

Abstract
Kaposi's sarcoma-associated herpesvirus (KSHV) expresses ∼20 viral microRNAs (miRNAs) in latently infected cells. We have previously shown that two of these miRNAs function as mimics of the cellular miRNAs miR-155 and miR-142-3p. Two additional KSHV miRNAs, miR-K3+1 and miR-K3, share perfect and offset 5′ homology with cellular miR-23, respectively. Here, we report a single nucleotide polymorphism that causes miR-K3+1 expression in a subset of KSHV-infected primary effusion lymphoma cell lines as a consequence of altered processing of the primary transcript by the Microprocessor complex. We confirm that miR-K3+1 regulates miR-23 targets, which is expected because these miRNAs share the entire seed region (nucleotides 2 to 8). Surprisingly, we found that miR-K3 also regulates miR-23 targets, despite offset seed sequences. In addition, the offset homology of miR-K3 to miR-23 likely allows this viral miRNA to expand its target repertoire beyond the targets of miR-23. Because miR-23 is highly expressed in endothelial cells but expressed at only low levels in B cells, we hypothesize that miR-K3 may function to introduce miR-23-like activities into KSHV-infected B cells. Together, our data demonstrate that KSHV has evolved at least three distinct viral miRNAs to tap into evolutionarily conserved cellular miRNA-regulatory networks. Furthermore, our data allow fundamental insights into the generation and functional impact of miRNA 5′ end variation.
INTRODUCTION
MicroRNAs (miRNAs) are ∼21- to 23-nucleotide (nt)-long noncoding RNAs that function within Argonaute-containing miRNA-induced silencing complexes (miRISCs) to repress target mRNAs. miRNAs are processed in a stepwise fashion from one or both arms of imperfect hairpin structures contained within longer RNA polymerase II transcripts termed primary miRNAs (pri-miRNAs). Cleavage of the pri-miRNA hairpin by the Microprocessor complex, which is composed of the RNase III enzyme Drosha and its double-stranded RNA (dsRNA)-binding domain-containing cofactor DGCR8, is the first step in the canonical miRNA biogenesis pathway in animals (1–3). Drosha cleaves about one helical turn (∼11 bp) away from base of the stem-loop structure to liberate the apical ∼65-nt hairpin, called the precursor miRNA (pre-miRNA) (4). The pre-miRNA is exported to the cytoplasm, where Dicer excises an imperfect ∼20-bp duplex with 2-nt 3′ overhangs, one strand of which represents the mature miRNA strand that is loaded into the miRISC (5, 6). miRNAs guide the recognition of imperfect binding sites most commonly located in the 3′ untranslated regions (UTRs) of target mRNAs. Typically, the “seed” region (nt 2 to 7) of the miRNA is perfectly base paired, which is accompanied by a perfect base pair at nt 8 of the miRNA (2-8mer seed match) and/or an A across from position 1 of the miRNA (2-7A1mer seed match) in effective sites (Fig. 1A) (7).
Fig 1.
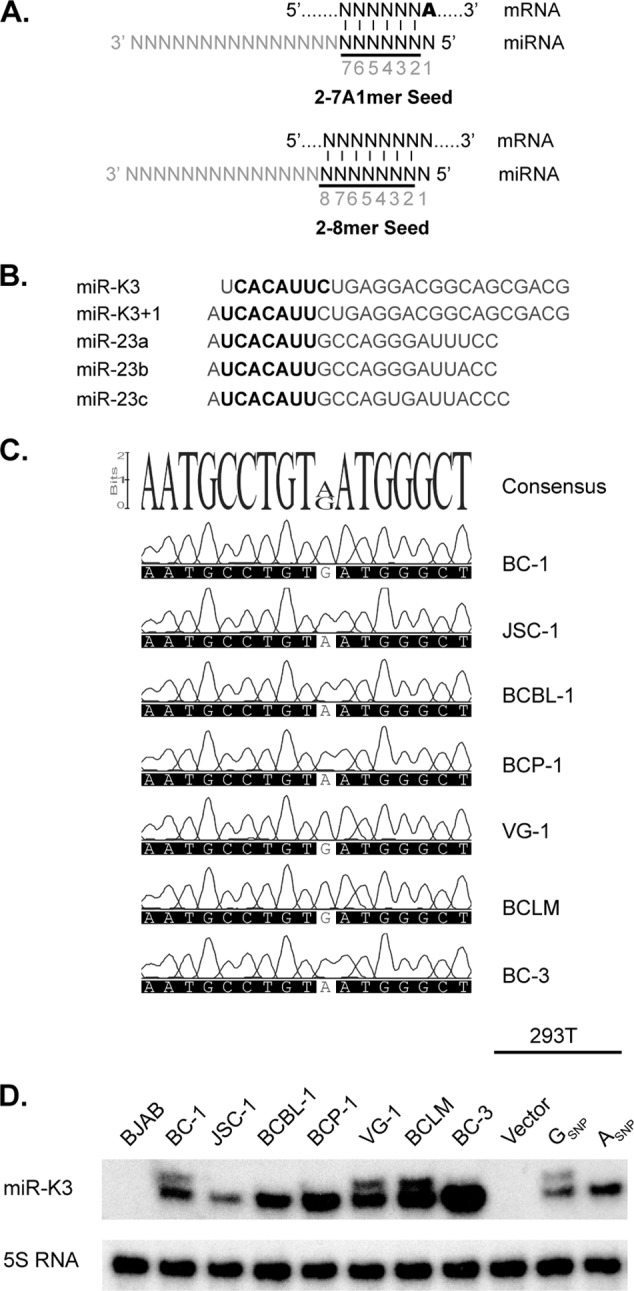
Expression of K3+1. (A) miRNAs commonly engage in seed interactions with mRNA targets, which minimally involve base pairing of nt 2 to 7 of the miRNA (“seed,” in black) together with an A residue across nt 1 of the miRNA (A in boldface, 2-7A1mer) or a Watson-Crick base pair at position 8 of the miRNA (2-8mer). “2-7A1mer” seed matches are commonly also designated “7mer1A” matches, while “2-8mer” sites are commonly designated “7mer-m8” sites (50–53). (B) Alignment of miR-K3, miR-K3+1, and the miR-23 family miRNAs. miR-K3+1 is a 5′ processing variant of miR-K3, which differs only by an additional 5′ nucleotide. Both miRNAs exhibit partial homology to the cellular miR-23 family at their 5′ ends. Seed sequences (nt 2 to 8) are shown in bold. (C) Partial sequencing chromatograms of 250-bp PCR products centered on mature miR-K3 from different PEL cell lines. Shown at the top is the consensus sequence of the aligned regions; the only detected polymorphism is highlighted by a white background. (D) Primer extension analysis of miR-K3 (lower band) and its variant miR-K3+1 (upper band) in the KSHV-negative BJAB cell line, KSHV-infected PEL cell lines (BC-1, JSC-1, BCBL-1, BCP-1, VG-1, BCLM, and BC-3), and 293T cells that were transfected with the parental lentiviral vector pLCE or with pLCE expressing a 250-bp fragment of pri-miR-K3 containing the GSNP or the ASNP. Primer extension of the 5S RNA served as a control for RNA input.
Herpesviruses stand out among virus families by often encoding multiple miRNAs in their genomes. Kaposi's sarcoma (KS)-associated herpesvirus (KSHV) contains one miRNA cluster with 12 pre-miRNAs, giving rise to a set of mature miRNAs called miR-K1 to miR-K12 (8–11). We and others have previously demonstrated that these miRNAs occupy hundreds of sites in the human transcriptome (12, 13). One major clue to the potential functions of the KSHV miRNAs is the identification of two of the KSHV miRNAs as viral analogs of the cellular miRNAs miR-155 and miR-142-3p (12, 14, 15). Several additional interactions and functions of the KSHV miRNAs have been identified (16–35); however, the overall impact these miRNAs have on host gene expression and their phenotypic consequences remain largely unknown.
