplasm and the nucleus. Recent studies of the nuclear import and nuclear export mechanisms of PABP1 have revealed an important role for RNA in both processes. PABP1 contains four RNA recognition motifs (RRMs) that are responsible for its RNA binding activity. It has recently been hypothesized that a nontraditional nuclear localization sequence (NLS) resides within the RRM. Kumar et al. demonstrated that the presence of poly(A) RNA antagonizes PABP1's ability to interact with nuclear import factors and a depletion of poly(A) RNA from the cytoplasm results in PABP1 relocalization to the nucleus (63). They propose a model in which poly(A) RNA binds PABP1, masking an NLS and thus preventing translocation into the nucleus. Conversely, when PABP1 is not bound by poly(A) RNA, it is translocation competent (63). It follows that, under normal conditions, when cytoplasmic levels of poly(A) RNA are relatively high, PABP1 remains predominantly cytoplasmic. Under conditions that alter poly(A) RNA levels, such as infection, this balance could be disrupted, allowing free PABP1 to accumulate in the nucleus. With the production of new mRNA being halted by RVFV NSs-mediated inhibition, the cytoplasmic pool of mRNA would become depleted due to normal mRNA degradation and turnover. Under such circumstances, one might expect PABP1 to accumulate in the nucleus, as is seen during infection.
Furthermore, the export of PABP1 from the nucleus has been linked to both ongoing transcription and mRNA export from the nucleus (53, 64). NSs shuts down host transcription and could thereby arrest nuclear export of PABP1. A potential increase in nuclear PABP1 import from an imbalance of poly(A) RNA coupled with a decrease in PABP1 nuclear export provides a likely explanation for PABP1 nuclear accumulation during infection. This is consistent with our observations that actinomycin D led to PABP1 nuclear accumulation.
Our study not only revealed that PABP1 relocalized to the nucleus of RVFV-infected cells, it also showed that PABP1 specifically accumulated in nuclear speckles, which are structures enriched in splicing proteins that are needed for processing pre-mRNA (65). These findings are consistent with previous observations that PABP1 can relocalize to speckles when transcription is inhibited (61) and that blocking transcription leads to the accumulation of poly(A) RNA in nuclear speckles (66). The accumulation of poly(A) RNA in nuclear speckles provides an attractive explanation for the accumulation of PABP1, a poly(A) RNA binding protein, in the speckles.
In summary, we show for the first time that RVFV infection results in disruption of the normal distribution of PABP1, resulting in a selective obstruction of host protein production while allowing viral protein production to proceed.
ACKNOWLEDGMENTS
We thank Shannon Taylor and Chris Hammerbeck for technical assistance, Stuart Nichol (Centers for Disease Control), Julie Costantino (USAMRIID), Sheli Radoshitzky (USAMRIID), Sina Buvari (USAMRIID), and Robert Doms (University of Pennsyvania) for generous gifts of plasmids, and Shinji Makino (University of Texas Medical Branch) for providing the MP-12rLuc virus.
This work was funded by the Defense Threat Reduction Agency and the National Academy of Sciences. The research was performed while Anna Maria Copeland held a National Academy of Sciences National Research Council Associateship.
The opinions, interpretations, conclusions, and recommendations are ours and are not necessarily endorsed by the U.S. Army or Department of Defense.
