

Abstract
Prion diseases are characterized by the conversion of the soluble protease-sensitive host-encoded prion protein (PrPC) into its aggregated, protease-resistant, and infectious isoform (PrPSc). One of the earliest events occurring in cells following exposure to an exogenous source of prions is the cellular uptake of PrPSc. It is unclear how the biochemical properties of PrPSc influence its uptake, although aggregate size is thought to be important. Here we show that for two different strains of mouse prions, one that infects cells (22L) and one that does not (87V), a fraction of PrPSc associated with distinct sedimentation properties is preferentially taken up by the cells. However, while the fraction of PrPSc and the kinetics of uptake were similar for both strains, PrPSc derived from the 87V strain was disaggregated more rapidly than that derived from 22L. The increased rate of PrPSc disaggregation did not correlate with either the conformational or aggregate stability of 87V PrPSc, both of which were greater than those of 22L PrPSc. Our data suggest that the kinetics of disaggregation of PrPSc following cellular uptake is independent of PrPSc stability but may be dependent upon some component of the PrPSc aggregate other than PrP. Rapid disaggregation of 87V PrPSc by the cell may contribute, at least in part, to the inability of 87V to infect cells in vitro.
INTRODUCTION
Transmissible spongiform encephalopathies (TSEs), or prion diseases, are a group of fatal neurological disorders, including Creutzfeldt-Jakob disease in humans, scrapie in sheep, and bovine spongiform encephalopathy (BSE) in cattle. A key event associated with prion diseases is the formation of a pathogenic, protease-resistant, and insoluble form of prion protein (PrPSc). PrPSc can propagate in an autocatalytic manner by binding the soluble and protease-sensitive host cellular prion protein (PrPC) and inducing its conversion into the PrPSc conformer (for a review, see reference 1). PrPSc aggregates are thought to be the main component of the causative agent (or prion) and eventually accumulate to pathogenic levels in the brain of infected individuals, leading to clinical prion disease.
The molecular mechanisms underlying prion replication have been investigated by using various cell models of persistent infection. These studies have shown that even though PrPC is essential for propagation of prion infectivity (2), not all cell lines expressing PrPC are susceptible to prion infection (for a comprehensive review, see reference 3). Thus, PrPC is not the only factor required for persistent prion infection in vitro (4). Furthermore, not all prion strains are able to infect cells. For example, the mouse scrapie strain 87V does not infect cell lines susceptible to other mouse-adapted scrapie strains, such as 22L or RML (5, 6), even though it can induce PrPSc formation in cells (6). However, it is not known what cellular factors other than PrPC, or what characteristics of PrPSc, may influence the efficiency of infection of cells by prions.
To understand why some prion strains can infect cells and others cannot, several studies have focused on the earliest events that occur in cells which have been exposed to an exogenous source of prions. A previous study employing cells expressing an epitope-tagged mutant of mouse PrPC demonstrated that, irrespective of the scrapie strain, the de novo (i.e., acute) formation of proteinase K (PK)-resistant prion protein can be detected by immunoblotting as early as 4 h after exposure to scrapie infectivity, suggesting that new PrPSc can be generated very rapidly following infection (6). However, even though acute PrPSc formation occurred regardless of the scrapie strain and cell type, the establishment of a persistent infection was dependent upon both the scrapie strain and the cell (6). These data suggested that the inability of a prion strain to infect cells was not necessarily due to an inability to convert the PrPC expressed by the cell but rather due to other events that occurred after an initial burst of PrPSc formation (6).
Another early event that must occur during prion infection is the association of PrPSc with the cell. The kinetics of PrPSc uptake appear to be largely independent of both prion strain and PrPC expression by the host cell (7, 8), although PrPC expression may influence PrPSc uptake for some prion species (9, 10). However, only some of the available PrPSc is internalized (8, 10), suggesting that there may be a specific population of PrPSc which is preferentially cell associated. PrPSc, even when purified, is a heterogeneous mixture of aggregates, and PrPSc aggregate size is known to influence both the specific infectivity of prion particles (11–13) and the association of PrPSc with the cell (7, 8). For example, large aggregates of purified, fluorescently labeled PrPSc associate with the cell surface and are broken down and internalized over a period of several days (7). In contrast, PrPSc in brain homogenates (10, 14) as well as smaller aggregates of purified, nonfluorescently labeled PrPSc (8) are not localized to the cell surface and are internalized within a few hours. Thus, PrPSc aggregate size appears to influence both the kinetics and localization of cell-associated PrPSc. These data suggest that qualitative differences in the available pool of PrPSc may influence the interaction of PrPSc with the cell and, potentially, the establishment of prion infection.
In the current study, we have used discontinuous sucrose gradient centrifugation to determine how PrPSc aggregates with different sedimentation properties interact with the cell. We show that for a mouse scrapie strain that can infect cells (22L) and one that cannot (87V), the same preferred fraction of PrPSc associates with the cells. However, over time, the cell-associated PrPSc becomes disaggregated by the cell, a process that is more rapid for 87V PrPSc than for 22L PrPSc. Rapid or efficient disaggregation of 87V PrPSc by the cell may explain, at least in part, the inability of 87V to establish persistent infection in vitro.
MATERIALS AND METHODS
Cells.
The generation of the PrP knockout cell line CF10 (8) and CF10 cells expressing mouse PrP containing the 3F4 epitope (CF10+Mo3F4 cells) have been described previously (15). The CF10 cells used for this study contain the retroviral expression vector pSFF but do not express PrPC. To generate CF10 cells with the pSFF vector, pSFF was packaged into a nonreplicating mouse retrovirus via transfection into the retroviral packaging cell lines ψ2 and PA317 (16). CF10 cells were transduced with the supernatant harvested from these cells, as described previously (17), and were not cloned. CF10+Mo3F4 cells express a PrP molecule derived from the short scrapie incubation time mouse allele Prnpa, which has been mutated to contain the epitope to the anti-PrP mouse monoclonal antibody 3F4 (15). These cells were also used uncloned. All CF10 cells were maintained in Opti-MEM (Invitrogen) supplemented with 10% fetal bovine serum (FBS), 100 U of penicillin, and 100 μg of streptomycin in a humidified incubator at 37°C with 5% CO2. Throughout the manuscript, cells expressing the pSFF vector are referred to as CF10 cells, while cells expressing mouse PrP with the 3F4 antibody epitope are referred to as CF10+Mo3F4 cells.
Preparation of inoculum.
All animal experimental protocols were reviewed and approved by the Rocky Mountain Laboratories Animal Care and Use Committee (animal protocols 2003-06 and 2006-03). The Rocky Mountain Laboratories are fully accredited by the American Association for Laboratory Animal Care, and this study was carried out in strict accordance with the recommendations of the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (18).
C57BL/10 mice infected with 22L scrapie and VMDK mice infected with 87V scrapie were culled at the clinical stage of disease, and their brains were harvested and stored at −80°C until use. Brain tissues were prepared as 10% or 1% (wt/vol) homogenates in phosphate-buffered saline (PBS) by using a Mini-BeadBeater-8 instrument (Biospec). The homogenized samples were cleared by centrifugation at 500 × g for 3 min and stored at −20°C until use. On the day of the experiment, the prepared brain homogenates were thawed and diluted in prewarmed Opti-MEM without FBS or antibiotics to final concentrations of either 0.5% or 0.25% (wt/vol) homogenates. Brain homogenates were not sonicated when initially prepared or prior to use.