All of the KSHV-positive primary effusion lymphoma (PEL) cell lines analyzed to date express miR-K3. Our recent analysis of KSHV miRNA expression in PEL cell lines revealed that a subset of PEL cell lines coexpressed miR-K3 together with a novel miR-K3 variant carrying an additional 5′ adenosine (A) (12). We refer to this novel miRNA variant as miR-K3+1 (Fig. 1B). The additional 5′ nucleotide of miR-K3+1 causes the seed sequence of miR-K3+1 to differ from that of miR-K3, which is expected to result in an altered target range. Interestingly, nt 2 to 8 of miR-K3+1 are identical to nt 2 to 8 of the miR-23 family miRNAs (Fig. 1B). This 7mer seed homology is reminiscent of the relationship between KSHV miR-K11 and cellular miR-155, which also share nt 2 to 8 but differ outside the seed region. In the case of miR-K11, we and others have demonstrated that this homology is sufficient to allow regulation of miR-155 seed targets by miR-K11 (12, 14, 15, 36, 37). Therefore, we initially hypothesized that miR-K3+1 may function as a mimic of cellular miR-23. Here we characterize the mechanisms underlying the expression of miR-K3+1 and the functional relationships among miR-K3+1, miR-K3, and miR-23. We mapped a single nucleotide polymorphism (SNP) responsible for altered processing of the miR-K3 hairpin by Drosha and the consequent production of miR-K3+1. In addition, we demonstrate that not just miR-K3+1, but also miR-K3 itself, is an effective mimic of miR-23. Finally, we demonstrate that miR-K3 is likely to engage at least a small number of binding sites that are not shared with the miR-23 miRNAs.
MATERIALS AND METHODS
Cell culture.
293T and HeLa cells were maintained in Dulbecco's modified Eagle's medium containing 4.5 g/liter glucose and l-glutamine (Sigma-Aldrich, St. Louis, MO) and supplemented with 10% fetal bovine serum (FBS; Gibco, Life Technologies, Carlsbad, CA). B cell lines were cultured in RPMI containing l-glutamine (Sigma-Aldrich) and supplemented with 10 to 20% FBS and 0.05 mM β-mercaptoethanol (Bio-Rad, Hercules, CA). Endothelial cells were grown in EBM2-MV (Lonza), and primary cells were used before passage 7.
RNA secondary-structure prediction.
Prediction of the secondary structure of the “pri-miRNAs” was done with Geneious Pro v5.5.6 (Biomatters), which applied the Vienna RNAfold package (38).
Cloning procedures.
The pLCE/miR-K3 (GSNP) expression vector containing 250 bp of pri-miR-K3 centered on the pre-miR-K3 stem-loop from BC-1 cells has been described previously (12). To analyze the corresponding 250-bp segment of other KSHV genomes, we PCR amplified this region from genomic DNA prepared from a panel of six additional PEL cell lines (JSC-1, BCBL-1, BCP-1, VG-1, BCLM, and BC-3). The resulting PCR products were sequenced. pLCE/miR-K3 (ASNP) was derived by cloning the PCR product from BCBL-1 between the XhoI and NotI sites of pLCE and confirmed by sequencing. A 250-bp fragment containing the miR-23a stem-loop at a central location was amplified from BCBL-1 genomic DNA, cloned into pLCE by using the XhoI and EcoRI sites, and confirmed by sequencing.
In vitro Drosha cleavage assays.
In vitro Drosha cleavage assays were carried out essentially as previously described, with minor modifications (39, 40). T7 RNA polymerase promoter-added templates for the in vitro transcription of “pri-miR-K3” RNAs were amplified from BC-1 or BCBL-1 genomic DNA by using forward primer 5′-TAATACGACTCACTATAGGCGCAACAGCTACAATGCCTG-3′) and reverse primer 5′-CGGTTCGTCGCTTGGACC-3′. The underlined nucleotides make up the minimal T7 promoter. In vitro transcription was performed in the presence of [α-32P]CTP with the AmpliScribe T7 High Yield Transcription kit (Epicentre Biotechnologies, Madison, WI). Each 20-μl transcription reaction mixture contained 120 ng PCR product; 5 mM (each) ATP, GTP, and UTP; 0.5 mM CTP; 10 mM dithiothreitol (DTT); 0.5 μl of RiboGuard RNase inhibitor; 2 μl of AmpliScribe T7 enzyme solution; and 20 μCi of [α-32P]CTP (3,000 Ci/mmol; PerkinElmer, Waltham, MA). Reaction mixtures were incubated at 37°C for 90 min. The reaction products were purified with Micro Bio-Spin P6 columns (Bio-Rad, Hercules, CA). Labeled transcripts were further purified from 10% Tris-borate-EDTA (TBE)-urea polyacrylamide gels (PAGs). 293T cells (107) were cotransfected with a total of 25 μg of Drosha-Flag and DGCR8-Flag expression vectors at a 9:1 ratio or pLCE (negative control). After 48 h, cells were collected by scraping into 5 ml ice-cold phosphate-buffered saline. Cell pellets were resuspended in 1 ml lysis buffer (20 mM HEPES [pH 7.6], 100 mM KCl, 0.1% IGEPAL CA-630). Cells were sonicated on ice with six pulses of 10 s each. The cleared lysates were incubated with 2.5 μg mouse anti-FLAG M2 monoclonal antibody (Sigma-Aldrich, St. Louis, MO) bound to 10 μl Dynabeads Protein G beads (Life Technologies, Grand Island, NY) at 4°C for 1.5 h with vigorous shaking. Magnetic beads were washed seven times with wash buffer (20 mM HEPES [pH 7.6], 300 mM KCl, 0.1% IGEPAL CA-630). Beads were again washed twice with reaction buffer (20 mM HEPES [pH 7.6], 100 mM KCl, 5% glycerol, 6.4 mM MgCl2, 0.5 mM DTT). At the last wash, the slurry was split into two tubes. Beads were resuspended in 15 μl reaction buffer, and ∼105 cpm of labeled pri-miRNA was added. Cleavage reaction mixtures were incubated on a Thermomixer for 90 min at 37°C at 1,200 rpm. Reaction products were extracted with acid phenol and ethanol precipitated. RNA pellets were resuspended in 60 μl water. To analyze cleavage products, 10-μl samples were resolved on either a 12.5% TBE-urea PAG or a 10% TBE-urea sequencing PAG and visualized by autoradiography.
Primer extension analysis.
Primer extension assays were done with the Primer Extension System (Promega) as previously described (12). A 1.5-μg sample of total RNA was used per reaction mixture. miR-23 probes have the following sequences: miR-23a, 5′-TTGGAAATCCCTGGCAA-3′; miR-23b, 5′-GTGGTAATCCCTGGCAA-3′; miR-23c, 5′-GGGTAATCACTGGCAA-3′. The miR-K3 and 5S probes were previously published (12). Primer extension products were resolved on 15% TBE-urea acrylamide gels and visualized by autoradiography.
3′ UTR reporter vectors and indicator assays.
The 3′ UTRs of putative target genes were amplified from PEL genomic DNA and cloned into the firefly luciferase reporter vector pLSG (14). Primer sequences and detailed cloning procedures are available on request and posted on the Gottwein lab webpage (http://bugs.mimnet.northwestern.edu/labs/gottweinlab/index.html). To mutate seed sequences, overlap extension PCRs were performed with the same outer primers and mutated inner primer pairs. In each case, the seed match was mutated from 5′-AATGTGA-3′ to 5′-AAAGTCA-3′.
Reporter assays with pLCE-based miRNA expression vectors were performed essentially as previously described (12, 14). For reporter assays with RNA mimics, 293T cells were seeded at 1.2 × 105/well of a 24-well plate. After ∼18 h, 5 pmol of synthetic mirVana miRNA mimics (Life Technologies) was cotransfected with 3 μl of Lipofectamine 2000 reagent (Life Technologies) with 2.5 ng of the 3′ UTR reporter vector, 2.5 ng of the internal control plasmid pLSR, and 0.3 μg of pLCE to serve as a carrier DNA. Dual luciferase activities were read after 48 h with the Dual Luciferase Reporter assay system (Promega).
Quantitative Western blotting.
