

Abstract
The deoxynucleoside triphosphohydrolase SAMHD1 restricts retroviral replication in myeloid cells. Human immunodeficiency virus type 2 (HIV-2) and a simian immunodeficiency virus from rhesus macaques (SIVmac) encode Vpx, a virion-packaged accessory protein that counteracts SAMHD1 by inducing its degradation. SAMHD1 is thought to work by depleting the pool of intracellular deoxynucleoside triphosphates but has also been reported to have exonuclease activity that could allow it to degrade the viral genomic RNA or viral reverse-transcribed DNA. To induce the degradation of SAMHD1, Vpx co-opts the cullin4a-based E3 ubiquitin ligase, CRL4. E3 ubiquitin ligases are regulated by the covalent attachment of the ubiquitin-like protein Nedd8 to the cullin subunit. Neddylation can be prevented by MLN4924, a drug that inhibits the nedd8-activating enzyme. We report that MLN4924 inhibits the neddylation of CRL4, blocking Vpx-induced degradation of SAMHD1 and maintaining the restriction. Removal of the drug several hours postinfection released the block. Similarly, Vpx-containing virus-like particles and deoxynucleosides added to the cells more than 24 h postinfection released the SAMHD1-mediated block. Taken together, these findings support deoxynucleoside triphosphate pool depletion as the primary mechanism of SAMHD1 restriction and argue against a nucleolytic mechanism, which would not be reversible.
INTRODUCTION
Mammalian cells express antiviral proteins that restrict the replication of viruses like HIV-1 and other lentiviruses. One such restriction factor is SAMHD1, a deoxynucleoside triphosphohydrolase that blocks retrovirus infection at reverse transcription in nondividing myeloid cells, such as macrophages and dendritic cells (1–3). SAMHD1 is also expressed in T cells, where it blocks the infection of resting T cells but has little effect on activated T cells (4, 5). It is thought to work by depleting the pool of intracellular deoxynucleoside (dN) triphosphates (dNTPs) to a level below that which supports reverse transcription (1–3, 6, 7), although other mechanisms have been proposed (8, 9). Viruses have evolved various means of counteracting antiviral host proteins, most notably by encoding accessory proteins (reviewed in reference 10). Lentiviruses, such as human immunodeficiency virus type 2 (HIV-2) and a simian immunodeficiency virus from rhesus macaques (SIVmac), encode Vpx, a virion-packaged accessory protein that is released into the target cell postentry (11). Upon its release, Vpx associates with the E3 ubiquitin ligase CRL4 and recruits SAMHD1 to the complex, inducing its proteasomal degradation (12–14). The degradation of SAMHD1 and subsequent rise in dNTP levels occur within 8 h postinfection, after which reverse transcription resumes (15). SAMHD1 localizes to the nucleus of the cell by an amino-terminal nuclear localization sequence (16, 17). Deletion of the nuclear localization sequence relocalizes SAMHD1 to the cytoplasm and causes it to be resistant to Vpx-induced degradation. The treatment of cells with leptomycin B, a drug that prevents nuclear export, does not interfere with Vpx-induced degradation of SAMHD1, suggesting that the degradation occurs in the nucleus (17).
Vpx induces the degradation of SAMHD1 by interacting with the E3 ubiquitin ligase CRL4, a complex that consists of DDB1, RBX1, Cullin4A (CUL4A), and DCAF1. In the complex, CUL4A serves as a scaffold that tethers DCAF1 and the adaptor DDB1 to the RING domain protein RBX1 (2, 12). Vpx associates with the substrate receptor DCAF1 and with SAMHD1 at its carboxy terminus (14, 18). The ubiquitin ligase activity of cullin-RING ligase complexes like CRL4 is regulated by the covalent attachment of the 9-kDa ubiquitin-like modifier Nedd8 (19). The conjugation of Nedd8 is mediated by the Nedd8-activating enzyme (NAE) in a multistep pathway in which ATP is used to generate a Nedd8-adenylate adduct (20, 21). Nedd8 then forms a thioester bond with a cysteine residue of NAE and is subsequently transferred by Ubc12 to a specific lysine of cullin 1-4 or by UBE2F to cullin 5. The resulting neddylated cullin is the active form of the E3 ubiquitin ligase complex. CRL4 complexes are negatively regulated by the COP9 signalosome, which removes the Nedd8 to inactivate the ubiquitin ligase activity (22, 23). MLN4924 is an adenosine sulfamate analog that specifically inhibits cullin neddylation (24). The drug forms an adduct with Nedd8 that prevents the formation of Nedd8-adenylate (25). The MLN4924-Nedd8 adduct binds to NAE but cannot form the thioester bond, preventing cullin neddylation.
SAMHD1 was proposed to restrict the replication of diverse retroviruses by diminishing the pool of intracellular dNTPs to a level below that which is required to support reverse transcription, based on several lines of evidence (6, 7, 15, 26, 27). The expression of SAMHD1 in differentiated U937 cells reduced dNTP levels by nearly 100-fold, and the addition of exogenous dN partially alleviated the restriction (7). Two recent reports, however, have suggested that nucleotide pool depletion does not fully account for SAMHD1-mediated restriction (9, 28). This was shown by mutation of SAMHD1 at T592, a site of CDK1-mediated phosphorylation. A mutation of T592 to A (T592A), which prevents phosphorylation, maintained the ability to restrict HIV-1 and to deplete the dNTPs. In contrast, T592E, which mimics the phosphorylated form, maintained the ability to deplete dNTPs but lost restriction activity (9). These findings suggested that SAMHD1 may restrict retroviruses by an alternative mechanism. Recombinant SAMHD1 was reported to have 3′-to-5′ exonuclease activity on single-stranded RNA and DNA, raising the possibility that SAMHD1 blocks infection by targeting the viral RNA or newly reverse-transcribed DNA (8).
In this study, we used MLN4924 to show that Vpx-mediated degradation of SAMHD1 is dependent upon the neddylation of CUL4A. MLN4924 prevented the Vpx-dependent infection of myeloid cells and the degradation of SAMHD1. The MLN4924-mediated block to Vpx function was reversed upon removal of the drug several hours postinfection, relieving the block to infection. The reversibility of the block argues against degradation of the viral RNA as the primary mechanism of SAMHD1-mediated restriction.
MATERIALS AND METHODS
Cell culture.