Footnotes
Published ahead of print 21 August 2013
REFERENCES
- 1.Daubney R, Hudson JR, Garnham PC. 1931. Enzootic hepatitis or Rift Valley fever. An undescribed virus disease of sheep cattle and man from East Africa. J. Pathol. Bacteriol. 34:545–579 [Google Scholar]
- 2.Coetzer JA. 1982. The pathology of Rift Valley fever. II. Lesions occuring in field cases in adult cattle, calves and aborted fetuses. Onderstepoort J. Vet. Res. 49:11–17 [PubMed] [Google Scholar]
- 3.Erasmus B, Coetzer J. 1981. The symptomatology and pathology of Rift Valley fever in domestic animals. Contr. Epidem. Biostatist. 3:77–82 [Google Scholar]
- 4.Abd el-Rahim I, Abd el-Hakin U, Hussein M. 1999. An epizootic of Rift Valley fever in Egypt in 1997. Rev. Sci. Tech. 18:741–748 [DOI] [PubMed] [Google Scholar]
- 5.Faye O, Diallo M, Diop D, Bezeid O, Ba H, Niang M, Dia I, Ould Mohamed S, Ndiaye K, Diallo D, Ogo Ly P, Diallo B, Nabeth P, Simon F, Lo B, Diop O. 2007. Rift Valley fever outbreak with East-Central African virus lineage in Mauritania, 2003. Emerg. Infect. Dis. 13:1016–1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Laughlin L, Meegan J, Strausbaugh L, Morens D, Watten R. 1979. Epidemic Rift Valley fever in Egypt: observations of the spectrum of human illness. Trans. R. Soc. Trop. Med. Hyg. 73:630–633 [DOI] [PubMed] [Google Scholar]
- 7.van Velden D, Meyer J, Olivier J, Gear J, McIntosh B. 1977. Rift Valley fever affecting humans in South Africa: a clinicopathological study. S. Afr. Med. J. 51:867–871 [PubMed] [Google Scholar]
- 8.Rice RM, Erlick BJ, Rosato RR, Eddy GA, Mohanty SB. 1980. Biochemical characterization of Rift Valley fever virus. Virology 105:256–260 [DOI] [PubMed] [Google Scholar]
- 9.Muller R, Argentini C, Bouloy M, Prehaud C, Bishop DHL. 1992. Completion of the genome sequence of Rift Valley fever phlebovirus indicates that the L RNA is negative sense or ambisense and codes for a putative transcriptase-replicase. Nucleic Acids Res. 20:6440. 10.1093/nar/20.23.6440-a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Reference deleted.
- 11.Muller R, Poch O, Delarue M, Bishop DHL, Bouloy M. 1994. Rift valley fever virus L segment: correction of the sequence and possible functional role of newly identified regions conserved in RNA-dependent polymerases. J. Gen. Virol. 75:1345–1352 [DOI] [PubMed] [Google Scholar]
- 12.Collett MS, Purchio AF, Keegan K, Frazier S, Hays W, Anderson DK, Parker MD, Schmaljohn C, Schmidt J, Dalrymple JM. 1985. Complete nucleotide sequence of the M RNA segment of rift valley fever virus. Virology 144:228–245 [DOI] [PubMed] [Google Scholar]
- 13.Kakach LT, Wasmoen TL, Collett MS. 1988. Rift Valley fever virus M segment: use of recombinant vaccinia viruses to study phlebovirus gene expression. J. Virol. 62:826–833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Piper ME, Sorenson DR, Gerrard SR. 2011. Efficient cellular release of Rift Valley fever virus requires genomic RNA. PLoS One 6:e18070. 10.1371/journal.pone.0018070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bird BH, Albarino CG, Nichol ST. 2007. Rift Valley fever virus lacking NSm proteins retains high virulence in vivo and may provide a model of human delayed onset neurologic disease. Virology 362:10–15 [DOI] [PubMed] [Google Scholar]
- 16.Gerrard SR, Bird BH, Albarino CG, Nichol ST. 2007. The NSm proteins of Rift Valley fever virus are dispensable for maturation, replication and infection. Virology 359:459–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Won S, Ikegami T, Peters CJ, Makino S. 2006. NSm and 78-kilodalton proteins of Rift Valley fever virus are nonessential for viral replication in cell culture. J. Virol. 80:8274–8278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Won S, Ikegami T, Peters CJ, Makino S. 2007. NSm protein of Rift Valley fever virus suppresses virus-induced apoptosis. J. Virol. 