Scrapie infection of cells and cellular uptake of PrPSc.
CF10+Mo3F4 cells were exposed to a 1:10 dilution of either 22L or 87V mouse scrapie-infected brain homogenate or normal brain homogenate, as described previously (15). After 4 h of incubation at 37°C, the homogenate was removed, and the cells were passaged at least 10 times. After 10 passages, cells were analyzed for PrPSc by immunoblotting using the 3F4 monoclonal antibody, as described below.
Uptake of PrPSc into CF10 cells was done as described previously (8), with minor modifications. Briefly, in order to analyze the amount of unfractionated PrPSc associated with CF10 cells, cells were plated at a density of 3.2 × 106 cells per well in 6-well plates, followed by an overnight incubation at 37°C. After removal of the medium, cells were overlaid with 750 μl of the prepared inoculum and incubated at 37°C. After an 8-h incubation, 1.5 ml of Opti-MEM containing 10% FBS, 100 U of penicillin, and 100 μg of streptomycin was added to the cells, and the cells were incubated for another 16 h. For some experiments, the brain homogenate was removed after 2 h, and the cells were rinsed 3 to 4 times in medium. Following the addition of fresh medium, the cells were then incubated at 37°C for another 24 h. Cells were removed from the plate by trypsin-EDTA treatment at 2, 8, or 24 h postinfection and were recovered by centrifugation at 5,200 × g for 5 min at 4°C. The cells were lysed in 200 μl lysis buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 0.5% Triton X-100, 0.5% sodium deoxycholate, and 5 mM EDTA). Insoluble cellular debris was removed by low-speed centrifugation, and supernatants were saved for the analysis of PrPSc by Western blotting.
Cell lysates used for separation of PrPSc by discontinuous sucrose gradient centrifugation were prepared as follows. Cells were plated at a density of either 3.2 × 106 cells per well in 6-well plates or 8 × 106 cells per 25-cm2 flask and incubated overnight at 37°C. After removal of the medium, 22L or 87V brain homogenate derived from a mouse with clinical scrapie was added to the cells (750 μl/well in a 6-well plate or 2 ml/25-cm2 flask), and the cells were incubated at 37°C. After 8 h, 1.5 ml (6-well plates) or 4 ml (25-cm2 flasks) of Opti-MEM with 10% FBS was added to the cells. Cells were collected as described above and then resuspended in 500 μl of lysis buffer (2% n-octyl-β-d-glucopyranoside [NOG] in PBS). Cell lysates were cleared by centrifugation at 3,800 × g for 1 min at 4°C and supernatants were saved for fractionation by velocity sedimentation.
In vitro conversion of Mo3F4 PrPC.
Cell homogenates from CF10+Mo3F4 cells and CF10 cells containing the empty pSFF vector, as well as brain homogenates from mice infected with either 22L or 87V scrapie, were prepared in PBS–0.5% Triton X-100–0.05% SDS as previously described (19). For the in vitro conversion reaction, 9 μl of brain homogenate was mixed with 60 μl of cell lysate, and the reaction mixture was incubated with agitation at 37°C for 4 days. Samples were then treated with 100 μg/ml of PK at 37°C for 1 h, followed by the addition of 5 μl of 0.1 M phenylmethylsulfonyl fluoride (PMSF) to stop the PK reaction. Half of the reaction mixture was analyzed for newly made protease-resistant PrP by Western blotting using anti-PrP mouse monoclonal antibody 3F4 conjugated to biotin (Covance) at a dilution of 1:10,000 followed by streptavidin-conjugated horseradish peroxidase (Cell Signaling Technologies), at a dilution of 1:250,000. Ten microliters was used to assay the total amount of PrPSc in the reaction mixture by Western blotting using anti-PrP mouse monoclonal antibody 6D11 (Covance) at a dilution of 1:15,000 followed by a 1:100,000 dilution of horseradish peroxidase-conjugated sheep anti-mouse secondary antibody (GE Healthcare Life Sciences). Blots were developed with the Super Signal West Femto kit (Thermo Scientific) according to the manufacturer's instructions.
Discontinuous sucrose gradient centrifugation.
Velocity sedimentation in sucrose step gradients was carried out as described previously (20), with minor modifications. Briefly, the brain homogenates prepared in PBS were mixed with an equal volume of 4% NOG in PBS and then incubated for 30 min in ice. Insoluble debris was removed by centrifugation at 3,800 × g for 1 min. Brain or cell lysates, prepared as described above, were incubated for 30 min on ice in the presence of 1% Sarkosyl and then loaded on top of a 10 to 60% sucrose step gradient also made in 1% Sarkosyl. The fractionation of PrP was achieved by centrifuging samples for 1 h at 4°C at 46,000 rpm (gaverage = 200,000 × g) in an SW55 rotor (Beckmann). Eleven fractions of 450 μl were collected from the top of the gradient and stored at −20°C until use.
Western blot analysis.
Samples were digested with 25 μg/ml PK for 1 h at 37°C, followed by the addition of Pefabloc (1 mM final concentration) to stop PK activity. PK-digested cell lysates were mixed with NuPAGE sample buffer (Invitrogen) to a final concentration of 1× (141 mM Tris base, 106 mM Tris HCl, 2% lithium dodecyl sulfate, 0.5 mM EDTA, 10% glycerol, 0.22 mM SERVA blue G250, and 0.175 mM phenol red) and then boiled for 10 min. For samples derived from sucrose gradient fractions, the volume of the entire sample in each fraction was brought to 1 ml by adding 550 μl of PBS containing 1% Sarkosyl. Subsequently, 500 μl of 2-butanol and methanol (5:1, vol/vol) was added, and the mixture was centrifuged at 20,800 × g for 10 min as described previously (21). Pellets were resuspended in 20 μl of 2× NuPAGE sample buffer (282 mM Tris base, 212 mM Tris HCl, 4% lithium dodecyl sulfate, 1.02 mM EDTA, 20% glycerol, 0.44 mM SERVA blue G250, and 0.35 mM phenol red [Invitrogen]) and boiled at 100°C for 10 min.
Protein analysis was performed by using the NuPAGE Novex gel system (Invitrogen) as previously described (22). Briefly, proteins were separated in 10% Bis-Tris NuPAGE gels and then transferred onto polyvinylidene difluoride (PVDF) Immobilon-P membranes (Millipore). After membranes were blocked with 5% (wt/vol) dry milk in TBST (10 mM Tris HCl [pH 8.0], 150 mM NaCl, and 0.05% Tween 20), the membranes were probed with a 1:5,000 dilution of mouse monoclonal antibody 6D11, followed by a 1:40,000 dilution of horseradish peroxidase-conjugated sheep anti-mouse secondary antibody. The blots were developed on film by using ECL Plus reagent (GE Healthcare Life Sciences). The developed films were scanned, and quantitative analysis of the blots was carried out by using ImageQuant software (version 5.2; Molecular Dynamics).
PrPSc stability assay.