293T cells were cultured in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS). U937 and THP1 cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium with 10% FBS. U937 and THP1 cells were differentiated for 20 h with 30 ng/ml phorbol 12-myristate 13-acetate (PMA). The peripheral blood mononuclear cells (PBMC) of anonymous healthy donors were obtained from the New York Blood Center and used to prepare buffy coats by Ficoll density gradient centrifugation. To generate monocyte-derived macrophages (MDM), the immature monocytes were purified from the buffy coat lymphocytes by adherence to plastic and cultured for 5 days in RPMI 1640 containing 10 mM HEPES, 24 μg/ml gentamicin, and 5% heat-inactivated pooled human serum supplemented with 116 U/ml granulocyte-macrophage colony-stimulating factor (GM-CSF; Invitrogen, Inc.). To generate monocyte-derived dendritic cells (MDDC), the medium was supplemented with GM-CSF and 300 U/ml interleukin-4 (IL-4; R&D systems). The medium and cytokines were replaced every other day. U937 cells that stably express hemagglutinin (HA)-tagged SAMHD1 (U937-HA.SAMHD1) and THP1 cells that stably express SAMHD1 small hairpin RNA (shRNA) (THP1-SAMHD1.shRNA) have been described previously (17, 26). HeLa cells stably expressing Flag-/HA-tagged SAMHD1 were generated by transduction of HeLa cells with pOZ.F/H.SAMHD1 (3).
Virus and VLP production.
To produce vesicular stomatitis virus glycoprotein (VSV-G)-pseudotyped HIV-1 green fluorescent protein (HIV.GFP) reporter virus that contained packaged Vpx, 293T cells were cotransfected by calcium phosphate coprecipitation with HIV.GFP containing the p6 Vpx-packaging motif of SIVmac (29), pcVSV-G (30), and pcVpx.mycHis (29) or pcDNA6/myc-His A (Invitrogen) at a mass ratio of 22:5:3. After 48 h, virus-containing supernatant was harvested, passed through a 0.45-μm filter, concentrated 10-fold by ultracentrifugation for 90 min through a 20% sucrose cushion at 30,000 rpm at 4°C, and frozen at −80°C. The virus titers were determined on 293T cells as the number of GFP-positive (GFP+) cells per milliliter of virus by flow cytometry. Luciferase reporter virus was produced by cotransfecting 293T cells with pNL43.Luc.ΔenvΔvpr, pSIVmac.luc.Δenv or pSIVmac.luc.ΔenvΔvpx (31), and pcVSV-G at a mass ratio of 5:1. The virus was normalized for luciferase activity on 293T cells by infecting 1.0 × 104 293T cells with 50 μl of virus and, after 72 h, measuring the luciferase activity with the SteadyLite Plus reporter gene assay substrate (PerkinElmer). Titers were typically 1 × 106 to 3 × 106 cps per milliliter. Virus-like particles (VLP) were produced by cotransfecting 293T cells with pSIV3+ (VLP Vpx+) or pSIV3+ Δvpx (VLP Vpx−) and pVSV-G at mass ratio of 2:1. Two days posttransfection, the VLP-containing cell culture supernatant was harvested, passed through a 0.45-μm filter, and concentrated 10-fold by ultracentrifugation through a 20% sucrose cushion.
Infections.
MDDC (2.0 × 105), U937 and U937-HA.SAMHD1 cells (5.0 × 104), and THP1 and THP1-SAMHD1.shRNA (5.0 × 104) cells were plated in a 96-well culture plate, and the U937 and THP1 cells were differentiated for 20 h with 30 ng/ml PMA. Vpx-containing or control VLP were added, and the cells were then infected with HIV.GFP at a multiplicity of infection (MOI) of 1 or with luciferase reporter virus (1.0 × 106 cps). Two hours prior to infection, MLN4924 (Active Biochem) was added at 0.05 μM, 0.25 μM, 1.0 μM, or 2.0 μM. A well was included that contained 0.1% dimethyl sulfoxide (DMSO), corresponding to the concentration present in the well that contained the drug at the highest concentration. After 24 h, the supernatant was removed and replaced with fresh culture medium containing the same amount of MLN4924 or 25.0 μM azidothymidine (AZT) (Sigma-Aldrich). Three days postinfection, the GFP+ cells were quantified on an LSRII (Becton, Dickinson) flow cytometer and analyzed with FlowJo software. Luciferase reporter virus-infected cells were harvested 3 days postinfection, and luciferase activity was measured using the SteadyLite Plus reagent (PerkinElmer). MDM (1.0 × 105) in 96-well plates were treated with VLP at specified time points between 24 h prior to and 48 h after infection with wild-type or Δvpx SIV luciferase reporter virus (3.0 × 105 cps). The medium was replaced 8 h postinfection, and 4 days postinfection, the luciferase activity was assayed. Deoxynucleosides (dN) were added at a concentration of 2 mM to MDDC infected with HIV.GFP. dN were washed out after 24 h for each time point. Three days postinfection, the GFP+ cells were quantified on an LSRII (Becton, Dickinson) flow cytometer and analyzed with FlowJo software. All infections were in triplicate, and the results are presented as the averages with standard deviations. Cell viability was determined using the CellTiter 96 aqueous one solution cell proliferation assay (Promega).
SAMHD1 degradation assay.
U937-HA.SAMHD1 cells and THP1 cells (2.0 × 105) were differentiated overnight with 30 ng/ml PMA in a 24-well plate. MDDC (4.0 × 105) were differentiated with GM-CSF and IL-4 for 5 days in 24-well plates. MLN4924 was added together with VLP. The amount of Vpx-containing VLP added was that which induced the nearly complete degradation of SAMHD1 in U937-HA.SAMHD1 cells. Control VLP were added with a corresponding amount of p27. The following day, the cells were lysed in buffer containing 50 mM HEPES, 150 mM KCl, 2.0 mM EDTA, 0.5% NP-40, and Halt protease inhibitor cocktail (ThermoScientific). For time course experiments, MLN4924 was added at time points before or after the VLP. The cells were harvested 20 h later, and lysates were analyzed on an immunoblot. HA.SAMHD1 was detected with anti-HA monoclonal antibody (MAb) HA.11 (Covance). Endogenous SAMHD1 was detected with anti-SAMHD1 MAb (Origene). The immunoblots were probed with anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) MAb (Ambion) as a loading control. Bound antibody was detected using goat anti-mouse horseradish peroxidase-conjugated antibody (Pierce). The signal was developed with Super Signal West Pico chemiluminescent substrate and visualized on an Odyssey FC imager (LI-COR).
qRT-PCR.
MDDC (2.0 × 106) in 6-well plates were pretreated with 0.05, 0.25, 1.0, or 2.0 μM MLN4924 for 2 h. Vpx-containing or control VLP were added, and the cells were then infected with HIV.GFP reporter virus at an MOI of 2 using virus that had been treated for 1 h with 50 U/ml Benzonase (Novagen) to remove plasmid DNA. To control for residual plasmid DNA, 25 μM AZT was added to control wells 14 h prior to infection. At 24 h postinfection, total DNA was isolated using a DNeasy kit (Qiagen), and the viral DNA copies were quantified by quantitative real-time PCR (qRT-PCR) using 250 ng DNA in an ABI Prism 7300 with SYBR green reagent (Roche). The primer pair used to detect late reverse transcription products (32) was MH531 (5′-TGTGTGCCCGTCTGTTGTGT) and MH532 (5′-GAGTCCTGCGTCGAGAGAGC). A standard curve was generated by amplification of proviral plasmid that was serially diluted in genomic DNA of the same uninfected MDDC donor.