For Western blot assays performed with 293T cells, cells were seeded at 4 × 105/well of a six-well plate. Cells were transfected with 50 pmol of miRNA mimics and 1.5 μg of pLCE with Lipofectamine 2000. For Western blot assays performed with BJAB cells, 50-pmol samples of miRNA mimics were electroporated into 4 × 105 cells with the Neon Transfection System (Life Technologies) with the recommended parameters of 1,350 V and a 40-ms pulse width for the 10-μl tip. After 48 h, cells were lysed with 20 mM Tris-Cl (pH 7)–100 mM NaCl–1% Triton X-100–10% glycerol–1 mM EDTA–1 mM NaF–1 mM sodium orthovanadate–complete protease inhibitor cocktail (Roche, Mannheim, Germany). Lysates were cleared by centrifugation at 16,000 × g for 20 min. Protein concentrations were determined with the BCA Protein Assay kit (Pierce). Ten micrograms of protein extract per lane was resolved on Bolt 4 to 12% Bis-Tris Plus gels (Life Technologies) and immunoblotted onto a nitrocellulose membrane with anti-HMGB2 rabbit antibody (Sigma-Aldrich) and anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mouse monoclonal IgG1 0411 (Santa Cruz Biotechnology, Santa Cruz, CA). Primary antibodies were detected with infrared dye IRDye 800CW-conjugated goat anti-rabbit or anti-mouse IgG (LI-COR Biosciences, Lincoln, NE). Immunoblot assays were imaged and quantified with the Odyssey Fc Dual-Mode Imaging System (LI-COR Biosciences).
RESULTS
A SNP near the base of the miR-K3 hairpin drives miR-K3+1 biogenesis.
We have previously shown that the PEL cell lines BC-1, VG-1, and BCLM express miR-K3+1 along with miR-K3, while the PEL cell lines JSC-1, BCBL-1, BCP-1, and BC-3 express only miR-K3 (12). To define the mechanism underlying miR-K3+1 expression, we PCR amplified a 250-bp region centered on the pre-miR-K3 stem-loop from the viral genome of each of these cell lines. Sanger sequencing of the resulting PCR products revealed only one difference between samples; i.e., cell lines that expressed miR-K3+1 had a G at position 121617 (GSNP) of the BC-1 genome (GenBank accession number U75698.1), and cell lines that expressed only miR-K3 had an A at the corresponding position (Fig. 1C). To directly test whether this SNP is responsible for the differential processing of the miR-K3 precursor, we cloned a PCR product containing either the ASNP or the GSNP from BCBL-1 or BC-1, respectively, into a miRNA expression vector (pLCE) (41). Primer extension analysis was used to visualize the 5′ end of the miR-K3 miRNAs and confirmed our hypothesis that the GSNP variant expressed both miR-K3+1 and miR-K3, while a vector carrying the ASNP variant expressed only miR-K3 (Fig. 1D). We therefore conclude that the presence of a G at this position is responsible for the expression of miR-K3+1. Primer extension analysis also confirmed miR-K3+1 expression in BC-1, VG-1, and BCLM cells, as previously reported (12). Interestingly, this SNP has been reported previously to be present in 3 of 19 analyzed KS patients, 13 of 23 KSHV-infected patients diagnosed with multicentric Castleman's disease (MCD), and 4 of 7 patients with KSHV-associated inflammatory cytokine syndrome, but its consequences for KSHV miRNA expression had not been explored (39, 42, 43).
The miR-K3 SNP allows insights into Drosha cleavage site selection.
miR-K3/K3+1 is expressed from the 5′ arm of the pre-miR-K3 hairpin, and therefore, its 5′ end is generated by Drosha processing (Fig. 2A). Determinants of 5′ variant (“iso-miR”) production by Drosha remain unexplored, and the polymorphism identified might give insights into the mechanisms underlying iso-miR production. Structural requirements for recognition by the Microprocessor are relatively well defined and include a hairpin structure with an imperfect stem of about three helical turns, a terminal loop and single-stranded flanking sequences (3, 4, 44–46). It is thought that DGCR8 is critical in the recognition of the single-stranded RNA (ssRNA)–dsRNA junction at the base of the stem and positions the dual catalytic sites of Drosha to introduce a staggered cut into both RNA strands ∼11 bp (about one helical turn) away from the base of the stem-loop structure. In addition to these structural requirements, microprocessing in human cells is also influenced by primary sequence determinants (47). Interestingly, the SNP mapped to a location well outside the pre-miR-K3 stem-loop in the basal stem region of the miR-K3 hairpin (Fig. 2B), which is consistent with a role in differential positioning of Drosha for alternative cleavage site selection.
Fig 2.
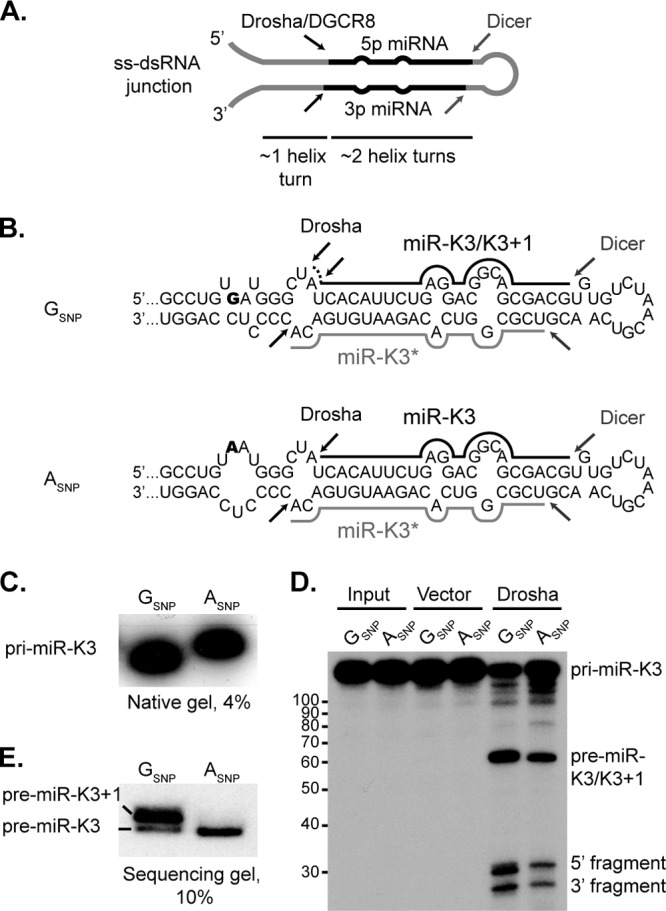
In vitro Drosha processing assay of pri-miR-K3. (A) Schematic representation of a typical pri-miRNA hairpin structure that serves as a substrate for the Microprocessor complex. Structural determinants of substrate recognition, including the ssRNA-dsRNA junction, the imperfect stem and the terminal loop, and Drosha and Dicer cleavage sites, are indicated. (B) Secondary-structure predictions of the pri-miR-K3 transcripts with the GSNP or the ASNP (SNP shown in bold). Black bars represent mature miR-K3, the dotted line indicates the additional 5′ A residue of miR-K3+1, while gray bars represent the passenger strand. Black and gray arrows represent sites of Drosha and Dicer cleavage, respectively. (C) Migration of in vitro-transcribed, 32P-labeled “pri-miR-K3” transcripts with the GSNP or the ASNP in a 4% native PAG. (D) In vitro Drosha processing assay of “pri-miR-K3” transcripts resolved in a denaturing urea-PAG. The tick marks on the left (showing numbers of nucleotides) correspond to the migration of the denatured Decade RNA marker (Life Technologies). (E) Migration of pre-miR-K3/K3+1 RNAs in a sequencing gel from the same Drosha processing reaction as in panel D.
To determine whether the SNP could alter the secondary structure of the pri-miRNA and thereby result in differential processing, we predicted the secondary structures of a 121-nt-long “pri-miR-K3” centered on the miRNA stem-loop with the Vienna package RNAfold tool (38). The algorithm predicts distinct structures for the two sequence variants that differ in the basal stem (Fig. 2B). The GSNP RNA, which expresses K3+1 in addition to K3, is predicted to exhibit additional base pairing compared to the ASNP RNA, which is predicted to have a larger internal loop within the lower stem. To directly test whether the SNP results in structural differences between the RNAs and whether this affects Drosha cleavage site selection, we transcribed a 121-nt RNA encompassing the entire pre-miRNA, 32 nt at the 5′ end, and 26 nt at the 3′ end, in vitro. The analysis of 32P-labeled RNA transcripts on a native acrylamide gel shows that the GSNP-containing transcript migrates faster than the ASNP-containing RNA (Fig. 2C), which is consistent with the hypothesis that the GSNP pri-miR-K3/K3+1 has a more compact structure.