Aliquots of 1% brain homogenates prepared in PBS were mixed with an equal volume of guanidine hydrochloride (GdnHCl), leading to a range of final guanidine concentrations from 0 to 4 M. After 1 h of incubation at room temperature on a shaking platform, the final concentration of GdnHCl in each sample was adjusted to 0.4 M in lysis buffer (50 mM Tris HCl [pH 8.0], 150 mM NaCl, 5 mM EDTA, 0.5% sodium deoxycholate, and 0.5% Triton X-100). Subsequently, samples were digested with 25 μg/ml PK for 1 h at 37°C. After PK activity was stopped by adding Pefabloc to a final concentration of 1 mM, proteins were precipitated as described above, using a solution of 2-butanol and methanol (5:1, vol/vol). In some experiments, PK digestion was not done. Samples (250 μl) were layered over 250 μl of a 5% sucrose–lysis buffer cushion in a Beckman TL 100.1 centrifuge tube and centrifuged for 45 min at 20,000 rpm (17,400 × g) (23). Supernatants were carefully discarded, and pellets were resuspended in 20 μl of 2× NuPAGE sample buffer. After boiling for 10 min, proteins were analyzed by using the NuPAGE Novex gel system, as described above.
Immunofluorescence.
Analysis of cellular PrPSc uptake was performed as described previously (8), with minor modifications. Briefly, cells were plated into 8-well chamber slides at a density of 1 × 105cells per well and kept overnight at 37°C. After removal of the medium, 100 μl of a 1:10 dilution of 10% brain homogenate in Opti-MEM was added to each well. After 8 h, 200 μl of Opti-MEM containing 10% FBS, 100 U of penicillin, and 100 μg of streptomycin was added. At different time points postexposure, cells were washed three times in Dulbecco's PBS (DPBS), fixed in 3.7% formaldehyde for 30 min, and then permeabilized with 0.2% Triton X-100 in PBS for 5 min. Subsequently, cells were digested with PK (10 μg/ml; 200 μl per chamber) for 5 min at room temperature and then exposed to 3 M guanidine thiocyanate for 5 min. The cells were blocked with 3% bovine serum albumin (BSA) in PBS for 45 min. To detect PrP, cells were incubated for 1 h with anti-PrP monoclonal antibody 6D11 prepared at a dilution of 1:200 in blocking solution. Following three rinses in DPBS, the cells were incubated for 45 min with an Alexa Fluor 488-conjugated F(ab′)2 fragment of goat anti-mouse IgG (Invitrogen) prepared at a dilution of 1:500 in blocking solution. After three rinses in DPBS, the chamber walls were removed, and the coverslip was affixed with ProLong Gold Antifade with 4′,6-diamidino-2-phenylindole (DAPI) (Invitrogen). Stained cells were observed under fluorescent illumination on a BX51 microscope (Olympus), and images were obtained with a DP71 digital camera (Olympus) and processed with Microsuite 5 software (Olympus).
RESULTS
Association of 87V and 22L PrPSc with CF10 cells increases over time.
Brain homogenates prepared from either 22L- or 87V-infected mice were diluted in cell culture medium and incubated for up to 24 h with CF10 cells which contain the retroviral vector pSFF but do not express PrPC. At different time points postexposure, cells were lysed, and the lysates were digested with PK and analyzed by immunoblotting. For both 22L and 87V, PrPSc associated with cells as early as 2 h postinoculation, with the amount of cell-associated PrPSc increasing over time (Fig. 1A). By 24 h postexposure, the amount of PrPSc associated with CF10 cells was >10% of the input PrPSc added to the cells (Fig. 1A). These results are consistent with a previous report in which the uptake of PrPSc from three different mouse scrapie strains, including 22L, was examined by using CF10 cells (8). The kinetics of 87V PrPSc uptake, which has not been previously reported, was similar to that of 22L and did not require the expression of PrPC.
Fig 1.

Uptake of 22L PrPSc and 87V PrPSc by CF10 cells. (A) CF10 cells were exposed to either 22L or 87V scrapie brain homogenate for 2, 8, or 24 h, and the amount of cell-associated PrPSc was determined by Western blotting using anti-PrP mouse monoclonal antibody 6D11. The relative amount of cell-associated PrPSc was estimated by comparison to the PrPSc level in 10% of the input inoculum (10% input). Molecular mass markers in kilodaltons are indicated on the left. (B) CF10 cells were exposed to 22L or 87V scrapie brain homogenate at either 37°C or 4°C for 2 and 8 h, and the amount of PrPSc internalized by the cell was determined by Western blotting as described above for panel A. Molecular mass markers in kilodaltons are indicated on the left. (C) Cells were incubated with uninfected brain homogenates (Mock) or 22L or 87V scrapie-infected brain homogenates for 2 to 24 h. After removal of the homogenate, cells were fixed, permeabilized, digested with PK, and treated with 3 M guanidine thiocyanate. PrPSc was detected by using mouse monoclonal antibody 6D11 and Alexa Fluor 488-labeled goat anti-mouse secondary antibody (green). Nuclei were stained with DAPI (blue).
PrPSc from 22L and 87V is internalized by cells.
In order to determine whether the PrPSc associated with the CF10 cells was internalized by the cells, CF10 cells were exposed to 22L and 87V mouse scrapie brain homogenates and incubated at either 37°C or 4°C, a temperature known to block the endocytosis of PrPSc (10). As shown in Fig. 1B, the amount of cell-associated PrPSc in CF10 cells exposed to scrapie brain homogenates at 4°C was greatly reduced compared to that in cells incubated at 37°C. When the cells were examined by fluorescence microscopy following immunostaining, PrPSc was detectable by as early as 2 h postexposure in the perinuclear and/or cytoplasmic regions of the cells (Fig. 1C). Collectively, these data suggested that the majority of cell-associated 22L and 87V PrPSc was being internalized by the cells.
CF10 cells preferentially internalize a distinct fraction of aggregated PrPSc.
Previously, we and others suggested that PrPSc aggregate size may influence the efficiency of PrPSc internalization (7, 8). In order to determine if cellular uptake of PrPSc was influenced by the size of the PrPSc aggregate, we compared the sedimentation properties of 22L scrapie PrPSc derived from scrapie-infected mouse brain to that of PrPSc taken up by the cell. Brain lysates from 22L-infected mice were fractionated by sucrose step gradient ultracentrifugation as detailed in Materials and Methods, the samples were digested with PK, and the sedimentation profile of PrPSc was analyzed by immunoblotting. As shown in Fig. 2A, brain-derived 22L PrPSc was found largely in the bottom three fractions of the gradient, with approximately 20% being detectable in the intermediate fractions (fractions 6 to 8) (Fig. 2C). In contrast, PrPSc recovered from cells following a 2-h exposure to the 22L brain homogenate was found almost exclusively in fraction 9 of the gradient (Fig. 2B and C). At 8 and 24 h, PrPSc was detectable in the bottom three fractions, with the majority still sedimenting in fraction 9 (Fig. 2B and C). Notably, however, at 8 and 24 h, detectable amounts of PrPSc were also present in intermediate fractions 4 to 6, a trend most evident at the 24-h time point (Fig. 2B). These results were consistent over multiple experiments (Fig. 2C) and suggested not only that there is a population of PrPSc in fraction 9 that is preferentially taken up by cells but also that PrPSc internalized by CF10 cells is gradually disaggregated over time.