81:13335–13345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Crabtree MB, Kent Crockett RJ, Bird BH, Nichol ST, Erickson BR, Biggerstaff BJ, Horiuchi K, Miller BR. Infection and transmission of Rift Valley fever viruses lacking the NSs and/or NSm genes in mosquitoes: potential role for NSm in mosquito infection. PLoS Negl. Trop. Dis. 6:e1639. 10.1371/journal.pntd.0001639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Giorgi C, Accardi L, Nicoletti L, Gro MC, Takehara K, Hilditch C, Morikawa S, Bishop DHL. 1991. Sequences and coding strategies of the S RNAs of Toscana and Rift Valley fever viruses compared to those of Punta Toro, Sicilian sandfly fever, and Uukuniemi viruses. Virology 180:738–753 [DOI] [PubMed] [Google Scholar]
- 21.Lopez N, Muller R, Prehaud C, Bouloy M. 1995. The L protein of Rift Valley fever virus can rescue viral ribonucleoproteins and transcribe synthetic genome-like RNA molecules. J. Virol. 69:3972–3979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bouloy M, Janzen C, Vialat P, Khun H, Pavlovic J, Huerre M, Haller O. 2001. Genetic evidence for an interferon-antagonistic function of Rift Valley fever virus nonstructural protein NSs. J. Virol. 75:1371–1377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ikegami T, Won S, Peters CJ, Makino S. 2006. Rescue of infectious Rift Valley fever virus entirely from cDNA, analysis of virus lacking the NSs gene, and expression of a foreign gene J. Virol. 80:2933–2940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Muller R, Saluzzo J-F, Lopez N, Dreier T, Turell M, Smith J, Bouloy M. 1995. Characterization of clone 13, a naturally attenuated avirulent isolate of Rift Valley fever virus, which is altered in the small segment. Am. J. Trop. Med. Hyg. 53:405–411 [DOI] [PubMed] [Google Scholar]
- 25.Kalveram B, Lihoradova O, Ikegami T. 2011. NSs protein of Rift Valley fever virus promotes posttranslational downregulation of the TFIIH subunit p62. J. Virol. 85:6234–6243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Le May N, Dubaele S, De Santis LP, Billecocq A, Bouloy M, Egly J-M. 2004. TFIIH transcription factor, a target for the Rift Valley hemorrhagic fever virus. Cell 116:541–550 [DOI] [PubMed] [Google Scholar]
- 27.Billecocq A, Spiegel M, Vialat P, Kohl A, Weber F, Bouloy M, Haller O. 2004. NSs protein of Rift Valley fever virus blocks interferon production by inhibiting host gene transcription. J. Virol. 78:9798–9806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Habjan M, Pichlmair A, Elliott RM, Overby AK, Glatter T, Gstaiger M, Superti-Furga G, Unger H, Weber F. 2009. NSs protein of Rift Valley fever virus induces the specific degradation of the double-stranded RNA-dependent protein kinase. J. Virol. 83:4365–4375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ikegami T, Narayanan K, Won S, Kamitani W, Peters CJ, Makino S. 2009. Rift Valley fever virus NSs protein promotes post-transcriptional downregulation of protein kinase PKR and inhibits eIF2alpha phosphorylation. PLoS Pathog. 5:e1000287. 10.1371/journal.ppat.1000287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Le May N, Mansuroglu Z, Leger P, Josse T, Blot G, Billecocq A, Flick R, Jacob Y, Bonnefoy E, Bouloy M. 2008. A SAP30 complex inhibits IFN-beta expression in Rift Valley fever virus infected cells. PLoS Pathog. 4:e13. 10.1371/journal.ppat.0040013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blobel G. 1973. A protein of molecular weight 78,000 bound to the polyadenylate region of eukaryotic messenger RNAs. Proc. Natl. Acad. Sci. U. S. A. 70:924–928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Imataka H, Gradi A, Sonenberg N. 1998. A newly identified N-terminal amino acid sequence of human eIF4G binds poly(A)-binding protein and functions in poly(A)-dependent translation. EMBO J. 17:7480–7489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Le H, Tanguay RL, Balasta ML, Wei C-C, Browning KS, Metz AM, Goss DJ, Gallie DR. 1997. Translation initiation factors eIF-iso4G and eIF-4B interact with the poly(A)-binding protein and increase its RNA binding activity. J. Biol. Chem. 272:16247–16255 [DOI] [PubMed] [Google Scholar]