To directly confirm that this change in the basal stem portion causes differential cleavage by Drosha, we used the labeled pri-miRNAs as substrates for in vitro Drosha assays (40). The ASNP RNA produces three distinct bands that are products of complete Drosha cleavage: the pre-miR-K3 (63 nt), a 5′ fragment (32 nt), and a 3′ fragment (26 nt) (Fig. 2D). The GSNP RNA, on the other hand, yielded a doublet (31 and 32 nt) in the region where the 5′ fragment is expected to migrate. This doublet is consistent with the expected different by-products from the differential cleavage of the pre-miRNA by Drosha. The corresponding pre-miRNA doublet, however, could not be resolved properly in this gel. Thus, we reran the same samples through a longer sequencing gel. This enabled us to clearly separate pre-miR-K3 (63 nt) from pre-miR-K3+1 (64 nt) (Fig. 2E). Thus far, we have collectively shown that the biogenesis of miR-K3+1 is due to the differential Drosha processing of an alternative pri-miRNA structure caused by the GSNP found in some PEL cell lines.
PAR-CLIP-predicted targetomes of miR-K3 and miR-K3+1.
As a first step in comparing the targets of miR-K3, miR-K3+1, and miR-23, we inspected the binding sites that were predicted for these miRNAs in our recent miRNA targetome analysis of PEL cell lines. In this analysis, we had used photoactivatable-ribonucleoside-enhanced cross-linking and Ago-2 immunoprecipitation (Ago2-PAR-CLIP) in combination with Illumina sequencing of protected fragments to identify miRNA binding sites (12, 48). Resulting fragments were assigned to the expressed miRNAs by seed matching. Here, we used only data from BC-1 cells, which express miR-K3 and miR-K3+1 (12) (Fig. 1D). Bioinformatically, seed targets of miR-K3+1 and miR-23 are indistinguishable; however, the miR-23 miRNAs together are expressed at ∼10-fold lower levels in this cell line than miR-K3+1 (12) (see below), suggesting that many of the identified target sites are occupied by miR-K3+1. This data set identified 190 candidate binding sites of miR-K3+1 and/or miR-23 and 158 candidate binding sites with seed matches to miR-K3. To our surprise, ∼94% of the sites with predicted binding to miR-K3 could also be assigned to miR-K3+1 and/or miR-23 (Fig. 3A). There are two reasons for this initially unexpectedly large overlap. (i) The second nucleotide of miR-K3+1 is a U, and therefore, 2-7A1mer matches to miR-K3 are also called as 2-8mer matches to miR-K3+1 (Fig. 3B), and (ii) more than 90% of the miR-K3 candidate target sites identified in BC-1 cells were 2-7A1mer sites. While these binding site assignments are not unambiguous, they raise the intriguing question of whether miR-K3 has a functional overlap with miR-23, despite the offset seed sequence.
Fig 3.
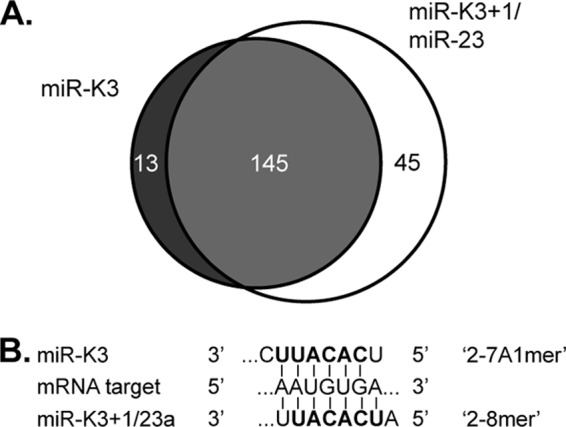
Predicted overlap between miR-K3 and miR-K3+1/miR-23 binding sites. (A) Venn diagram summarizing the numbers of shared (light gray) and distinct (dark gray and white) PAR-CLIP-identified binding sites of miR-K3 and miR-K3+1/miR-23 in the PEL cell line BC-1. (B) Schematic of seed interactions responsible for recognition of the same miRNA-binding sites by either miR-K3 or miR-K3+1/miR-23, despite the offset seed sequences of these miRNAs.
miR-23a expression in cell types relevant to KSHV infection.
If miR-K3 indeed mimics miR-23 functions, we wanted to establish in which cell types this may be relevant. Our previously published data suggested that the miR-23 miRNAs are expressed at only very low levels in PEL cell lines (12). We have also performed small-RNA deep sequencing with primary human dermal lymphatic endothelial cells (LECs) and LECs infected with rKSHV.219 at 72 h after infection (rK-LECs; not shown). Interestingly, miR-23 was among the top 15 most highly expressed miRNAs in both LECs and rK-LECs, with no indication of altered miR-23 expression in the context of KSHV infection. The full data set will be presented elsewhere. To confirm low expression of miR-23 in B and PEL cell lines and high expression in endothelial cells, we performed primer extension analysis of miR-23 family miRNA expression on total RNA prepared from telomerase-immortalized microvascular endothelial cells (iHMVEC) (49), LECs, the PEL cell lines BC-1 and BC-3, and the KSHV-negative Burkitt's lymphoma cell line BJAB. This analysis also included 293T and HeLa cells, because we commonly use 293T for 3′ UTR reporter assays and several targets of miR-23 have previously been validated in HeLa cells (50). These experiments confirmed that both miR-23a and miR-23b are well expressed in endothelial cells (Fig. 4). On the other hand, these miRNAs were detected at much lower levels in B cells. Primer extension for miR-23c was also performed but did not show significant expression in any of our samples, in agreement with the deep-sequencing data (12) (data not shown). Finally, these data confirmed that HeLa cells express substantial levels of miR-23a and -b while 293T cells express very low levels of miR-23a and -b.
Fig 4.
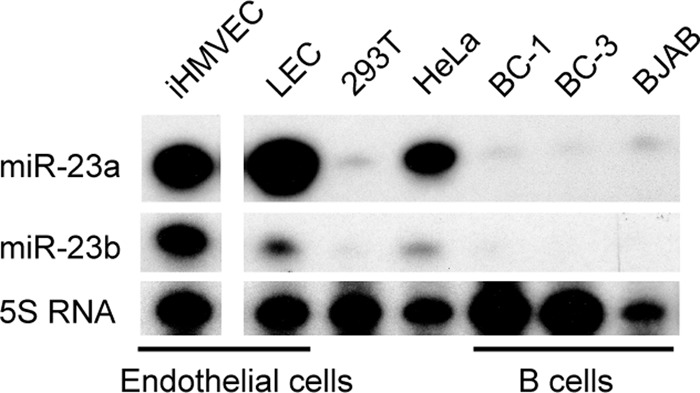
Endogenous expression of miR-23 family miRNAs in endothelial and B cells. Primer extension analysis was performed for miR-23a and miR-23b in telomerase-iHMVEC, primary dermal microvascular LECs, 293T cells, HeLa cells, the B cell line BJAB, and PEL cell lines BC-1 and BC-3. All reactions were performed in the same experiment and run on the same gel. One irrelevant lane was removed from the autoradiograph, as indicated by the gap. Primer extension for miR-23c was also performed, but all samples had little detectable expression (not shown).
Both miR-K3 and K3+1 regulate targets of miR-23a.
3′ UTR reporter assays directly measure binding site usage, if predicted sites are mutated. We selected a subset of PAR-CLIP-identified candidate targets of miR-K3 and/or miR-K3+1/miR-23 for validation (Tables 1 and 2). We also included a number of miR-23 targets that were predicted by TargetScan v6.2 (50–53) but not identified by PAR-CLIP because our PAR-CLIP data set may have missed valid miR-23 binding sites because of limited sequencing depth and the low expression of the miR-23 miRNAs in PEL cell lines. This initial set of 3′ UTR reporters also included several candidates for targets that we predicted to be differentially regulated by miR-K3 and miR-23. A relatively large number of 3′ UTR reporters was constructed, because miR-23a has previously been reported to have poor targeting proficiency, a consequence of its relatively weak predicted seed-pairing stability (50). This property caused a smaller subset of reporters than typical for other miRNAs to be repressed by miR-23a (50). We first tested each of the reporter constructs for regulation by lentiviral vectors expressing miR-23a or miR-K3 alone (ASNP), in the absence of miR-K3+1 (Fig. 5A). Only miR-23a was tested, because the miR-23 miRNAs are expected to have identical seed targets and miR-23a is the most abundant miR-23 miRNA in endothelial cells. Of the 21 candidate targets we screened, only 7 reporters were specifically repressed upon cotransfection with miR-23a and/or miR-K3 (Tables 1 and 2 and Fig. 5B to G and 6B). Our results therefore confirm the previously reported poor proficiency of miR-23a for target regulation and further suggest that miR-K3 may share this property with miR-23a. These results are in sharp contrast to the much higher success rate (>75%) for validation of PAR-CLIP-predicted binding sites we have reported previously and typically observe for other KSHV miRNAs (12, 54) (data not shown).