Fig 2.

A limited population of 22L PrPSc aggregates is preferentially taken up and disaggregated by CF10 cells. (A and B) The brain homogenate from a 22L scrapie-infected mouse (A) or cell lysates from CF10 cells exposed to the 22L brain homogenate for up to 24 h (B) were fractionated by discontinuous sucrose gradient centrifugation. Eleven fractions (fractions 1 to 11) were collected from the top of the gradient, digested with PK, and analyzed by Western blotting using anti-PrP mouse monoclonal antibody 6D11. In panels A and B, molecular mass markers in kilodaltons are indicated on the left. (C) Quantitation of the relative amount of PrPSc in individual sucrose gradient fractions derived from either the 22L scrapie-infected brain homogenate or CF10 cell lysates after exposure to the 22L scrapie brain homogenate for 2 h, 8 h, and 24 h. The values represent averages ± standard errors of the percentage of PrPSc in each fraction for 4 experiments (brain homogenate) or 4 to 5 experiments (cell lysate).
Cellular disaggregation of PrPSc following uptake.
Our data suggested that over time, the cells were disaggregating the specific fraction of PrPSc that they had internalized. However, it was also possible that the gradual appearance of smaller PrPSc aggregates in the cell was due to the delayed internalization of less aggregated PrPSc species that either were present in the scrapie brain homogenate or had already been disaggregated by enzymes in the cell culture medium. In order to determine the origin of the PrPSc species found in cellular sucrose fractions 6 to 8 (Fig. 2B), we recovered the 22L brain homogenate incubated with the cells for 2, 8, and 24 h and analyzed the remaining PrPSc using discontinuous sucrose gradient centrifugation. As shown in Fig. 3A, PrPSc in the cellular supernatants from all three time points was detected only in the bottom three fractions of the sucrose gradient. These data suggested that the PrPSc species found in the intermediate cellular sucrose fractions at 8 and 24 h following exposure to the brain homogenate were not the result of delayed uptake of either slowly migrating PrPSc species from the homogenate or PrPSc that had been disaggregated in the cell supernatant.
Fig 3.

Smaller PrPSc aggregates present in the cell after 24 h are not derived from the supernatants of cells exposed to 22L scrapie-infected brain homogenate. (A) Cellular supernatants containing unfractionated 22L scrapie-infected brain homogenates were collected after continuous incubation with CF10 cells for up to 24 h and were fractionated by discontinuous sucrose gradient centrifugation. Eleven fractions (fractions 1 to 11) were collected from the top of the gradient, digested with PK, and analyzed by Western blotting using anti-PrP mouse monoclonal antibody 6D11. Molecular mass markers in kilodaltons are indicated on the left. (B) CF10 cells were incubated for 2 h in 22L scrapie-infected brain homogenates. After 2 h, the homogenate was completely removed, and the cells were incubated for an additional 24 h in medium alone. Cells were lysed, and the lysate was fractionated by discontinuous sucrose gradient centrifugation. Eleven fractions (fractions 1 to 11) were collected from the top of the gradient, digested with PK, and analyzed by Western blotting using anti-PrP mouse monoclonal antibody 6D11. The relative amount of PrPSc in each fraction was then quantified. The values shown represent averages ± standard errors of the percentage of PrPSc in each fraction for 4 experiments.
As a further test to determine the origin of the intermediate 22L PrPSc species in the cells after 24 h, CF10 cells were exposed to the 22L brain homogenate for 2 h, after which the homogenate was fully removed and the cells were incubated in medium for a further 24 h. After 2 h, the majority of cell-associated PrPSc migrated in fraction 9 of the sucrose gradient (Fig. 3B). However, after 24 h, the amount of PrPSc in fraction 9 had decreased substantially, and PrPSc was once again present in the intermediate fractions of the gradient (Fig. 3B). These results are consistent with the above-described experiments where the cells were exposed to brain homogenate for the full 24-h incubation period (Fig. 2). Taken together, the data strongly suggested that the smaller PrPSc species present after 24 h were not derived from the input brain homogenate but were the result of the fraction of 22L PrPSc taken up by the cells being disaggregated over time.
Cellular uptake and disaggregation of 87V PrPSc.
Scrapie strain 22L can easily infect various types of cells (3), including CF10 cells expressing a 3F4 epitope-tagged mouse PrP molecule (CF10+Mo3F4) (Fig. 4A) (15). In contrast, scrapie strain 87V can trigger PrPSc formation in cells that are susceptible to other scrapie prion strains (6) but is unable to establish a persistent infection in either those cells or CF10+Mo3F4 cells (Fig. 4A). The reasons for this difference in the abilities of different scrapie strains to infect cells are unknown, but PrPC sequence, PrPSc conversion efficiency, PrPSc aggregate size, and PrPSc stability could all be contributing factors.
Fig 4.
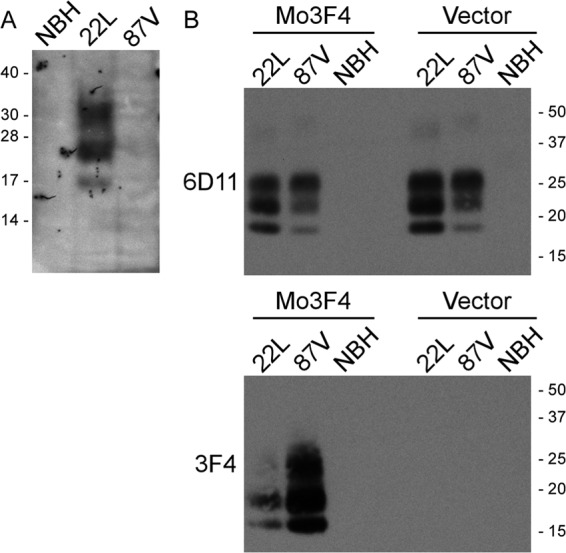
87V mouse scrapie does not induce persistent PrPSc formation in CF10+Mo3F4 cells but can efficiently convert Mo3F4 PrPC to protease resistance. (A) CF10+Mo3F4 cells (15) were exposed to 1% normal mouse brain homogenate (NBH) or 1% brain homogenates from mice infected with scrapie strains 22L and 87V. After 12 passages (22L and 87V) or 13 passages (normal mouse brain homogenate), the cells were analyzed by immunoblotting for PrPSc using anti-PrP mouse monoclonal antibody 3F4. Only cells exposed to the 22L mouse scrapie brain homogenate were positive for PrPSc. Molecular mass markers in kilodaltons are shown on the left. (B) Brain homogenates from mice infected with 22L or 87V scrapie as well as normal brain homogenate were mixed with homogenates from CF10+Mo3F4 cells (Mo3F4) or CF10 cells (Vector). After 4 days, the reaction mixtures were treated with PK and analyzed for total PrPSc by using mouse monoclonal antibody 6D11 (top) and for newly formed PrPSc by using mouse monoclonal antibody 3F4 (bottom). Molecular mass markers in kilodaltons are shown on the right.