Cullin4A immunoblots.
To visualize neddylated CUL4A, MDDC were lysed in buffer containing 6.0 M urea, 50 mM NaHPO4, and 150 mM NaCl, and the proteins were separated on a 4 to 12% SDS-PAGE gel. The proteins were transferred to a polyvinylidene difluoride (PVDF) membrane and probed with anti-CUL4A rabbit MAb clone EPR3198 (Abcam). The bands were quantified on an Odyssey FC imaging system (LI-COR) using Image Studio software (LI-COR).
MTS assay.
PMA-differentiated U937 and U937-HA.SAMHD1 cells (5.0 × 104) were incubated with increasing amounts of MLN4924 for 24 h or 72 h. Cell viability was determined by a 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay (CellTiter 96 aqueous one solution cell proliferation assay; Promega), an assay that measures the activity of cellular enzymes that reduce the tetrazolium dye. The viability of untreated cells was set to 100%, and cells treated with Triton X-100 served as a control for dead cells.
RESULTS
MLN4924 blocks the ability of Vpx to enhance the infection of MDDC.
The ubiquitin ligase activity of cullin-based E3 ubiquitin ligases is regulated by neddylation. We reasoned that if that was the case for CRL4, then MLN4924, a drug which prevents neddylation of CUL4A, would interfere with Vpx-dependent infection of MDDC by lentiviruses. To determine whether MLN4924 would prevent Vpx from counteracting SAMHD1, we incubated MDDC from four healthy donors with MLN4924 and then infected the cells with Vpx-containing or control HIV.GFP reporter virus. Because HIV-1 does not encode Vpx, we used an engineered HIV-1 that has the Vpx packaging motif of SIVmac incorporated into p6 (29). The viruses were produced as VSV-G pseudotypes with and without Vpx and containing an HIV-1 genome in which a GFP cassette under the control of a cytomegalovirus (CMV) promoter was placed in env. Three days postinfection, we quantified the infected cells by flow cytometry. The results showed that, as the concentration of MLN4924 increased, the number of infected cells decreased (Fig. 1A). There was variability between donors, but overall, the drug worked on all of the donor samples tested, causing an average 8-fold reduction in the number of infected cells.
Fig 1.

MLN4924 inhibits Vpx function in cells that express SAMHD1. (A) MLN4924 (0.05, 0.25, 1.0, or 2.0 μM) or DMSO was added to cultures of MDDC isolated from four donors. The cells were infected with Vpx-containing or control HIV.GFP reporter virus, and after 3 days, the number of infected cells was quantified by flow cytometry. The data shown are representative of three experiments. FSC, forward scatter. (B) MLN4924 was added to MDDC from two donors at the same concentrations as listed in the legend to panel A. The cells were infected with an HIV.GFP reporter virus, and after 24 h, DNA was isolated. Newly synthesized late reverse transcripts were quantified by qRT-PCR using the DNA as the template. To control for contaminating plasmid DNA, a control was included in which AZT was added 14 h prior to infection. VLP X−, VLP lacking Vpx; VLP X+, VLP containing Vpx. (C) MLN4924 was added, as described in the legend to panel A, to PMA-differentiated U937-HA.SAMHD1 or parental U937 cells. After 2 h, Vpx-containing or control VLP were added and the cells were infected with an HIV.GFP reporter virus. After 3 days, the number of infected cells was quantified by flow cytometry. The results shown are the averages of triplicate infections. (D) Differentiated U937 and U937.HA-SAMHD1 cells were incubated with increasing amounts of MLN4924 for 24 h or 72 h. Metabolic activity was then measured by MTS assay. Cells treated with Triton X-100 (TX-100) served as a control for dead cells. Error bars show standard deviations.
To determine whether the drug blocked the infection at reverse transcription, as is the case for SAMHD1, we quantified the viral reverse transcripts generated in MDDC infected in the presence or absence of MLN4924 by qRT-PCR. We found that, as the concentration of MLN4924 increased, the number of late reverse transcripts decreased to a level similar to that of the AZT control (Fig. 1B), a number that was similar to that of cells infected by Vpx-deficient virus. We therefore concluded that the drug caused a block at reverse transcription.
To determine whether the effect of MLN4924 on infection was dependent upon SAMHD1, we used U937 cells that stably expressed a transduced SAMHD1 gene (U937-HA.SAMHD1) and parental U937 cells that do not express endogenous SAMHD1. Upon differentiation with PMA, U937-HA.SAMHD1 cells resist lentiviral infection (7, 17). The cells were treated with MLN4924 and control and Vpx-containing VLP and were then infected with HIV.GFP. Analysis 3 days postinfection by flow cytometry showed that increasing concentrations of MLN4924 rendered the U937-HA.SAMHD1 cells resistant to HIV-1 but had no effect on the Vpx-induced HIV infection of the U937 cells (Fig. 1C). The U937 cells showed a higher level of infectivity than U937-HA.SAMHD1 cells infected with HIV.GFP in the presence of Vpx. An analysis by MTS assay of the viability of the drug-treated cells 24 h and 72 h posttreatment showed that there had been no effect on cell viability, ruling out the possibly that the results were caused by drug toxicity (Fig. 1D). Taken together, these findings suggested that MLN4924 inhibited the infection by its effect on SAMHD1.
Vpx-induced degradation of SAMHD1 is blocked by MLN4924.
If MLN4924 inhibits CRL4 function, the drug should block the Vpx-induced degradation of SAMHD1. To test whether this was the case, we treated MDDC from two donors with MLN4924, added Vpx-containing or control VLP and, after 20 h, quantified the amount of SAMHD1 in the cells on an immunoblot. We found that, in both donor cell preparations, with increasing concentrations of MLN4924, SAMHD1 became increasingly resistant to Vpx-induced degradation (Fig. 2A). Similarly, Vpx caused the partial degradation of SAMHD1 expressed in stable U937-HA.SAMHD1 cells (Fig. 2B, left). Increasing amounts of MLN4924 prevented the Vpx-induced degradation of SAMHD1 in these cells. Vpx did not degrade all of the SAMHD1, and as a result, Vpx did not fully restore the infectivity of HIV.GFP on the U937-HA.SAMHD1 cells. In THP1 cells, degradation of the endogenous SAMHD1 was nearly complete (Fig. 2B, right). The addition of MLN4924 prevented the degradation in a dose-dependent manner, although the highest concentration of the drug did not completely prevent degradation. In the U937-HA.SAMHD1 and THP1 cells treated with control VLP that lacked Vpx, MLN4924 had no effect on the amount of SAMHD1. We also tested the effect of the drug on SAMHD1 in dividing cells. For this, we used HeLa.SAMHD1 cells, a cell line in which SAMHD1 is expressed but does not restrict infection. In these cells, MLN4924 caused a small but reproducible increase in the steady-state level of SAMHD1 (Fig. 2C). This result suggested that in actively dividing cells, the amount of SAMHD1 is regulated by a cullin-based E3 ubiquitin ligase independently of Vpx.