- 34.Borman AM, Michel YM, Kean KM. 2000. Biochemical characterisation of cap-poly(A) synergy in rabbit reticulocyte lysates: the eIF4G-PABP interaction increases the functional affinity of eIF4E for the capped mRNA 5′-end. Nucleic Acids Res. 28:4068–4075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kahvejian A, Svitkin YV, Sukarieh R, M'Boutchou M-N, Sonenberg N. 2005. Mammalian poly(A)-binding protein is a eukaryotic translation initiation factor, which acts via multiple mechanisms. Genes Dev. 19:104–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Luo Y, Goss DJ. 2001. Homeostasis in mRNA Initiation: wheat germ poly(A)-binding protein lowers the activation energy barrier to initiation complex formation. J. Biol. Chem. 276:43083–43086 [DOI] [PubMed] [Google Scholar]
- 37.Wei C-C, Balasta ML, Ren J, Goss DJ. 1998. Wheat germ poly(A) binding protein enhances the binding affinity of eukaryotic initiation factor 4F and (iso)4F for Cap analogues. Biochemistry 37:1910–1916 [DOI] [PubMed] [Google Scholar]
- 38.Joachims M, Van Breugel PC, Lloyd RE. 1999. Cleavage of poly(A)-binding protein by enterovirus proteases concurrent with inhibition of translation in vitro. J. Virol. 73:718–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kuyumcu-Martinez NM, Van Eden ME, Younan P, Lloyd RE. 2004. Cleavage of poly(A)-binding protein by poliovirus 3C protease inhibits host cell translation: a novel mechanism for host translation shutoff. Mol. Cell. Biol. 24:1779–1790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dobrikova E, Shveygert M, Walters R, Gromeier M. 2010. Herpes simplex virus proteins ICP27 and UL47 associate with polyadenylate-binding protein and control its subcellular distribution. J. Virol. 84:270–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Salaun C, MacDonald AI, Larralde O, Howard L, Lochtie K, Burgess HM, Brook M, Malik P, Gray NK, Graham SV. 2010. Poly(A)-binding protein 1 partially relocalizes to the nucleus during herpes simplex virus type 1 infection in an ICP27-independent manner and does not inhibit virus replication. J. Virol. 84:8539–8548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Harb M, Becker MM, Vitour D, Baron CH, Vende P, Brown SC, Bolte S, Arold ST, Poncet D. 2008. Nuclear localization of cytoplasmic poly(A)-binding protein upon rotavirus infection involves the interaction of NSP3 with eIF4G and RoXaN. J. Virol. 82:11283–11293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Piron M, Vende P, Cohen J, Poncet D. 1998. Rotavirus RNA-binding protein NSP3 interacts with eIF4GI and evicts the poly(A) binding protein from eIF4F. EMBO J. 17:5811–5821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee YJ, Glaunsinger BA. 2009. Aberrant herpesvirus-induced polyadenylation correlates with cellular messenger RNA destruction. PLoS Biol. 7:e1000107. 10.1371/journal.pbio.1000107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pattnaik AK, Abraham G. 1983. Identification of four complementary RNA species in Akabane virus-infected cells. J. Virol. 47:452–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ulmanen I, Seppala P, Pettersson RF. 1981. In vitro translation of Uukuniemi virus-specific RNAs: identification of a nonstructural protein and a precursor to the membrane glycoproteins. J. Virol. 37:72–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Blakqori G, van Knippenberg I, Elliott RM. 2009. Bunyamwera orthobunyavirus S-segment untranslated regions mediate poly(A) tail-independent translation. J. Virol. 83:3637–3646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hart TJ, Kohl A, Elliott RM. 2009. Role of the NSs protein in the zoonotic capacity of orthobunyaviruses. Zoonoses Public Health 56:285–296 [DOI] [PubMed] [Google Scholar]