In order to determine if 22L and 87V PrPSc could similarly convert Mo3F4 PrPC to protease resistance, brain homogenates from mice infected with either 87V or 22L were incubated with cell homogenates from either CF10+Mo3F4 or CF10 cells, and the formation of protease-resistant Mo3F4 was assayed. As shown in Fig. 4B, 87V PrPSc was able to convert more Mo3F4 PrPC to protease-resistant PrP than 22L PrPSc (Fig. 4B, bottom), even though the amount of PrPSc in the 22L brain homogenate was greater than that in the 87V brain homogenate (Fig. 4B, top). The specificity of the reaction for Mo3F4 PrPC was confirmed by the lack of protease-resistant PrP in reaction mixtures where the homogenate from CF10 cells was used as the substrate (Fig. 4B, vector lanes). Thus, the inability of 87V to infect CF10+Mo3F4 cells does not appear to be due to an inefficient conversion of Mo3F4 PrPC by 87V PrPSc. These data suggest that either unknown host factors or 87V strain-specific characteristics, such as PrPSc uptake, aggregate size, or stability, could be influencing whether or not 87V infects CF10+Mo3F4 cells.
In order to determine whether the same fraction of 87V PrPSc is taken up and disaggregated by the cells as 22L PrPSc, we compared the sedimentation properties of 87V PrPSc derived from scrapie-infected mouse brain to that of PrPSc taken up by the cells. The sedimentation profile of brain-derived 87V PrPSc was similar to that of 22L, with the majority of PrPSc being found in the bottom three fractions of the gradient (Fig. 5A). As with 22L PrPSc, the majority of the 87V PrPSc taken up by the cells after 2 h sedimented in fraction 9 of the sucrose gradient (Fig. 5B). However, unlike 22L PrPSc, where the majority of the PrPSc remained in the bottom three fractions after 24 h, 87V PrPSc taken up by the cells after 24 h was relatively evenly distributed throughout the gradient, with only the lightest fractions, fractions 1 to 3, being negative (Fig. 5B). These results were consistent over multiple experiments (Fig. 5C) and demonstrated that, on average, approximately 50% of the 87V PrPSc taken up by the CF10 cells was disaggregated after 24 h (Fig. 5C), versus 30% of the 22L PrPSc (Fig. 2C). Thus, while both 22L PrPSc and 87V PrPSc were similar in that the same fraction of PrPSc was preferentially taken up by cells and gradually disaggregated over time, the two strains were distinguishable based upon the extent of PrPSc disaggregation.
Fig 5.

A limited population of 87V PrPSc aggregates is preferentially taken up and rapidly disaggregated by CF10 cells. (A and B) Brain homogenates from an 87V scrapie-infected mouse (A) or cell lysates from CF10 cells exposed to the 87V brain homogenate for up to 24 h (B) were fractionated by discontinuous sucrose gradient centrifugation. Eleven fractions (fractions 1 to 11) were collected from the top of the gradient, digested with PK, and analyzed by Western blotting using anti-PrP mouse monoclonal antibody 6D11. In panels A and B, molecular mass markers in kilodaltons are indicated on the left. (C) Quantitation of the relative amount of PrPSc in individual sucrose gradient fractions derived from either the 87V scrapie-infected brain homogenate or CF10 cell lysates after exposure to the 87V scrapie brain homogenate for 2 h, 8 h, and 24 h. The values represent averages ± standard errors of the percentage of PrPSc in each fraction for 3 experiments.
87V PrPSc is more stable than 22L PrPSc.
The observation that 87V PrPSc became disaggregated more rapidly in CF10 cells than 22L PrPSc suggested that it might be conformationally less stable than 22L PrPSc. We therefore determined the relative stabilities of 87V PrPSc and 22L PrPSc using a conformational stability assay. Aliquots of 87V and 22L brain homogenates were incubated with concentrations of guanidine hydrochloride (GdnHCl) ranging from 0 to 4 M and then digested with PK. Contrary to our expectations based upon the CF10 cell data, 87V PrPSc was found to be more resistant to guanidine-induced denaturation than 22L PrPSc (Fig. 6A). While 22L PrPSc became largely susceptible to proteolysis following incubation with 1.75 M GdnHCl, as much as 40% of 87V PrPSc was still resistant to proteolysis even after being exposed to 3.5 M GdnHCl (Fig. 6B).
Fig 6.

22L PrPSc is conformationally less stable than 87V PrPSc. (A) Brain homogenates obtained from either 22L or 87V scrapie-infected mice were prepared in PBS at a concentration of 1% (wt/vol) and incubated with increasing concentrations of GdnHCl for 1 h. After the concentration of GdnHCl was adjusted to 0.4 M, samples were digested with PK, and proteins were precipitated. Protein pellets were examined by Western blotting using anti-PrP mouse monoclonal antibody 6D11. Molecular mass markers in kilodaltons are indicated on the left. (B) Graph showing the amount of PrPSc remaining in the sample following GdnHCl denaturation and treatment with PK. The values represent the average values ± standard errors for 3 independent experiments.
PrPSc stability based on PK resistance may vary depending upon the conformation of the protein, which in turn will influence how accessible the protein is to different antibodies (21, 24). Furthermore, guanidine hydrochloride-based conformational assays do not necessarily distinguish between the stability of the tertiary structure of PrPSc (i.e., protein conformation) and the stability of the quaternary structure of PrPSc particles (i.e., aggregate stability) (25). Thus, we measured the resistance of PrP aggregates in 87V and 22L brain homogenates to GdnHCl solubilization. Aliquots of 87V and 22L brain homogenates were incubated with concentrations of GdnHCl ranging from 0 to 4 M and then centrifuged without PK treatment in order to recover the detergent-insoluble fraction of PrP. Similar to the results obtained from the conformational stability assay shown in Fig. 6, PrP aggregates in the 87V brain homogenate were more resistant to GdnHCl denaturation than those in the 22L brain homogenate (Fig. 7A). While ∼30% of the PrP aggregates in the 22L brain homogenate were detergent insoluble following incubation with up to 2 M GdnHCl, almost 100% of the PrP aggregates in the 87V brain homogenate were still detergent insoluble after incubation with up to 3 M GdnHCl (Fig. 7B). 87V PrPSc was therefore more stable than 22L PrPSc in terms of both conformation, as determined by its resistance to PK digestion with increasing concentrations of GdnHCl, and aggregation, as determined by its relative detergent insolubility with increasing concentrations of GdnHCl. Thus, PrPSc stability did not correlate with the increased disaggregation of 87V PrPSc in CF10 cells.
Fig 7.

PrP aggregates in 22L scrapie-infected brain are less stable than those in 87V scrapie-infected brain. (A) Brain homogenates from either 22L or 87V scrapie-infected mice were prepared in PBS at a concentration of 1% (wt/vol) and incubated with increasing concentrations of GdnHCl for 1 h. After the concentration of GdnHCl was adjusted to 0.4 M, samples were centrifuged for 45 min at 17,400 × g. Protein pellets were analyzed by Western blotting using anti-PrP mouse monoclonal antibody 6D11. Molecular mass markers in kilodaltons are indicated on the left. (B) Graph showing the amount of aggregated PrP remaining in the sample following GdnHCl denaturation and centrifugation. The values represent the average values ± standard errors for 2 independent experiments.