Fig 2.

MLN4924 prevents the Vpx-induced degradation of SAMHD1. (A) MLN4924 (0.05, 0.25, 1.0, or 2.0 μM) was added to MDDC from two donors. After 2 h, Vpx-containing (VLP X+) or control VLP (VLP X−) were added. The next day, cell lysates were prepared and SAMHD1 was visualized on an immunoblot probed with anti-SAMHD1 MAb and anti-GAPDH antibody as a loading control. (B) Differentiated U937-HA.SAMHD1 (left) and THP1 (right) cells were pretreated with MLN4924, as described in the legend to panel A, for 2 h, and Vpx-containing or control VLP were added. The next day, lysates were prepared and the SAMHD1 was visualized on an immunoblot probed with an anti-HA MAb (U937.HA-SAMHD1) or an anti-SAMHD1 MAb (THP1) and with anti-GAPDH antibody as a loading control. SAMHD1 levels were quantified relative to the GAPDH level, and the ratio of the DMSO control treated with control VLP was set to 1. (C) HeLa cells stably expressing FLAG-/HA-tagged SAMHD1 were incubated with DMSO or MLN4924 for 16 h or 40 h. Untreated cells and cells treated for 16 h with MG132 served as controls. Lysates were prepared, and SAMHD1 was visualized on an immunoblot probed with anti-HA MAb and anti-GAPDH MAb as a loading control.
Our findings suggest that CUL4A in the E3 ubiquitin ligase must be neddylated for Vpx to induce the degradation of SAMHD1. We considered the possibility that Vpx could induce the neddylation of CUL4A or could stabilize neddylated CUL4A complexes, protecting them from the COP9 signalosome. To test these possibilities, we added Vpx-containing and control VLP to THP1 and THP1-SAMHD1.shRNA cells in which SAMHD1 was stably knocked down by shRNA. Over 24 h, we determined the ratios of neddylated to unneddylated CUL4A by immunoblot analysis. We found that Vpx did not affect the ratio of neddylated to unneddylated CUL4A, suggesting that Vpx does not induce the neddylation of CUL4A and does not stabilize neddylated CRL4 (Fig. 3).
Fig 3.

Vpx does not induce the neddylation of CUL4A. Vpx-containing and control VLP were added to differentiated THP1 and THP1-SAMHD1.shRNA cells. At the indicated time points, the cells were lysed in urea-containing lysis buffer. Neddylated and unneddylated CUL4A was detected on an immunoblot probed with anti-CUL4A antibody. The ratios of neddylated to unneddylated CUL4A at the indicated time points are shown below the lanes. SAMHD1 was detected on an immunoblot probed with anti-SAMHD1 antibody and with anti-GAPDH antibody as a loading control.
Neddylated CUL4A complexes are short-lived, and high levels of Vpx cannot overcome the MLN4924-induced block to degradation.
The neddylation of cullin-RING ligases is a dynamic process in which the cullin is neddylated by NAE and deneddylated by the COP9 signalosome (22, 23). We reasoned that, upon treatment with MLN4924, neddylated CRL4 complexes decay at a rate determined by their half-life and that, once the number of neddylated complexes falls to a low level, Vpx function fails. To understand the dynamics of the process, we varied the time of addition of MLN4924 with respect to the addition of Vpx-containing VLP, adding the drug 2 h before, at the same time as, or 4 h after adding VLP. After 20 h, we detected SAMHD1 by immunoblot analysis. The results showed that MLN4924 prevented Vpx-mediated degradation of SAMHD1 most effectively when added 2 h prior to the VLP (Fig. 4A). The earlier the MLN4924 was added, the lower was the concentration of the drug required to block SAMHD1 degradation. When added 2 h before the VLP, 0.25 μM drug was sufficient; when added at the same time, 1.0 μM MLN4924 was required; when the drug was added 4 h after the VLP, MLN4924 failed to block SAMHD1 degradation.
Fig 4.
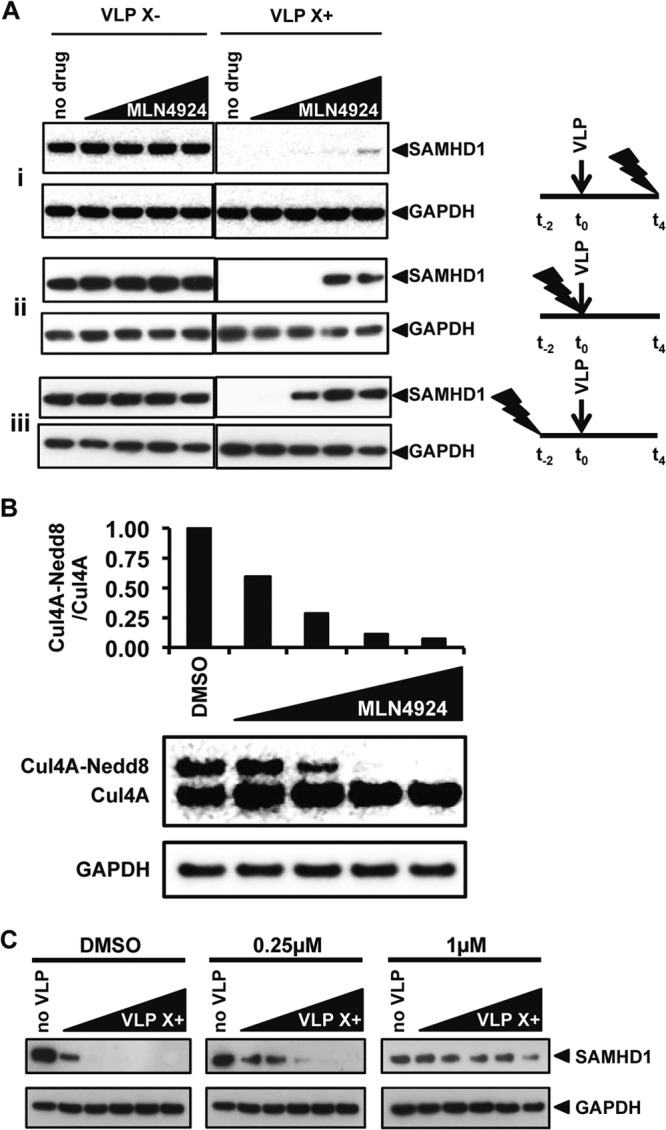
Neddylated CUL4A complexes are short lived, and high levels of Vpx cannot overcome the MLN4924-induced block to degradation. (A) MLN4924 (0.05, 0.25, 1.0, or 2.0 μM) was added to the MDDC 4 h after, at the same time as, or 2 h before the addition of VLP. After 20 h of exposure to the VLP, cell lysates were prepared and SAMHD1 was detected on an immunoblot. GAPDH was used as a loading control. The arrows denote the time of addition of the VLP, and the lightning bolts denote the time of addition of MLN4924. (B) MDDC were incubated with MLN4924, as described in the legend to panel A, for 20 h, after which the cells were lysed in urea-containing lysis buffer and CUL4A was visualized on an immunoblot probed with an anti-CUL4A antibody, with GAPDH serving as a loading control. The ratios of neddylated to unneddylated CUL4A were quantified. (C) MLN4924 was added to MDDC at the concentrations shown. After 2 h, increasing amounts of Vpx-containing or control VLP were added. After 20 h, the cells were lysed and SAMHD1 was visualized on an immunoblot probed with anti-SAMHD1 MAb and with anti-GAPDH antibody as a loading control.