Table 1.
Summary of functional miR-K3/miR-23 binding sites validated in this studya
| Gene product | PAR-CLIP targetb | Prediction by TargetScanb | Seed type |
||
|---|---|---|---|---|---|
| miR-K3 | miR-K3+1 | miR-23 | |||
| CASP3 | Y | N | 2-7A1mer | 2-8A1mer | 2-8A1mer |
| CASP7 | Y | Y | 2-7A1mer | 2-8A1mer | 2-8A1mer |
| HMGB2 site 1 | N | N | 2-8mer | No site | No site |
| HMGB2 site 2 | Y | Y | 2-9A1mer | 2-10A1mer | 2-8A1mer |
| JARID2 | Y | Y | 2-8A1mer | 2-9A1mer | 2-8A1mer |
| TNFRSF10B | Y | N | 2-8A1mer | 2-9mer | 2-8mer |
| HMMR | Y | N | No site | 2-7A1mer | 2-7A1mer |
| RBL2 | N | Y | 2-7A1mer | 2-8A1mer | 2-9A1mer |
Each seed match was isolated by mutation. Whether these sites were identified by PAR-CLIP and/or predicted by TargetScan as targets of miR-23 is indicated. The predicted seed types for the interactions with miR-K3, miR-K3+1, and miR-23 are also indicated. Nucleotide numbers refer to the nucleotides from the 5′ end of the miRNA that are base paired. A1 indicates the presence of an A across from nt 1 of the miRNA. Additional candidate targets were tested but were not inhibited by any of the miRNAs tested (see Table 2; total n = 21).
Y, yes; N, no.
Table 2.
Summary of indicator vectors that did not respond to miR-K3 or miR-23aa
| Gene product | Prediction by: |
Seed type |
Note(s) | |||
|---|---|---|---|---|---|---|
| PAR-CLIPb | TargetScanb | miR-K3 | miR-K3+1 | miR-23 | ||
| KLHDC5 | Y | N | 2-8mer | No site | No site | No significant inhibition |
| TBL1XR1 | Y | N | 2-8mer | No site | No site | No significant inhibition |
| GATAD2B | Y | N | 2-9mer | No site | No site | No significant inhibition |
| SMC1A | Y | N | No site | 2-7A1mer | 2-7A1mer | No significant inhibition |
| RAD21 | Y | N | No site | 2-7A1mer | 2-7A1mer | No significant inhibition |
| WEE1 | Y | N | No site | 2-7A1mer | 2-7A1mer | No significant inhibition |
| PHF17 | N | Y | No site | 2-7A1mer | 2-7A1mer | No significant inhibition |
| PPP3CA | N | Y | No site | 2-7A1mer | 2-7A1mer | No significant inhibition |
| TPD52 | N | Y | No site | 2-7A1mer | 2-7A1mer | No significant inhibition |
| RAB4A | Y | N | 2-9mer | No site | No site | No significant inhibition; not rescued by seed mutant |
| MTUS1 | Y | N | 2-8mer | No site | No site | No significant inhibition; not rescued by seed mutant |
| CREBBP | Y | Y | No site | 2-7A1mer | 2-7A1mer | No significant inhibition; not rescued by seed mutant |
| CARD8 | Y | N | No site | 2-7A1mer | 2-7A1mer | Unexpected repression by miR-K3; seed match not mutated |
| SOS1 | N | Y | 2-7A1mer | 2-8A1mer | 2-9A1mer | Only miR-23a inhibits; seed site not mutated |
Whether these sites were identified by PAR-CLIP and/or predicted by TargetScan as targets of miR-23 is indicated. The predicted seed types for the interactions with miR-K3, miR-K3+1, and miR-23 are also indicated. Nucleotide numbers refer to the nucleotides from the 5′ end of the miRNA that are base paired. A1 indicates the presence of an A across from nt 1 of the miRNA. The last column summarizes the results from at least three independent reporter assays. The relevant seed match in all clones obtained for CARD8 contained a SNP compared to the reference sequences, which alters the sequence from AATGTGAA to CATGTGAA.
Y, yes; N, no.
Fig 5.

Reporter assays confirm the overlapping target range of miR-K3 or miR-23a. (A) Primer extension analysis of miR-23a (top) and miR-K3 (middle) expression in 293T cells transfected with pLCE miRNA expression vectors for miR-23a and miR-K3 from BCBL-1 (ASNP). Note that miR-K3+1 is not expressed from the expression vector used. HeLa cells and the PEL cell lines BC-1 and BC-3 serve as positive controls for miR-23a and miR-K3/K3+1, respectively. The unextended probe for miR-K3 is also shown in the middle and migrates as a single band. (B to G) Reporter assays for different candidate miR-K3 and miR-23a 3′ UTR targets identified from PAR-CLIP and/or TargetScan analyses. 3′ UTR reporters for the following candidate targets were tested: caspase 3 (CASP3), caspase 7 (CASP7), TRAIL receptor (TNFRSF10B), retinoblastoma-like 2 (p130, RBL2), JARID2, and HMMR. Firefly luciferase reporter activities were normalized to Renilla luciferase (ReL) signals from the cotransfected control plasmid pLSR, control reaction mixtures transfected with empty firefly luciferase vector, control reaction mixtures transfected with pLCE (no miRNA), and the corresponding mutant (mut) construct containing a 2-nt mutation in the seed region. Statistical analysis (two-way unpaired Student t test) was performed in GraphPad Prism. P values are indicated where significant (P < 0.05). Error bars represent standard deviations from three or more independent experiments. (H) The predicted noncanonical interaction of miR-K3 with the miR-23a-binding site in the HMMR 3′ UTR.
Fig 6.

Targeting of HMGB2 by miR-K3, miR-K3+1, and miR-23a. (A) Schematic of miR-K3, miR-K3+1, and miR-23a binding sites in the HMGB2 3′ UTR showing the expected seed type for each miRNA. Not drawn to scale. Numbers indicate positions of the seed matches in HMGB2 RefSeq NM_002129. (B) Reporter assay for the HMGB2 3′ UTR with synthetic miRNA mimics in 293T cells. Firefly luciferase reporter activities were normalized to Renilla luciferase (ReL) signals from the cotransfected control plasmid pLSR, control reaction mixtures transfected with an empty firefly luciferase vector, control reaction mixtures transfected with a negative-control mimic, and the double mutant 3′ UTR containing 2-nt mutations to disrupt both seed matches. Statistical analysis (two-way unpaired Student t test) was performed in GraphPad Prism. P values are indicated where significant (P < 0.05). Error bars represent standard deviations from three or more independent experiments. (C, D) Quantitative Western immunoblot (IB) assay of 293T (C) and BJAB (D) cells transfected with miRNA mimics and probed for HMGB2 and GAPDH (left panel). Band intensities from at least three independent experiments were quantified with the Odyssey FC Imaging System (LI-COR Biosciences) and are graphed on the right. HMGB2 signals were normalized to GAPDH levels and to the control mimic-treated sample. The two-way paired Student t test was performed in GraphPad Prism. Error bars represent standard deviations from independent experiments, and P values are shown.
We next focused our attention on six of the candidate targets for which we observed specific and comparable repression following miR-K3 and/or miR-23a expression compared to the empty-vector control (Fig. 5B to G). This set included reporters for CASP3 and CASP7, which had previously been shown to be repressed by miR-K3 and miR-23a, respectively (34, 55).To verify that miR-K3 and miR-23a indeed exert their effect through the same PAR-CLIP- and/or TargetScan-predicted binding sites in these 3′ UTRs, we introduced 2-nt mutations into each of the candidate seed matches. Results from these experiments confirmed that miR-K3 and miR-23a indeed regulate their targets through the same predicted sites (Fig. 5B to G). One surprising finding was that the hyaluronan-mediated motility receptor (HMMR) reporter, which was specifically repressed by miR-23a through a 2-7A1mer site, was also repressed by miR-K3 alone in a manner dependent on the same site, although only nt 2 to 6 of miR-K3 can pair to this site (Table 1). miR-K3 could potentially also engage this site in the HMMR 3′ UTR with nonseed interactions (Fig. 5H); however, mutation of 4 nt upstream of the seed match did not rescue reporter activity (not shown). This interaction could therefore present an unusual case where a very minimal seed interaction could be sufficient for repression. Alternatively, nonseed interactions other than those tested could contribute to the observed regulation.