DISCUSSION
One of the earliest events occurring in cells following exposure to scrapie brain homogenates is the cellular uptake of PrPSc. This process can be independent of scrapie strain and cell type (8) but may be influenced by the size of the PrPSc particle (7, 8). In this study, we have shown that PrPSc particles with distinct sedimentation properties were taken up preferentially by scrapie-susceptible cells. The fractions of PrPSc taken up by the cells were similar for scrapie strains 22L and 87V, even though only 22L is able to infect CF10 cells in vitro (Fig. 4) (15). Thus, while particle size appears to influence the population of PrPSc that enters the cell, it does not appear to correlate with the ability of a scrapie strain to infect cells.
PrPSc particle size influences the converting activity of hamster PrPSc, with the highest converting activity per unit mass associated with PrPSc particles of 17 to 27 nm (11). Similarly, the amount of PrPSc made in neuroblastoma cells exposed to sonicated mouse PrPSc increases more rapidly with decreasing PrPSc particle size (12), although prion particles below a certain size poorly induce the conversion of PrPC to PrPSc (11–13). In contrast, mouse PrPSc that has been disaggregated by sonication actually takes longer to induce disease in vivo, even though the PrPSc particle size is smaller (12), and some data have suggested that the relationship between PrPSc particle size and infectivity may be strain dependent (26). Our data suggest one possible explanation for why smaller PrPSc aggregates may be less efficient at triggering infection (12), even though they can be more efficient at converting PrPC to PrPSc (11). Smaller PrPSc particles from the upper fractions of our sucrose gradients were not taken up efficiently by the cells (Fig. 2 and 5), suggesting that they might be less able to induce infection. Thus, fragmentation of PrPSc into smaller particles could lead to inefficient infection of susceptible cell types and an increase in disease incubation times in vivo. Similarly, our results may also explain why PrPSc in brain homogenates associates with the cell more efficiently than purified PrPSc or PrPSc in microsomes (8), either of which may contain less of the population of PrPSc aggregates preferentially internalized by the cell. When taken together, the data suggest that perhaps only a subfraction of PrPSc particles is capable of both efficiently infecting cells and efficiently triggering the conversion of PrPC into PrPSc.
While PrPSc in scrapie-infected cells and mice can have a half-life of ≥24 h (27–29), multiple studies have shown that cells are in fact capable of breaking it down. Brain homogenate-derived PrPSc is degraded over time following uptake by cells (14, 30–34), and PrPSc in scrapie-infected cells can be degraded by cocultivation with dendritic cells (35, 36). Our data demonstrate that cells disaggregate PrPSc within the first 24 h following infection with scrapie. Importantly, despite the fact that the PrPSc particle size was decreasing, the overall level of PrPSc in the cells continued to increase over the same 24-h time period. These results suggest that disaggregation of PrPSc was preceding PrPSc degradation. It is possible that PrPSc disaggregation, which likely occurs in an acidic environment such as the lysosome (33, 36, 37), is needed to increase the sensitivity of PrPSc to cellular proteases prior to its degradation by the cell. A similar mechanism has been proposed to explain how myeloid dendritic cells are able to rapidly degrade PrPSc (35).
Our data show that 87V PrPSc was disaggregated into smaller particles more rapidly than 22L PrPSc, suggesting that there may be prion strain-specific differences in how cells process PrPSc following uptake. Conformational stability of PrPSc has been suggested to contribute significantly to prion disease incubation times by influencing the rate at which PrPSc is broken down into smaller aggregates (25, 38–40). However, as with the relationship between PrPSc particle size and infectivity (26), the correlation between increased stability and increased incubation time may also be prion strain dependent (41). Thus, PrPSc stability might explain the strain-specific difference in PrPSc disaggregation kinetics between 22L and 87V in our experiments. Our results, in which intracellular disaggregation appears to be more rapid for 87V PrPSc than for 22L PrPSc, even though its stability is greater, are consistent with the results of Ayers et al., which suggest that conformationally stable PrPSc may be more prone to fragmentation (41).
Another possibility is that disaggregation may be related less to PrPSc stability than to the presence of other molecules in the PrPSc aggregate. No strain-specific proteins have been found to be associated with PrPSc from different mouse scrapie strains (42). However, multiple nonprotein components, including fatty acids (43), polysaccharides (44, 45), sphingolipids (46), and nucleic acids (47, 48), have been found in association with PrPSc, while glycosaminoglycans (49–51) and phospholipids (52, 53) are known to influence PrPSc formation. Some of these molecules may act as a molecular scaffold for the conversion of PrPC to PrPSc, possibly by enabling the ordered addition of newly formed PrPSc into a growing aggregate (12, 44, 45, 51, 54, 55). Strain-specific differences in these nonprotein cofactors could explain why PrPSc molecules from 87V mouse scrapie may be more prone to disaggregation than PrPSc molecules from 22L mouse scrapie. Their removal by cellular enzymes could lead to the breakdown of PrPSc into smaller aggregates, yielding PrPSc products that are more unstable and/or more sensitive to proteolysis and thus potentially less efficient at converting PrPC to PrPSc. This may also help to explain why, contrary to previous studies using purified PrPSc which demonstrated that smaller PrPSc aggregates have the highest converting activity per unit mass (11), disaggregation of brain-derived 87V PrPSc into smaller aggregates did not lead to a more efficient infection of CF10 cells.
The formation of PrPSc in the first 96 h after exposure of cells to scrapie-infected brain homogenates does not necessarily lead to persistent infection, and it remains unclear why some scrapie strains cannot infect cells susceptible to other scrapie strains (6). We previously hypothesized that the inability to persistently infect cells in vitro may be dependent upon events occurring after an initial burst of PrPSc formation (6). These events would likely involve scrapie strain- and host-specific factors that enable the continued production and/or cell-to-cell transmission of PrPSc. We found strain-specific differences between 87V and 22L both in their ability to convert Mo3F4 PrPC to protease resistance and in the rate at which PrPSc is disaggregated. Given that PrPSc from the 87V mouse scrapie strain converts Mo3F4 PrPC more efficiently than 22L PrPSc (Fig. 4B), it is unlikely that conversion efficiency is a primary factor in the resistance of CF10+Mo3F4 cells to 87V infection. However, it is possible that the increased rate of 87V PrPSc disaggregation that we observed could influence its ability to establish persistent infection in vitro. The ability of 87V PrPSc to efficiently convert Mo3F4 PrPC could be negated if the disaggregation of 87V PrPSc led to particles that were too small to efficiently infect cells or induce the conversion of PrPC to PrPSc (11–13) or that were more easily degraded and cleared by the cell. As such, the production of new PrPSc may not be able to keep up with cell division (56), and persistent infection might not be established. Thus, it is possible that the rapid disaggregation of 87V PrPSc by the cell may contribute, at least in part, to the inability of 87V to infect cells in vitro.
ACKNOWLEDGMENTS
We thank Byron Caughey and Karin Peterson for critical readings of the manuscript and Anita Mora, Austin Athman, and Heather Murphy for graphical assistance.
This research was supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases, National Institutes of Health.