If MLN4924 interferes with the Vpx-mediated degradation of SAMHD1 by inhibiting the neddylation of CUL4A, the concentration of the drug that blocks CUL4A neddylation should block the degradation of SAMHD1. To determine how much MLN4924 is required to block CUL4A neddylation, we added a range of concentrations of the drug to MDDC. After 20 h, the ratio of neddylated to unmodified CUL4A was determined by immunoblot analysis. To prevent the deneddylation of CUL4A in the cell lysates, we used lysis buffer that contained 6 M urea. We found that 1.0 μM MLN4924 blocked CUL4A neddylation in MDDC (Fig. 4B). We then added increasing amounts of Vpx-containing VLP to MDDC that were pretreated for 2 h with 0, 0.25 μM, or 1 μM MLN4924. In the absence of drug, SAMHD1 was detected only at the smallest amount of VLP. Increasing the amount of VLP resulted in degradation of SAMHD1 to an undetectable level (Fig. 4C). The addition of 0.25 μM MLN4924 SAMHD1 largely blocked the degradation, and 1.0 μM completely blocked the degradation. These results show that Vpx is dependent upon neddylated CRL4 to degrade SAMHD1.
SAMHD1 restriction is reversible.
A nuclease-dependent mechanism of virus restriction by SAMHD1 acting on the viral genomic RNA or newly synthesized reverse transcripts has been proposed based on the analysis of CDK1 phosphorylation site SAMHD1 mutants (9). The relative importance of a nuclease-dependent mechanism is not known. An essential difference between nucleolytic degradation and nucleotide pool depletion is that degradation of the viral genome cannot be undone. Nucleotide pool depletion would block reverse transcription, but in principle, the block could be relieved by the restoration of the nucleotide pool. To distinguish between these mechanisms, we tested whether SAMHD1-mediated restriction could be reversed. To do this, we added increasing concentrations of MLN4924 and Vpx-containing or control VLP to MDDC and then infected them with HIV.GFP. After 24 h, we removed the drug and added fresh medium with or without AZT, and after 2 days, analyzed the cells by flow cytometry, as diagrammed in Fig. 5A. AZT was added to inhibit reverse transcription of viral RNA that may have remained in the cytoplasm. The results showed that removal of the drug after 24 h restored viral infectivity (Fig. 5B and C, top). The addition of AZT at the time of drug removal resulted in a dose-dependent restriction to Vpx-mediated HIV-1 infection (Fig. 5B and C, bottom). These results suggested that SAMHD1-mediated restriction can be reversed, inconsistent with a mechanism involving degradation of the viral RNA.
Fig 5.

Removal of MLN4924 reverses the block to infection. (A) Experimental timeline. (B) MLN4924 (0.05, 0.25, 1.0, or 2.0 μM) was added to MDDC which were then infected with an HIV.GFP reporter virus in the presence of Vpx-containing or control VLP. After 24 h, the VLP- and drug-containing viral supernatant was replaced with fresh medium with or without AZT. Infectivity was measured 72 h postinfection by flow cytometry. (C) Graphical display of the results of the infection experiment shown in panel B. The data are averages and standard deviations of triplicate infections. (D and E) Differentiated THP1 and THP1-SAMHD1.shRNA cells were infected with an HIV luciferase reporter virus. The times of VLP and MLN4924 (0.05, 0.25, 1.0, or 2.0 μM) addition and removal were as shown in panel A. At 24 h, the medium was replaced by fresh medium without AZT (D) or with AZT (E). Luciferase activity was measured at 72 h postinfection. The data are averages and standard deviations of triplicate infections.
To determine whether the reversibility of the block was dependent upon SAMHD1, we repeated the drug removal experiment on PMA-differentiated THP1 and THP1-SAMHD1.shRNA cells infected with a luciferase reporter virus. The results showed that the parental THP1 cell line was more than 10-fold less infectible by a virus that lacked Vpx than the shRNA-expressing cells (Fig. 5D and E). Vpx did not affect the infection of the shRNA-expressing cells, but it elevated infectivity in THP1 cells by 15-fold (Fig. 5D and E). Removal of the drug after 24 h rescued the infection of THP1 cells, but infection was blocked when AZT was added at the time of MLN4924 removal (Fig. 5D and E). Replacing the MLN4924 with AZT resulted in restriction of infection only in THP1 cells that expressed SAMHD1 but had no influence on the infectivity of SAMHD1 knockdown cells (Fig. 5E).
The reversibility of MLN4924 inhibition requires that Vpx is still active upon the removal of the drug. To determine whether this was the case, we tested whether, upon removal of the drug, Vpx induced the degradation of SAMHD1. We added MLN4924 to MDDC and then added Vpx-containing or control VLP, and after 24 h, we removed the drug. We then harvested the cells immediately or after 48 h. The results showed that upon removal of the drug, SAMHD1 was degraded (Fig. 6A). At 48 h, the drug did not entirely prevent SAMHD1 degradation, either because the half-life of the drug is limited or through a minor alternative pathway of degradation.
Fig 6.