Not all binding sites are shared between miR-K3 and miR-K3+1/miR-23.
The sites validated thus far were predicted to be targeted by both miR-K3 and miR-23a. To test whether these miRNAs could also have at least a small number of differentially regulated targets owing to the 1-nt offset, we performed additional reporter assays with the HMGB2 3′ UTR. HMGB2 has a PAR-CLIP-identified seed match for miR-K3, miR-K3+1, and miR-23a (site 2, Fig. 6A). In addition, a manual examination of its 3′ UTR revealed a second potential seed match for miR-K3, but not for miR-K3+1 or miR-23a, just 28 nt upstream of the PAR-CLIP-identified site (site 1). The proximity of these sites, within the distance observed for cooperative miRNA interactions (52, 56), suggests that both sites might be used by miR-K3. To achieve a quantitative comparison of HMGB2 repression by miR-K3 and miR-23a and to confirm that miR-K3+1 does indeed have the same targeting potential as miR-23a, we cotransfected wild-type (WT) and seed mutant HMGB2 3′ UTR reporters together with equimolar amounts of a control mimic or mimics of mature miR-K3, miR-K3+1, or miR-23a. Repression of the WT HMGB2 3′ UTR by miR-K3+1 was comparable to that by miR-23a, as expected from the direct seed homology of these two miRNAs (Fig. 6B). In contrast, miR-K3 resulted in significantly more robust repression of the WT HMGB2 3′ UTR than miR-K3+1 and miR-23a, which is consistent with repression through both candidate miR-K3 binding sites. Indeed, mutation of site 1 partially rescued inhibition by miR-K3 (P < 0.01) to levels similar to the inhibition by miR-K3+1 or miR-23a, which was unaffected by this mutation. Mutation of site 2 also partially rescued inhibition by miR-K3 and completely rescued inhibition by miR-K3+1 or miR-23a. A double mutant was not repressed by any of the miRNAs. From these data, we conclude that miR-K3 has at least one binding site that is not shared with miR-K3+1 and miR-23a. miR-K3 is therefore likely to function only as a partial mimic of miR-23a, with some targets that are unique to either miR-K3 or miR-23a.
We finally sought to extend our observations to the regulation of an endogenous miRNA target. For this, 293T cells were transfected with a control mimic or individual mimics of miR-K3, miR-K3+1, or miR-23a and HMGB2 protein expression was analyzed by quantitative Western blotting. As expected, all three miRNAs significantly (P < 0.01) downregulated endogenous HMGB2 protein expression (Fig. 6C). To test whether miR-K3 could indeed function to introduce miR-23-like activity into B cells, which are physiological targets of KSHV infection and express low levels of miR-23, we also measured the regulation of endogenous HMGB2 protein in the B cell line BJAB following transfection with a control mimic or individual mimics of miR-K3, miR-K3+1, and miR-23a. These experiments revealed that both miR-K3 and miR-23 indeed significantly downregulated HMGB2 protein (Fig. 6D). However, the relative differences in the targeting of HMGB2 by miR-K3 and miR-K3+1/miR-23a suggested by the indicator assays in Fig. 6B were not resolved in these experiments. Taken together, our data clearly show that miR-K3+1 is a functional analog of miR-23a. In addition, miR-K3 itself largely acts as a mimic of miR-23a, despite offset seed sequences.
DISCUSSION
KSHV stands out among viruses through its extensive “molecular piracy” of cellular factors and pathways (57). KSHV encodes, for example, viral homologs of cyclin D2, interleukin-6, and a G protein-coupled receptor, among others (58). Interestingly, not only have these viral proteins evolved to fulfill functions of their cellular homologs but they also generally differ from their cellular counterparts, for example, through an extended range of activities and to avoid negative regulation. Our identification of three viral miRNAs that have functions that strikingly overlap those of the cellular miRNAs miR-155, miR-142-3p, and miR-23 suggests that KSHV has also evolved multiple miRNAs to tap into evolutionarily conserved miRNA-regulatory networks (12, 14, 15, and this report). These viral miRNAs share 5′ seed sequence homologies with the cellular miRNAs but differ from them in their 3′ ends and precursor sequences, suggesting that these viral miRNAs have independently evolved functions that are analogous to those of the cellular miRNAs.
We and others have demonstrated that KSHV uses viral miR-K11 to repress seed targets of the cellular oncogenic miRNA miR-155 (14, 15), presumably leading to the functional mimicry of miR-155 by miR-K11 in mouse models of miRNA-induced B cell expansion (36, 37). In addition, the viral miR-K10a miRNAs share at least a subset of their targets with the cellular miR-142-3p miRNAs (12). One functional consequence of this mimicry is the damping of transforming growth factor β signaling (23). A second known function of miR-K10a is to inhibit the expression of the cytokine receptor tumor necrosis factor-like weak inducer of apoptosis receptor, TWEAKR, but it is unknown if this function is shared with miR-142-3p (17). Here, we add to the known repertoire of viral analogs of cellular miRNAs. We have mapped a SNP that leads to the coexpression of miR-K3 with the additional 5′ processing variant miR-K3+1, which is expressed by a subset of KSHV isolates. miR-K3+1 shares 7mer seed identity (nt 2 to 8) with the cellular miR-23 miRNAs (miR-23a, -b, and -c), reminiscent of the well-validated mimicry between miR-K11 and miR-155. As expected, miR-K3+1 and miR-23a expression resulted in comparable target repression, confirming that these miRNAs share their seed targets. miR-K3+1 expression is not conserved among all KSHV isolates, however, implying that miR-K3+1 is dispensable for the normal KSHV life cycle and also for the development of KS, MCD, PEL, and KSHV-associated inflammatory cytokine syndrome (39, 42, 43). We therefore explored the relationship between miR-23 and miR-K3, which is always expressed by KSHV. Despite the offset seed sequence of these two miRNAs, miR-K3 was able to effectively repress a number of miR-23 candidate targets that we predicted to be shared by the two miRNAs. The offset seed homology of miR-K3 to miR-23, however, allows KSHV to also expand the target repertoire of miR-K3 beyond the targets that are shared with miR-23. This is illustrated by the upstream miR-K3 binding site in the 3′ UTR of HMGB2, which is not shared by miR-23. By engaging two adjacent sites, miR-K3 is able to inhibit HMGB2 expression ∼20% more than miR-23, as shown by indicator assays (Fig. 6B). While we were unable to resolve this difference by Western blotting, it is conceivable that even one such differential binding site that extends the target range of miR-K3 compared to that of miR-23 could provide a selective advantage for KSHV.
Our validation experiments for PAR-CLIP- or TargetScan-predicted candidate targets of miR-K3 and/or miR-23 showed a strikingly lower-than-typical percentage of functional interactions (∼38% validation rate). This is in contrast to an ∼75% PAR-CLIP validation rate of other cellular or viral miRNA-binding sites (12, 54, and unpublished results). Thus, this low success rate is likely unique to miR-K3/K3+1 and miR-23. miR-23 has previously been reported to have one of the lowest targeting proficiencies among cellular miRNAs owing to its weak predicted seed interactions (50). In this previous study, only 33% of the reporters tested were inhibited by miR-23a, which is in close agreement with our validation rate. The observed low targeting proficiency of miR-K3, however, does not necessarily imply a lack of functional significance of this miRNA to KSHV infection, as KSHV may benefit from the repression of a small number of key proteins by miR-K3, such as HMGB2.
While this study has characterized the functional relationship between the miR-K3 miRNAs and miR-23, future studies will have to address the functional role of miR-K3 in KSHV infection. We hypothesize that one role of miR-K3 could be to introduce miR-23-like activities into B cells, which show only very low miR-23 expression. In contrast, miR-K3 is likely largely redundant with highly expressed miR-23 in endothelial cells, the cell type thought to drive the development of KS. In this cellular compartment, however, miR-K3 could still serve to access some targets that are not shared with miR-23 or, potentially, to modestly overexpress miR-23-like activities.