- 49.Szemiel AM, Failloux A-B, Elliott RM. 2012. Role of Bunyamwera orthobunyavirus NSs protein in infection of mosquito cells. PLoS Negl. Trop. Dis. 6:e1823. 10.1371/journal.pntd.0001823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Caplen H, Peters CJ, Bishop DHL. 1985. Mutagen-directed attenuation of Rift Valley fever virus as a method for vaccine development. J. Gen. Virol. 66:2271–2277 [DOI] [PubMed] [Google Scholar]
- 51.Keegan K, Collett MS. 1986. Use of bacterial expression cloning to define the amino acid sequences of antigenic determinants on the G2 glycoprotein of Rift Valley fever virus. J. Virol. 58:263–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vaughn V, Streeter C, Miller D, Gerrard S. 2010. Restriction of Rift Valley fever virus virulence in mosquito cells. Viruses 2:655–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Burgess HM, Richardson WA, Anderson RC, Salaun C, Graham SV, Gray NK. 2011. Nuclear relocalisation of cytoplasmic poly(A)-binding proteins PABP1 and PABP4 in response to UV irradiation reveals mRNA-dependent export of metazoan PABPs. J. Cell Sci. 124(Pt 19):3344–3355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schmidt EK, Clavarino G, Ceppi M, Pierre P. 2009. SUnSET, a nonradioactive method to monitor protein synthesis. Nat. Meth. 6:275–277 [DOI] [PubMed] [Google Scholar]
- 55.Struthers JK, Swanepoel R. 1982. Identification of a major non-structural protein in the nuclei of Rift Valley fever Virus-infected cells. J. Gen. Virol. 60:381–384 [DOI] [PubMed] [Google Scholar]
- 56.Struthers JK, Swanepoel R, Shepherd SP. 1984. Protein synthesis in Rift Valley fever virus-infected cells. Virology 134:118–124 [DOI] [PubMed] [Google Scholar]
- 57.Swanepoel R, Blackburn NK. 1977. Demonstration of nuclear immunofluorescence in Rift Valley fever infected cells. J. Gen. Virol. 34:557–561 [DOI] [PubMed] [Google Scholar]
- 58.Parker MD, Smith JF, Dalrymple JM. 1984. Rift Valley fever virus intracellular RNA: a functional analysis, p 21–28 In Compans R, Bishop D. (ed), Segmented negative strand viruses. Arenaviruses, Bunyaviruses, and Orthomyxoviruses. Academic Press, Orlando, FL [Google Scholar]
- 59.Pennington TH, Pringle CR, McCrae MA. 1977. Bunyamwera virus-induced polypeptide synthesis. J. Virol. 24:397–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schmaljohn C, Dalrymple JM. 1984. Biochemical characterization of Hantaan virus, p 117–124 In Compans RW, Bishop DHL. (ed), Segmented Negative Strand Viruses. Arenaviruses, Bunyaviruses, and Orthomyxoviruses. Academic Press, Orlando, FL [Google Scholar]
- 61.Afonina E, Stauber R, Pavlakis GN. 1998. The human poly(A)-binding protein 1 shuttles between the nucleus and the cytoplasm. J. Biol. Chem. 273:13015–13021 [DOI] [PubMed] [Google Scholar]
- 62.Ma S, Bhattacharjee RB, Bag J. 2009. Expression of poly(A)-binding protein is upregulated during recovery from heat shock in HeLa cells. FEBS J. 276:552–570 [DOI] [PubMed] [Google Scholar]
- 63.Kumar GR, Shum L, Glaunsinger BA. 2011. Importin α-mediated nuclear import of cytoplasmic poly(A) binding protein occurs as a direct consequence of cytoplasmic mRNA depletion. Mol. Cell. Biol. 31:3113–3125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Khacho M, Mekhail K, Pilon-Larose K, Pause A, Côté J, Lee S. 2008. eEF1A is a novel component of the mammalian nuclear protein export machinery. Mol. Biol. Cell 19:5296–5308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Spector DL, Lamond AI. 2011. Nuclear Speckles. Cold Spring Harb. Perspect. Biol. 3:a000646. 10.1101/cshperspect.a000646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Girard C, Will CL, Peng J, Makarov EM, Kastner B, Lemm I, Urlaub H, Hartmuth K, Lührmann R. 2012. Post-transcriptional spliceosomes are retained in nuclear speckles until splicing completion. Nat. Commun. 3:994. [DOI] [PubMed] [Google Scholar]