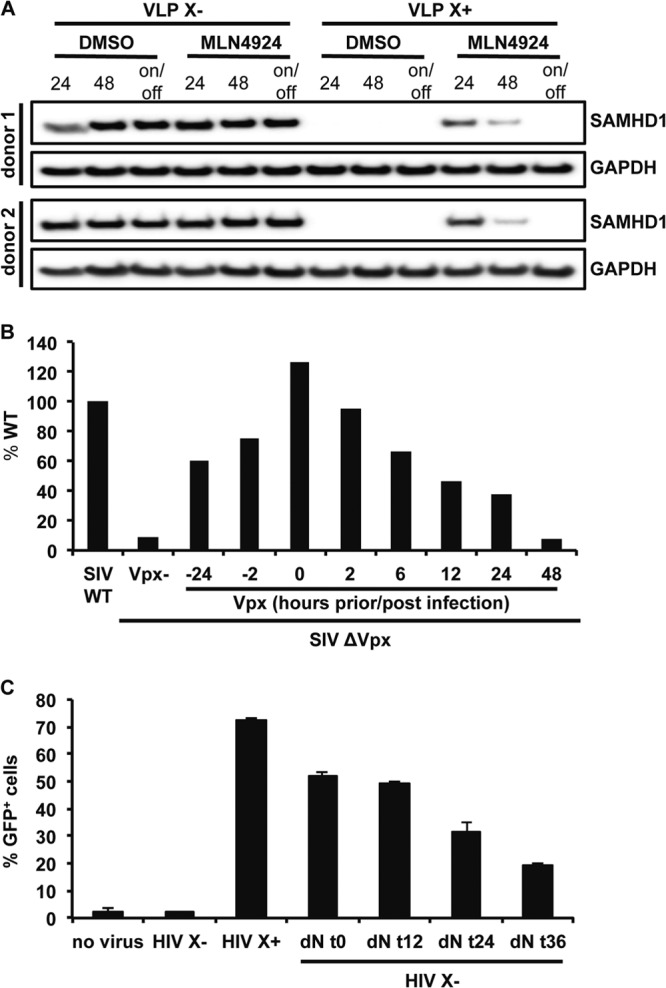
The block to infection is reversed by Vpx-mediated degradation of SAMHD1. (A) MDDC were pretreated with MLN4924 or DMSO for 2 h, and then Vpx-containing or control VLP were added and washed away after 24 h. The drug was kept on the cells for 24 h or 48 h or for 24 h and then replaced by fresh medium that was cultured for another 24 h (on/off). SAMHD1 was detected on an immunoblot probed with a monoclonal anti-SAMHD1 antibody. GAPDH was detected as a loading control. (B) Vpx-containing VLP were added to MDM at the indicated time points relative to infection with wild-type or Δvpx SIVmac luciferase reporter virus. Results are shown as luciferase activity of Δvpx SIVmac infection relative to that of WT virus infection. The results shown are representative of 3 independent donors tested. (C) MDDC were infected with HIV.GFP reporter virus containing (HIV X+) or lacking (HIV X−) Vpx. dNs were added to the cells infected with HIV X− and then removed at the indicated time points. After 3 days, the number of infected cells was quantified by flow cytometry. Results shown are averages for triplicate infections with standard deviations. The results shown are representative of 3 independent donors tested.
To determine whether the reversibility of SAMHD1-mediated restriction could occur independent of MLN4924 and with a different lentivirus, we tested whether the delayed introduction of Vpx in cells in which infection was initially blocked by SAMHD1 could rescue the infection. For this, we infected MDM with Δvpx SIV luciferase reporter virus and then added Vpx-containing VLP at various times postinfection. We measured luciferase activity 4 days postinfection and plotted the results as the percentage of luciferase compared to that in cells infected with wild-type SIV. VLP were effective when added 24 h before or at the same time as the virus (Fig. 6B). The VLP were also able to overcome the restriction when added 12 h or 24 h postinfection, restoring the infectivity to approximately 50% of that of wild-type virus. These results further argue against a nucleolytic mechanism of SAMHD1 restriction.
These results suggested that the delayed removal of SAMHD1 could release the block to infection. To determine whether this could have been due to restoration of the dNTP levels in the cells, we tested whether simply adding dNs to the cell culture medium at increasing time points postinfection would relieve the block to infection. To test this possibility, we infected MDDC with HIV.GFP that contained or lacked Vpx and then added dNs to the culture medium at increasing time points postinfection. Analysis of the cells 3 days postinfection by flow cytometry showed that the addition of the dNs at the time of infection or 12 h postinfection fully relieved the block to infection (Fig. 6C). Addition of the dNs at 24 h and 36 h postinfection also restored the infectivity of HIV.GFP in MDDC, although to a lesser extent. The ability of dNs to restore infectivity several hours postinfection further supports the reversibility of the SAMHD1-mediated block and confirms dNTP depletion as the major mechanism of SAMHD1-mediated restriction.
DISCUSSION
We show here that Vpx-induced degradation of SAMHD1 requires neddylation of the scaffold protein of the CRL4 E3 ubiquitin ligase, CUL4A. Blocking the neddylation with MLN4924 reduced the ability of Vpx-containing virus to infect SAMHD1-restricted nondividing myeloid cells by preventing the degradation of SAMHD1. The findings suggest that it is not sufficient for Vpx to bind to SAMHD1 but that SAMHD1 must be degraded to relieve the restriction. The drug was most effective when added prior to infection, allowing for the previously neddylated CUL4A complexes to become deneddylated by the COP9 signalosome. This occurred within 20 min of MLN4924 treatment, suggesting that the CUL4A modification is a dynamic process (data not shown). The drug had no effect on the infectibility of cells that did not express SAMHD1 and was not toxic. Vpx-induced ubiquitination of SAMHD1 has not been reported, but the findings imply that this is likely to occur. Removal of the drug from recently infected cells allowed Vpx to degrade SAMHD1 and infection to proceed. The reversibility of SAMHD1-mediated restriction is consistent with dNTP pool depletion as the mechanism of restriction but inconsistent with a nucleolytic mechanism.
The mechanisms that regulate the abundance of SAMHD1 during normal cellular metabolism have not been explored. Regulation of the size of the dNTP pool during the course of the cell cycle is critical for cell viability (33). It would seem likely that SAMHD1 is cell cycle regulated to prevent the depletion of deoxynucleotides when they are needed in S phase. In dividing cells, MLN4924 caused the amount of SAMHD1 to increase, suggesting that SAMHD1 levels are regulated in normal cell metabolism by a cullin-based E3 ubiquitin ligase. CRL4 regulates cellular proteins that regulate the cell cycle or that are involved in DNA repair (reviewed in reference 34), and thus, it might regulate SAMHD1 independently of Vpx. It is equally possible that SAMHD1 is regulated by another cullin-RING E3 ubiquitin ligase during normal cellular metabolism. An interesting possibility is that the neddylation of CUL4A is regulated by the cell cycle. Cell cycle regulation of CRL4 activity would determine the ability of Vpx to induce SAMHD1 degradation at different phases of the cell cycle.
Nucleotide pool depletion as the mechanism of SAMHD1-mediated restriction was proposed based on the phosphohydrolase activity of the recombinant protein in vitro and the finding that transduction of U937 cells with a SAMHD1 lentiviral expression vector caused a precipitous drop in the dNTP levels (6, 7, 15, 27). Several findings further support this mechanism. SAMHD1 restricts a wide range of retroviruses, with the exception of foamy virus, which reverse transcribes prior to infection (26). SAMHD1 also restricts vaccinia virus and herpes simplex virus (35), DNA viruses that rely on the cellular dNTP pool for DNA synthesis. Moreover, vaccinia virus with a mutation in the viral thymidine kinase gene is less infectious in MDM, and the infectivity is restored by the addition of Vpx-containing VLP (35). Inhibition of ribonucleotide reductase, the enzyme that converts NTPs to dNTPs, with hydroxyurea accentuates SAMHD1-mediated restriction (7), further supporting a role for dNTP levels.