Footnotes
Published ahead of print 21 August 2013
REFERENCES
- 1.Priola SA, Vorberg I. 2006. Molecular aspects of disease pathogenesis in the transmissible spongiform encephalopathies. Mol. Biotechnol. 33:71–88 [DOI] [PubMed] [Google Scholar]
- 2.Sailer A, Bueler H, Fischer M, Aguzzi A, Weissmann C. 1994. No propagation of prions in mice devoid of PrP. Cell 77:967–968 [DOI] [PubMed] [Google Scholar]
- 3.Grassmann A, Wolf H, Hofmann J, Graham J, Vorberg I. 2013. Cellular aspects of prion replication in vitro. Viruses 5:374–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Solassol J, Crozet C, Lehmann S. 2003. Prion propagation in cultured cells. Br. Med. Bull. 66:87–97 [DOI] [PubMed] [Google Scholar]
- 5.Nishida N, Harris DA, Vilette D, Laude H, Frobert Y, Grassi J, Casanova D, Milhavet O, Lehmann S. 2000. Successful transmission of three mouse-adapted scrapie strains to murine neuroblastoma cell lines overexpressing wild-type mouse prion protein. J. Virol. 74:320–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vorberg I, Raines A, Priola SA. 2004. Acute formation of protease-resistant prion protein does not always lead to persistent scrapie infection in vitro. J. Biol. Chem. 279:29218–29225 [DOI] [PubMed] [Google Scholar]
- 7.Magalhaes AC, Baron GS, Lee KS, Steele-Mortimer O, Dorward D, Prado MA, Caughey B. 2005. Uptake and neuritic transport of scrapie prion protein coincident with infection of neuronal cells. J. Neurosci. 25:5207–5216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Greil CS, Vorberg IM, Ward AE, Meade-White KD, Harris DA, Priola SA. 2008. Acute cellular uptake of abnormal prion protein is cell type and scrapie-strain independent. Virology 379:284–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morel E, Andrieu T, Casagrande F, Gauczynski S, Weiss S, Grassi J, Rousset M, Dormont D, Chambaz J. 2005. Bovine prion is endocytosed by human enterocytes via the 37 kDa/67 kDa laminin receptor. Am. J. Pathol. 167:1033–1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Paquet S, Daude N, Courageot MP, Chapuis J, Laude H, Vilette D. 2007. PrPc does not mediate internalization of PrPSc but is required at an early stage for de novo prion infection of Rov cells. J. Virol. 81:10786–10791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Silveira JR, Raymond GJ, Hughson AG, Race RE, Sim VL, Hayes SF, Caughey B. 2005. The most infectious prion protein particles. Nature 437:257–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weber P, Reznicek L, Mitteregger G, Kretzschmar H, Giese A. 2008. Differential effects of prion particle size on infectivity in vivo and in vitro. Biochem. Biophys. Res. Commun. 369:924–928 [DOI] [PubMed] [Google Scholar]
- 13.Weber P, Giese A, Piening N, Mitteregger G, Thomzig A, Beekes M, Kretzschmar HA. 2006. Cell-free formation of misfolded prion protein with authentic prion infectivity. Proc. Natl. Acad. Sci. U. S. A. 103:15818–15823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krejciova Z, Pells S, Cancellotti E, Freile P, Bishop M, Samuel K, Barclay GR, Ironside JW, Manson JC, Turner ML, De Sousa P, Head MW. 2011. Human embryonic stem cells rapidly take up and then clear exogenous human and animal prions in vitro. J. Pathol. 223:635–645 [DOI] [PubMed] [Google Scholar]
- 15.McNally KL, Ward AE, Priola SA. 2009. Cells expressing anchorless prion protein are resistant to scrapie infection. J. Virol. 83:4469–4475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Priola SA, Caughey B, Race RE, Chesebro B. 1994. Heterologous PrP molecules interfere with accumulation of protease-resistant PrP in scrapie-infected murine neuroblastoma cells. J. Virol. 68:4873–4878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chesebro B, Wehrly K, Caughey B, Nishio J, Ernst D, Race R. 1993. Foreign PrP expression and scrapie infection in tissue culture cell lines. Dev. Biol. Stand. 80:131–140 [PubMed] [Google Scholar]
- 18.National Research Council 2011. Guide for the care and use of laboratory animals, 8th ed. National Academies Press, Washington, DC [Google Scholar]
- 19.Vorberg I, Priola SA. 2002. Molecular basis of scrapie strain glycoform variation. J. Biol. Chem. 277:36775–36781 [DOI] [PubMed] [Google Scholar]
- 20.Tzaban S, Friedlander G, Schonberger O, Horonchik L, Yedidia Y, Shaked G, Gabizon R, Taraboulos A. 2002. Protease-sensitive scrapie prion protein in aggregates of heterogeneous sizes. Biochemistry 41:12868–12875 [DOI] [PubMed] [Google Scholar]
- 21.Shindoh R, Kim CL, Song CH, Hasebe R, Horiuchi M. 2009. The region approximately between amino acids 81 and 137 of proteinase K-resistant PrPSc is critical for the infectivity of the Chandler prion strain. J. Virol. 83:3852–3860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yull HM, Ritchie DL, Langeveld JP, van Zijderveld FG, Bruce ME, Ironside JW, Head MW. 2006. Detection of type 1 prion protein in variant Creutzfeldt-Jakob disease. Am. J. Pathol. 168:151–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Priola SA, Chesebro B. 1998. Abnormal properties of prion protein with insertional mutations in different cell types. J. Biol. Chem. 273:11980–11985 [DOI] [PubMed] [Google Scholar]
- 24.Kocisko DA, Lansbury PT, Jr, Caughey B. 1996. Partial unfolding and refolding of scrapie-associated prion protein: evidence for a critical 16-kDa C-terminal domain. Biochemistry 35:13434–13442 [DOI] [PubMed] [Google Scholar]
- 25.Legname G, Nguyen HO, Peretz D, Cohen FE, Dearmond SJ, Prusiner SB. 2006. Continuum of prion protein structures enciphers a multitude of prion isolate-specified phenotypes. Proc. Natl. Acad. Sci. U. S. A. 103:19105–19110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tixador P, Herzog L, Reine F, Jaumain E, Chapuis J, Le Dur A, Laude H, Beringue V. 2010. The physical relationship between infectivity and prion protein aggregates is strain-dependent. PLoS Pathog. 6:e1000859. 10.1371/journal.ppat.1000859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Borchelt DR, Scott M, Taraboulos A, Stahl N, Prusiner SB. 1990. Scrapie and cellular prion proteins differ in their kinetics of synthesis and topology in cultured cells. J. Cell Biol. 110:743–752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Caughey B, Raymond GJ. 1991. The scrapie-associated form of PrP is made from a cell surface precursor that is both protease- and phospholipase-sensitive. J. Biol. Chem. 266:18217–18223 [PubMed] [Google Scholar]
- 29.Safar JG, Dearmond SJ, Kociuba K, Deering C, Didorenko S, Bouzamondo-Bernstein E, Prusiner SB, Tremblay P. 2005. Prion clearance in bigenic mice. J. Gen. Virol. 86:2913–2923 [DOI] [PubMed] [Google Scholar]