The identities of the miR-K3 targets validated here or those of previously established miR-23 targets could provide clues to understanding the roles of miR-K3 in KSHV infection and KSHV-associated malignancies. Although pathway analyses in this case have to be considered with caution, given our low validation rate for miR-K3 targets, one pathway with a statistically enrichment for miR-K3 and/or miR-K3+1 candidate targets was programmed cell death (GO:0012501∼programmed cell death, P < 0.024) (59, 60). Validated targets of miR-K3 with roles in the induction of apoptosis include the previously reported interactions of miR-K3 or miR-23 with the mRNAs encoding caspase 3 and caspase 7 and a novel interaction with tumor necrosis factor receptor superfamily member 10B (TNFRSF10B), which can be activated by TRAIL. It is therefore possible that miR-K3 may contribute to the inhibition of apoptosis by KSHV also through targets other than caspase 3 (34).
ACKNOWLEDGMENTS
Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under awards U54CA143869 and R00CA137860 and by the Zell Family Foundation and the Robert H. Lurie Comprehensive Cancer Center.
The content is solely our responsibility and does not necessarily represent the official views of the National Institutes of Health, the Zell Family Foundation, or the Robert H. Lurie Comprehensive Cancer Center.
We thank Eleonora Forte for helpful discussions and comments on the manuscript.
Footnotes
Published ahead of print 28 August 2013
REFERENCES
- 1.Gregory RI, Yan KP, Amuthan G, Chendrimada T, Doratotaj B, Cooch N, Shiekhattar R. 2004. The Microprocessor complex mediates the genesis of microRNAs. Nature 432:235–240 [DOI] [PubMed] [Google Scholar]
- 2.Han J, Lee Y, Yeom KH, Kim YK, Jin H, Kim VN. 2004. The Drosha-DGCR8 complex in primary microRNA processing. Genes Dev. 18:3016–3027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J, Lee J, Provost P, Radmark O, Kim S, Kim VN. 2003. The nuclear RNase III Drosha initiates microRNA processing. Nature 425:415–419 [DOI] [PubMed] [Google Scholar]
- 4.Han J, Lee Y, Yeom KH, Nam JW, Heo I, Rhee JK, Sohn SY, Cho Y, Zhang BT, Kim VN. 2006. Molecular basis for the recognition of primary microRNAs by the Drosha-DGCR8 complex. Cell 125:887–901 [DOI] [PubMed] [Google Scholar]
- 5.Grishok A, Pasquinelli AE, Conte D, Li N, Parrish S, Ha I, Baillie DL, Fire A, Ruvkun G, Mello CC. 2001. Genes and mechanisms related to RNA interference regulate expression of the small temporal RNAs that control C. elegans developmental timing. Cell 106:23–34 [DOI] [PubMed] [Google Scholar]
- 6.Hutvágner G, McLachlan J, Pasquinelli AE, Balint E, Tuschl T, Zamore PD. 2001. A cellular function for the RNA-interference enzyme Dicer in the maturation of the let-7 small temporal RNA. Science 293:834–838 [DOI] [PubMed] [Google Scholar]
- 7.Bartel DP. 2009. MicroRNAs: target recognition and regulatory functions. Cell 136:215–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cai X, Lu S, Zhang Z, Gonzalez CM, Damania B, Cullen BR. 2005. Kaposi's sarcoma-associated herpesvirus expresses an array of viral microRNAs in latently infected cells. Proc. Natl. Acad. Sci. U. S. A. 102:5570–5575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grundhoff A, Sullivan CS, Ganem D. 2006. A combined computational and microarray-based approach identifies novel microRNAs encoded by human gamma-herpesviruses. RNA 12:733–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pfeffer S, Sewer A, Lagos-Quintana M, Sheridan R, Sander C, Grasser FA, van Dyk LF, Ho CK, Shuman S, Chien M, Russo JJ, Ju J, Randall G, Lindenbach BD, Rice CM, Simon V, Ho DD, Zavolan M, Tuschl T. 2005. Identification of microRNAs of the herpesvirus family. Nat. Methods 2:269–276 [DOI] [PubMed] [Google Scholar]
- 11.Samols MA, Hu J, Skalsky RL, Renne R. 2005. Cloning and identification of a microRNA cluster within the latency-associated region of Kaposi's sarcoma-associated herpesvirus. J. Virol. 79:9301–9305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gottwein E, Corcoran DL, Mukherjee N, Skalsky RL, Hafner M, Nusbaum JD, Shamulailatpam P, Love CL, Dave SS, Tuschl T, Ohler U, Cullen BR. 2011. Viral microRNA targetome of KSHV-infected primary effusion lymphoma cell lines. Cell Host Microbe 10:515–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haecker I, Gay LA, Yang Y, Hu J, Morse AM, McIntyre LM, Renne R. 2012. Ago HITS-CLIP expands understanding of Kaposi's sarcoma-associated herpesvirus miRNA function in primary effusion lymphomas. PLoS Pathog. 8:e1002884. 10.1371/journal.ppat.1002884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gottwein E, Mukherjee N, Sachse C, Frenzel C, Majoros WH, Chi JT, Braich R, Manoharan M, Soutschek J, Ohler U, Cullen BR. 2007. A viral microRNA functions as an orthologue of cellular miR-155. Nature 450:1096–1099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Skalsky RL, Samols MA, Plaisance KB, Boss IW, Riva A, Lopez MC, Baker HV, Renne R. 2007. Kaposi's sarcoma-associated herpesvirus encodes an ortholog of miR-155. J. Virol. 81:12836–12845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abend JR, Ramalingam D, Kieffer-Kwon P, Uldrick TS, Yarchoan R, Ziegelbauer JM. 2012. KSHV microRNAs target two components of the TLR/IL-1R signaling cascade, IRAK1 and MYD88, to reduce inflammatory cytokine expression. J. Virol. 86:11663–11674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abend JR, Uldrick T, Ziegelbauer JM. 2010. Regulation of tumor necrosis factor-like weak inducer of apoptosis receptor protein (TWEAKR) expression by Kaposi's sarcoma-associated herpesvirus microRNA prevents TWEAK-induced apoptosis and inflammatory cytokine expression. J. Virol. 84:12139–12151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bellare P, Ganem D. 2009. Regulation of KSHV lytic switch protein expression by a virus-encoded microRNA: an evolutionary adaptation that fine-tunes lytic reactivation. Cell Host Microbe 6:570–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dölken L, Malterer G, Erhard F, Kothe S, Friedel CC, Suffert G, Marcinowski L, Motsch N, Barth S, Beitzinger M, Lieber D, Bailer SM, Hoffmann R, Ruzsics Z, Kremmer E, Pfeffer S, Zimmer R, Koszinowski UH, Grasser F, Meister G, Haas J. 2010. Systematic analysis of viral and cellular microRNA targets in cells latently infected with human gamma-herpesviruses by RISC immunoprecipitation assay. Cell Host Microbe 7:324–334 [DOI] [PubMed] [Google Scholar]
- 20.Gottwein E, Cullen BR. 2010. A human herpesvirus microRNA inhibits p21 expression and attenuates p21-mediated cell cycle arrest. J. Virol. 84:5229–5237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hansen A, Henderson S, Lagos D, Nikitenko L, Coulter E, Roberts S, Gratrix F, Plaisance K, Renne R, Bower M, Kellam P, Boshoff C. 2010. KSHV-encoded miRNAs target MAF to induce endothelial cell reprogramming. Genes Dev. 24:195–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lei X, Bai Z, Ye F, Xie J, Kim CG, Huang Y, Gao SJ. 2010. Regulation of NF-kappaB inhibitor IkappaBalpha and viral replication by a KSHV microRNA. Nat. Cell Biol. 12:193–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lei X, Zhu Y, Jones T, Bai Z, Huang Y, Gao SJ. 2012. A KSHV microRNA and its variants target TGF-beta pathway to promote cell survival. J. Virol. 86:11698–116711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liang D, Gao Y, Lin X, He Z, Zhao Q, Deng Q, Lan K. 2011. A human herpesvirus miRNA attenuates interferon signaling and contributes to maintenance of viral latency by targeting IKKepsilon. Cell Res. 21:793–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin HR, Ganem D. 2011. Viral microRNA target allows insight into the role of translation in governing microRNA target accessibility. Proc. Natl. Acad. Sci. U. S. A. 108:5148–5153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lin X, Liang D, He Z, Deng Q, Robertson ES, Lan K. 2011. miR-K12-7-5p encoded by Kaposi's sarcoma-associated herpesvirus stabilizes the latent state by targeting viral ORF50/RTA. PLoS One 6:e16224. 