In spite of these findings supporting a central role for nucleotide pools in SAMHD1-mediated restriction, White et al. have shown that T592E SAMHD1 maintains phosphohydrolase activity but fails to restrict HIV-1 infection (9). Beloglazova et al. reported that recombinant SAMHD1 has 3′-to-5′ exonuclease activity on single-stranded RNA and DNA, raising the possibility that SAMHD1 directly attacks the nucleic acid of the incoming virion (8). While such a mechanism is difficult to imagine given that reverse transcription occurs largely in the cytoplasm and SAMHD1 localizes to the nucleus, a fraction of SAMHD1 may be in the cytoplasm, where it could attack the incoming virion prior to nuclear import. To distinguish the relative importance of these mechanisms, we tested the reversibility of SAMHD1-mediated restriction. The restriction could be reversed by removing MLN4924 from the cells. Removal of the drug caused the rapid neddylation of CUL4A and degradation of SAMHD1, releasing the block to infection. In addition, Vpx-containing VLP relieved the restriction when added up to 24 h postinfection. Similarly, dNs added as late as 36 h postinfection were still effective at relieving the restriction. Taken together, these findings suggest that SAMHD1-mediated restriction is reversible, a finding that is consistent with dNTP pool depletion but not with nucleolytic degradation as the mechanism of restriction. dNTP pool depletion would cause reverse transcription to stall. If the stalled reverse transcription complex were to remain stable for several hours, upon restoration of the dNTP pool, reverse transcription would complete. In contrast, a nucleolytic mechanism cannot be reversed.
The finding that Vpx-induced degradation of SAMHD1 requires CUL4A neddylation raises the possibility that neddylation of CUL4A regulates the ability of Vpx to relieve the restriction. Vpx may be more effective at relieving SAMHD1-mediated restriction in some cells than in others. In addition, control over CUL4A neddylation could serve as a defense against Vpx. Whether the ability of MLN4924 to block Vpx function has clinical application is not clear. HIV-1 does not have Vpx, and therefore, the drug would not be useful in the treatment of HIV-1 infection. HIV-2 does have Vpx, and blocking its function in infected individuals could therefore be beneficial. The drug may also be of interest in nonhuman primate studies to investigate the role of Vpx in viral pathogenesis. The findings reported here provide further insight into the complexity with which lentiviruses co-opt cellular processes to accomplish their replication.
ACKNOWLEDGMENTS
We thank Michele Pagano for MLN4924 and advice.
This work was supported by the National Institutes of Health (AI058864 and AI067059), the NYU CTSA program (UL1TR000038), a Grunenbaum AIDS Research Scholarship (T.D.N.), and a Vilcek Endowment Fund fellowship (H.H.).
Footnotes
Published ahead of print 28 August 2013
REFERENCES
- 1.Berger A, Sommer AF, Zwarg J, Hamdorf M, Welzel K, Esly N, Panitz S, Reuter A, Ramos I, Jatiani A, Mulder LC, Fernandez-Sesma A, Rutsch F, Simon V, Konig R, Flory E. 2011. SAMHD1-deficient CD14+ cells from individuals with Aicardi-Goutieres syndrome are highly susceptible to HIV-1 infection. PLoS Pathog. 7:e1002425. 10.1371/journal.ppat.1002425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hrecka K, Hao C, Gierszewska M, Swanson SK, Kesik-Brodacka M, Srivastava S, Florens L, Washburn MP, Skowronski J. 2011. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature 474:658–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Laguette N, Sobhian B, Casartelli N, Ringeard M, Chable-Bessia C, Segeral E, Yatim A, Emiliani S, Schwartz O, Benkirane M. 2011. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature 474:654–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baldauf H, Pan X, Erikson E, Schmidt S, Daddacha W, Burggraf M, Schenkova K, Ambiel I, Wabnitz G, Gramberg T, Panitz S, Flory E, Landau NR, Sertel S, Rutsch F, Lasitschka F, Kim B, Konig R, Fackler O, Keppler O. 2012. SAMHD1 restricts HIV-1 infection in resting CD4+ T cells. Nat. Med. 18:1682–1687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Descours B, Cribier A, Chable-Bessia C, Ayinde D, Rice G, Crow Y, Yatim A, Schwartz O, Laguette N, Benkirane M. 2012. SAMHD1 restricts HIV-1 reverse transcription in quiescent CD4(+) T-cells. Retrovirology 9:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goldstone DC, Ennis-Adeniran V, Hedden JJ, Groom HC, Rice GI, Christodoulou E, Walker PA, Kelly G, Haire LF, Yap MW, de Carvalho LP, Stoye JP, Crow YJ, Taylor IA, Webb M. 2011. HIV-1 restriction factor SAMHD1 is a deoxynucleoside triphosphate triphosphohydrolase. Nature 480:379–382 [DOI] [PubMed] [Google Scholar]
- 7.Lahouassa H, Daddacha W, Hofmann H, Ayinde D, Logue EC, Dragin L, Bloch N, Maudet C, Bertrand M, Gramberg T, Pancino G, Priet S, Canard B, Laguette N, Benkirane M, Transy C, Landau NR, Kim B, Margottin-Goguet F. 2012. SAMHD1 restricts the replication of human immunodeficiency virus type 1 by depleting the intracellular pool of deoxynucleoside triphosphates. Nat. Immunol. 13:621. 10.1038/ni0813-877a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beloglazova N, Flick R, Tchigvintsev A, Brown G, Popovic A, Nocek B, Yakunin AF. 2013. Nuclease activity of the human SAMHD1 protein implicated in the Aicardi-Goutieres syndrome and HIV-1 restriction. J. Biol. Chem. 288:8101–8110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.White TE, Brandariz-Nunez A, Valle-Casuso JC, Amie S, Nguyen LA, Kim B, Tuzova M, Diaz-Griffero F. 2013. The retroviral restriction ability of SAMHD1, but not its deoxynucleotide triphosphohydrolase activity, is regulated by phosphorylation. Cell Host Microbe 13:441–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kirchhoff F. 2010. Immune evasion and counteraction of restriction factors by HIV-1 and other primate lentiviruses. Cell Host Microbe 8:55–67 [DOI] [PubMed] [Google Scholar]