- 30.Luhr KM, Nordstrom EK, Low P, Kristensson K. 2004. Cathepsin B and L are involved in degradation of prions in GT1-1 neuronal cells. Neuroreport 15:1663–1667 [DOI] [PubMed] [Google Scholar]
- 31.Mohan J, Hopkins J, Mabbott NA. 2005. Skin-derived dendritic cells acquire and degrade the scrapie agent following in vitro exposure. Immunology 116:122–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rybner-Barnier C, Jacquemot C, Cuche C, Dore G, Majlessi L, Gabellec MM, Moris A, Schwartz O, Di Santo J, Cumano A, Leclerc C, Lazarini F. 2006. Processing of the bovine spongiform encephalopathy-specific prion protein by dendritic cells. J. Virol. 80:4656–4663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gilch S, Schmitz F, Aguib Y, Kehler C, Bulow S, Bauer S, Kremmer E, Schatzl HM. 2007. CpG and LPS can interfere negatively with prion clearance in macrophage and microglial cells. FEBS J. 274:5834–5844 [DOI] [PubMed] [Google Scholar]
- 34.Elhelaly AE, Inoshima Y, Ishiguro N. 2013. Alteration of cell responses to PrP(Sc) in prolonged cell culture and its effect on transmission of PrP(Sc) to neural cells. Arch. Virol. 158:651–658 [DOI] [PubMed] [Google Scholar]
- 35.Luhr KM, Wallin RP, Ljunggren HG, Low P, Taraboulos A, Kristensson K. 2002. Processing and degradation of exogenous prion protein by CD11c(+) myeloid dendritic cells in vitro. J. Virol. 76:12259–12264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Luhr KM, Nordstrom EK, Low P, Ljunggren HG, Taraboulos A, Kristensson K. 2004. Scrapie protein degradation by cysteine proteases in CD11c+ dendritic cells and GT1-1 neuronal cells. J. Virol. 78:4776–4782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dron M, Moudjou M, Chapuis J, Salamat MK, Bernard J, Cronier S, Langevin C, Laude H. 2010. Endogenous proteolytic cleavage of disease-associated prion protein to produce C2 fragments is strongly cell- and tissue-dependent. J. Biol. Chem. 285:10252–10264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peretz D, Scott MR, Groth D, Williamson RA, Burton DR, Cohen FE, Prusiner SB. 2001. Strain-specified relative conformational stability of the scrapie prion protein. Protein Sci. 10:854–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Legname G, Nguyen HO, Baskakov IV, Cohen FE, Dearmond SJ, Prusiner SB. 2005. Strain-specified characteristics of mouse synthetic prions. Proc. Natl. Acad. Sci. U. S. A. 102:2168–2173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ghaemmaghami S, Watts JC, Nguyen HO, Hayashi S, Dearmond SJ, Prusiner SB. 2011. Conformational transformation and selection of synthetic prion strains. J. Mol. Biol. 413:527–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ayers JI, Schutt CR, Shikiya RA, Aguzzi A, Kincaid AE, Bartz JC. 2011. The strain-encoded relationship between PrP replication, stability and processing in neurons is predictive of the incubation period of disease. PLoS Pathog. 7:e1001317. 10.1371/journal.ppat.1001317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moore RA, Timmes A, Wilmarth PA, Priola SA. 2010. Comparative profiling of highly enriched 22L and Chandler mouse scrapie prion protein preparations. Proteomics 10:2858–2869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stahl N, Borchelt DR, Hsiao K, Prusiner SB. 1987. Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell 51:229–240 [DOI] [PubMed] [Google Scholar]
- 44.Appel TR, Dumpitak C, Matthiesen U, Riesner D. 1999. Prion rods contain an inert polysaccharide scaffold. Biol. Chem. 380:1295–1306 [DOI] [PubMed] [Google Scholar]
- 45.Dumpitak C, Beekes M, Weinmann N, Metzger S, Winklhofer KF, Tatzelt J, Riesner D. 2005. The polysaccharide scaffold of PrP 27-30 is a common compound of natural prions and consists of alpha-linked polyglucose. Biol. Chem. 386:1149–1155 [DOI] [PubMed] [Google Scholar]
- 46.Klein TR, Kirsch D, Kaufmann R, Riesner D. 1998. Prion rods contain small amounts of two host sphingolipids as revealed by thin-layer chromatography and mass spectrometry. Biol. Chem. 379:655–666 [DOI] [PubMed] [Google Scholar]
- 47.Akowitz A, Sklaviadis T, Manuelidis EE, Manuelidis L. 1990. Nuclease-resistant polyadenylated RNAs of significant size are detected by PCR in highly purified Creutzfeldt-Jakob disease preparations. Microb. Pathog. 9:33–45 [DOI] [PubMed] [Google Scholar]
- 48.Diringer H, Beekes M, Ozel M, Simon D, Queck I, Cardone F, Pocchiari M, Ironside JW. 1997. Highly infectious purified preparations of disease-specific amyloid of transmissible spongiform encephalopathies are not devoid of nucleic acids of viral size. Intervirology 40:238–246 [DOI] [PubMed] [Google Scholar]
- 49.Gabizon R, Meiner Z, Halimi M, Ben-Sasson SA. 1993. Heparin-like molecules bind differentially to prion-proteins and change their intracellular metabolic fate. J. Cell. Physiol. 157:319–325 [DOI] [PubMed] [Google Scholar]
- 50.Caughey B, Raymond GJ. 1993. Sulfated polyanion inhibition of scrapie-associated PrP accumulation in cultured cells. J. Virol. 67:643–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Priola SA, Caughey B. 1994. Inhibition of scrapie-associated PrP accumulation. Probing the role of glycosaminoglycans in amyloidogenesis. Mol. Neurobiol. 8:113–120 [DOI] [PubMed] [Google Scholar]
- 52.Deleault NR, Piro JR, Walsh DJ, Wang F, Ma J, Geoghegan JC, Supattapone S. 2012. Isolation of phosphatidylethanolamine as a solitary cofactor for prion formation in the absence of nucleic acids. Proc. Natl. Acad. Sci. U. S. A. 109:8546–8551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Deleault NR, Walsh DJ, Piro JR, Wang F, Wang X, Ma J, Rees JR, Supattapone S. 2012. Cofactor molecules maintain infectious conformation and restrict strain properties in purified prions. Proc. Natl. Acad. Sci. U. S. A. 109:E1938–E1946. 10.1073/pnas.1206999109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wong C, Xiong LW, Horiuchi M, Raymond L, Wehrly K, Chesebro B, Caughey B. 2001. Sulfated glycans and elevated temperature stimulate PrP(Sc)-dependent cell-free formation of protease-resistant prion protein. EMBO J. 20:377–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Taylor DR, Whitehouse IJ, Hooper NM. 2009. Glypican-1 mediates both prion protein lipid raft association and disease isoform formation. PLoS Pathog. 5:e1000666. 10.1371/journal.ppat.1000666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ghaemmaghami S, Phuan PW, Perkins B, Ullman J, May BC, Cohen FE, Prusiner SB. 2007. Cell division modulates prion accumulation in cultured cells. Proc. Natl. Acad. Sci. U. S. A. 104:17971–17976 [DOI] [PMC free article] [PubMed] [Google Scholar]