10.1371/journal.pone.0016224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin YT, Sullivan CS. 2011. Expanding the role of Drosha to the regulation of viral gene expression. Proc. Natl. Acad. Sci. U. S. A. 108:11229–11234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu Y, Sun R, Lin X, Liang D, Deng Q, Lan K. 2012. KSHV-encoded miR-K12-11 attenuates transforming growth factor beta signaling through suppression of SMAD5. J. Virol. 86:1372–1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lu CC, Li Z, Chu CY, Feng J, Sun R, Rana TM. 2010. MicroRNAs encoded by Kaposi's sarcoma-associated herpesvirus regulate viral life cycle. EMBO Rep. 11:784–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lu F, Stedman W, Yousef M, Renne R, Lieberman PM. 2010. Epigenetic regulation of Kaposi's sarcoma-associated herpesvirus latency by virus-encoded microRNAs that target Rta and the cellular Rbl2-DNMT pathway. J. Virol. 84:2697–2706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nachmani D, Stern-Ginossar N, Sarid R, Mandelboim O. 2009. Diverse herpesvirus microRNAs target the stress-induced immune ligand MICB to escape recognition by natural killer cells. Cell Host Microbe 5:376–385 [DOI] [PubMed] [Google Scholar]
- 32.Qin Z, Dai L, Defee M, Findlay VJ, Watson DK, Toole BP, Cameron J, Peruzzi F, Kirkwood K, Parsons C. 2013. Kaposi's sarcoma-associated herpesvirus suppression of DUSP1 facilitates cellular pathogenesis following de novo infection. J. Virol. 87:621–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qin Z, Freitas E, Sullivan R, Mohan S, Bacelieri R, Branch D, Romano M, Kearney P, Oates J, Plaisance K, Renne R, Kaleeba J, Parsons C. 2010. Upregulation of xCT by KSHV-encoded microRNAs facilitates KSHV dissemination and persistence in an environment of oxidative stress. PLoS Pathog. 6:e1000742. 10.1371/journal.ppat.1000742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Suffert G, Malterer G, Hausser J, Viiliainen J, Fender A, Contrant M, Ivacevic T, Benes V, Gros F, Voinnet O, Zavolan M, Ojala PM, Haas JG, Pfeffer S. 2011. Kaposi's sarcoma herpesvirus microRNAs target caspase 3 and regulate apoptosis. PLoS Pathog. 7:e1002405. 10.1371/journal.ppat.1002405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ziegelbauer JM, Sullivan CS, Ganem D. 2009. Tandem array-based expression screens identify host mRNA targets of virus-encoded microRNAs. Nat. Genet. 41:130–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boss IW, Nadeau PE, Abbott JR, Yang Y, Mergia A, Renne R. 2011. A Kaposi's sarcoma-associated herpesvirus-encoded ortholog of microRNA miR-155 induces human splenic B-cell expansion in NOD/LtSz-scid IL2Rgammanull mice. J. Virol. 85:9877–9886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dahlke C, Maul K, Christalla T, Walz N, Schult P, Stocking C, Grundhoff A. 2012. A microRNA encoded by Kaposi sarcoma-associated herpesvirus promotes B-cell expansion in vivo. PLoS One 7:e49435. 10.1371/journal.pone.0049435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hofacker IL. 2003. Vienna RNA secondary structure server. Nucleic Acids Res. 31:3429–3431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gottwein E, Cai X, Cullen BR. 2006. A novel assay for viral microRNA function identifies a single nucleotide polymorphism that affects Drosha processing. J. Virol. 80:5321–5326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee Y, Kim VN. 2007. In vitro and in vivo assays for the activity of Drosha complex. Methods Enzymol. 427:89–106 [DOI] [PubMed] [Google Scholar]
- 41.Zhang J, Jima DD, Jacobs C, Fischer R, Gottwein E, Huang G, Lugar PL, Lagoo AS, Rizzieri DA, Friedman DR, Weinberg JB, Lipsky PE, Dave SS. 2009. Patterns of microRNA expression characterize stages of human B-cell differentiation. Blood 113:4586–4594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marshall V, Martro E, Labo N, Ray A, Wang D, Mbisa G, Bagni RK, Volfovsky N, Casabona J, Whitby D. 2010. Kaposi sarcoma (KS)-associated herpesvirus microRNA sequence analysis and KS risk in a European AIDS-KS case control study. J. Infect. Dis. 202:1126–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ray A, Marshall V, Uldrick T, Leighty R, Labo N, Wyvill K, Aleman K, Polizzotto MN, Little RF, Yarchoan R, Whitby D. 2012. Sequence analysis of Kaposi sarcoma-associated herpesvirus (KSHV) microRNAs in patients with multicentric Castleman disease and KSHV-associated inflammatory cytokine syndrome. J. Infect. Dis. 205:1665–1676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yeom KH, Lee Y, Han J, Suh MR, Kim VN. 2006. Characterization of DGCR8/Pasha, the essential cofactor for Drosha in primary miRNA processing. Nucleic Acids Res. 34:4622–4629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zeng Y, Cullen BR. 2003. Sequence requirements for micro RNA processing and function in human cells. RNA 9:112–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zeng Y, Yi R, Cullen BR. 2005. Recognition and cleavage of primary microRNA precursors by the nuclear processing enzyme Drosha. EMBO J. 24:138–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Auyeung VC, Ulitsky I, McGeary SE, Bartel DP. 2013. Beyond secondary structure: primary-sequence determinants license pri-miRNA hairpins for processing. Cell 152:844–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M, Jr, Jungkamp AC, Munschauer M, Ulrich A, Wardle GS, Dewell S, Zavolan M, Tuschl T. 2010. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 141:129–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shao R, Guo X. 2004. Human microvascular endothelial cells immortalized with human telomerase catalytic protein: a model for the study of in vitro angiogenesis. Biochem. Biophys. Res. Commun. 321:788–794 [DOI] [PubMed] [Google Scholar]
- 50.Garcia DM, Baek D, Shin C, Bell GW, Grimson A, Bartel DP. 2011. Weak seed-pairing stability and high target-site abundance decrease the proficiency of lsy-6 and other microRNAs. Nat. Struct. Mol. Biol. 18:1139–1146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Friedman RC, Farh KK, Burge CB, Bartel DP. 2009. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 19:92–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Grimson A, Farh KK, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP. 2007. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol. Cell 27:91–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lewis BP, Burge CB, Bartel DP. 2005. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120:15–20 [DOI] [PubMed] [Google Scholar]
- 54.Skalsky RL, Corcoran DL, Gottwein E, Frank CL, Kang D, Hafner M, Nusbaum JD, Feederle R, Delecluse HJ, Luftig MA, Tuschl T, Ohler U, Cullen BR. 2012. The viral and cellular microRNA targetome in lymphoblastoid cell lines. PLoS Pathog. 8:e1002484. 10.1371/journal.ppat.1002484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ruan W, Xu JM, Li SB, Yuan LQ, Dai RP. 2012. Effects of down-regulation of microRNA-23a on TNF-alpha-induced endothelial cell apoptosis through caspase-dependent pathways. Cardiovasc. Res. 93:623–632 [DOI] [PubMed] [Google Scholar]
- 56.Saetrom P, Heale BS, Snove O, Jr, Aagaard L, Alluin J, Rossi JJ. 2007. Distance constraints between microRNA target sites dictate efficacy and cooperativity. Nucleic Acids Res. 35:2333–2342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Moore PS, Chang Y. 1998. Antiviral activity of tumor-suppressor pathways: clues from molecular piracy by KSHV. Trends Genet. 14:144–150 [DOI] [PubMed] [Google Scholar]
- 58.Ganem D. 2007. Kaposi's sarcoma-associated herpesvirus, p 2847–2888 In Knipe DM, Howley PM. (ed), Fields virology, 5th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 59.Huang da W, Sherman BT, Lempicki RA. 2009. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4:44–57 [DOI] [PubMed] [Google Scholar]
- 60.Huang da W, Sherman BT, Tan Q, Kir J, Liu D, Bryant D, Guo Y, Stephens R, Baseler MW, Lane HC, Lempicki RA. 2007. DAVID Bioinformatics Resources: expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 35:W169–W175 [DOI] [PMC free article] [PubMed] [Google Scholar]