- 11.Accola MA, Bukovsky AA, Jones MS, Gottlinger HG. 1999. A conserved dileucine-containing motif in p6(gag) governs the particle association of Vpx and Vpr of simian immunodeficiency viruses SIV(mac) and SIV(agm). J. Virol. 73:9992–9999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ahn J, Hao C, Yan J, DeLucia M, Mehrens J, Wang C, Gronenborn AM, Skowronski J. 2012. HIV/simian immunodeficiency virus (SIV) accessory virulence factor Vpx loads the host cell restriction factor SAMHD1 onto the E3 ubiquitin ligase complex CRL4DCAF1. J. Biol. Chem. 287:12550–12558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sharova N, Wu Y, Zhu X, Stranska R, Kaushik R, Sharkey M, Stevenson M. 2008. Primate lentiviral Vpx commandeers DDB1 to counteract a macrophage restriction. PLoS Pathog. 4:e1000057. 10.1371/journal.ppat.1000057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Srivastava S, Swanson SK, Manel N, Florens L, Washburn MP, Skowronski J. 2008. Lentiviral Vpx accessory factor targets VprBP/DCAF1 substrate adaptor for cullin 4 E3 ubiquitin ligase to enable macrophage infection. PLoS Pathog. 4:e1000059. 10.1371/journal.ppat.1000059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim B, Nguyen LA, Daddacha W, Hollenbaugh JA. 2012. Tight interplay among SAMHD1 protein level, cellular dNTP levels, and HIV-1 proviral DNA synthesis kinetics in human primary monocyte-derived macrophages. J. Biol. Chem. 287:21570–21574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brandariz-Nunez A, Valle-Casuso JC, White TE, Laguette N, Benkirane M, Brojatsch J, Diaz-Griffero F. 2012. Role of SAMHD1 nuclear localization in restriction of HIV-1 and SIVmac. Retrovirology 9:49. 10.1186/1742-4690-9-49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hofmann H, Logue EC, Bloch N, Daddacha W, Polsky SB, Schultz ML, Kim B, Landau NR. 2012. The Vpx lentiviral accessory protein targets SAMHD1 for degradation in the nucleus. J. Virol. 86:12552–12560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Laguette N, Rahm N, Sobhian B, Chable-Bessia C, Munch J, Snoeck J, Sauter D, Switzer WM, Heneine W, Kirchhoff F, Delsuc F, Telenti A, Benkirane M. 2012. Evolutionary and functional analyses of the interaction between the myeloid restriction factor SAMHD1 and the lentiviral Vpx protein. Cell Host Microbe 11:205–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Petroski MD, Deshaies RJ. 2005. Function and regulation of cullin-RING ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 6:9–20 [DOI] [PubMed] [Google Scholar]
- 20.Gong L, Yeh ET. 1999. Identification of the activating and conjugating enzymes of the NEDD8 conjugation pathway. J. Biol. Chem. 274:12036–12042 [DOI] [PubMed] [Google Scholar]
- 21.Huang DT, Schulman BA. 2005. Expression, purification, and characterization of the E1 for human NEDD8, the heterodimeric APPBP1-UBA3 complex. Methods Enzymol. 398:9–20 [DOI] [PubMed] [Google Scholar]
- 22.Kato JY, Yoneda-Kato N. 2009. Mammalian COP9 signalosome. Genes Cells 14:1209–1225 [DOI] [PubMed] [Google Scholar]
- 23.Wei N, Serino G, Deng XW. 2008. The COP9 signalosome: more than a protease. Trends Biochem. Sci. 33:592–600 [DOI] [PubMed] [Google Scholar]
- 24.Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, Brownell JE, Burke KE, Cardin DP, Critchley S, Cullis CA, Doucette A, Garnsey JJ, Gaulin JL, Gershman RE, Lublinsky AR, McDonald A, Mizutani H, Narayanan U, Olhava EJ, Peluso S, Rezaei M, Sintchak MD, Talreja T, Thomas MP, Traore T, Vyskocil S, Weatherhead GS, Yu J, Zhang J, Dick LR, Claiborne CF, Rolfe M, Bolen JB, Langston SP. 2009. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 458:732–736 [DOI] [PubMed] [Google Scholar]
- 25.Brownell JE, Sintchak MD, Gavin JM, Liao H, Bruzzese FJ, Bump NJ, Soucy TA, Milhollen MA, Yang X, Burkhardt AL, Ma J, Loke HK, Lingaraj T, Wu D, Hamman KB, Spelman JJ, Cullis CA, Langston SP, Vyskocil S, Sells TB, Mallender WD, Visiers I, Li P, Claiborne CF, Rolfe M, Bolen JB, Dick LR. 2010. Substrate-assisted inhibition of ubiquitin-like protein-activating enzymes: the NEDD8 E1 inhibitor MLN4924 forms a NEDD8-AMP mimetic in situ. Mol. Cell 37:102–111 [DOI] [PubMed] [Google Scholar]
- 26.Gramberg T, Kahle T, Bloch N, Wittmann S, Mullers E, Daddacha W, Hofmann H, Kim B, Lindemann D, Landau NR. 2013. Restriction of diverse retroviruses by SAMHD1. Retrovirology 10:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.St Gelais C, de Silva S, Amie SM, Coleman CM, Hoy H, Hollenbaugh JA, Kim B, Wu L. 2012. SAMHD1 restricts HIV-1 infection in dendritic cells (DCs) by dNTP depletion, but its expression in DCs and primary CD4+ T-lymphocytes cannot be upregulated by interferons. Retrovirology 9:105. 10.1186/1742-4690-9-105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cribier A, Descours B, Valadao AL, Laguette N, Benkirane M. 2013. Phosphorylation of SAMHD1 by cyclin A2/CDK1 regulates its restriction activity toward HIV-1. Cell Rep. 3:1036–1043 [DOI] [PubMed] [Google Scholar]
- 29.Sunseri N, O'Brien M, Bhardwaj N, Landau NR. 2011. Human immunodeficiency virus type 1 modified to package simian immunodeficiency virus Vpx efficiently infects macrophages and dendritic cells. J. Virol. 85:6263–6274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, Naldini L. 1998. A third-generation lentivirus vector with a conditional packaging system. J. Virol. 72:8463–8471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gramberg T, Sunseri N, Landau NR. 2010. Evidence for an activation domain at the amino terminus of simian immunodeficiency virus Vpx. J. Virol. 84:1387–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Butler SL, Hansen MS, Bushman FD. 2001. A quantitative assay for HIV DNA integration in vivo. Nat. Med. 7:631–634 [DOI] [PubMed] [Google Scholar]
- 33.D'Angiolella V, Donato V, Forrester FM, Jeong YT, Pellacani C, Kudo Y, Saraf A, Florens L, Washburn MP, Pagano M. 2012. Cyclin F-mediated degradation of ribonucleotide reductase M2 controls genome integrity and DNA repair. Cell 149:1023–1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kerzendorfer C, Hart L, Colnaghi R, Carpenter G, Alcantara D, Outwin E, Carr AM, O'Driscoll M. 2011. CUL4B-deficiency in humans: understanding the clinical consequences of impaired Cullin 4-RING E3 ubiquitin ligase function. Mech. Ageing Dev. 132:366–373 [DOI] [PubMed] [Google Scholar]
- 35.Hollenbaugh JA, Gee P, Baker J, Daly MB, Amie SM, Tate J, Kasai N, Kanemura Y, Kim DH, Ward BM, Koyanagi Y, Kim B. 2013. Host factor SAMHD1 restricts DNA viruses in non-dividing myeloid cells. PLoS Pathog. 9:e1003481. 10.1371/journal.ppat.1003481 [DOI] [PMC free article] [PubMed] [Google Scholar]